

Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited condition caused by PKD1 and PKD2 mutations. Complete analysis of both genes is typically required in each patient. In this study, we explored the utility of High‐Resolution Melt (HRM) as a tool for mutation analysis of the PKD2 gene in ADPKD families.
Methods
HRM is a mismatch‐detection method based on the difference of fluorescence absorbance behavior during the melting of the DNA double strand to denatured single strands in a mutant sample as compared to a reference control. Our families were previously screened by linkage analysis. Subsequently, HRM was used to characterize PKD2‐linked families. Amplicons that produced an overlapping profile sample versus wild‐type control were not further evaluated, while those amplicons with profile deviated from the control were consequently sequenced.
Results
We analyzed 16 PKD2‐linked families by HRM analysis. We observed ten different variations: six single‐nucleotide polymorphisms and four mutations. The mutations detected by HRM and confirmed by sequencing were as follows: 1158T>A, 2159delA, 2224C>T, and 2533C>T. In particular, the same haplotype block and nonsense mutation 2533C>T was found in 8 of 16 families, so we suggested the presence of a founder effect in our province.
Conclusions
We have developed a strategy for rapid mutation analysis of the PKD2 gene in ADPKD families, which utilizes an HRM‐based prescreening followed by direct sequencing of amplicons with abnormal profiles. This is a simple and good technique for PKD2 genotyping and may significantly reduce the time and cost for diagnosis in ADPKD.
Keywords: High‐Resolution Melt, ADPKD, screening methods, mutation scanning, founder effect
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent renal cystic disease with an incidence between 1:400 and 1:1,000 1. ADPKD is a systemic disorder involving many organs 2; it is characterized by abnormal proliferation of renal tubular epithelial cells, which manifests as cysts that increase gradually in both size and number, leading to massive kidney enlargement and progressive decline in kidney function 3, 4. The disease is genetically heterogeneous with two different genes identified: PKD1 (16p13.3) and PKD2 (4q21) 5, 6, 7. PKD1 is the major locus, accounting for approximately 85% of families 8. PKD1 gene consists of 46 exons and encodes a large protein, polycystin‐1 (4,303 amino acids) 9, 10. Exons 1 to 33 lie in a complex genomic region that is reiterated approximately six times further proximally on chromosome 16 6. Similarity between PKD1 and these pseudogenes means that locus‐specific amplification methods are required to analyze PKD1 11. PKD2 consists of 15 exons and encodes polycystin‐2 (968 amino acids) 12. A high level of allelic heterogeneity is found for both genes, with a total of 1,443 different variations reported for PKD1 and 241 for PKD2 13.
The genetic diagnosis of ADPKD is challenging because of the genetic heterogeneity and marked allelic heterogeneity at ADPKD genes. In fact, numerous studies have demonstrated a high degree of allelic heterogeneity in PKD1 and PKD2 with no single mutation found to account for more than 2% of affected families; in the majority of families, ADPKD is caused by a unique and private mutation 14. Although several indirect screening methodologies have been used to characterize the PKD genes 4, 11, direct sequencing of exon and flanking intronic regions from genomic DNA is now the gold standard for diagnostis proposed and mutation analysis; but it is extremely time‐consuming, expensive, and technically complex 15.
Hence, complete analysis of both genes is typically needed in each patient and diagnostic screening of a new family required sequencing of all PKD1 and PKD2 exons with the correlated cost.
In this study, we explored the utility, robustness and accuracy of High‐Resolution Melt (HRM) as a tool for mutation analysis of the PKD2 gene in ADPKD families and showed the results of an HRM screening strategy to identify sequence variants in PKD2 gene in 16PKD2‐linked families.
MATERIALS AND METHODS
Subjects
Fifty‐four patients from the Nephrology Center of Vicenza were screened for sequence variants in PKD2 gene; HRM analysis was used to characterize our PKD2‐linked families previously screened by linkage analysis 16.
Twenty healthy individuals were the control group. They were from the same area and ethnic group as patients.
The study was performed with the approval of the Ethics Committee of the San Bortolo Hospital in Vicenza. Informed consent was obtained from each patient before genetic testing.
Blood samples were obtained from all subjects and genomic DNA was extracted from peripheral blood lymphocytes. DNA working solutions were diluted in ultrapure water to obtain the correct dilution, after quantification and quality test of all experimental DNA samples.
Mutational Screening
Real‐time PCR and HRM conditions
HRM analysis was used to characterize PKD2‐linked families. The coding regions and intron–exon junctions, which are important in order to identify variants affecting mRNA splicing, were screened for mutation in PKD2 gene using this method. Seventeen primer sets were designed to cover the complete coding region plus flanking intronic sequence of the PKD2 gene. Exon 1 was divided into three fragments: only two amplicons of three could be screened by HRM, the rest was directly sequenced. We designed exon 1 primers in order to yield a series of overlapping fragments spanning the entire first coding region.
The HRM approach is based on real‐time DNA amplification in the presence of a double‐stranded DNA (dsDNA) intercalating fluorescent dye, and on monitoring the melting curves of each amplicon. HRM technique detects the change in fluorescence intensity induced by the release of the intercalating dye from a dsDNA to its two single‐stranded DNAs (ssDNA) as it denatures at high temperatures.
Amplification and HRM were performed in 25 μl volumes by Rotor Gene 6000 (Qiagen, S.A.S. Milan, Italy). The amplification mixture included 50 ng of genomic as template; 1.25 μl of Eva Green (Biotium, Hayward, CA); 5 μl of Real‐Time PCR buffer, magnesium free (TakaRa, Shiga, Japan); appropriate concentration of magnesium (TakaRa; 0.5 μl of dNTP mixture (TakaRa, Shiga, Japan); 5 μM of each primer; and 0.3 μl of Takara Ex Taq R‐PCR (TakaRa, Shiga, Japan).
Each run included an activation step at 95°C for 3 min followed by 45 amplification cycles of 40‐sec denaturation, 30‐sec annealing, and 40‐sec elongation, and a final elongation step at 72°C for 5 min, with fluorescence acquired on the green channel. The PCR products were heated to 95°C and cooled for heteroduplex formation, and melting was monitored by fluorescence emission using appropriate denaturation range. Annealing temperatures and HRM denaturation range differed according to each fragment (Table 1).
Table 1.
HRM Analysis Conditions for PKD2 Amplicons: Temperature of Annealing and HRM Range
| Amplicon | Annealing temperature (°C) | HRM range (°C) |
|---|---|---|
| Ex 1a | 58 | 89–99 |
| Ex 1c | 60 | 77–90 |
| Ex 2 | 58 | 73–85 |
| Ex 3 | 59 | 75–88 |
| Ex 4 | 60 | 79–89 |
| Ex 5 | 62 | 78‐ 88 |
| Ex 6 | 60 | 75–85 |
| Ex 7 | 55 | 70–86 |
| Ex 8 | 56 | 74–87 |
| Ex 9 | 59 | 71–84 |
| Ex 10 | 59 | 73–83 |
| Ex 11 | 59 | 73–87 |
| Ex 12 | 60 | 74–87 |
| Ex 13 | 60 | 76–89 |
| Ex 14 | 58 | 79–91 |
| Ex 15 | 58 | 70–85 |
HRM analysis
To ensure reproducibility of the melt curves, each sample was performed in triplicate and each wild‐type control in triplicate, thus avoiding errors and any misinterpretation of the results.
For HRM scoring, particular wild‐type samples (negative control with confirmed sequence) were set as reference samples. Results were obtained by software Rotor‐Gene 6000 series version 1.7.87, comparing the concordance between unknown genotype samples and selected references.
For HRM analysis, the melting curves were normalized and the temperature shifted (temp‐shifted) so that samples were comparable. The normalized and temp‐shifted melting curves corresponded to the final curve after normalization. In this study, normalization was manually adjusted. Normalized melting curves, melt curves, and difference graphs were used to distinguish between the wild‐type and mutated/deviated samples. Amplicons that produced an overlapping profile sample versus wild‐type control were not further evaluated. On the contrary, in case of an amplicon sequence variation, the normalized and temp‐shifted melting curve had a different shape from wild‐type amplicon. All amplicons with a different derived, normalized, and temp‐shifted curve from wild‐type control were consequently sequenced.
Post‐HRM sequence analysis
To verify and identify the PKD2 gene alterations presented by HRM, all de novo investigated samples were directly sequenced. Note that only mutant/polymorphism‐positive samples were further analyzed by sequencing analysis using the same forward and reverse PCR primers used for the real‐time amplification. Sequencing analysis was performed by direct Sanger sequencing method. The sequencing process included purification of PCR products using ExoSAP‐IT (GE Healthcare Life Science, Buckinghamshine, UK); cycling sequencing reaction using BigDye Terminator v1.1 kit (Applied Biosystem, Forster City, CA), and final purification with Centrisep Columns (Princeton Separations, Freehold, NJ), according to operational manuals. The capillary electrophoresis was performed by automated ABI PRISM 3130 Genetic Analyzer (Applied Biosystem, Forster City, CA), according to the manufacturer's instructions.
Sequence Variation Analysis and Classification
Reference sequence of PKD2 was obtained from NCBI RefSeq and all sequences were compared to this following PKD2 reference sequence: NM_000297.2. The standard nomenclature recommended by Human Genome Variation Society (HGVS; http://www.hgvs.org/mutnomen/) was used to number nucleotides and name mutations or variants 17. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG traslantion initiation codon (codon 1) in the reference sequence.
For previously described sequence variants, we reported the clinical significance assessed in the Polycystic Kidney Disease Mutation Database 18 and/or NCBI/Ensemble databases.
Whenever a mutation was confirmed in a proband, first‐ and second‐degree relatives were tested for the identified variant in order to check the cosegregation with the disease in the family.
RESULTS AND DISCUSSION
The extensive allelic heterogeneity in PKD1 and PKD2, combined with the fact that no single mutation accounts for >2% of ADPKD‐affected families, requires mutation screening methods for accurate molecular diagnosis of ADPKD 14. Several indirect methods have been developed to screen mutations in ADPKD subjects for research and clinical purposes, including single‐strand confirmation polymorphism (SSCP) 19, 20, 21, denaturing high‐performance liquid chromatography (DHPLC) 11, denaturing gradient gel electrophoresis (DGGE) 22, 23, 24, RT‐PCR 25, the protein truncation test 26, 27, Transgenomics SURVEYOR Nuclease and WAVE Nucleic Acid High‐Sensitivity Fragment Analysis 4, and HRM analysis 28. However, each of these methods has limitations for detecting certain types of gene variations and direct sequencing is still considered the method of choice for mutation detection in many laboratories.
The current work describes the evaluation of the HRM performance and its utility as screening method in PKD2 gene. This genetic study was performed in search of mutations in PKD2 gene in 16 PKD2‐linked families, previously analyzed by linkage analysis.
For the first time, Bataille et al. described the use of HRM technique in ADPKD. They evidenced a good diagnosis rate of HRM in the identification of a mutation in their cohort (including definitely pathogenic and probably pathogenic mutations) 28.
With this method, only mutation/polymorphism‐positive samples need to be further analyzed, while others can be considered wild type. HRM results were used as the criterion to distinguish between positive and negative samples, considering their concordance by normalized and derivative plots, where a particular wild‐ type sample was set as the reference and all other samples were compared against this sample 29.
HRM demonstrated high reproducibility as evidence by the large number of different wild‐type samples and duplicates of the same samples that are clustered tightly in unique melting curves. More than 85% of amplicons suspected to have a sequence variant after HRM analysis carried one after sequencing, highlighting the specificity of this technology. Overall, less than 10% of the PKD2 fragments analyzed by HRM were sequenced. Moreover, we screened in a blind procedure 11 patients with known variants previously identified by direct Sanger sequencing and HRM analysis recognized all of them. Using this approach, all samples revealed absolute conformity with direct sequencing, and sequence variants were 100% distinguished. Thus, HRM is efficient, sensitive, and specific for mutation screening in PKD2 gene. Furthermore, HRM analysis reduced drastically the need for systematic sequencing and the time of sequence analysis to identify a sequence variation.
Additionally, HRM is the only method that is performed in the same tube and in the same run used for PCR amplification (without exposure to the environment, thus avoiding the risk of contamination). HRM method does not require post‐PCR manipulation of samples as in DNA sequencing technologies and conventional gel‐ or HPLC‐based scanning methods. Following real‐time amplification, the PCR products are melted and analyzed in the same HRM instrument. Scanning of samples only takes the time of a PCR plus approximately 10–15 min for melting and analysis, so each sample only costs the price of a PCR reaction plus the costs of intercalating fluorescent DNA‐binding dye, which are cheaper than conventional scanning and sequencing chemistries 30: HRMA is more effective than full gene sequencing even for genes with small amounts of exons 31.
Finally, the fact that the HRM dye does not interfere during sequencing 32 accelerates the whole process, and the entire assay (containing direct sequencing of mutation/polymorphism‐positive samples) can be performed within a working day.
The quality of the HRM result is highly dependent on the quality of DNA extraction and real‐time amplification. Therefore, the preparation of pure products is crucial and a necessary prerequisite for correct HRM analysis. In fact, complexity of genomic DNA as well as concentration of salts has been reported to affect HRM 33, 34. It is, therefore, recommended to use DNA samples and DNA controls that have been prepared using a common extraction procedure.
We observed ten different variations: six single‐nucleotide polymorphisms (83G>C, 180G>A, IVS1–16C>T, IVS3–22G>A, 1459T>C, 1830G>A) and four mutations. The mutations detected by HRM analysis and confirmed by sequencing were as follows: 1158T>A, 2159delA, 2224C>T, and 2533C>T. All of the mutations and polymorphisms that were found had been previously reported. We found the polymorphism 83G>C, resulting in the substitution Arg28Pro, in four screened subjects; the 180G>A synonymous substitution; the IVS1–16C>T polymorphism; and the 1830G>A synonymous substitution one time. Twelve subjects were heterozygous for the polymorphism IVS3–22G>A, and two were homozygous for less‐frequented allele. In two subjects from the same family, we found the 1459T>C substitution, which implies a change in the tyrosine (Tyr) to histidine (His) in position 487 (Tyr487His).
The no‐overlapping HRM profile in exon 5, found in affected members of one of our PKD2 families, was caused by a T‐to‐A transversion producing a stop mutation in codon 386 (1158T>A, Y386X; Fig. 1).
Figure 1.
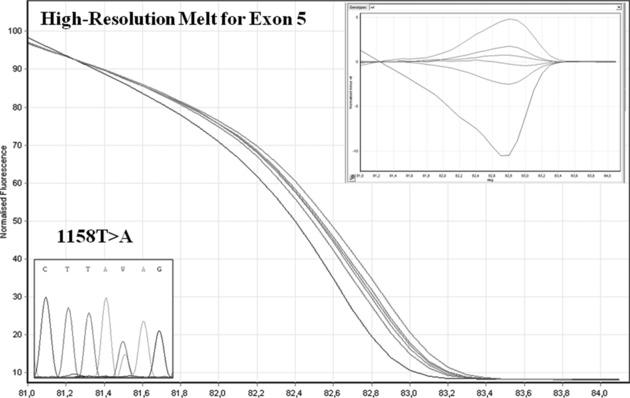
HRM analysis of exon 5 (HRM curve and sequencing); the no overlapping HRM profile was caused by 1158T>A mutation.
We observed two strange HRM profiles in exon 11 (Fig. 2): electropherograms of these fragments shown, in one case, a single adenosine deletion of 2159 nucleotide in a polyadenosine tract, which results in a frameshift with premature translational termination of polycystin‐2 and a C‐to‐T transition on 2224 nucleotide results in nonsense substitution results in truncated C‐terminus domain. This mutation causes the lack of polycystin‐1 interacting region, in fact polycystin‐2 interacts through its cytoplasmic carboxyl‐terminal region with a coiled‐coil motif in the cytoplasmic tail of polycystin‐1 35.
Figure 2.
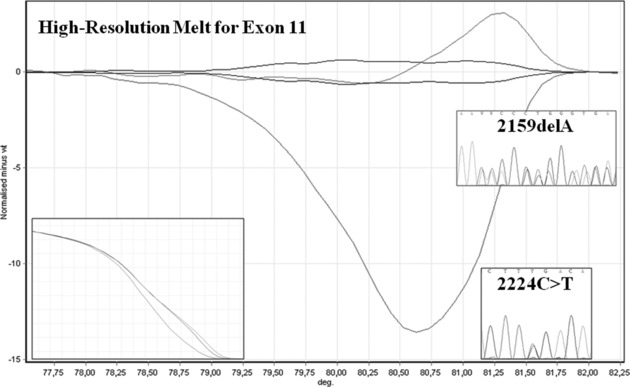
HRM analysis of exon 11 (HRM curve and sequencing); we found two abnormal profiles and we identified two different mutations: 2159delA and 2224C>T.
In particular, the nonsense mutation 2533C>T in exon 14, resulting in R845X, was found in 8 of 16 families for a total of 20 subjects (Fig. 3). These eight families were also characterized by the same disease haplotype (CEN‐ VG2 (TG)22, VG3 (TA+GA)33, VG4 (CA)13, 2929 (AC)27, 1563 (TG)21‐TEL) and came from the same area in the Veneto region. In our opinion, the presence of a common haplotype block and mutation in these apparently unrelated families is compatible with the presence of a common ancestral founder and the hypothesis of a founder effect in Vicenza province 36.
Figure 3.
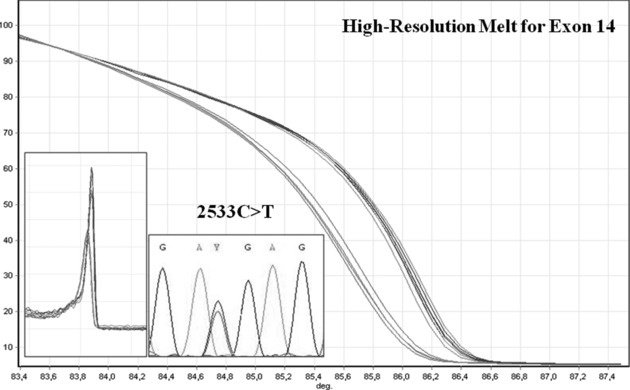
HRM analysis of exon l4 (Melt graph, HRM curve, and sequencing); we found 2533C>T in 16 subjects from 8 of 16 PKD2 families analyzed.
Our hypothesis is also based on historical and geographical context: this province is characterized and limited by several isolated valleys that in past were not connected with other nearby zones. It could be responsible for consanguinity. If the ADPKD mutation is present in original family, it is inherited subsequently. As these families remained in the same geographical area for long period many of the affected members may have originated from the same native family. These data suggest that the specific mutation 2533C>T segregates in this geographically isolated population.
By HRM analysis and linkage analysis, we excluded ADPKD in 16 young subjects.
No pathogenic mutations were detected in 5 of 16 of our families, although ADPKD was confirmed in all subjects by clinical and imaging criteria. The unidentified ADPKD mutations may involve genetic alterations that are not detected with our predominantly exon‐based screening methods, such as changes in promoters and other distantly located regulatory regions 37 as well as changes in regulatory elements, intronic changes that generate cryptic splice sites or large rearrangements and large deletions of gene segments. In addition, when there is somatic mosaicism, there may be different levels of a mutant allele in different tissues. Therefore, a determination in peripheral blood may not be descriptive of what is happening in the kidneys 38. Lastly, some variants, that is missense, that were classified as nonpathogenic by the current scoring system may in fact represent hypomorphic variants: such variants alone may result only in mild phenotype, but two such variants in trans can cause typical to severe disease 39.
CONCLUSION
We have developed a strategy for rapid molecular mutation analysis of the PKD2 gene in ADPKD families, which utilizes an HRM‐based prescreening followed by direct Sanger sequencing of amplicons with abnormal melting profiles. This is a simple and good approach for PKD2 genotyping and dramatically reduces the number of samples that need to be sequenced, significantly decreasing the amount of time needed to analyze the samples and decreasing the overall financial costs for molecular diagnosis in ADPKD.
Future developments of this work include extending the workflow to the major ADPKD gene, PKD1. In addition, HRM technology allows genomic qPCR screening to be performed for large deletions/duplications that represent 4% of ADPKD pathogenic variants 38. This will improve molecular diagnosis in ADPKD by directly identifying large deletions/duplications that have not been identified by HRM analysis or with sequencing.
Therefore, HRM analysis could prove to be a highly integrated molecular diagnosis tool.
ACKNOWLEDGMENTS
This study was approved by the Ethics Committee of San Bortolo Hospital (N. 47/07). This study was performed in accordance with the Declaration of Helsinki.
REFERENCES
- 1. Gabow PA. Polycystic kidney disease: Clues to pathogenesis. Kidney Int 1991;40(6):989–996. [DOI] [PubMed] [Google Scholar]
- 2. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med 1993;329(5):332–342. [DOI] [PubMed] [Google Scholar]
- 3. Torres VE. Treatment of polycystic liver disease: One size does not fit all. Am J Kidney Dis 2007;49(6):725–728. [DOI] [PubMed] [Google Scholar]
- 4. Tan YC, Michael A, Blumenfeld S, et al. Novel method for genomic analysis of PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease. Hum Mutat 2009;30(2):264–273. [DOI] [PubMed] [Google Scholar]
- 5. Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell 1995;81(2):289–298. [DOI] [PubMed] [Google Scholar]
- 6. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell 1994;77(6):881–894. [DOI] [PubMed] [Google Scholar]
- 7. Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996;272(5266):1339–1342. [DOI] [PubMed] [Google Scholar]
- 8. Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006;17(11):3013–3019. [DOI] [PubMed] [Google Scholar]
- 9. Rossetti S, Harris, PC . Genotype‐phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2007;18(5):1374–1380. [DOI] [PubMed] [Google Scholar]
- 10. Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10(2):151–160. [DOI] [PubMed] [Google Scholar]
- 11. Rossetti S, Chauveau D, Walker D, et al. A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int 2002;61(5):1588–1599. [DOI] [PubMed] [Google Scholar]
- 12. Hayashi T, Hochizuki T, Reynolds OM, Wu G, Cai Y, Somb S. Characterization of the exon structure of the polycystic kidney disease 2 gene (PKD2). Genomics 1997;44(1):131–136. [DOI] [PubMed] [Google Scholar]
- 13. Autosomal Dominant Polycystic Kidney Disease Mutation Database: PKDB. Available from: http://pkdb.mayo.edu/.
- 14. Harris PC, Rossetti, S . Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010;6(4):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan YC, Blumenfeld J, Rennert H. Autosomal dominant polycystic kidney disease: Genetics, mutations and microRNAs. Biochim Biophys Acta 2011;1812(10):1202–1212. [DOI] [PubMed] [Google Scholar]
- 16. Corradi V, Gastaldon F, Virzi GM, et al. Clinical pattern of adult polycystic kidney disease in a northeastern region of Italy. Clin Nephrol 2009;72(4):259–267. [DOI] [PubMed] [Google Scholar]
- 17. den Dunnen JT, Antonarakis SE. Mutation nomenclature. Curr Protoc Hum Genet 2003. Chapter 7: Unit 7.13. [DOI] [PubMed] [Google Scholar]
- 18. Gout AM, Martin NC, Brown AF, Rauine D. PKDB: Polycystic Kidney Disease Mutation Database—A gene variant database for autosomal dominant polycystic kidney disease. Hum Mutat 2007;28(7):654–659. [DOI] [PubMed] [Google Scholar]
- 19. Zhang S, Mei C, Zhang D, et al. Mutation analysis of autosomal dominant polycystic kidney disease genes in Han Chinese. Nephron Exp Nephrol 2005;100(2):e63–e76. [DOI] [PubMed] [Google Scholar]
- 20. Veldhuisen B, Sans JJ, de Haij S, et al. A spectrum of mutations in the second gene for autosomal dominant polycystic kidney disease (PKD2). Am J Hum Genet 1997;61(3):547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reiterová J, Stekrouà J, Peters DJ, et al. Four novel mutations of the PKD2 gene in Czech families with autosomal dominant polycystic kidney disease. Hum Mutat 2002;19(5):573. [DOI] [PubMed] [Google Scholar]
- 22. Peters DJ, Ariyurek Y, van Dijk M, Breuning MH. Mutation detection for exons 2 to 10 of the polycystic kidney disease 1 (PKD1)‐gene by DGGE. Eur J Hum Genet 2001;9(12):957–960. [DOI] [PubMed] [Google Scholar]
- 23. Perrichot R, Mercier B, Carne A, Cledes J, Ferec C. Identification of 3 novel mutations (Y4236X, Q3820X, 11745+2 ins3) in autosomal dominant polycystic kidney disease 1 gene (PKD1). Hum Mutat 2000;15(6):582. [DOI] [PubMed] [Google Scholar]
- 24. Perrichot RA, Mercier B, Simon PM, Whebe B, Cleoles J, Ferec C. DGGE screening of PKD1 gene reveals novel mutations in a large cohort of 146 unrelated patients. Hum Genet 1999;105(3):231–239. [DOI] [PubMed] [Google Scholar]
- 25. Burtey S, Berland Y. Polycystic kidneys. Rev Prat 2006;56(18):2053–2058. [PubMed] [Google Scholar]
- 26. Roelfsema JH, Spwit L, Saris JJ, et al. Mutation detection in the repeated part of the PKD1 gene. Am J Hum Genet 1997l;61(5):1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rossetti S, Strmecki L, Gamble V, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet 2001;68(1):46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bataille S, Berland Y, Fontes M, Burtey S. High Resolution Melt analysis for mutation screening in PKD1 and PKD2. BMC Nephrol 2011;12:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reed GH, Kent JO, Wittwer CT. High‐resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics 2007;8(6):597–608. [DOI] [PubMed] [Google Scholar]
- 30. Olsen RK, Dobrowolski SF, Kjeldsen M, et al. High‐resolution melting analysis, a simple and effective method for reliable mutation scanning and frequency studies in the ACADVL gene. J Inherit Metab Dis 2010;33(3):247–260. [DOI] [PubMed] [Google Scholar]
- 31. Vossen RH, Aten E, Roos A, den Dunnen JT, et al. High‐resolution melting analysis (HRMA): More than just sequence variant screening. Hum Mutat 2009;30(6):860–866. [DOI] [PubMed] [Google Scholar]
- 32. Poláková KM, Lopotová T, Klamová H, Moravcová J. High‐resolution melt curve analysis: Initial screening for mutations in BCR‐ABL kinase domain. Leuk Res 2008;32(8):1236–1243. [DOI] [PubMed] [Google Scholar]
- 33. Seipp MT, Durtschi JD, Liew MA, et al. Unlabeled oligonucleotides as internal temperature controls for genotyping by amplicon melting. J Mol Diagn 2007;9(3):284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gundry CN, Vandersteen JG, Reed GH, Pryor RJ, Chen J, Wittwer CT. Amplicon melting analysis with labeled primers: A closed‐tube method for differentiating homozygotes and heterozygotes. Clin Chem 2003;49(3):396–406. [DOI] [PubMed] [Google Scholar]
- 35. Xu GM, González‐Perrett S, Essafi M, et al. Polycystin‐1 activates and stabilizes the polycystin‐2 channel. J Biol Chem 2003;278(3):1457–1462. [DOI] [PubMed] [Google Scholar]
- 36. Corradi V, Gastaldon F, Virzi GM, et al. Epidemiological and molecular study of autosomal dominant polycystic kidney disease (ADPKD) in the province of Vicenza, Italy: Possible founder effect? G Ital Nefrol 2010;27(6):655–663. [PubMed] [Google Scholar]
- 37. Lantinga‐van Leeuwen IS, Leonhard WN, Dauwerse H, et al. Common regulatory elements in the polycystic kidney disease 1 and 2 promoter regions. Eur J Hum Genet 2005;13(5):649–659. [DOI] [PubMed] [Google Scholar]
- 38. Consugar MB, Wong WC, Lundquist PA, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 2008;74(11):1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int 2009;75(8):848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]