

Abstract
Background
Iron overload is a major complication in patients with hemoglobin H (Hb H) disease and causes damage of tissues.
Methods
We investigated 26 Hb H patients and 75 controls to evaluate their oxidative stress and antioxidant statuses.
Results
There were significantly increased levels of superoxide anion in leucocytes, nitrite (N), and malondialdehyde (MDA) in plasma, and activities of superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GRx) and oxidized glutathione (GSSG) in erythrocytes, decreased levels of nitrate (N) and vitamin C in plasma, and reduced glutathione (GSH) in erythrocytes, in addition to the abnormal iron status in the patients when compared with those in the controls. Meanwhile, levels of serum ferritin were positively correlated with serum iron, plasma MDA, and erythrocyte SOD in the patients. In addition, the activities of SOD were positively correlated with those of GPx and GRx, and the levels of GSSG and MDA, but negatively correlated with those of GSH. Furthermore, the levels of MDA were negatively correlated those of vitamin C.
Conclusions
These results demonstrate the presence of oxidative stress and decreased levels of antioxidants; moreover, the related metabolic antioxidant pathway is active in Hb H patients with iron overload.
Keywords: Hb H disease, iron overload, oxidative stress, antioxidant status, malondialdehyde, superoxide dismutase
INTRODUCTION
α‐Thalassemia is the most common inherited genetic disease involving α‐globin genes and the cause of a major public health problem in Asia 1. Hemoglobin (Hb) H disease, with three dysfunctional loci of the four α‐globin genes, is usually characterized as moderate anemia, marked microcytosis and hypochromia with an excess of β‐globin chains within erythrocytes 2. The patients with Hb H disease may require sporadic packed red blood cell (RBC) transfusions for symptomatic anemia, and moderate iron overload develops with increase of age 3.
Increased oxidative stress may arise from excess iron deposition, nonheme iron, and imbalanced α‐ or β‐globin chains in the erythrocytes 4. The excess iron deposition is from transfused RBCs, hemolysis, and increased absorption of dietary iron resulting from anemia 1. Unpaired globin chains are relatively unstable and can be oxidized to form intracellular precipitates that can cause oxidative damage, cell membrane dysfunction, intramedullary early erythroid cell death, ineffective erythropoiesis, and shortened red cell life 5. The presence of excess iron can react with reactive oxygen species (ROS) to generate cytotoxic hydroxyl radicals that cause oxidative cell damage via lipid peroxidation, DNA hydroxylation, and carbohydrate and protein oxidation 6, 7. The organs that accumulate excess iron, especially the liver, pituitary gland, pancreas, and heart in patients with severe iron‐overloaded thalassemia ultimately progress to function failure or lead to fatality 1.
There is much research about increased oxidative stress and excess iron deposition in β‐thalassemia, but only very little about Hb H disease. The pathophysiology is different in these two diseases. In Hb H disease, there is a deficiency of α‐globin mRNA and α‐globin chains. While in β‐thalassemia, there is a deficiency of β‐globin mRNA and β‐globin chains. This study explores the iron status, oxidative stress, and antioxidant status in patients with Hb H disease. Our findings will be helpful in understanding the pathway in the altered redox statuses and in planning the treatment strategy for these patients.
PATIENTS AND METHODS
Participants and Study Design
Twenty‐six patients with Hb H disease and 75 healthy age‐matched individuals as controls were enrolled in this study (Table 1). These patients were diagnosed by hemoglobin electrophoresis or gene mutation. They received none or sporadic packed RBC transfusions according to the severity of anemia and clinical manifestation.
Table 1.
Clinical Features of the Patients and the Controlsa
| Hb H disease (n = 26) | Controls (n = 75) | |
|---|---|---|
| Variables | Median value (IQR)b | Median value (IQR) |
| Age, years | 18 (12) | 22 (4) |
| Sex, M:F | 15:11 | 36:39 |
| Serum iron, μmol/l | 24.6 (16.8)* | 16.3 (8.2) |
| Transferrin, μmol/l | 2.07 (0.70) | 2.66 (0.62) |
| TIBC, μmol/l | 51.7 (17.4)* | 65.1 (15.3) |
| Ferritin, pmol/l | 1,724 (2,066)* | 144 (304) |
| Transferrin saturation, % | 44.5 (46.6)* | 25.4 (14.9) |
| AST, U/l | 31 (18.5)* | 20 (4.5) |
| ALT, U/l | 16 (16.8) | 14 (10) |
| CK, U/l | 57 (54.3)* | 82 (57.5) |
| GGT, U/l | 24 (16.3) | 24 (7.3) |
| TG, mmol/l | 3.79 (2.66)* | 2.48 (1.09) |
| Cholesterol, mmol/l | 4.34 (1.11)* | 5.56 (1.32) |
| HDL‐C, mmol/l | 1.42 (0.24)* | 2.12 (0.62) |
| LDL‐C, mmol/l | 2.43 (0.90)* | 3.31 (1.23) |
The independent sample data used for the Mann–Whitney U test are expressed as a median value (IQR).
*P < 0.05 when compared with those of the controls.
TIBC, total iron bind capacity; AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase; GGT, gamma‐glutamyl transpeptidase; TG, triglyceride; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol.
Laboratory Analyses
Blood samples obtained from fasting subjects with and without anticoagulants (heparin and ethylenediamine tetraacetic acid) were collected after informed consent was obtained. The serum, plasma, erythrocytes, and white blood cells (WBCs) were separated by centrifugation and divided into several aliquots that were either analyzed immediately or stored at −80°C and analyzed within 72 hr. The samples with hemolysis were excluded from this study.
Clinical biochemistry tests for iron, liver function, and lipid profiles were performed by using commercial analytical kits (Beckman Coulter, Brea, CA). Serum transferrin was determined by a turbidimetric method (Beckman Coulter, Brea, CA) and ferritin was measured by the ARCHITECT ferritin assay (Abbott Diagnostics Division, North Chicago, IL). All chemicals and reagents used in the study were of analytical grade. Lucigenin, thiobarbituric acid, boric acid, metaphosphoric acid, reduced glutathione (GSH), oxidized glutathione (GSSG), trichloroacetic acid, phorbol 12‐myristate 13‐acetate (PMA), and tricine were obtained from Sigma (St. Louis, MO).
Superoxide () generation was measured from 103 WBC with the exclusion of nucleated RBC by lucigenin‐based chemiluminescence and analyzed by an ultraweak chemiluminescence analyzer with a high‐sensitivity detector (3.3 × 10−15 W/cm2 count) from Jye Horn Co. (Taipei, Taiwan). Photon emission was counted at 37℃ for 10 min before (resting) and after (stimulated) the addition of PMA. The levels of nitrite (N) and nitrate (N) and vitamin C in plasma, and GSH in erythrocytes were assayed by a P/ACE‐MDQ capillary electrophoresis system (Beckman Coulter) equipped with a fixed wavelength detector. Data were quantified on the basis of corrected peak areas with migration time. Commercial kits for superoxide dismutase (SOD), glutathione peroxidase (GPx), and glutathione reductase (GRx) measurements were obtained from Randox Laboratories (Crumlin, Country Antrim, UK). The activities of SOD, GPx, and GRx in the erythrocytes were measured by an autoanalyzer (Synchron CX 7, Beckman Coulter, Brea, CA), and the results were expressed as units per gram of Hb. Malondialdehyde (MDA) in the plasma was estimated by the thiobarbituric acid method. MDA, which is a stable end product of fatty acid peroxidation, reacts with thiobarbituric acid in an acidic condition to form a complex that has a maximum absorbance at 532 nm and is measured by spectrophotometry.
Analyses of Statistics
Statistical analysis was conducted by using the SPSS version 19.0 software (SPSS Inc., Chicago, IL). All statistical tests were two sided, and statistical significance was assumed at P < 0.05. Comparisons between the patient and control groups were performed by the nonparametric Mann–Whitney U test to identify the significance of differential expression. The data used for the Mann–Whitney U test were expressed as a median value and an interquartile range (IQR). Correlation studies were performed by using Pearson's correlation statistics.
RESULTS
Clinical Features of the Patients and the Controls (Table 1)
The levels of serum iron and ferritin were significantly increased, and the levels of transferrin and total iron bind capacity (TIBC) were decreased in the patients when compared with those in the controls. The activities of creatine kinase (CK), and levels of total cholesterol, high‐density lipoprotein cholesterol (HDL‐C) and low‐density lipoprotein cholesterol (LDL‐C) were decreased while the levels of triglyceride (TG) increased in the patients when compared with those in the controls.
Comparisons of the Parameters Related to Oxidative Stress and the Antioxidant Status between the Patients and the Controls (Table 2)
Table 2.
Comparisons of the Parameters Related to Oxidative Stress and the Antioxidant Status between the Patients and the Controlsa
| Hb H disease (n = 26) | Controls (n = 75) | |
|---|---|---|
| Variables | Median value (IQR)b | Median value (IQR) |
| (−PMA), counts/ 10 sec × 103 WBC | 592 (316)* | 208 (169) |
| (+PMA), counts/ 10 sec × 103 WBC | 6,302 (3,789)* | 2,858 (2,163) |
| Nitrite, μmol/l | 22.0 (20.1)* | 13.3 (9.1) |
| Nitrate, μmol/l | 55.9 (25.1)* | 68.8 (27.3) |
| Total NO, μmol/l | 81.8 (31.0) | 80.5 (30.2) |
| GSH, μmol/l | 1,153 (401)* | 1,338 (344) |
| GSSG, μmol/l | 93.6 (56.0)* | 113.0 (50.6) |
| SOD, U/g Hb | 1,067 (260)* | 574 (95) |
| GPx, U/g Hb | 163 (50.9)* | 50.5 (10.4) |
| GRx, U/g Hb | 13.2 (5.6)* | 10.6 (3.0) |
| MDA, μmol/l | 4.2 (1.5)* | 1.4 (0.2) |
| Vitamin C, μmol/l | 25.6 (35.0)* | 43.6 (38.9) |
The independent sample data used for the Mann–Whitney U test are expressed as a median value (IQR).
*P < 0.05 when compared with those of the controls.
, superoxide anion; PMA, phorbol 12‐myristate 13‐acetate; MDA, malondialdehyde, GSH, reduced glutathione; GSSG, oxidized glutathione; SOD, superoxide dismutase; GPx, glutathione peroxidase; GRx, glutathione reductase.
There were significantly increased levels of resting or PMA‐stimulated in leucocytes, increased activities of SOD, GPx, and GRx, and levels of GSSG in erythrocytes, and the levels of nitrite and MDA in plasma, but decreased plasma levels of nitrate and vitamin C, and erythrocyte GSH in the patients when compared with those in the controls.
Correlation Studies Between Iron Status and Analytical Parameters
The levels of serum ferritin were positively correlated with the levels of serum iron, serum ALT, and plasma MDA, and the activities of erythrocyte SOD, and negatively correlated with the activities of serum CK in the patients (Fig. 1). In addition, the activities of erythrocyte SOD were positively correlated with those of erythrocyte GPx and GRx, and the levels of GSSG and plasma MDA, but negatively correlated with those of GSH (Fig. 2). Furthermore, the levels of plasma MDA were negatively correlated with those of plasma vitamin C (Fig. 2). Figure 3 demonstrates the metabolic pathway related to oxidative stress in the patients with Hb H disease.
Figure 1.
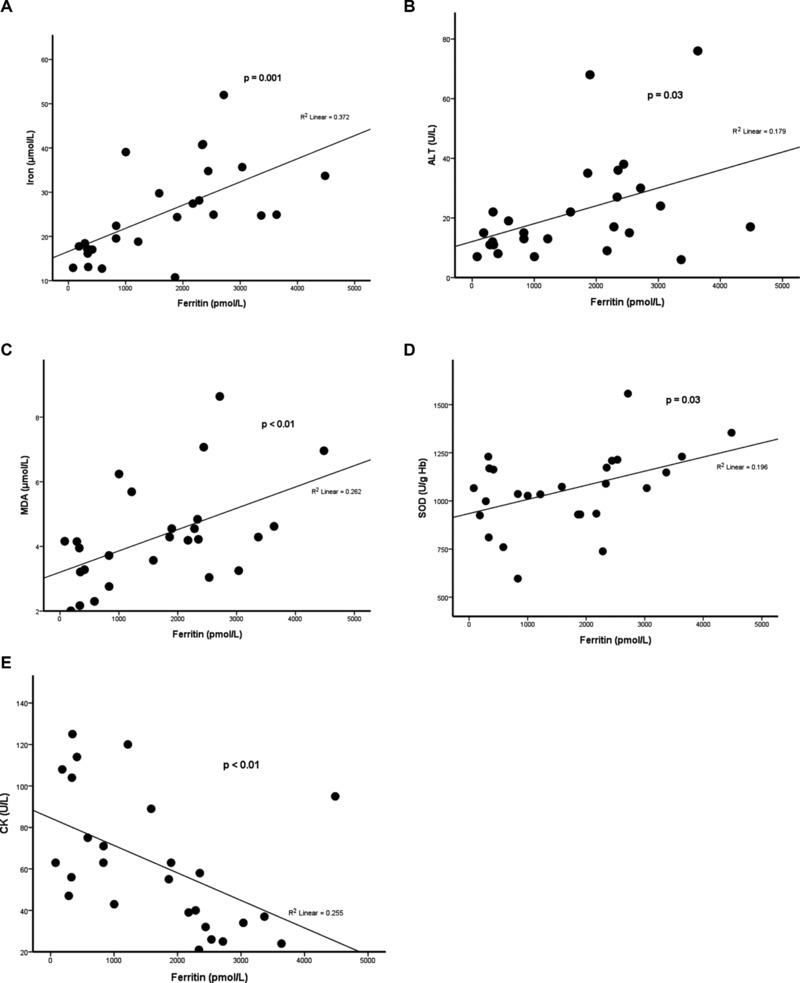
Positive correlation of serum ferritin with serum iron (A), serum ALT (B), plasma MDA (C), and erythrocyte SOD (D), and negative correlation with serum CK (E) in patients with Hb H disease.
Figure 2.
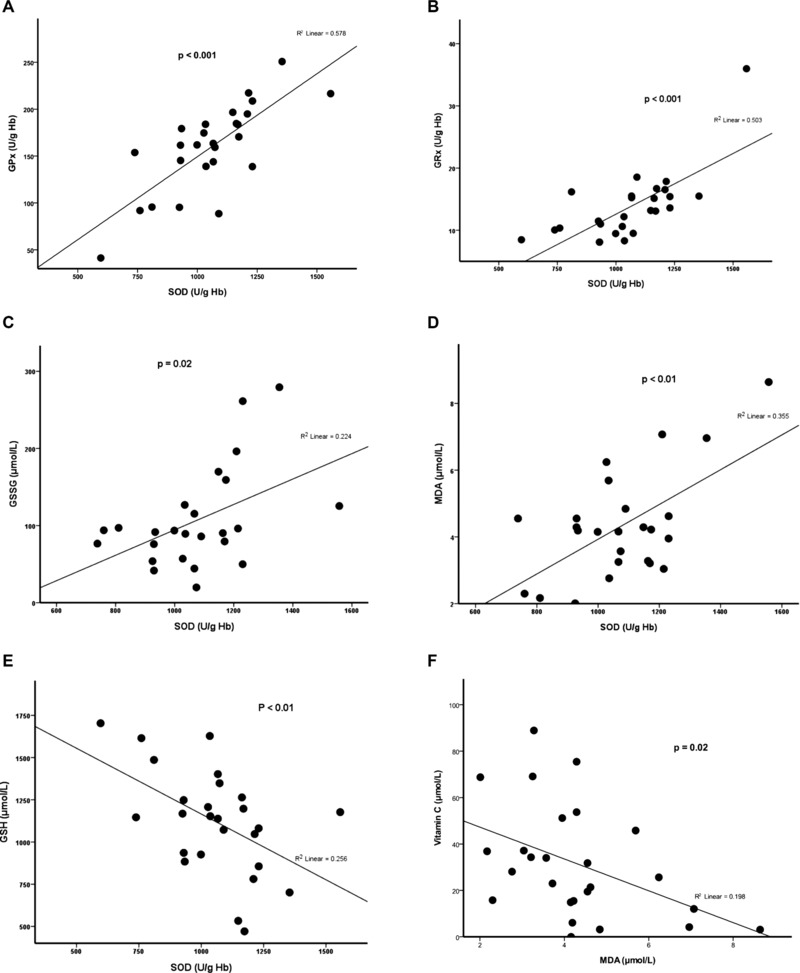
Positive correlation of erythrocyte SOD with erythrocyte GPx (A), GRx (B), GSSG (C), and plasma MDA (D), but negative correlation with erythrocyte GSH (E); negative correlation between plasma MDA and vitamin C (F) in the patients with Hb H disease.
Figure 3.
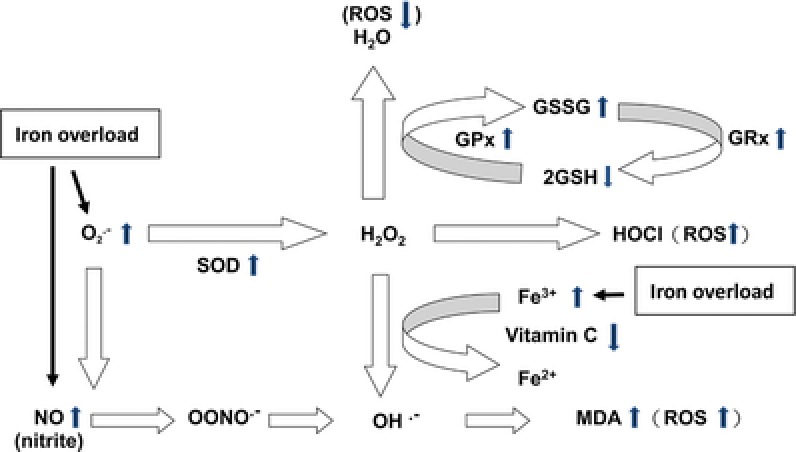
The possible metabolic pathway related to oxidative stress in patients with Hb H disease with iron overload. Iron overload causes the increase of superoxide () and nitric oxide (NO) in patients with Hb H disease. Increase of leads to compensatory increase of superoxide dismutase (SOD), and causes increase of hydrogen peroxide (H2O2) The increased hydrogen peroxides activate regulatory antioxidant system in the erythrocytes, resulting increased glutathione peroxidase (GPx), glutathione reductase (GRx), decreased reduced glutathione (GSH) and increased oxidized glutathione (GSSG) to reduce the excessive ROS. Furthermore, highly toxic, reactive hydroxyl radicals (OH.−) can be formed due to excessive free radicals (, NO, or H2O2), and leads to lipid peroxidation and generation of malondialdehyde (MDA). In addition, plasma vitamin C may be depleted due to iron overload and formation of excessive radicals (H2O2).
DISCUSSION
In patients with Hb H disease, iron overload increases with age, but is not related to the patients’ transfusion history or genotypes (3). Additional iron absorption from the intestine due to anemia passes through the liver putting it at increased risk for further exposure and subsequent oxidative injury 8. Iron, catalyzing electron transfer to O2, plays a major role in the metabolism and overproduction of free radicals in biological systems when overloaded 7. Marked iron overload exceeds the binding capacity of plasma transferrin leading to a significant increase of nontransferrin bound iron that can generate ROS through the Fenton and Haber–Weiss reaction to form cytotoxic hydroxyl radicals and cause cellular injury 6, 9. Iron overload and accumulation in the liver can overwhelm the liver's storage capacity and result in hepatotoxicity and hepatocellular injury due to Fe++‐initiated peroxide damage 10. Iron overload in moderate and severe patients is still the major reason for long‐term, life‐threatening complications even though such patients have undergone intense iron‐chelating treatment. More severe liver damage with higher ALT, GGT, TG, and lower HDL‐C, LDL‐C was found in the patients with β‐thalassemia major 11. Several mechanisms, including accelerated erythropoiesis, cholesterol consumption, liver dysfunction, iron overload, and oxidative stress, have been described for alterations of lipid and lipoprotein levels in β‐thalassemia patients 12. In this study, the levels of ferritin were positively correlated with the levels of ALT and MDA. Meanwhile, the lower levels of total cholesterol, HDL‐C and LDL‐C, and higher levels of TG were found in the patient group, which indicated liver damage from iron overload and oxidation stress. These findings have not been described in patients with Hb H disease.
Two free radicals, and nitric oxide (NO), are considered as major precursors of all ROS and nitrogen species formed in living organisms and are involved in numerous pathological disorders 13, 14. WBC, killing microbial organisms by generating ROS, can also produce a respiratory burst when they are exposed to PMA in vitro 15. Our data showed significantly increased levels of generation in the WBC in the patient group, which indicated that these patients were under high oxidative stress. Moreover, a tenfold increase of levels induced by PMA indicates that the damage from free radicals could be more severe than previously assumed, especially after exogenous stimulation. Generation of has been reported in β‐thalassemia patients 16. This is the first report describing the generation of in the WBCs with or without PMA stimulation in patients with Hb H disease. Iron overload may lead to an aggravation of the clinical course of infectious diseases due to modulation of cellular immunity and impaired phargocyte and neutrophil function 17, 18. Overproduction of ROS from WBC during the infectious process might be associated with more advanced cellular or tissue damages. However, very limited data are available in the literature. These patients deserve more careful monitoring for oxidative damage especially after exogenous stimulations such as bacterial or fungal infections.
NO is a nonstable radical and will convert to nitrite and nitrate in a very short time. Nitrite is physiologically recycled in blood and tissues to form bioactive nitrogen oxides as an important alternative source of NO, in particular, in hypoxic states 19. In the present study, nitrite/nitrate was measured as the major end product of NO metabolism. NO can react with to form reactive hydroxyl radicals and peroxynitrite and lead to mitochondrial damage, apoptosis, and mediate the cytotoxic effects in liver pathologies 14. Enhanced NO production has been shown in β‐thalassemia, but not in Hb H disease 16. In this study, the enhanced nitrite production could be detected in our patient group in spite of evidence of an increase in antioxidant defenses, which indicated that reactive nitrogen species was another pathogenic pathway to cell damage in thalassemia patients.
MDA is a low molecular weight end product formed via the decomposition of certain primary and secondary lipid peroxidation products and is considered as a marker of tissue injury and lipid peroxidation from oxidative stress. Increased MDA levels have been demonstrated in patients with β‐thalassemia major and Hb H disease, as in this study 20. Levels of MDA have been reported to be correlated with iron concentrations in the livers of thalassemia patients 21. In this study, there was a positive correlation between levels of MDA and serum ferritin, and a negative correlation between levels of MDA and vitamin C in the patients with Hb H disease.
Different antioxidant systems, including nonenzymatic antioxidants such as GSH and vitamin C, and various antioxidant enzymes, will defend against free radical attacks. The activities of SOD have been reported to be elevated or reduced in the RBCs of patients with β‐thalassemia major 20. This upregulation of SOD may be a compensatory mechanism to the formation of in check to combat oxidative stress. This is the first report in regard to the increased activities of SOD in the patients with Hb H disease. GSH can help the redox cycling of antioxidants such as ascorbate and α‐tocopheral. GSH depletion has been shown to increase NO‐induced oxidative stress in primary rat hepatocyte cultures, which was related to low molecular weight iron 22. In this study, the increase of activities of SOD, GPx, and GRx and reduced levels of GSH indicate that the patients with Hb H disease have intense antioxidant responses. Meanwhile, the activities of SOD were strongly correlated with the levels of ferritin, GSH, and GSSG, and the activities of GPx and GRx indicate that the antioxidant pathway (SOD and GPx/GSSG/GRx/GSH) was active in response to iron overload and oxidative stress in the patients with Hb H disease.
As an antioxidant, vitamin C works along with vitamin E and GPx to stop free radical chain reactions 23. A considerable ascorbic acid deficiency has been reported in patients with idiopathic hemochromatosis or in secondary iron overload due to chronic transfusion and has been suggested to be the result of oxidation by iron 24. The reduced levels of vitamin C in our patient group indicate persistent production of oxidative radicals and consumption of antioxidants in these patients. CK may function as temporal energy buffering and regulation of oxidative phosphorylation and the transport of chemical potential between sites of ATP production and energy utilization. The combination of NO and inhibited CK more potently than either of these agents alone. Peroxynitrite, formed in the reaction between and NO, is a potent oxidant of GSH forming GSSG and a rapid and irreversible inhibitor of CK activity. CK is a key enzyme of energy metabolism, and is of central importance to cellular energy homeostasis. CK deficiency is associated with heart failure and may cause a decline of more than 70% in ATP delivery to myofibrils, which leads to a blunted contractile reserve 25. The activities of CK were significantly lower in the patient group than in the controls, and they were negatively correlated with serum ferritin levels. This indicates the persistent production of oxidative radicals and consumption of antioxidants in these patients, which has not been reported in Hb H diseases. There is a trend of increasing cardiac abnormality with increasing serum ferritin levels. Finding suitable antioxidants to reduce toxicities from oxidative damage and to recover cellular function is an important therapy issue for Hb H patients.
These results demonstrate the presence of oxidative stress, and the active metabolic antioxidant pathway, SOD, and GPx/GSSG/GRx/GSH, in patients with Hb H disease is related to iron overload (Fig. 3). High levels of MDA, low levels of vitamin C and GSH, and low activities of CK indicate that there were not enough antioxidants to counteract the generation of reactive radical species in the patients with Hb H disease with iron overload. Many complications related to iron overload and oxidation stress were reported in Hb H disease that is not considered as a benign disease. Thalassemic erythrocytes were also found to be sensitive to oxidative stress, which led to an accelerated apoptosis and shortening of the life span of the defective erythrocytes 26. Therefore, early therapeutic intervention, including iron‐chelating therapy and supplementation of useful antioxidants, is important for patients with excess free radical production and iron overload. Monitoring oxidant and antioxidant statuses are necessary for the efficacy of antioxidant supplementation and the prevention of complications from free radical damage especially during infection.
Grant sponsor: Chi‐Mei Medical Center and Kaohsiung Medical University; Grant number: 99CM‐KMU‐13.
REFERENCES
- 1. Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol 2002;9(2):123–126. [DOI] [PubMed] [Google Scholar]
- 2. Chui DHK, Fucharoen S, Chan V. Hemoglobin H disease: Not necessarily a benign disorder. Blood 2003;101(3):791–800. [DOI] [PubMed] [Google Scholar]
- 3. Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med 2000;343(8):544–550. [DOI] [PubMed] [Google Scholar]
- 4. Scott MD, van den Berg JJ, Repka T, et al. Effect of excess alpha‐hemoglobin chains on cellular and membrane oxidation in model beta‐thalassemic erythrocytes. J Clin Invest 1993;91(4): 1706–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yuan J, Bunyaratvej A, Fucharoen S, Fung C, Shinar E, Schrier S. The instability of the membrane skeleton in thalassemic red blood cells. Blood 1995;86(10):3945–3950. [PubMed] [Google Scholar]
- 6. Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 1995;18(2):321–336. [DOI] [PubMed] [Google Scholar]
- 7. Schaible UE, Kaufmann SHE. Iron and microbial infection. Nat Rev Micro 2004;2(12):946–953. [DOI] [PubMed] [Google Scholar]
- 8. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol 2007;69(1):69–85. [DOI] [PubMed] [Google Scholar]
- 9. Anderson GJ. Mechanisms of iron loading and toxicity. Am J Hematol 2007;82(S12):1128–1131. [DOI] [PubMed] [Google Scholar]
- 10. Ozment CP, Turi JL. Iron overload following red blood cell transfusion and its impact on disease severity. Biochim Biophys Acta 2009;1790(7):694–701. [DOI] [PubMed] [Google Scholar]
- 11. Madani H, Rahimi Z, Manavi‐Shad M, et al. Plasma lipids and lipoproteins in children and young adults with major β‐thalassemia from western Iran: Influence of genotype. Mol Biol Rep 2011;38(4):2573–2578. [DOI] [PubMed] [Google Scholar]
- 12. Ricchi P, Ammirabile M, Spasiano A, et al. Hypocholesterolemia in adult patients with thalassemia: A link with the severity of genotype in thalassemia intermedia patients. Eur J Haematol 2009;82(3): 219–222. [DOI] [PubMed] [Google Scholar]
- 13. Afanas'ev IB. Signaling functions of free radicals superoxide & nitric oxide under physiological & pathological conditions. Mol Biotechnol 2007;37(1):2–4. [DOI] [PubMed] [Google Scholar]
- 14. Afanas'ev IB. Superoxide and nitric oxide in pathological conditions associated with iron overload. The effects of antioxidants and chelators. Curr Med Chem 2005;12:2731–2739. [DOI] [PubMed] [Google Scholar]
- 15. Roos D, van Bruggen R, Meischl C. Oxidative killing of microbes by neutrophils. Microbes Infect 2003;5(14):1307–1315. [DOI] [PubMed] [Google Scholar]
- 16. Korkina L, De Luca C, Deeva I, et al. L1 effects on reactive oxygen (ROS) and nitrogen species (RNS) release, hemoglobin oxidation, low molecular weight antioxidants, and antioxidant enzyme activities in red and white blood cells of thalassemic patients. Transfus Sci 2000;23(3):253–254. [DOI] [PubMed] [Google Scholar]
- 17. Wiener E. Impaired phagocyte antibacterial effector functions in β‐thalassemia: A likely factor in the increased susceptibility to bacterial infections. Hematol 2003;8(1):35–40. [DOI] [PubMed] [Google Scholar]
- 18. Pieracci FM, Barie PS. Iron and the risk of infection. Surg Infect (Larchmt) 2005;6(s1):s41–s46. [DOI] [PubMed] [Google Scholar]
- 19. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate‐nitrite‐nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 2008;7(2):156–167. [DOI] [PubMed] [Google Scholar]
- 20. Naithani R, Chandra J, Bhattacharjee J, Verma P, Narayan S. Peroxidative stress and antioxidant enzymes in children with beta‐thalassemia major. Pediatr Blood Cancer 2006;46(7): 780–785. [DOI] [PubMed] [Google Scholar]
- 21. Walter PB, Fung EB, Killilea DW, et al. Oxidative stress and inflammation in iron‐overloaded patients with β‐thalassaemia or sickle cell disease. Br J Haematol 2006;135(2):254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinbandhit‐Tricot S, Cillard J, Chevanne M, Morel I, Cillard P, Sergent O. Glutathione depletion increases nitric oxide‐induced oxidative stress in primary rat hepatocyte cultures: Involvement of low‐molecular‐weight iron. Free Radic Biol Med 2003;34(10):1283–1294. [DOI] [PubMed] [Google Scholar]
- 23. Padayatty SJ, Katz A, Wang Y, et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J Am Coll Nutr 2003;22(1):18–35. [DOI] [PubMed] [Google Scholar]
- 24. Young IS, Trouton TG, Torney JJ, McMaster D, Callender ME, Trimble ER. Antioxidant status and lipid peroxidation in hereditary haemochromatosis. Free Radic Biol Med 1994;16(3):393–397. [DOI] [PubMed] [Google Scholar]
- 25. Lygate CA, Fischer A, Sebag‐Montefiore L, Wallis J, ten Hove M, Neubauer S. The creatine kinase energy transport system in the failing mouse heart. J Mol Cell Cardiol 2007;42(6):1129–1136. [DOI] [PubMed] [Google Scholar]
- 26. Lang KS, Roll B, Myssina S, et al. Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose‐6‐phosphate dehydrogenase deficiency. Cell Physiol Biochem 2002;12(5–6):365–372. [DOI] [PubMed] [Google Scholar]