

Abstract
Background
Respiratory pathogens are a leading cause of hospital admission and traditional detection methods are time consuming and insensitive. Multiplex molecular detection methods have recently been investigated in hope of replacing these traditional techniques with rapid panel‐based testing.
Objectives
This study evaluated the FilmArray® Respiratory Panel ([FARP], Idaho Technology Inc., Salt Lake City, UT) as a replacement for direct fluorescent antibody (DFA) testing in a pediatric hospital.
Methods
Eleven of the 21 FARP analytes (Adenovirus, Bordetella pertussis, human Metapneumovirus, Influenza A, Influenza A H1N1 2009, Influenza B, Parainfluenza [1, 2, & 3], Respiratory Syncytial Virus, and rhinovirus) were evaluated using nasopharyngeal specimens. Positive samples were pooled in groups of 5. Samples identified by reference methods as positive for respiratory pathogens were used for the majority of positive samples. DFA was the reference method for ten analytes; LuminexTM xTAG Respiratory Virus Panel (RVP) was the reference method for rhinovirus. Discrepant results were resolved by positive culture and fluorescent antibody stain and/or laboratory‐developed real‐time polymerase chain reaction (PCR) assays (LDT).
Results
The agreement for most analytes was in concordance with the established reference methods with the exception of Adenovirus. Additionally, the FARP detected several pathogens not previously detected by DFA, and most were confirmed by LDT. Several DFA‐positive analytes were confirmed as true‐negatives by the FARP and LDT.
Conclusion
FARP overall performed better than DFA with the exception of Adenovirus, making the FARP an attractive alternative to laboratories looking to replace DFA with a rapid, user‐friendly, multiplex molecular assay. J. Clin. Lab. Anal. 27:148–154, 2013. © 2013 Wiley Periodicals, Inc.
Keywords: DFA, multiplex PCR, respiratory virus
Abbreviations
- FARP
FilmArray® Respiratory Panel
- RVP
LuminexTM xTag Respiratory Virus Panel
- PCR
Polymerase Chain Reaction
- LDT
Laboratory Developed real‐time PCR
- DFA
Direct Fluorescent Antibody stain
- ELISA
enzyme linked immunosorbent assay
- LOD
Limit of Detection
- Adeno
Adenovirus
- RSV
Respiratory Syncytial Virus
- hMPV
Human Metapneumovirus
- FluA
Influenza A
- 2009
FluA H1N1 Influenza A H1N1 2009 pandemic
- FluB
Influenza B
- Rhino
Rhinovirus
- Para1
Parainfluenza 1
- Para2
Parainfluenza 2
- Para3
Parainfluenza 3
- Bpert
Bordetella pertussis
INTRODUCTION
Respiratory infections represent a leading cause of acute hospital admission and often lead to serious complications in patients with chronic immunomodulatory conditions. In the general population, pediatric and elderly patients are particularly at risk for these infections and complications. Historically, culture and detection by direct fluorescence antibody staining and ELISA have been the traditional method for diagnosis of respiratory infections. Culture for many organisms is notoriously insensitive due to fastidious growth requirements, and DFAs and ELISA are labor‐intensive and insensitive due to low‐level infections that fall below the practical sensitivity of these methodologies. DFAs are also subject to technical error and subjective/inconsistent result interpretation. Each of these methodologies has also been shown to be less sensitive than polymerase chain reaction (PCR)‐based detection methods for viral respiratory pathogens 1, 2, 3, 4, 5, 6, 7. Additionally, influenza A & B and RSV can be rapidly tested using chromatographic immunoaasays in the ambulatory care setting; however, the influenza assays in particular have displayed unacceptable sensitivities 8, 9. Taken together, these various limitations can lead to a delay in or lack of pathogen identification, as well as inappropriate use of antimicrobial therapy 10.
Molecular platforms capable of detecting respiratory pathogens are currently available as cleared by FDA and laboratory‐developed PCR assays. Unfortunately, these technologies typically require advanced molecular expertise and often target only one pathogen in a labor‐intensive assay 11. The FARP from Idaho Technology Inc. allows simultaneous detection of respiratory pathogens in a single, user‐friendly pouch format. This technology combines extraction and purification of nucleic acid, nested, and real‐time PCR, with fluorescence‐based automated reporting in a single pouch. The closed assay format and limited hands‐on time drastically reduce the chances for contamination. The assay provides routine microbiology laboratories with the tools to perform panel‐based molecular detection of respiratory pathogens, a function typically limited to reference laboratories or large hospital systems. This comparison study serves as an independent evaluation for laboratories interested in the FARP as a candidate to replace viral DFA stains.
This study aimed to evaluate selected analytes of the FARP for use in a clinical laboratory using nasopharyngeal samples from an exclusively pediatric population. This study investigated the performance of 11 analytes (Adenovirus [Adeno], Respiratory Syncytial Virus [RSV], human Metapneumovirus [hMPV], Influenza A [FluA], Influenza A H1N1 2009 pandemic [2009 FluA H1N1], Influenza B [FluB], Rhinovirus [Rhino], Parainfluenza 1 [Para1], Parainfluenza 2 [Para2], Parainfluenza 3 [Para3], and Bordetella pertussis [Bpert]), using a pre‐FDA approved version of the assay that also included Bocavirus. The goal was to evaluate this assay's performance as a candidate to replace traditional culture/DFA‐based laboratory techniques employed by many potential users of the FARP.
MATERIALS AND METHODS
Specimens
Nasopharyngeal aspirates in M4 transport media previously tested by DFA at Primary Children's Medical Center (Salt Lake City, UT) or DFA and RVP ([specifically for rhinovirus], Intermountain Central Laboratory, Murray, UT) were analyzed for this validation study in accordance with our institutional review board. The FDA approved specimen type for the FARP is nasopharyngeal swabs, which were not utilized at our institution for pediatrics. Specimens were obtained from children between the ages of 0–19 years. The clinical specimens were initially tested by DFA, and negative specimens were then reflexed to RVP for further testing. Thirty positive and 30 negative clinical samples were selected for testing. To obtain a minimum of 30 positive samples for analytes with limited clinical isolates, a subset of cultured organisms were spiked into previously frozen clinical nasopharyngeal specimens and screened by fluorescent antibody after the initial culture. The virally infected cells used for spiking were used at approximate densities of that seen in clinical positive DFAs. A total of 393 positive and 370 negative specimens were tested. The specific breakdown of samples tested is listed in Table 1. Most primary patient specimens were frozen at –70°C for 1–3 years; however, four fresh specimens were included in the study on the day of DFA testing. FluA specimens were chosen from a distribution of three consecutive flu seasons.
Table 1.
Total Specimens Tested for Each Analyte by FARP and Reference Methods Including the Specific Breakdown of Positive Specimens
| Specimens tested | ||
|---|---|---|
| Reference method positive | Reference method | |
| Analyte | total (clinical/spiked) | negative |
| Adeno | 39 (21/18) | 30 |
| Bpert | 30 (1/29) | 60 |
| FluA | 61 (29/32) | 60 |
| 2009 FluA H1N1 | 30 (30/0) | 30 |
| FluB | 41 (19/22) | 30 |
| hMPV | 30 (30/0) | 30 |
| Para1 | 30 (30/0) | 30 |
| Para2 | 42 (21/21) | 30 |
| Para3 | 30 (30/0) | 30 |
| Rhino | 30 (30/0) | 10 |
| RSV | 30 (30/0) | 30 |
| Total | 393 (271/122) | 370 |
FilmArray® & Run set‐up
The FilmArray® platform and respiratory panels used in this study were identical to the platform evaluated for the FDA submission. The software from the pre‐FDA approved system reported the results of all analytes tested, whereas the FDA approved panel will release the results of all analytes except Bocavirus.
Each FilmArray® machine can analyze one pouch at a time and in order to maximize efficiency and minimize cost, analytes were pooled in groups of five unique analytes per run. The pooled sample of 325 μl was injected into the FARP pouch for analysis. Extraction, amplification, detection, and analysis were completely automated within the pouch. Results for the assay were only provided by the software if the quality control reactions were appropriately detected. Each pouch included internal run controls for both the primary amplification and the analyte specific amplification stages. All set up was performed in a biosafety cabinet.
Discrepant Analysis
Discrepant results were resolved by culture with fluorescent antibody stain and/or LDT. For FARP false‐negative results (excluding Rhino), culture positive results were considered for discrepant analysis and negative culture was subsequently interrogated by LDT. The schematic algorithm for discrepant resolution is shown in Figure 1. LDT assays were performed at Associated Regional and University Pathologists (ARUP) Laboratories (Salt Lake City, UT). LDT for FluA, FluB, and RSV were performed as a multiplex assay. The remaining analytes were performed as singleplex reactions. Nucleic acids for these LDTs were obtained by extracting 220 μl on the Qiagen BioRobot 9604 per the established procedure. Resolution was achieved when culture with fluorescent antibody stain, and/or PCR results correlated to either the initial DFA/RVP result or the FARP result.
Figure 1.
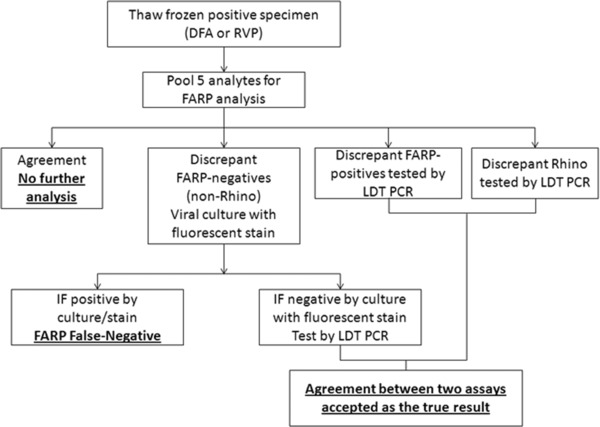
Schematic flow chart for specimen preparation, testing, and discrepant analysis.
Limit of Detection Determination
Clinical samples from each cohort of analytes were quantified by ARUP Laboratories using standard curves generated with log dilutions of plasmids or genomic DNA (Bpert and Adeno group C) or RNA transcripts (RNA viruses). Serial five‐fold dilutions of the quantified clinical samples were then prepared in M4 media and pooled in groups of four analytes. For determination of the limit of detection (LoD), specimens were refrigerated at 4–6°C for less than 7 days before testing by LDT. The LoD was determined and the theoretical concentration (copies/ml) was calculated for each dilution.
Reproducibility and Analytical Specificity
Each analyte was tested four times on a single day in three concentrations: 3X LoD, 1X LoD, and LoD/10. The samples were run four times over 5 days. The analytical specificity of the assay was determined by spiking cultured organisms into M4 transport media and running the samples in the absence of analytes targeted by the FARP.
RESULTS
The positive and negative percent agreements for each analyte were established for the FARP (Table 2). Agreement of 95% versus the current laboratory methods of DFA or RVP (rhinovirus only) was desired for the purpose of effectively replacing DFA methods. Several of the analytes had 100% agreement in the initial phase of the validation including: Bpert, FluA, and hMPV. The remaining analytes required further investigation by culture with fluorescent antibody and/or LDT to resolve discrepant results. Several samples were resolved by culture and fluorescent antibody stain and did not require PCR analysis. However, because the specimens were stored in a –70°C freezer, negative culture could not be considered absolute resolution. These samples were then resolved by LDT interrogation. After reanalysis, each discrepant sample for Para2 and FluB was resolved to complete agreement between the two methods (Table 2). For the remaining analytes, all but Adeno, 2009 FluA H1N1, and Para1 attained greater than 95% positive and negative agreement versus DFA (Table 2). The positive percent agreement for Adeno fell below 83%, while Para1 was 94%.
Table 2.
Analyte‐Specific Results and Agreement Values (Positive Percent and Negative Percent Agreement) for 11 Tested Analytes Before and After Resolution of Discrepant Specimens
| Positive | Negative | ||||||
|---|---|---|---|---|---|---|---|
| Analyte | Dataset | FARP+/REF+ | FARP+/REF‒ | FARP‒/REF+ | FARP‒/REF‒ | agreement (%) | agreement (%) |
| Adeno | Initial | 32 | 1 | 7 | 29 | 82 | 97 |
| – | Resolved | 33 | 0 | 7 | 29 | 83 | 100 |
| – | – | – | – | – | – | – | – |
| Bpert | No further analysis | 30 | 0 | 0 | 60 | 100 | 100 |
| – | – | – | – | – | – | – | – |
| FluA | No further analysis | 61 | 0 | 0 | 60 | 100 | 100 |
| – | – | – | – | – | – | – | – |
| 2009 FluA H1N1 | Initial | 28 | 0 | 2 | 30 | 93 | 100 |
| – | Resolved | 28 | 0 | 2 | 30 | 93 | 100 |
| – | – | – | – | – | – | – | – |
| FluB | Initial | 30 | 0 | 11 | 30 | 73 | 100 |
| – | Resolved* | 30 | 0 | 0 | 38 | 100 | 100 |
| – | – | – | – | – | – | – | – |
| hMPV | No further analysis | 30 | 0 | 0 | 30 | 100 | 100 |
| – | – | – | – | – | – | – | – |
| Para1 | Initial | 27 | 1 | 3 | 29 | 90 | 97 |
| – | Resolved | 30 | 0 | 2 | 28 | 94 | 100 |
| – | – | – | – | – | – | – | – |
| Para2 | Initial | 31 | 0 | 11 | 30 | 74 | 100 |
| – | Resolved* | 31 | 0 | 0 | 37 | 100 | 100 |
| – | – | – | – | – | – | – | – |
| Para3 | Initial | 29 | 3 | 1 | 27 | 97 | 90 |
| – | Resolved | 31 | 1 | 0 | 28 | 100 | 97 |
| – | – | – | – | – | – | – | – |
| Rhino | Initial | 29 | 0 | 1 | 10 | 97 | 100 |
| – | Resolved | 29 | 0 | 1 | 10 | 97 | 100 |
| – | – | – | – | – | – | – | – |
| ‐RSV | Initial | 30 | 1 | 0 | 29 | 100 | 97 |
| – | Resolved | 30 | 1 | 0 | 29 | 100 | 97 |
REF, Reference method (DFA or LuminexTM xTAG RVP [Rhino only]).
a) Four specimens for Para2 and three specimens for FluB could not be resolved due to the lack of specimen or the lack of consensus between three methodologies (LDT, FARP, and culture with fluorescent antibody stain).
The pooled specimens were intended to contain five analytes; however, seven analytes were detected in several pools. For these pools, each individual specimen was interrogated individually on the FARP to confirm these identifications. Each sample was also confirmed by LDT, and it was determined that there were in fact seven distinct analytes present; two deemed “unexpected positive.” Several other analytes were detected that were not part of the evaluation, and therefore were not resolved (Bocavirus n = 16, Coronavirus n = 6, Parainfluenza 4 n = 2, Chlamydophila pneumoniae n = 1).
The LoD for each analyte was determined for the FARP using LDTs. Each LoD sample was quantified by real‐time PCR and the analyte concentration was determined (Table 3). The dilution series was also tested by LDT to compare the LoD of the LDTs to the FARP. The LDT LoD values were consistently lower than the FARP with the exception of RSV. Because the LDT LoD for RSV exceeded the value originally established for that assay by greater than 1 log, sequencing of the target region of the test virus was performed to check for polymorphisms potentially affecting PCR efficiency 12. Sequencing revealed two previously undescribed polymorphisms that likely account for this difference. These results are consistent with LDT and FARP having comparably low LoDs for RSV strains not containing these polymorphisms. Two analytes with comparatively high LoD values on the FARP (Adeno and Bpert) were interrogated individually to determine whether the pooling of analytes had any negative effect on the LoD values. Neither analyte showed a change in LoD values when assayed individually on the FARP.
Table 3.
Relative Limit of Detection Values for the FARP and Laboratory‐Developed Real‐Time PCR Assays
| FilmArray LoD | ARUP LoD | Log | |
|---|---|---|---|
| Organism | (Log copies/ml) | (Log copies/ml) | difference |
| Adeno | 5.78 | 3.08 | 2.70 |
| Bpert | 5.70 | 2.30 | 3.40 |
| FluA | 5.44 | 3.04 | 2.40 |
| 2009 FluA H1N1 | 6.33 | 4.63 | 1.70 |
| FluB | 4.89 | 4.49 | 0.40 |
| hMPV | 5.57 | 5.18 | 0.40 |
| Para1 | 3.76 | 2.36 | 1.40 |
| Para2 | 5.16 | 4.46 | 0.70 |
| Para3 | 4.24 | 3.94 | 0.30 |
| Rhino | 5.16 | 2.59 | 2.57 |
| RSV | 2.38 | 3.99 | –1.60 |
The FARP performed well in reproducibility studies. Each analyte was detected consistently at the LoD and at three times the LoD. As would be expected, the analytes showed varying performance for samples diluted to 1/10 the LoD. The LoD determination studies also provided additional reproducibility data since each sample was detected in triplicate on a single day at the established LoD.
To challenge the analytical specificity of the FARP, cultured organisms were spiked in M4 media and interrogated on the FARP. The organisms were chosen based on their potential to be encountered in the nasopharyx. None of the tested organisms cross reacted on the FARP including Candida albicans, Streptococcus pneumoniae, Streptococcus pyogenes, Haemophilus influenzae type B, Staphylococcus aureus (methicillin resistant), Moraxella catarrhalis, Aspergillus sp., and Bordetella bronchiseptica.
DISCUSSION
Panel‐testing for respiratory pathogens is currently achieved by DFA in many laboratories; however, this is neither rapid nor sensitive for many of the suspected pathogens. As a result, industry efforts toward developing rapid, sensitive, panel‐based molecular diagnostics for respiratory pathogens have been aggressively pursued in recent years, and many are now commercially available 11. One such assay, the LuminexTM xTAG RVP, was shown to be a cost effective option for respiratory pathogen detection and its use resulted in shorter hospital stays overall in Canadian hospitals 13. This technology however is labor‐intensive and requires proficiency in molecular techniques as well as a dedicated amplicon room. For laboratories unable to practically adopt a complex multiplex assay, but interested in replacing viral DFA with molecular multiplex testing, the FARP is an attractive alternative. The major advantages to this system are the fully enclosed platform and the overall simplicity of the setup and performance. A separate molecular work area and molecular expertise are not required as a result of these attributes. Several recent studies have compared the FARP head‐to‐head with other real‐time PCR platforms for overall agreement, and one study performed a clinical comparison in real‐time for immunocompromised patients; however, this study provides a fully resolved comparison to DFA, the methodology likely to be displaced by FARP in most clinical laboratories adopting this technology 14, 15, 16, 17.
The FARP performed very well overall in this comparison study for all of the parameters tested. In fact, most of our agreement values were greater than those reported by Idaho Technology Inc 18. Consistent with the findings of Rand et al., less than 2% of our runs failed, despite running the machines almost 24 h at a time; making the platform very robust for testing in the urgent care setting 16. Several of these failures were due to malfunction of one FilmArray® machine that was replaced without further failures. After resolution of the discrepant subset, many analytes were 100% in concordance with DFA results. The majority of unexpected negatives were resolved as “true negatives” by LDT, misinterpreted DFAs, or clerical errors in the freezer database. For many of the discrepant samples, the FARP detected unexpected positives that were not previously detected by DFA. Most unexpected positive results were verified by LDT, making the FARP a more favorable assay for all analytes except Adeno. There were three unexpected positive analytes detected in the Rhino‐positive specimen cohort that are not reflected in the calculations in Tables 2 and 3 since the reference method for these other analytes (DFA) was negative during patient testing. Because the specimens were negative by DFA, they were reflexed to RVP testing; however, the RVP detected Adeno in two samples and 2009 H1N1 in one sample, none of which were detected by FARP. LDTs confirmed the presence of all three analytes. The concentration of virus in the 2009 H1N1 specimen was close to the calculated LoD of the FARP, which likely explains why this specimen was not detected.
Nine Adeno “false negatives” were positive by both DFA/LDT (n = 7) or RVP/LDT (n = 2). This suggests that the FARP may not detect each serotype of Adeno with equal sensitivity, because several of the specimens were clearly positive by culture/DFA and measured up to 2.5 × 1010 copies/ml by the LDT. Though the concentration of some samples fell below the FARP LoD for Adeno, several of the samples were within range of the established LoD, which further suggests that the target nucleic acid sequence was not amplified. In fact, the FARP does not detect all Adeno subtypes (personal communication, Idaho Technology Inc.), a limitation shared by the RVP 19. However, the agreement values for Adeno were consistent with those reported by Idaho Technology Inc. 18. This analyte is troubling in its current performance, and we have adopted an algorithm that includes reflex to Adeno‐DFA for all FARP negative specimens to allow for comprehensive detection of this pathogen. This approach would be appropriate for laboratories considering the FARP as a replacement for DFA. Alternatively, negative specimens could be submitted to a reference laboratory for LDT testing specifically for Adeno, though the turn‐around‐time is no longer favorable in this scenario.
Two unresolved specimens were excluded from the FluB cohort due to a lack of consensus (DFA = FluB, LDT = FluA, FARP = Negative). There was no evidence of mislabeling to account for this discrepant result. Six specimens (four Para2, one FluB, and one 2009 FluA H1N1) were also excluded due to lack of specimen for repeat culture/fluorescent antibody stain or real‐time PCR. For each of these discrepant results, the initial DFA result was not confirmed by the FARP. Thirteen samples showed lack of concordance between DFA or RVP and FARP after final resolution (Table 2). Two Para1 specimens were DFA and culture positive upon reanalysis, one Para3 specimen was determined negative by both DFA and LDT, and one 2009 FluA H1N1 specimen was detected by DFA and LDT. One unexpected RSV‐positive specimen was not confirmed when the pool and the individual specimens in the pool were tested by LDT. A single Rhino specimen that tested positive by RVP and LDT was negative by FARP.
Five FARP LoDs were within approximately 1.5 log of the LDTs (Table 3). The remaining analytes were greater than 1.5 log less sensitive than the corresponding LDTs, with Adeno, Rhino, and Bpert having LoD values over 2.5 log higher than the LDTs. Though these relative differences in LoD are striking, the assay was able to detect positives consistently in our cohort of clinical specimens with the exception of Adeno. Interestingly, the LoD values for RSV were comparably low for both LDT and FARP for viruses lacking polymorphisms in the targeted nucleic acid. Consistent with this low LoD, the FARP was previously shown to detect RSV samples that the RVP could not detect 16. This comparative LoD analysis has not previously been performed for the FARP, and this information provides adopting laboratories with information on the relative analytical sensitivity of the assay in measured units that are universally applicable in all molecular diagnostics.
Specimen pooling proved to be a robust application of the FARP for evaluation purposes. The run capacity of the FARP is approximately one pouch per 70 min; therefore, a significant time and cost investment would be required to individually evaluate all of the available analytes using robust sample sizes. We pooled analytes in order to achieve a high number of positive and negative specimens as well as test the overall capabilities of the multiplexing platform for “mixed infections.” Pooling was shown to have no deleterious effects on the FARPs ability to detect targeted analytes. As well, pooling did not have a negative impact on the assay's LoD for Bpert and Adeno. An inherent limitation of pooling clinical specimens for evaluation purposes is that failure to detect a target analyte may be masked by an unexpected positive of the same analyte from another member of that pool. For instance, if a pool includes FluA, FluB, RSV, Rhino, and hMPV, the FluB may be undetected in the intended patient positive; however, another patient could have an unexpected FluB co‐infection (previously undetected) that may be detected. In the final run output, this scenario would not be appreciated, and could skew agreement statistics. Due to our high numbers of discrepant samples, we ran multiple pools individually as single specimens for resolution purposes and only encountered this hypothetical scenario once with a Rhino specimen. Also, PCR inhibitors that may be present in a single specimen may be diluted in a pool, therefore specimens that would normally be missed may amplify, though this scenario was not documented.
As with any comparison study, the integrity of the specimens included is of utmost importance, however this is even more pronounced when pooling specimens. Though it is preferable to utilize patient specimens in order to maintain the appropriate matrix and clinically realistic analyte concentrations, commercially manufactured analytes may also be considered as a pooled specimen in order to achieve an appropriate cohort size. Commercially prepared samples are quality controlled, and as a result there should be little to no discrepant resolution required for the laboratory; an attractive attribute for laboratories that do not have access to individual LDTs. As well, rare analytes or analytes with no readily available reference method can be validated using commercially prepared analytes. We chose to use primary specimens from our pediatric population for this study to simulate the condition in which the assay would be normally operating, as this study also served as a validation of the FARP as a research use only assay (preceding FDA approval). As a result, we could not evaluate nine of the now FDA approved analytes including Coronavirus HKU1, Coronavirus NL63, Coronavirus 229E, Coronavirus OC43, Parainfluenza 4, Influenza A subtypes H1 seasonal and H3, C. pneumonia, and Mycoplasma pneumoniae. We also did not have confirmed specimens of Bocavirus (non‐FDA approved analyte), so it could not be evaluated, despite having identified this pathogen in several pools. It should also be noted that the pre‐market version of this platform provided identifications of “Rhinovirus”, however, the FDA approved product reports the analyte as “Rhinovirus/Enterovirus.”
The FARP is a powerful multiplex real‐time PCR platform that allows any moderately complex laboratory to perform molecular respiratory pathogen detection in a short amount of time. This assay could serve as a valuable tool for clinical decision making in terms of whether to admit high‐risk patients with respiratory symptoms presenting to an emergency department. More importantly, this report serves as a resource to laboratories interested in replacing DFA in favor of the FARP by demonstrating the overall performance of the various analytes with primarily clinical specimens, as well as providing justification for pooling analytes in order to balance time, expense, and robustness of sample size. Laboratories should consider a backup method for Adeno detection in light of the suboptimal performance of this analyte.
ACKNOWLEDGMENTS
MRC received an honorarium from Idaho Technology Inc. to present a subset of this data at an industry‐sponsored session at the 2011 Clinical Virology Symposium. For all other authors, there were no conflicts.
REFERENCES
- 1. Aguilar JC, Perez‐Brena MP, Garcia ML, Cruz N, Erdman DD, Echevarria JE. Detection and identification of human parainfluenza viruses 1, 2, 3, and 4 in clinical samples of pediatric patients by multiplex reverse transcription‐PCR . J Clin Microbiol 2000;38(3):1191–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balada‐Llasat JM, LaRue H, Kelly C, Rigali L, Pancholi P. Evaluation of commercial ResPlex II v2.0, MultiCode‐PLx, and xTAG respiratory viral panels for the diagnosis of respiratory viral infections in adults. J Clin Virol 2011;50(1):42–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liao RS, Tomalty LL, Majury A, Zoutman DE. Comparison of viral isolation and multiplex real‐time reverse transcription‐PCR for confirmation of respiratory syncytial virus and influenza virus detection by antigen immunoassays. J Clin Microbiol 2009;47(3):527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mahony J, Chong S, Merante F, et al. Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead‐based assay. J Clin Microbiol 2007;45(9):2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marshall DJ, Reisdorf E, Harms G, et al. Evaluation of a multiplexed PCR assay for detection of respiratory viral pathogens in a public health laboratory setting. J Clin Microbiol 2007;45(12):3875–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nolte FS, Marshall DJ, Rasberry C, et al. MultiCode‐PLx system for multiplexed detection of seventeen respiratory viruses. J Clin Microbiol 2007;45(9):2779–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schindera C, Kraemer AL, Regamey N, Aebi C, Gorgievski‐Hrisoho M, Barbani MT. Immunofluorescence versus xTAG multiplex PCR for the detection of respiratory picornavirus infections in children. J Clin Virol 2010;48(3):223–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evaluation of rapid influenza diagnostic tests for detection of novel influenza A (H1N1) Virus – United States, 2009. MMWR Morb Mortal Wkly Rep 2009;58(30):826–829. [PubMed] [Google Scholar]
- 9. Cruz AT, Cazacu AC, Greer JM, Demmler GJ. Rapid assays for the diagnosis of influenza A and B viruses in patients evaluated at a large tertiary care children's hospital during two consecutive winter seasons. J Clin Virol 2008;41(2):143–147. [DOI] [PubMed] [Google Scholar]
- 10. Cooper RJ, Hoffman JR, Bartlett JG, et al. Principles of appropriate antibiotic use for acute pharyngitis in adults: background. Ann Intern Med 2001;134(6):509–517. [DOI] [PubMed] [Google Scholar]
- 11. Mahony JB. Detection of respiratory viruses by molecular methods. Clin Microbiol Rev 2008;21(4):716–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hymas WC, Hillyard DR. Evaluation of nanogen MGB alert detection reagents in a multiplex real‐time PCR for influenza virus types A and B and respiratory syncytial virus. J Virol Methods 2009;156(1–2):124–128. [DOI] [PubMed] [Google Scholar]
- 13. Mahony JB, Blackhouse G, Babwah J, et al. Cost analysis of multiplex PCR testing for diagnosing respiratory virus infections. J Clin Microbiol 2009;47(9):2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Loeffelholz MJ, Pong DL, Pyles RB, et al. Comparison of the FilmArray Respiratory Panel and Prodesse Real‐Time PCR assays for detection of respiratory pathogens. J Clin Microbiol 2011;49(12):4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pierce VM, Elkan M, Leet M, McGowan KL, Hodinka RL. Comparison of the idaho technology FilmArray system to real‐time PCR for detection of respiratory pathogens in children. J Clin Microbiol 2011;50(2):364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rand KH, Rampersaud H, Houck HJ. Comparison of two multiplex methods for detection of respiratory viruses: FilmArray RP and xTAG RVP . J Clin Microbiol 2011;49(7):2449–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammond SP, Gagne LS, Stock SR, et al. Respiratory virus detection in immunocompromised patients with FilmArray respiratory panel compared to conventional methods. J Clin Microbiol 2012;50(10):3216–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Idaho Technology Incorporated . 2010. FilmArray(R) Respiratory Panel Information sheet (MRKT‐PRT‐0229–01).
- 19. Luminex Molecular Diagnostics Incorporated . 2008. xTAG TM RVP for use with Luminex (R) LX100/200 Systems.