

Abstract
Objectives
To develop a clinically significant and practical enzyme‐linked immunosorbent assay (ELISA) for the detection of MxA protein in human whole blood, a biological marker of viral infection.
Design and Methods
A sandwich ELISA suitable for the measurement of human MxA protein in whole blood was developed using mouse monoclonal antibodies (mAbs) raised against the GTP‐binding domain of human MxA protein. Prior to the assay, the whole blood sample was treated with special buffer to extract the MxA protein, improve its stability, and avoid interference from hemoglobin.
Results
This ELISA meets all the requirements for use in routine clinical assays, especially in terms of sensitivity (detection limit: 1.3 ng/ml whole blood), accuracy (recovery: 93.0–100.0%), and rapidity (<1.5 h). The present ELISA had a sensitivity of100% and a specificity of 100% for viral infection when compared to samples from healthy control and 87.1% and 90.9% when compared to samples from the bacterial infection group.
Conclusion
We have developed a new ELISA for measuring MxA protein in human whole blood using mAbs specific for the GTP‐binding domain of MxA. This ELISA has analytical performance enough for routine clinical assay and can be used in detecting the possibility of viral infection. J. Clin. Lab. Anal. 26:174‐183, 2012. © 2012 Wiley Periodicals, Inc.
Keywords: bacterial infection; interferon α, high sensitivity; high stability; rapid assay
INTRODUCTION
Interferons are produced in response to viral infections and contribute to host defense by establishing an antiviral state in target cells 1, 2. Directly measuring the interferon levels in plasma is technically difficult since interferon is rapidly turned over in vivo 3. Furthermore, the circulating level of interferon does not reflect the biologically active interferon effects on target cells 2, 4.
The antiviral activity of the interferon is mediated by the induction of unique proteins and more than 30 different intracellular proteins are known to be induced by interferon 5. By measuring interferon‐induced proteins, the presence of biologically active interferons can be detected more consistently 6. Among the interferon‐induced proteins, MxA protein is remarkable for its high levels of expression that may reach 1% of the total cytosolic protein 7 and that can be specifically induced in a dose‐dependent manner by interferons both in vivo and in vitro 7, 8. MxA protein is rapidly induced after interferon‐treatment (1–2 h), detectable within 2–4 h after interferon exposure, both in vitro and in vivo, and reaches a maximum level within 36 h 7, 8, 9, 10. Moreover, cellular induction of MxA protein is not subject to feedback inhibition 8. Since interferons are the sole inducers of MxA protein 11, levels of MxA protein reflect the biologically active interferon present during host cell defense mechanisms. Therefore, elevated levels of MxA protein could be an indicator of endogenous interferon production mediated by an unknown viral activation 4 and so, the MxA protein levels could be used as a general marker of viral infection. It has been reported that MxA protein expression in peripheral blood mononuclear cells is a highly specific and reliable marker for interferon bioactivity 12 and that elevated leukocyte MxA protein levels reflect endogenous interferon production 13. Accordingly, the rapid determination of MxA protein from patient samples could be considered a useful test for the diagnosis of viral disease 14, 15.
Recent advances in the understanding of the structure and function of human MxA protein have revealed the presence of a GTP‐binding domain and GTPase activity, which are now regarded as important factors in relation to its antiviral activity against a wide variety of viruses 16, 17, 18, 19, 20. Detailed elucidation of the relationship between the structure and function of MxA protein may make the accurate diagnosis of viral infectious conditions possible.
Here, we report the development of a clinically useful enzyme‐linked immunosorbent assay (ELISA) for the detection of human MxA protein in whole blood, using mouse mAb that recognize a GTP‐binding domain, which meet the requirements for a routine clinical assay.
MATERIALS, SAMPLES, AND METHODS
Materials and Chemicals
Reagents were obtained from the following sources: bovine serum albumin (BSA) (Miles, Inc., Kankakee, IL); 3,3′,5,5′‐tetramethylbenzidine (TMBZ) solution (TMBlue, TSI Co., Milford, MA); oleamide diethanolamide (NOF Corporation, Tokyo, Japan), N‐succinimidyl‐6‐maleimidohexanoate and 3‐((3‐Cholamidopropyl)dimethylammonio] propanesulfonate) (Dojindo Laboratories, Kumamoto, Japan); horseradish peroxidase (HRP) (Toyobo Co., Ltd., Osaka, Japan); BLOCK ACE® (DS pharma Biomedical Co., Ltd., Osaka, Japan); all other reagents were of analytical grade.
Clinical Samples
Whole blood samples from children who visited the outpatient clinic of Toyama University Hospital (Toyama, Japan) for acute onset fever were collected. Seventeen healthy children who had been without symptoms of infectious diseases during the previous two weeks served as controls. The study was conducted according to the ethical standards of the University of Toyama, which require informed consent from the parents of each subject.
Children with acute onset fever were subdivided into three groups according to the final diagnoses: (1) etiologically diagnosed viral infection, (2) clinically diagnosed viral infection, and (3) bacterial infection. Individuals with etiologically diagnosed viral infection were identified by positive results using the following virus‐detection kits: Adenovirus GSA (Meridian Bioscience, Cincinnati, OH) for adenovirus, ROTA‐ADENO DRY (Orion Diagnostica, Espoo, Finland) for rotavirus, Capilia Flu A, B (Nippon Becton Dickinson, Tokyo, Japan) for influenza viruses, and Directigen RSV (Becton Dickinson, Cockeyville, MD) for respiratory syncytial virus. Children with mild symptoms of upper respiratory infection or gastroenteritis were classified into the clinically diagnosed viral infection group. Their symptoms resolved spontaneously even though antibiotics were not used. Bacterial infections were diagnosed based on positive culture results and all the child patients with bacterial infections were treated successfully with antibiotics.
Whole blood samples were freshly prepared from EDTA anticoagulated blood. When whole blood was used for the ELISA, the sample was diluted immediately with the dilution buffer (0.1 mol/l Tris‐HCl buffer [pH8.5] containing 2.5% [w/v] 3‐[(3‐Cholamidopropyl) dimethylammonio] propanesulfonate, 1.2% [w/v] oleamide diethanolamide, 0.1 mol/l NaCl, 4.0% [w/v] BLOCK ACE®, 0.1% [w/v] BSA, and 0.1% [w/v] NaN3), stored at 4°C and was measured within 24 h after the collection. When the sample was stored for a long time, the diluted sample was stored at –80°C and was thawed just before ELISA assay.
Serum samples were prepared from blood allowed to clot at 37°C for 30 min, by centrifugation at 3,000 rpm for 10 min.
Preparation of MxA Protein
An expression vector pET‐14b (Merck Biosciences, Darmstadt, Germany) containing full‐length MxA protein‐encoding cDNA was introduced into Escherichia coli (E. coli) cells 21, and were cultured at 37 °C for 4 h in 200 ml of LB medium containing 50 μg/ml of ampicillin, followed by the addition of 0.4 mmol/l of isopropylthiogalactoside and subsequently cultured at 37°C for 2 h.
E. coli was collected from 200 ml of the cultured broth by centrifugation at 3,000 rpm for 15 min, washed with phosphate‐buffered saline (PBS), and was suspended in 20 mmol/l Tris‐HCl buffer (pH7.9) containing 5 mmol/l imidazole and 0.5 mol/l NaCl, and then lysed by ultrasonic treatment. After the suspension was centrifuged, the supernatant was discarded, and the residue was added to the binding buffer (20 mmol/l Tris‐HCl buffer (pH7.9) containing 6 mol/l urea, 5 mmol/l imidazole, and 0.5 mol/l NaCl) and Ni‐NTA His‐Bind resin (EMD Bioscience, Inc., Milwaukee, WI) to purify full‐length human MxA protein with a N‐terminal His 6 tag from the E. coli inclusion bodies. The suspension was mixed by gentle rolling at 4°C for 2 h to bind MxA protein on the resin and the resin was recovered by centrifuge. The recovered resin was washed with the binding buffer and was eluted using 20 mmol/ml Tris‐HCl buffer (pH7.9) containing 6 mol/l urea, 1 mol/l imidazole, and 0.5 mol/l NaCl to produce crude MxA protein. Using this method, 1.4 mg of MxA protein was recovered from 250 ml of the E. coli culture broth.
Immunization, Cell Fusion, and Cloning
BALB/C mice (six weeks old, male) were immunized with the recombinant human MxA protein. The monoclonal hybridoma cells were made by fusion with spleen cells derived from the immunized mouse and 2 × 107 P3×63‐Ag.8.U1 cells, as described previously 22. Cell culture supernatants were tested for antibody activity by ELISA using a MxA protein‐coated plate as described previously 22. Cells from positive wells were further selected on the basis of specificity and subclass, and were cloned twice by limiting dilution. Stable hybridoma clones were propagated as ascites tumors in BALB/C mice. Antibodies from monoclonal hybridoma cells were purified from the ascites by affinity chromatography using Protein‐A Sepharose fast flow (GE healthscience Japan, Tokyo, Japan) according to the manufacturer's instructions.
The subclass of the monoclonal antibody was determined by a Zymed mouse mAb isotyping kit (Zymed Laboratories, Inc., San Francisco, CA). The dissociation constant of the antibody was determined by the method of Djavadi‐Ohaniance et al. 23.
Western Blotting
One microgram of MxA protein derived from E. coli was fractionated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and then blotted on a polyvinylidene difluoride (PVDF) membrane. After blocking with a BSA solution, the membrane was incubated with 10 μg/ml of each of the purified anti‐human MxA protein mAb for 2 h at room temperature. After thorough washing with PBS containing 0.1% (w/v) Tween 20, a HRP‐labeled anti‐mouse immunoglobulin antibody (DAKO Japan, Kyoto, Japan) was incubated at room temperature for 1 h. After thorough washing, color development was performed using the HAP‐Color Development Reagent substrate solution (DAKO Japan).
Immnocytostaining
A suspension of human glioma T98G (ATCC CRL 1690) cells was dispensed in 500 μl portions to separate wells of an eight‐well chamber slide (2 × 104 cells/well), stimulated with interferon α (1,000 U/ml) and cultured at 37°C. After washing with PBS, 500 μl of freshly prepared 4% (v/v) paraformaldehyde was added into each well and the cells were fixed at room temperature for 30 min.
The fixed cells were treated with 0.2% (v/v) Triton X‐100 for 5 min to permeablize the cell membrane, incubated with 10% (v/v) normal horse serum for 30 min for blocking, and then with 10 μg/ml of each of the purified anti‐human MxA protein mAb for 30 min at room temperature. After washing with PBS, 200 μl of fluorescein isothiocyanate (FITC)‐labeled anti‐mouse immunoglobulin antibody (Wako Pure Chemical Industries, Osaka, Japan) was added into each well at room temperature for 30 min in the dark. After thorough washing with PBS, the cells were sealed with glycerol on a glass slide and observed under a fluorescence microscope.
T98G infected with influenza virus were obtained from the Tokyo Institute of Technology and cultured at 37°C. The T98G were fixed with 4% (v/v) paraformaldehyde and were used for immunocytostaining using the same method as described above.
Determination of the mAb‐Binding Domain
The MxA protein‐encoding DNA was digested with restriction enzymes BstXI, Pstl, Nael, and BamHI following established procedures, and was subjected to in vitro transcription in the presence of T7 RNA polymerase to synthesize several mRNA samples having different lengths. Next, in vitro translation was carried out using these mRNA samples in the presence of 35S‐methionine to synthesize 35S‐methionine‐labeled protein fragments that were successively shortened from the C‐terminal end. The in vitro translation was carried out using reticulocyte lysate (#L416A, E. Y. Labo Inc., San mateo, CA) in accordance with the manufactured instruction manual. As a result, labeled protein fragments corresponding to amino acids 10–662, amino acids 10–468, amino acids 10–297 and amino acids 10–220, were synthesized. Each of these labeled protein fragments were allowed to react with each of the anti‐human MxA protein mAb in a reaction buffer (50 mmol/l Tris‐HCl buffer (pH 8.0) containing 1% (v/v) NP‐40 and 0.2% (w/v) BSA) at 4°C for 1 h. The reaction solution was mixed with 40 μl of secondary antibody‐labeled beads (AG‐005, E. Y. Labo Inc.) and the reaction incubated at 4°C for 1 h with shaking for immunoprecipitation. The reaction mixture was subjected to centrifugation at 10,000 × g at 4°C for 15 s and the precipitate obtained was separated by SDS‐PAGE and the molecular weight of the precipitated protein was determined by autoradiography.
ELISA System
Sensitization of plates
KM1135‐coated polystyrene 96‐well microtiter plates (Maxisorp Immunoplates, NUNC, Rosklide, Denmark) were prepared as described previously 24. The plates were sealed in an aluminum‐coated pack with a drying agent (Hi‐sheet dry, Marutani chemical Plant & Engineering Co., Ltd., Tokyo, Japan) and stored at 4°C until use. Under these storage conditions, the sensitized plates were stable for at least 1 year.
Antibody‐HRP conjugate
The KM1124‐HRP conjugate was prepared as described previously 24. The average number of HRP molecules introduced per IgG molecule was 4.3.
The conjugate produced was diluted to 50 ng/ml with 50 mmol/l Bis‐Tris buffer (pH 7.0) containing 0.1% (w/v) BSA, 50 mmol/l NaCl, 0.01% (w/v) 4‐aminoantipyrine, 0.1% (v/v) polyoxyethylene polyoxypropylene glycol, 0.035% (v/v) Proclin 300® (Sigma‐Aldrich Japan Co, Tokyo, Japan), and 100 mg/l mouse IgG (Roche Diagnostics GmbH, Mannheim, Germany), divided into 11 ml each in clear bottles and stored at 4°C until use. Under these storage conditions, the conjugate was stable for at least 1 year.
Standard preparation
For the standard in the ELISA, a recombinant MxA protein was prepared from E. coli cells transformed with an expression vector containing a MxA protein‐encoding cDNA as described above. The recombinant MxA protein was furthermore purified by gel filtration column chromatography using Superose 12HR10/30 (GE healthscience Japan) eluted with 20 mmol/ml Tris‐HCl buffer (pH7.9) containing 6 mol/l urea, 0.5 mmol/l imidazole, 0.1 mmol/l dithiothreitol, and 0.5 mol/l NaCl at 0.5 ml/min. The highly purified MxA protein was confirmed to be monomeric and homogeneous by high performance liquid chromatography (HPLC) and SDS‐PAGE, respectively. Amino acid sequence analysis confirmed the highly purified protein as human MxA protein.
The protein concentration of the MxA protein was determined by the Bradford method 25 using the Bio‐Rad DC Protein Assay Kit (Bio‐Rad Laboratories, CA) with BSA as the standard, and was found to be 0.257 mg/ml.
The MxA protein solution was diluted to 24 ng/ml with the dilution buffer, divided into 1 ml each in a brown vial and lyophilized at –50°C for 48 h. After displacement of the atmospheric gases in the vial by N2, the vial was sealed and the lyophilized MxA protein was stored at 4°C until use. When used in the ELISA, 1 ml of distilled water was added to the vial to resuspend the MxA protein, and the concentration of this MxA protein was determined to be 24 ng/ml.
To prepare the standards, 24 ng/mL of MxA protein was serially diluted with the dilution buffer and 0.38, 0.75, 1.50, 3.00, 6.00, 12.00, and 24.00 ng/ml of standards were prepared. The results of the ELISA were expressed in ng/ml of whole blood as determined by the standards.
Enzyme‐linked immunosorbent assay
Twenty‐five microliter of whole blood was diluted with 225 μl of the dilution buffer for cell lysis. One hundred microliter of the diluted sample or standard was added to each well of the antibody‐coated plates and the plate was then incubated for 30 min at room temperature. After washing the plate five times with the washing buffer (PBS containing 0.05% [v/v] Tween‐20), 100 μl of HRP‐labeled KM1124 conjugate was added to each well, and the plate was incubated for 30 min at room temperature. After washing the plate five times with the washing buffer, 100 μl of TMBZ solution was added to each well, and the plate was then incubated for 10 min at room temperature. To stop the enzyme reaction, 50 μl of 0.5 mol/l H2SO4 was added, and the absorbance at 450 nm was measured with an MTP‐120 plate reader (Corona Electric Co. Ltd., Tokyo, Japan).
Interference Study
Interference by lipids, direct bilirubin, indirect bilirubin, hemoglobin, and various anticoagulants was examined by the addition of the test metabolite to aliquots of whole blood. Metabolites and chemicals to be tested for interference were obtained from commercial sources at the highest purity available. Interference by lipids was tested using Intralipid® (Kabivitrum, Inc., California). Interference by heterophilic antibodies was tested using HAMA Sera® (Roche Diagnostics GmbH).
Statistical Analysis
Data are expressed as mean ± SD or median. Differences were examined by the Student's t‐test or the Mann–Whitney U test. Receiver operating characteristic (ROC) analyses were done using the method of Hanley and McNeil 26. Computer analysis was performed with the Statcel package (OMS, Ltd., Saitama, Japan).
RESULTS
Mouse Hybridomas
Several hundred mouse hybridomas were screened by ELISA for the presence of antibodies against human recombinant MxA protein. Consequently, 14 mAb‐producing lines, designated as KM1122∼1135 were selected for expansion and used to produce ascites. mAb purified from ascites were used in all subsequent experiments to detect MxA protein employing the ELISA.
Characterization of mAb
As shown in Table 1, western blot analysis demonstrated all the mAb selected recognized the 76‐kDa MxA protein. In addition, KM 1123, 1128, 1130–1135 also reacted with a 30‐kDa band, a degradation product of the MxA protein. Immunocytostaining study said that KM1124, KM1126, KM 1132, and KM1135 could recognize the MxA protein from interferon α‐stimulated cells but not unstimulated cells and additionally, KM1124, KM1132, and 1135 also reacted the MxA protein from the influenza virus‐infected cells. The binding site of each mAb was determined based on the measured molecular weight. As a result, it was revealed that the anti‐human MxA protein mAb KM1126 and KM1135 have a binding site at amino acids 10–220, KM1124 at amino acids 220–297, and KM1132 at amino acids 468–662, counting from the amino terminus of the MxA protein.
Table 1.
Properties of Anti‐MxA Monoclonal Antibodies
| Western blotting | Immunocytostaining | Immunoprecipitation | ||||||||
| KM No. | Type | 76 kDa | 30 kDa | IFNα | Inf | 10–662 | 10–468 | 10–297 | 10–220 | Kd |
| 1122 | IgG1 | + | − | − | ND | ND | ND | ND | ND | |
| 1123 | IgG1 | + | + | − | ND | ND | ND | ND | ND | ND |
| 1124 | IgG1 | + | − | + | + | + | + | + | − | 1.9 × 10−6 |
| 1125 | IgG1 | + | − | − | ND | ND | ND | ND | ND | ND |
| 1126 | IgG1 | + | − | + | − | + | + | + | + | 1.8 × 10−6 |
| 1127 | IgG1 | + | − | − | ND | ND | ND | ND | ND | ND |
| 1128 | IgG2a | + | + | − | ND | ND | ND | ND | ND | ND |
| 1129 | IgG1 | + | − | ND | ND | ND | ND | ND | ND | |
| 1130 | IgG2a | + | + | − | ND | ND | ND | ND | ND | |
| 1131 | IgG1 | + | + | − | ND | ND | ND | ND | ND | ND |
| 1132 | IgG2a | + | + | + | + | + | − | − | – | ND |
| 1133 | IgG1 | + | + | − | ND | ND | ND | ND | ND | ND |
| 1134 | IgG2a | + | + | − | ND | ND | ND | ND | ND | ND |
| 1135 | IgG1 | + | + | + | + | + | + | + | + | 1.8 × 10−7 |
ND, not done; IFNα, interferon α‐stimulated cells; inf, influenza virus‐infected cells.
+: positive result; −: negative result.
KM1124 and KM1135 were chosen for ELISA because of its binding site and reactivity against the cell induced by interferon α and viral infection. These antibodies had a suitable dissociation constant (Kd) to develop the ELISA system for measurement of MxA protein in whole blood.
ELISA for Detection of MxA Protein
A sandwich ELISA for whole blood MxA protein was performed with a 96‐well microtiter plate as the solid phase, KM1135 antibody as the capture antibody, KM1124‐HRP conjugate as the second enzyme‐labeled antibody, and TMBZ solution as the enzyme substrate. Figure 1 is a typical calibration curve generated by plotting the absorbance at 450 nm vs. the concentration of each standard.
Figure 1.
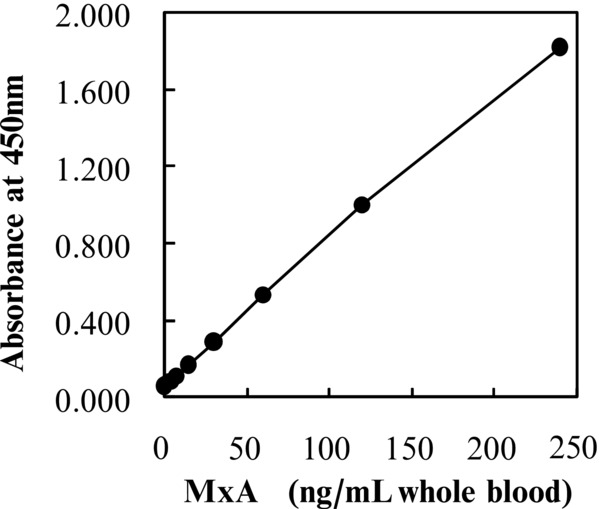
Standard curve for the MxA protein‐specific ELISA. Each point represents the mean of two measurements.
Specificity
To confirm that the MxA protein‐specific ELISA was clinically useful, T98G cells were stimulated with interferon α, and the MxA protein in the cell lysate was measured between 0 and 50 h. By immunocytostaining with fluorescent‐labeled antibody, MxA protein was confirmed in the cytoplasm of interferon α‐stimulated T98G cells (Fig. 2A, B, and C). This ELISA detected MxA protein in the cell lysate concomitant with the appearance of MxA protein in the cytoplasm of interferon α‐stimulated cells (Fig. 2D).
Figure 2.
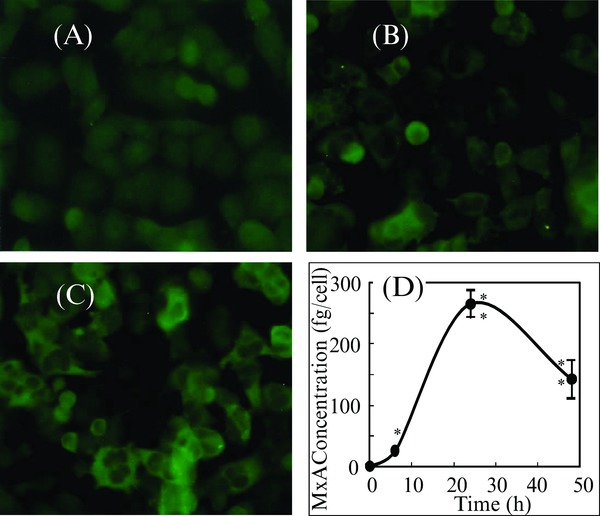
Immunocytostaining and expression of MxA protein in interferon α‐stimulated 98TG cells. (A) Immunocytostaining of MxA protein in 98TG cells before stimulation with interferon α, (B) after 2‐h stimulation, (C) after 6‐h stimulation, (D) concentration of MxA protein in 98TG cells stimulated with interferon α at 0 h. Each point represents the mean ± SD of three cultures. * and **: Significantly different from the value at 0 h, P < 0.05 and P < 0.001, respectively.
Dilution Analysis
Whole blood samples serially diluted gave results close to linearity (Fig. 3), indicating that the assay was quantitative and confirming parallelism between the standard and whole blood.
Figure 3.

Dilution curve for MxA protein in whole blood.
Interference
Whole blood samples containing low (47 ng/ml), medium (105 ng/ml), and high (193 ng/ml) MxA protein concentrations were supplemented with potential interfering agents at various concentrations. There was no substantial interference from lipids up to 5 g/l (in terms of total glycerol), from hemoglobin up to 15 g/l, from direct and indirect bilirubin up to 200 mg/l, heparin up to 400 mg/l, NaF up to 4 g/l, EDTA up to 2 g/l, and sodium citrate up to 10 g/l. Human anti‐mouse antibody positive serum was serially added to normal human whole blood and the whole blood was used for testing for interference of heterophilic antibodies. No false‐positive signals were observed.
Detection Limit and Practical Assay Range of MxA Protein in Human Whole Blood
The detection limit of MxA protein in human whole blood by the ELISA was determined. Human whole blood containing MxA protein was serially diluted and used for testing. A significant difference of each serially diluted sample, compared to 0 ng/ml was confirmed by a Student's t‐test (n = 5, P < 0.001). Figure 4 shows that 1.3 ng/ml of MxA protein in whole blood could be detected within 90 min by this assay. With the standard as the test sample, 0.13 ng/ml could be detected without pretreatment with the dilution buffer. The working range of the assay was established by calculating the coefficients of variation (CV) of each standard (range between 1.3 and 240 ng/ml) in whole blood was <10% (Fig. 4).
Figure 4.
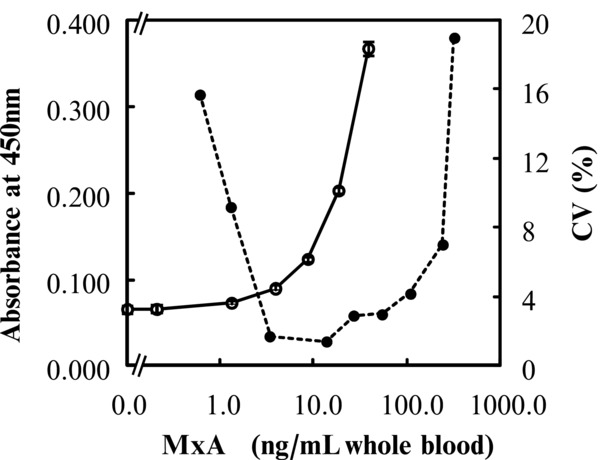
Detection limit and practical assay range of MxA protein by ELISA. Each point of the detection limit represents the mean ± 2 SD of five measurements (○). Each point of the practical assay range represents the coefficients of variation (CV) value of six measurements (●).
Assay Variation
The assay variation in the ELISA was examined using whole blood samples that contained MxA protein at three different concentrations (approximately 47 ng/ml, 105 ng/ml, and 193 ng/ml). The CV within assay and between assay were 2.0–5.5% (n = 12) and 2.2–7.2% (n = 5), respectively. To determine the analytical recovery, one volume of recombinant MxA protein (27 ng/ml and 114 ng/ml) was mixed with nine volumes of the whole blood samples containing 17, 37, and 81 ng/ml of MxA protein. Recoveries of MxA protein ranged from 93.0 to 95.0% (27 ng/ml, n = 2) and from 96.0 to 98.0% (114 ng/ml, n = 2), respectively.
Stability of MxA Protein
In this study, we used whole blood samples that had been freshly prepared. Figure 5 shows the stability of MxA protein in whole blood samples. Under refrigerated conditions, the MxA protein concentration in whole blood tended to decrease gradually. When untreated whole blood was frozen and then thawed, the MxA protein value in whole blood decreased beyond the limits of acceptability. On the other hand, when the whole blood was treated with the dilution buffer, MxA protein was stable even under refrigerated conditions. Additionally, MxA protein in diluted whole blood at –80°C was very stable for at least 3 years and freezing and thawing had no effects on the MxA protein value in the dilution buffer. Therefore, we suggest that samples for MxA protein assay are incubated with the dilution buffer immediately and then used for analysis or frozen at –80°C until needed.
Figure 5.
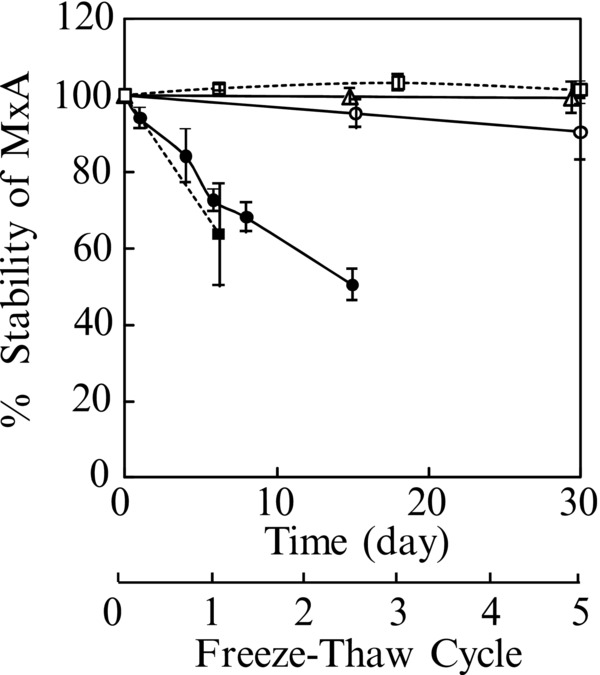
Stability of MxA protein in whole blood.
–●–, stored at 4°C; –○– , treated with the dilution buffer and stored at 4°C; △ , treated with the dilution buffer and then stored at –80°C; −‐−▪−‐−, frozen and thawed; −‐−□−‐−, treated with the dilution buffer and then frozen and thawed.
Clinical Samples
The clinical performance of the present ELISA was assessed by assaying MxA protein in the whole blood of healthy controls and patients with viral infection and bacterial infection. Table 2 shows a summary of the clinical characteristics of the subjects. Significant differences in MxA protein levels, white blood cells (WBC), and C‐reactive protein (CRP) were observed in the patients with viral infection compared to the healthy controls or the patients with bacterial infection. In all the groups, no significant correlations were observed between MxA protein and CRP.
Table 2.
Clinical Characteristics of the Child Patients with Infection and the Healthy Control
| Viral infection | Bacterial infection | Healthy control | ||||
|---|---|---|---|---|---|---|
| Etiologically diagnosed | Clinically diagnosed | Total | ||||
| Number | 17 | 14 | 31 | 11 | 18 | |
| Age | (Range) | 0‐10 | 0‐6 | 0‐10 | 0‐6 | 5‐11 |
| (Mean±SD) | 2.35±2.99a | 2.71±2.27a | 2.52±2.66a | 2.36±1.96a | 7.50±1.75 | |
| Gender | (Male/Female) | 6/11 | 8/6 | 14/17 | 8/3 | 12/6 |
| MxA | (Median) | 112.7c,d | 91.7c,d | 110.0c,d | 10.6a | 2.0 |
| WBC | (Mean±SD) | 10190±7175b | 7270±1825b | 8871±5575b | 16352±10032 | 6599±1556b |
| CRP | (Median) | 0.59b | 0.49b | 0.52b | 7.42 | 0.08b |
| Final Diagnoses | RSV10 | Gastroenteritis4 | URI5 | |||
| Adenovirus3 | URI10 | UTI3 | ||||
| Influenza virus A/B3 | Sepsis2 | |||||
| Rotavirus1 | Pneumonia1 | |||||
Significantly different from the value of healthy control, P < 0.05.
Significantly different from the value of bacterial infection, P < 0.05.
Significantly different from the value of healthy control, P < 0.0001.
Significantly different from the value of bacterial infection, P < 0.0001.
URI, upper respiratory infection; UTI, urinary tract infection.
Figure 6 shows the distribution of MxA protein values in the whole blood of the patients with viral infection, the patients with bacterial infection, and the healthy controls. ROC analysis was applied to the detection of the cut‐off limits and then, clinical sensitivities and specificities were calculated at the cut‐off limits. The MxA‐specific ELISA had a sensitivity of 100% and a specificity of 100% for viral infection against healthy control and at a cut‐off point of 36.7ng/ml, 87.1% and 90.9% for viral infection against bacterial infection, respectively.
Figure 6.
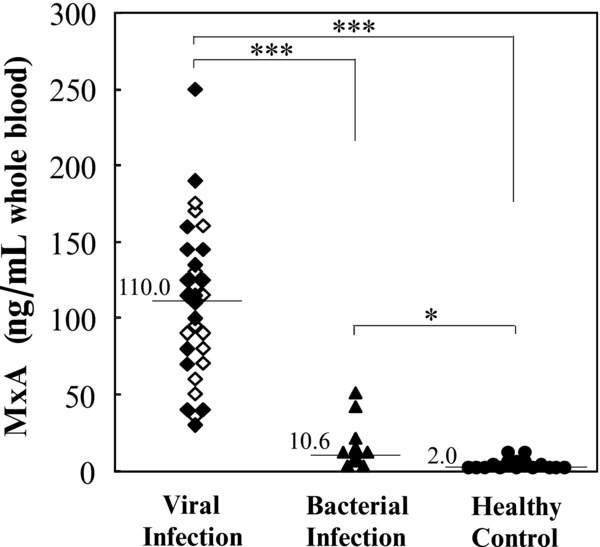
MxA protein levels in whole blood of the healthy controls and patients with infection. ◆, patients with etiologically diagnosed viral infection; ◇, Patients with clinically diagnosed viral infection; ▲, patients with bacterial infection; ●, healthy controls. Horizontal bars and values indicate the median for each group. *P < 0.05; ***P < 0.001.
DISCUSSION
The goal of this study was to develop an ELISA that met the requirements of a routine clinical assay for the measurement of MxA proteins in human whole blood.
The present ELISA is a sandwich‐type assay developed using the mouse mAb KM1135 and KM1124. KM1135 recognized an epitope corresponding to the amino acids sequence of 10–220 of the human MxA protein GTP‐binding domain and was used as the capture antibody. HRP‐labeled KM1124 that recognized an epitope corresponding to the amino acids sequence of 221–297 was used as the secondary antibody. We selected mAb that reacted with the MxA protein GTP‐binding domain, localized to the N‐terminus and displayed an antivirus activity, all of which determined the specificity of the ELISA. We also checked that the mAb in this ELISA detected MxA protein in cells that was stimulated not only by interferon α but also influenza virus.
A rapid determination of MxA protein could be considered as a useful test in the diagnosis of viral disease. Towbin et al. prepared five mAb using recombinant MxA protein and they divided them into those that recognized an epitope corresponding to amino acids 1–429, and those which recognized another epitope corresponding to amino acids 430–662. They also developed a simple two‐site immunometric enzyme assay with the mAb to measure MxA protein directly in blood 9. This method is simple and can measure intracellular MxA protein in whole blood, but took 20 h to perform the analysis. Oh et al. also developed a simple immunochemiluminescent assay for MxA protein 4 with the same mAb. They devised a method to reduce the time taken for analysis and to eliminate proteolytic degradation of MxA protein in whole blood lysate. However, the detection limit was 20 ng/ml in whole blood, which was insufficient to distinguish patients with viral infection from those with bacterial infection in the clinical setting.
Since MxA protein is highly susceptible to proteolysis, development of an assay system should consider eliminating any possibility of proteolytic degradation. Chieux et al. used denaturing media and heat‐shock treatment to eliminate proteolytic degradation of MxA protein in blood lysate 6. In the present ELISA, we used the whole blood as a sample to measure MxA protein with the intention of developing a clinically significant and practical ELISA. As MxA protein is induced in white blood cells, the cells must be lysed before measuring MxA protein. Normally, NP‐40 is used for the extraction of cytoplasmic proteins from cells. We used oleamide diethanolamide and 3‐[(3‐Cholamidopropyl) dimethylammonio] propanesulfonate in the dilution buffer to lyse the cells and to elute MxA protein from the cells. We checked the stability of MxA protein in various conditions by the ELISA, which detected the MxA protein GTP‐binding domain of human MxA protein and found that our dilution buffer has a powerful effect on stability of the MxA protein. By using the dilution buffer for pretreatment of the whole blood, the stability of MxA protein was remarkable without the need for freezing, denaturing processes, or heat‐shock treatment 6. Moreover, as freezing and thawing had also no effect on the MxA protein concentration in the dilution buffer, samples for MxA protein measurement by the ELISA can be stored for long term at –80°C. When the whole blood was used for measuring MxA protein, high concentrations of hemoglobin were present in the sample and could interfere with the immunoreaction. Pretreatment of the whole blood with the dilution buffer also removed the interference of the hemoglobin. Additionally, the dilution buffer reduced the influence of variations in assay temperature on the assay results (data not shown). As long as we pretreated the whole blood with the dilution buffer, we could accurately measured MxA protein value in whole blood without proteolytic degradation, influence of assay condition, or interference from hemoglobin.
In this report, we demonstrated that the measurement of MxA protein in whole blood by the ELISA may be useful in the diagnosis of acute viral diseases. This assay is highly sensitive with a detection limit of 1.3 ng/ml MxA in whole blood and at 0.13 ng/ml in solution, which is sufficient to distinguish patients with viral infection from those with bacterial infections in the clinical setting. Due to the high sensitivity of the ELISA for measuring MxA protein, a sample could be pretreated and diluted 1/10 with the dilution buffer before assay. This pretreatment may remove interference from blood cell‐based and plasma‐based proteins and make it suitable for the routine measurement of MxA protein in human whole blood. The reproducibility and the accuracy of the ELISA are sufficient to measure MxA protein in whole blood. A total CV value of between 2.0 and 7.2% suggests this assay is very reproducible. In addition, the accuracy, as determined by the recovery of added MxA protein, is high enough for the measurement of clinical samples. This ELISA has a wide range of detection, corresponding to a clinically useful range of 1.3–240 ng/ml in whole blood. Highly purified recombinant human MxA protein was used as a standard and the use of this standard produced quantitative results in whole blood sample assays (Fig. 3).
As expected, the level of MxA protein detected in whole blood derived from patients with viral infection was significantly higher than that from healthy controls and from patients with a bacterial infection (Table 2, Fig. 6). At a cut‐off point of 36.7 ng/ml, the present ELISA had a sensitivity of 87.1% and a specificity of 90.9% for viral infection against bacterial infection and the patients with viral infection were sharply distinguished from the healthy controls, that is, 100% of both a sensitivity and a specificity. The peripheral WBC count and serum CRP concentration have been used as markers to distinguish bacterial infection and viral infection. We observed that the peripheral WBC count and serum CRP concentration values were higher in the bacterial infection group compared to the viral infection group. However, the sensitivity and specificity of the MxA protein‐specific ELISA was far higher than that of other markers tested, suggesting that it would be useful for the discrimination of patients with a viral infection.
It has been reported that many subjects received antibiotic treatment, despite their infections being viral in origin, and this inappropriate use of antibiotics is considered to be the main cause of the spread of multidrug‐resistant bacteria 27. Several criteria have been proposed for identifying patients with bacterial infection 28, 29, 30. The present study shows that a whole blood assay for the MxA protein by the ELISA is useful for identifying the subjects whose illness is due to a viral infection. This ELISA might contribute toward a reduction in the number of unnecessary antibiotic prescriptions. However, the clinical study in this report was to assess the clinical performance of the MxA protein‐specific ELISA. Further studies are necessary to establish the clinical use of the whole blood assay for MxA protein. In order to make the MxA assay more easily accessible in a clinical setting, we are pursuing the development of a rapid detection kit for MxA protein, using the same mAb and immunochromatography technology. This can detect MxA protein in whole blood within 15 min without the need for special equipment and we hope the kit will soon to be ready for clinical use.
In conclusion, we have developed a new assay, suitable for the measurement of MxA protein in human whole blood, which meets the requirements for routine clinical assay. The whole blood assay for MxA protein by the ELISA is useful for the identification of subjects who have a viral infection.
REFERENCES
- 1. Taylor JL, Grossberg SE. Recent progress on interferon research: Molecular mechanisms of regulation, action, and virus circumvention. Virus Res 1990;15:1–26. [DOI] [PubMed] [Google Scholar]
- 2. Belardelli F. Role of interferon and other cytokines in the regulation of the immune response. APMIS 1995;103:161–179. [DOI] [PubMed] [Google Scholar]
- 3. Kuzrrock R, Rosenblum MG, Sherwin AA, et al. Pharmacokinetics, single‐dose tolerance, and biological activity of recombinant interferon‐gamma in cancer patients. Cancer Res 1985;45:2866–2872. [PubMed] [Google Scholar]
- 4. Oh S‐k, Luhowskyj S, Witt P, et al. Quantitation of interferon‐induced Mx protein in whole blood lysates by an immunochemiluminescent assay: Elimination of protease activity of cell lysates in toto. J Immunol Methods 1994;176:79–91. [DOI] [PubMed] [Google Scholar]
- 5. Sen CS, Lengyel P. A bird's eye view of its biochemistry. J Biol Chem 1992;267:5017–5020. [PubMed] [Google Scholar]
- 6. Chieux V, Hober D, Harvey J, et al. The MxA protein level in whole blood lysates of patients with various viral infections. J Virol Mehods 1998;70:183–191. [DOI] [PubMed] [Google Scholar]
- 7. Horisberger MA. Interferon‐induced human protein MxA is a GTPase which binds transiently to cellular proteins. Virology 1992;66:4705–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. von Wussow P, Jakschies D, Hochkeppel KH, Fibisch C, Penner L, Deicher H. The human intracellular Mx‐homologous protein is specifically induced by type I interferons. Eur J Immunol 1990;20:2015–2019. [DOI] [PubMed] [Google Scholar]
- 9. Towbin H, Schmitz A, Jackschies D, von Wussow P, Horisberger MA. A whole blood immunoassay for the interferon‐inducible human Mx protein. J Interferon Res 1992;12:67–74. [DOI] [PubMed] [Google Scholar]
- 10. Ronni T, Melen K, Malygin A, Julkunen I. Control of IFN‐inducible MxA gene expression in human cells. J Immunol 1993;150:1715–1718. [PubMed] [Google Scholar]
- 11. Simon A, Fäh J, Haller O, Staeheli P. Interferon‐regulated Mx genes are not responsive to interleukin‐1, tumor necrosis factor, and other cytokines. J Virol 1991;65:968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meier V, Mihm S, Ramadori G. MxA gene expression in peripheral blood mononuclear cells from patients infected chronically with hepatitis C virus treated with interferon‐alpha. J Med Virol 2000;62:318–326. [PubMed] [Google Scholar]
- 13. Halminen M, Ilonen J, Julkunen I, Ruuskanen O, Simell O, Mäkelä MJ. Expression of MxA protein in blood lymphocytes discriminates between viral and bacterial infections in febrile children. Pediatr Res 1997;41:647–650. [DOI] [PubMed] [Google Scholar]
- 14. Baca LM, Gems P, Kalvakolanu D, et al. Regulation of interferon‐α‐inducible cellular genes in human immunodeficiency virus‐infected monocytes. J Leukocyte Biol 1994;55:299–309. [DOI] [PubMed] [Google Scholar]
- 15. Jakschies D, Armbrust A, Clare KU, et al. Significant difference of the MxA‐protein expression in human PBL of patients with viral and bacterial infections. J Interferon Res 1994;14:S124. [Google Scholar]
- 16. Haller O, Kochs G. Interferon‐induced Mx proteins: Dynamin‐like GTPases with antiviral activity. Traffic 2002;3:710–717. [DOI] [PubMed] [Google Scholar]
- 17. Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: Constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 1986;44:147–158. [DOI] [PubMed] [Google Scholar]
- 18. Zhao H, De BP, Das T, Banerjee AK. Inhibition of human parainfluenza virus‐3 replication by interferon and human MxA . Virology 1996;220:330–338. [DOI] [PubMed] [Google Scholar]
- 19. Schnorr JJ, Schneider‐Schaulies S, Simon‐Jodicke A, Pavlovic J, Horisberger MA, ter Meulen V. MxA‐dependent inhibition of measles virus glycoprotein synthesis in a stably transfected human monocytic cell line. J Virol 1993;67:4760–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chieux V, Chehadeh W, Harvey J, Haller O, Wattre P, Hober D. Inhibition of coxsackievirus B4 replication in stably transfected cells expressing human MxA protein. Virology 2001;283:84–92. [DOI] [PubMed] [Google Scholar]
- 21. Horisberger MA, McMaster G, Zeller H, Wathelet M, Dellis MG, Content J. Cloning and sequence analysis of cDNAs for interferon‐ and virus‐induced human Mx‐proteins reveal that they contain putative guanine nucleotide‐binding site: Functional study of the corresponding gene promoter. J Virol 1990;64:1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kohno H, Sato S, Chiba H, et al. Monoclonal antibodies specific for 18‐hydroxycortisol and their use in an enzyme immunoassay for human urinary 18‐hydrocortisol for diagnosis of primary aldosteronism. Clin Biochem 1994;27:227–282. [DOI] [PubMed] [Google Scholar]
- 23. Djavadi‐Ohaniance L, Goldberg ME, Friguet B. Chapter 4. Measuring antibody affinity in solution In: McCafferty J, editor. Antibody Engineering. Oxford: IRL Press; 1996. p 77–97. [Google Scholar]
- 24. Kohno H, Sueshige N, Oguri K, et al. Simple and practical sandwich‐type enzyme immunoassay for human oxidatively modified low density lipoprotein using anti‐oxidized phosphatidylcholine monoclonal antibody and anti‐human apolipoprotein‐B antibody. Clin Biochem 2000;33:243–253. [DOI] [PubMed] [Google Scholar]
- 25. Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 26. Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 1983;148:839–843. [DOI] [PubMed] [Google Scholar]
- 27. Huang N, Morlock L, Lee CH, Chen LS, Chou YJ. Antibiotic prescribing for children with nasopharyngitis (common colds), upper respiratory infections, and bronchitis who have health‐professional parents. Pediatrics 2005;116:826–832. [DOI] [PubMed] [Google Scholar]
- 28. Baker MD, Bell LM, Avner JR. Outpatient management without antibiotics of fever in selected infants. N Eng1 J Med 1993;329:1437–1441. [DOI] [PubMed] [Google Scholar]
- 29. Baskin MN, O'Rourke EJ, Fleisher OR. 1992. Outpatient treatment of febrile infants 28 to 89 days of age with intramuscular administration of ceftriaxone. J Pediatr 120: 22–27. [DOI] [PubMed] [Google Scholar]
- 30. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection‐an appraisal of the Rochester criteria and implications for management. Febrile Infant Collaborative Study Group. Pediatrics 1994;94:390–396. [PubMed] [Google Scholar]