

Abstract
Patients who survive a myocardial infarction (MI) are at high risk for ventricular dilation and heart failure. While infarct size is an important determinant of post-MI remodeling, different patients with the same size infarct often display different levels of left ventricular (LV) dilation. The acute physiologic response to MI involves reflex compensation, whereby increases in heart rate (HR), arterial resistance, venoconstriction, and contractility of the surviving myocardium act to maintain mean arterial pressure (MAP). We hypothesized that variability in reflex compensation might underlie some of the reported variability in post-MI remodeling, a hypothesis that is difficult to test using experimental data alone because some reflex responses are difficult or impossible to measure directly. We, therefore, employed a computational model to estimate the balance of compensatory mechanisms from experimentally reported hemodynamic data. We found a strikingly wide range of compensatory reflex profiles in response to MI in dogs and verified that pharmacologic blockade of sympathetic and parasympathetic reflexes nearly abolished this variability. Then, using a previously published model of postinfarction remodeling, we showed that observed variability in compensation translated to variability in predicted LV dilation consistent with published data. Treatment with a vasodilator shifted the compensatory response away from arterial and venous vasoconstriction and toward increased HR and myocardial contractility. Importantly, this shift reduced predicted dilation, a prediction that matched prior experimental studies. Thus, postinfarction reflex compensation could represent both a source of individual variability in the extent of LV remodeling and a target for therapies aimed at reducing that remodeling.
Introduction
Over 800,000 Americans suffer a myocardial infarction (MI) every year [1]. Although about 85% now survive the initial event [1], survivors are at risk for a range of complications ranging from sudden cardiac death due to arrhythmia to chronic heart failure. Postinfarction heart failure is a particularly important problem because it is common, with an incidence of 16–30% [1,2], and it is difficult to treat, with a 1-year mortality of 31–45% [1,3].
The development of heart failure following MI is driven by a number of processes including neurohormonal activation, and drugs such as angiotensin converting enzyme inhibitors and beta-adrenergic blockers that target those pathways can help slow its onset [4,5]. Changes in regional mechanics following infarction also appear to play a central role, triggering a pattern of myocyte growth termed eccentric hypertrophy in which individual myocytes preferentially lengthen and the left ventricle (LV) dilates, while myocyte width and wall thickness remain relatively unchanged [6]. This process of eccentric hypertrophy increases wall stresses, myocardial oxygen consumption, and the likelihood of heart failure, but the underlying mechanisms that drive it are not fully understood.
Although a number of studies have shown that the extent of post-MI eccentric hypertrophy and dilation correlates with infarct size, there is a striking variability in the degree of hypertrophy observed in individual patients, particularly among those with moderately sized infarcts (15–25%) [7,8]. In this study, we focused on the potential role of compensatory reflexes that act to preserve mean arterial pressure (MAP) following infarction. These reflexes sense blood pressure through baroreceptors in the carotid arteries and trigger a suite of responses through the sympathetic nervous system, including an increase in heart rate (HR), an increase in contractility of the surviving myocardium, arterial vasoconstriction, and venous vasoconstriction. Sympathetic activation not only directly activates intracellular pathways that stimulate myocyte hypertrophy but also indirectly modulates hypertrophy by affecting the mechanical load placed on the myocardium. Thus, we hypothesize that postinfarction reflex compensation could represent both a source of individual variability in the extent of LV remodeling and a target for therapies aimed at reducing that ventricular growth and remodeling.
A number of different mathematical models—often termed growth laws—have been developed to predict ventricular growth from changes in the myocardial strain and/or stress [9–14]. We recently showed that most of these models reproduce some observed features of ventricular growth in response to changes in hemodynamic loading, and some can match a variety of data across multiple different experimental models of ventricular hypertrophy [15]. In those experiments, forcing the LV to pump higher volumes of blood at a relatively normal pressure (volume overload) induces lengthening of myocytes and dilation of the cavity, while increasing the pressure the LV must generate (pressure overload) produces thickening of myocytes and LV wall thickening. Here, we employ a strain-based growth model that we recently demonstrated can predict ventricular growth patterns following pressure overload, volume overload, and myocardial infarction [16].
Hemodynamic compensation has the potential to alter strains and stresses in the myocardium, thereby altering ventricular growth. However, studying postinfarction reflex compensation is complicated by the fact that only two of the four compensatory reflex responses listed above (changes in heart rate and arterial vasoconstriction) can be directly measured or calculated from routinely available hemodynamic data. Thus, in the present study, we first used a computational model to reinterpret published hemodynamic measurements to better understand the contributions and variability of the individual compensatory mechanisms following MI. We then employed our previously validated growth model to explore how variability in compensation might translate to variability in global LV remodeling.
Methods
We used a computational model to reinterpret published hemodynamic measurements from four published studies. First, we fitted the model to animal-specific hemodynamic data reported by Hood et al. [17] in order to examine the degree of variability in post-MI hemodynamic compensation. Next, we confirmed that variability in those fitted model parameters was likely due to post-MI hemodynamic compensation by fitting animal-specific data from a study by Fomovsky et al. [18] in which propranolol and atropine were administered to block reflex responses. Then, we demonstrated that differences in hemodynamic compensation could plausibly drive differences in hypertrophy using a published model of LV remodeling that correctly reproduces growth in response to pressure overload, volume overload, and myocardial infarction [16]. Finally, we tested the ability of our model to predict longer-term changes in hypertrophy induced by drugs that alter reflex compensation by fitting acute hemodynamic changes in response to post-MI administration of nitroglycerin [19], predicting subsequent ventricular hypertrophy, and comparing the predictions to an independent study of post-MI changes in ventricular dimensions with and without nitroglycerin treatment [20].
Compartmental Model of the Ventricles and Circulation.
We used our previously published model of the circulation and infarcted LV [16] to quantify compensatory responses post-MI. We used a time-varying elastance model of ventricular contraction and defined the left ventricular end-systolic and end-diastolic pressure–volume relationships as
| (1) |
| (2) |
respectively, where was the peak end-systolic elastance of the ventricle, was the unloaded volume of the ventricle, and and were coefficients describing the exponential shape of the end-diastolic pressure-volume relationship (EDPVR). We represented the systemic and pulmonary vessels using capacitors in parallel with resistors and simulated the valves with pressure sensitive diodes that permitted flow in only one direction. The complete set of equations describing the circulation model and valve opening/closing are presented by Burkhoff and Tyberg [21]. We simulated myocardial infarction using the approach of Sunagawa et al. [22], dividing the LV into a noninfarcted, contractile compartment and an infarcted, noncontractile compartment. At any time point during the cardiac cycle, the pressure in the LV compartments was the same and their volumes summed to the total LV volume. The left ventricular end-systolic and end-diastolic pressure–volume relationships for the noninfarcted and infarcted compartments were
| (3) |
| (4) |
and
| (5) |
respectively, where was the infarct size and was the peak end-systolic elastance of the noninfarcted compartment. Sunagawa showed that this simple approach replicates measured pressure–volume behavior across a wide range of infarct sizes remarkably well. As discussed in detail below, we varied systemic arterial resistance, stressed blood volume (SBV), heart rate, and the end-systolic elastance of the noninfarcted myocardium to match data from the studies we simulated, and then used the fitted values of each parameter as measures of the contributions of the individual compensatory mechanisms.
We used a single model EDPVR for all simulations since all studies utilized healthy dogs of similar weights and most did not provide enough information to fit study-specific or dog-specific EDPVR parameters. Fomovsky et al. [18] performed temporary inferior vena cava occlusions to vary preload in each dog to measure a range of LV end-diastolic pressures and volumes; the black circles in Fig. 1 show their average EDPVR for end-diastolic pressures between 6 and 12 mmHg. Because Fomovsky et al. computed LV volume from sonomicrometry measurements using crystals embedded in the LV wall, their computed volumes included some muscle mass. In order to subtract this additional wall volume, we shifted the fitted EDPVR from Fomovsky leftward until it matched pressure and volume data measured by Jugdutt and Khan [20] after acute coronary ligation using echocardiography (yellow dot in Fig. 1), which provides a better reflection of true cavity volume. Since many sets of parameters produced exponential relations that fitted the shifted EDPVR well, we further constrained . For consistency with our previously published growth model [16] and based on our previous analysis [15], we constrained by setting baseline end-diastolic stretch, defined as , to 1.44. The final fitted values of , , and , were 0.032 1/ml, 2.51 mmHg, and 18.3 ml, respectively, and Fig. 1 shows the model EDPVR (green line). To validate the model EDPVR, we overlaid measurements made both before and after coronary ligation in independent studies by Jugdutt et al. [23–25]. The model EDPVR was consistent with these end-diastolic points (blue boxes in Fig. 1 show mean ± 1 SD before and after coronary ligation).
Fig. 1.
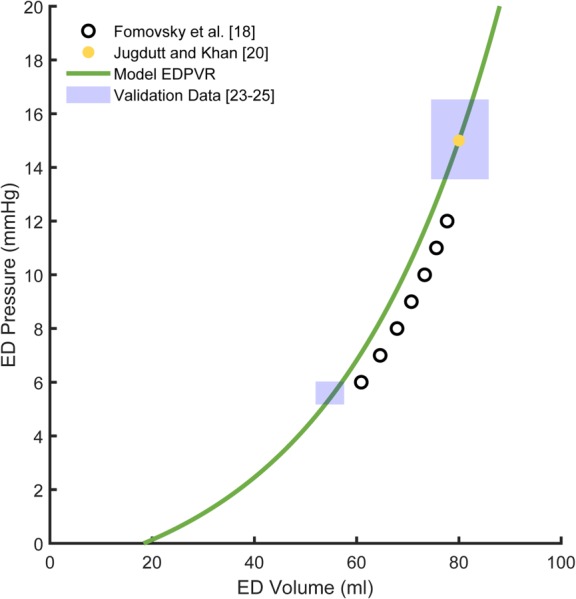
The end-diastolic pressure volume relationship (EDPVR) utilized in the model (green line) where A = 0.032 1/ml, B = 2.51 mmHg, and V0 = 18.3 ml. We shifted the EDPVR reported by Fomovsky et al. [18] (black circles) leftward to match the end-diastolic point reported postligation by Jugdutt and Khan [20] (yellow dot). Independent end-diastolic (ED) pressure and volume measurements from Jugdutt and colleagues [23–25] (shaded blue regions indicate mean ± 1 SD) were consistent with the model EDPVR.
We implemented the model in matlab as a series of differential equations for changes in the volume of each compartment or capacitor (noninfarcted left ventricle, infarcted left ventricle, systemic arteries, systemic veins, right ventricle, pulmonary arteries, and pulmonary veins) at 5000 time points over the cardiac cycle. We ran all simulations in matlab 2018a on a 16-GB RAM, 64-bit operating system, 3.4-GHz Intel Core i7-3770. After each acute intervention or growth step, we ran the model until the circulation reached steady-state, defined as the point at which the volumes within each compartment or capacitor at the beginning of a new cardiac cycle were within 0.1 ml of the volumes at the beginning of the current cardiac cycle.
Modeling the Compensatory Response Following Myocardial Infarction.
Four model parameters reflect the primary hemodynamic compensations that occur in response to myocardial infarction to maintain mean arterial pressure: , systemic vascular resistance (SVR), peak end-systolic elastance of the noninfarcted compartment , and , computed as the total volume contained in the circulatory capacitors plus ventricles. Heart rate was reported in all the studies simulated, and was therefore prescribed directly in the model. Systemic vascular resistance provides an index of the degree of arterial vasoconstriction. We estimated using the ratio of mean arterial pressure to cardiac output (CO). In cases where mean arterial pressure was not reported [18,19], we estimated arterial pressure by assuming it was the same as left ventricular pressure during ejection and linearly interpolating for the rest of the cardiac cycle. When we used this method to estimate MAP from the LV pressure data reported by Hood et al. [26] and compared it directly to MAP computed from their recorded arterial pressure curves, we found that the estimated MAP was within 5% of the reported value both before and after coronary occlusion. The contractility of an intact ventricle is typically quantified by the slope of its end-systolic pressure–volume relationship [27,28]. In an infarcted heart, however, this slope depends not only on the behavior of the surviving contractile myocardium but also on the size and behavior of the noncontractile infarct region. Thus, the peak end-systolic elastance of the noninfarcted compartment, , could not be measured directly from the pressure–volume behavior of the left ventricle. Changes in stressed blood volume provide important information on the extent of volume shifts due to venous vasoconstriction. True measurement of is not trivial, however, requiring circulatory arrest [29,30]. Therefore, we utilized the model to fit for and based on other reported metrics. All studies we simulated reported end-diastolic pressure as well as sufficient data to compute mean arterial pressure and cardiac output. Studies with acute hemodynamic measurements [17–19] also reported the maximum rate of LV pressure generation.
We performed a sensitivity analysis to determine whether the reported hemodynamic measures depended strongly enough on and to allow reliable estimation. We varied , ,, and from 50% to 200% of the values reported by Santamore and Burkhoff [31] and used the model to calculate values of experimentally available hemodynamic variables: LV end-diastolic pressure (EDP), maximum rate of LV pressure generation (dP/dt), MAP, and CO. LV EDP was very sensitive to and relatively insensitive to the other parameters, suggesting it should be possible to obtain a good estimate of from end-diastolic pressure (Fig. 2(a)). LV dP/dt depended on (Fig. 2(b)) as expected, but also had high sensitivity to and , suggesting that both and must be known to accurately estimate from dP/dt. Finally, MAP and CO varied with all four parameters (Figs. 2(c) and 2(d)), suggesting that these measures can provide additional useful information in a simultaneous multiparameter optimization scheme. When we simulated a 20% infarct, the sensitivity analysis exhibited the same trends.
Fig. 2.
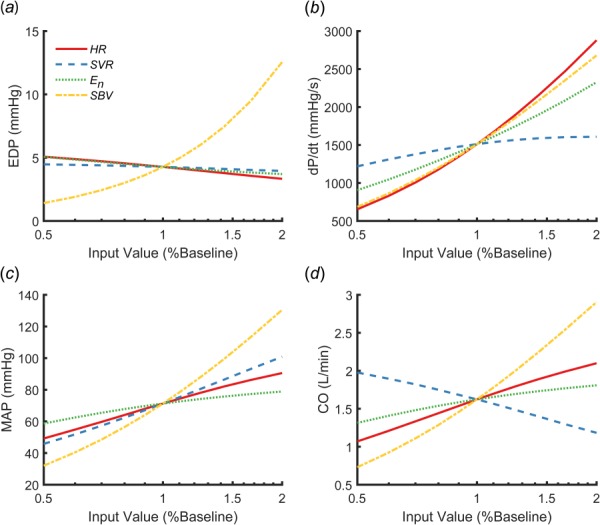
Sensitivity analysis showing how end-diastolic pressure (EDP) (a), maximum pressure generation (dP/dt) (b), mean arterial pressure (MAP) (c), and cardiac output (CO) (d) changed when heart rate (HR), systemic vascular resistance (SVR), peak end-systolic elastance (En), and stressed blood volume (SBV) were varied between 50% and 200% of the values reported by Santamore and Burkhoff [31]
When matching experimental data, we fitted for and simultaneously by minimizing the sum squared error (SSE) in LV end-diastolic pressure, the maximum derivative of LV pressure, and mean arterial pressure using the fminsearch function in matlab. Thus, SSE was defined as
Acute infarct size was set to the reported value for each animal from Hood et al. [17], to an estimated value of 20% for all animals studied by Fomovsky et al. [18], and to 12.2% for the mean data reported by Theroux et al. [19], based on similar studies by the same group [32]. To evaluate the precision of our fitted values for and within the solution space, we systematically varied them by 50–150% about their best-fit values. Figure 3 shows how SSE varies with and for one sham and one infarct animal from Hood et al. [17] ((a) and (b)) and one animal from Fomovsky et al. [18] both prior to and following infarction ((c) and (d)). SSE is indicated by the contour color and was ≤ 0.03 in all cases. When we varied or by more than 25% of their fitted values the sum squared error increased above 0.04, corresponding to an average error of greater than 10% in LV end-diastolic pressure, maximum derivative of LV pressure, and mean arterial pressure. At baseline, postocclusion, and postnitroglycerin the minimum sum squared error was also low (<0.03), corresponding to an average error below 9% in LV end-diastolic pressure, maximum derivative of LV pressure, and mean arterial pressure in all cases for the mean data from Theroux et al. [19] (not shown). Therefore, our optimization procedure produced well-determined estimates of and that matched reported hemodynamics well.
Fig. 3.
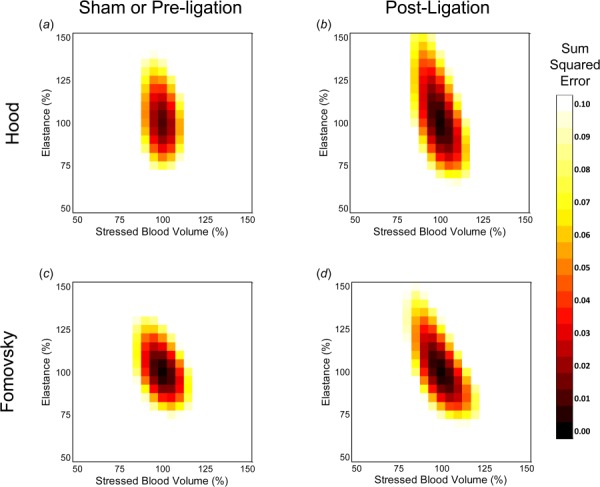
Sum squared error computed as peak end-systolic elastance of the noninfarcted compartment and stressed blood volume were varied between 50% and 150% of their optimized values for one sham (a) and one postligation (b) animal from Hood et al. [17] as well as one animal both before (c) and after ligation (d) from Fomovsky et al. [18]
Simulating Left Ventricular Growth.
To explore how variability in hemodynamic compensation could affect subsequent ventricular hypertrophy, we used our previously published model of postinfarction growth [16]. The left ventricle was treated as a thin-walled spherical pressure vessel with an initial unloaded radius and an initial unloaded thickness . Growth in the circumference of the ventricle was driven by changes in the maximum circumferential strain, which typically occurred at end diastole, and growth in the radial direction was driven by changes in the maximum radial strain, which typically occurred at end systole. Baseline or sham strains in the absence of infarction or drug treatment determined the homeostatic set points for each simulation. All other parameters within the growth law were set to their previously published values [16]. We assumed ventricular growth did not alter the intrinsic material properties of the myocardium and thus held the relationship between stress and stretch in the myocardium constant throughout growth using the strategy detailed in Witzenburg and Holmes [16]. After each day of simulated growth, the model was run until the circulation reached steady-state. We held circulatory parameters (e.g., heart rate, systemic vascular resistance, etc.) constant at their acute postinfarction values during growth.
Results
Compensatory Responses Following Myocardial Infarction.
To assess the variability in compensatory responses following myocardial infarction, we fitted individual data from 4 sham animals and 12 acutely infarcted animals originally published by Hood et al. [17]. There was a high degree of individual variability in the reported ( and ) and fitted ( and ) parameters reflecting the different compensatory mechanisms employed in response to myocardial infarction (Table 1). Note that we multiplied the sham elastance values by ) in order to compare how the contractility of only the noninfarcted compartment was altered by infarction. In one infarcted animal, none of the fitted parameters were greater than the values observed in the shams, and in another animal three of the four (systemic vascular resistance, myocardial elastance, and stressed blood volume) were elevated. Most animals employed one or two compensatory mechanisms, but in some cases heart rate, systemic vascular resistance, or myocardial elastance actually decreased in individual dogs even as they compensated with other mechanisms. Overall, there was no clear relationship between infarct size and the compensatory mechanisms employed.
Table 1.
The reported mean arterial pressure, heart rate, and systemic vascular resistance as well as the fitted ventricular elastance and stressed blood volume for each animal from Hood et al. [17]
| Animal | Infarct size (%) | Mean arterial pressure (mmHg) | Heart rate (beats/min) | Systemic vascular resistance (mmHg s/ml) | Elastance ×(1-infarct size) (mmHg/ml) | Stressed blood volume (ml) |
|---|---|---|---|---|---|---|
| Sham 1 | 0 | 119 | 148 | 2.9 | 11.6 | 244 |
| Sham 2 | 0 | 147 | 109 | 2.8 | 5.5 | 397 |
| Sham 3 | 0 | 100 | 87 | 3.2 | 8.0 | 335 |
| Sham 4 | 0 | 114 | 109 | 2.7 | 5.8 | 324 |
| Ligation 1 | 6 | 98 | 63 | 2.8 | 10.4 | 395 |
| Ligation 2 | 11 | 116 | 101 | 2.6 | 12.5 | 340 |
| Ligation 3 | 16 | 113 | 102 | 4.3 | 19.8 | 264 |
| Ligation 4 | 16 | 101 | 50 | 3.3 | 26.9 | 335 |
| Ligation 5 | 20 | 124 | 143 | 3.2 | 4.1 | 411 |
| Ligation 6 | 21 | 147 | 129 | 2.9 | 9.0 | 400 |
| Ligation 7 | 21 | 102 | 106 | 4.2 | 22.4 | 267 |
| Ligation 8 | 22 | 80 | 120 | 1.9 | 13.7 | 259 |
| Ligation 9 | 26 | 134 | 106 | 3.6 | 6.4 | 453 |
| Ligation 10 | 28 | 114 | 178 | 3.5 | 4.4 | 491 |
| Ligation 11 | 29 | 113 | 181 | 2.0 | 8.1 | 318 |
| Ligation 12 | 36 | 130 | 109 | 3.3 | 17.6 | 406 |
Highlighted values were above (dark red) or below (light blue) the sham range.
To confirm that the variability in fitted parameters we observed in the Hood dataset was likely due to variability in reflex hemodynamic compensation, we also fitted pre- and postinfarction data from a study by Fomovsky et al. [18] in which propranolol and atropine were administered prior to infarction to block sympathetic and parasympathetic reflex responses. As expected, we observed few large changes (>30%) in parameters following infarction, with half of the animals exhibiting no large change in any parameter (Table 2).
Table 2.
The reported heart rate, computed systemic vascular resistance the fitted ventricular elastance, and the fitted stressed blood volume for each animal from Fomovsky et al. [18]
| Animal | Infarct size (%) | Mean arterial pressure (mmHg) | Heart rate (beats/min) | Systemic vascular resistance (mmHg s/ml) | Elastance ×(1-infarct size) (mmHg/ml) | Stressed blood volume (ml) |
|---|---|---|---|---|---|---|
| Preligation 1 | 0 | 86 | 107 | 4.3 | 2.4 | 330 |
| Preligation 2 | 0 | 68 | 92 | 2.7 | 2.2 | 319 |
| Preligation 3 | 0 | 77 | 94 | 4.2 | 2.3 | 321 |
| Preligation 4 | 0 | 107 | 126 | 2.7 | 2.7 | 540 |
| Preligation 5 | 0 | 85 | 115 | 2.4 | 2.7 | 387 |
| Preligation 6 | 0 | 87 | 108 | 2.7 | 2.6 | 422 |
| Preligation 7 | 0 | 97 | 114 | 2.5 | 1.8 | 610 |
| Preligation 8 | 0 | 92 | 112 | 4.9 | 2.6 | 305 |
| Preligation 9 | 0 | 82 | 104 | 3.7 | 3.2 | 257 |
| Preligation 10 | 0 | 91 | 108 | 2.8 | 1.8 | 536 |
| Ligation 1 | 20 | 98 | 102 | 4.5 | 4.7 | 329 |
| Ligation 2 | 20 | 79 | 80 | 6.2 | 2.9 | 314 |
| Ligation 3 | 20 | 78 | 94 | 4.0 | 2.7 | 346 |
| Ligation 4 | 20 | 106 | 128 | 3.1 | 3.3 | 518 |
| Ligation 5 | 20 | 89 | 118 | 2.4 | 3.1 | 437 |
| Ligation 6 | 20 | 90 | 110 | 2.4 | 2.7 | 498 |
| Ligation 7 | 20 | 72 | 120 | 3.5 | 1.2 | 769 |
| Ligation 8 | 20 | 92 | 121 | 4.6 | 2.5 | 390 |
| Ligation 9 | 20 | 83 | 100 | 4.3 | 3.0 | 341 |
| Ligation 10 | 20 | 101 | 117 | 3.0 | 2.0 | 775 |
Highlighted values were 30% above (dark red) or below (light blue) the preligation value.
In order to test the ability of the model to assess changes in postinfarction hemodynamic compensation due to drug treatment, we simulated a study by Theroux et al. [19] in which nitroglycerin was administered following coronary occlusion. On average, dogs in that study compensated for acute ischemia primarily by increasing heart rate (Table 3). Treatment with the vasodilator nitroglycerin reduced systemic vascular resistance and to a lesser degree stressed blood volume, as expected. Following nitroglycerin administration, heart rate and myocardial contractility both increased, suggesting that animals relied more on these compensatory mechanisms when vasoconstriction was pharmacologically inhibited.
Table 3.
The reported heart rate, computed systemic vascular resistance, fitted ventricular elastance, and fitted stressed blood volume for the summary data from Theroux et al. [19]
| Animal | Infarct size (%) | Mean arterial pressure (mmHg) | Heart rate (beats/min) | Systemic vascular resistance (mmHg s/ml) | Elastance ×(1-infarct size) (mmHg/ml) | Stressed blood volume (ml) |
|---|---|---|---|---|---|---|
| Preligation no treatment | 0 | 109 | 70 | 2.8 | 10.3 | 428 |
| Ligation no treatment | 12.2 | 107 | 106 | 2.4 | 6.1 | 468 |
| Ligation with nitroglycerin | 12.2 | 94 | 140 | 1.9 | 8.1 | 350 |
Highlighted values were 30% above (dark red) or below (light blue) the preligation value.
Predicted Growth Following Myocardial Infarction.
We next explored whether the variability in hemodynamic compensation observed for Hood et al. [17] would lead to variability in predicted ventricular hypertrophy using our previously validated model of postinfarction growth. We simulated 6 weeks of ventricular growth for each infarcted animal, assuming hemodynamic parameters remained stable at their acute postinfarction values. Although the predicted change in end-diastolic volume generally increased with infarct size (Fig. 4(a)), there was considerable variability from animal to animal, due in part to the mix of compensatory mechanisms employed (Table 1). End-diastolic volume increased beyond the sham range for all animals with large infarcts (>25%), but only half of the animals with moderately sized infarcts (>15% and <25%). Interestingly, animals that compensated primarily by increasing contractility of the surviving myocardium with little change in heart rate or venoconstriction showed the lowest levels of predicted dilation. Only animals with large predicted increases in LV end-diastolic volume also exhibited increased predicted end-diastolic thickness, and in all cases dilation far exceeded thickening (Fig. 4(b)).
Fig. 4.
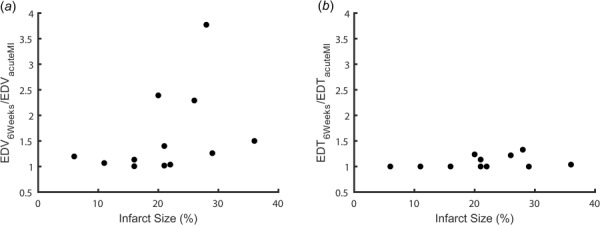
The predicted change in left ventricular end-diastolic volume (EDV) (a) and thickness (EDT) (b) 6 weeks following infarction for each animal from Hood et al. [17]
We also tested whether incorporating hemodynamic alterations induced by nitroglycerin would lead to growth predictions consistent with an independent study showing the effect of nitroglycerin on postinfarction remodeling in dogs. We simulated 6 weeks of ventricular growth utilizing the acute hemodynamic parameters shown in Table 3 and compared the predictions to data on the effect of nitroglycerin on postinfarction remodeling reported by Jugdutt and Khan [20]. In general, the model predicted the experimental trends fairly well. In the absence of nitroglycerin treatment, the model correctly predicted increases in LV volume (Fig. 5(a)) and the thickness of the surviving myocardium (Fig. 5(b)), as well as a slight drop in end-diastolic pressure with little change in mean arterial pressure (Figs. 5(c) and 5(d)). With infarction plus nitroglycerin treatment, the model predicted an acute drop in LV volume (Fig. 5(a)) but no subsequent dilation, consistent with the results reported by Jugdutt and Khan; model-predicted end-diastolic pressure fell within one standard deviation of the reported mean for the latter half of the simulated period for the nitroglycerin case. However, the model over-predicted the decrease in mean arterial pressure observed with nitroglycerin treatment and failed to predict a small amount of wall thinning observed by Jugdutt and Khan [20].
Fig. 5.
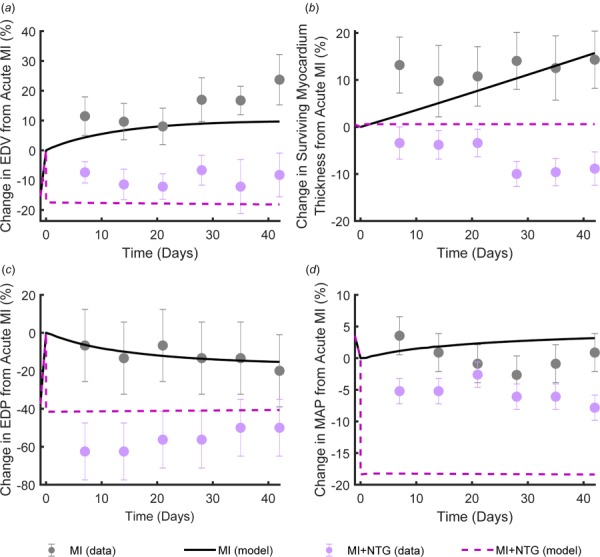
The change in end-diastolic volume (EDV) (a), thickness of the surviving myocardium (b), end-diastolic pressure (EDP) (c), and mean arterial pressure (MAP) (d) from acute infarction over 6 weeks of healing with and without therapeutic intervention with nitroglycerin. Dots (mean ± SE) indicate the experimental measurements from Jugdutt and Khan [20] and lines indicate simulation results following the hemodynamic changes from Theroux et al. [19].
Relationship Between Hemodynamic Compensation and Predicted Left Ventricle Dilation.
We also used our computational model to explore the potential growth implications of a much wider range of hemodynamic compensation patterns than reported in the experimental studies employed here. We began with the hemodynamic profile from each Hood sham animal and imposed a range of simulated infarct sizes. For each infarct size, we restored mean arterial pressure to within 90% of its control value by individually increasing heart rate, systemic vascular resistance, stressed blood volume, or end-systolic elastance of the surviving myocardium. If the value necessary to maintain mean arterial pressure was outside the range observed by Hood in any animal (for example, if heart rate exceeded 181 beats/min), the mechanism was considered saturated and simulations for larger infarcts were not performed. These simulations revealed that relatively modest increases (<60%) in heart rate, systemic vascular resistance and stressed blood volume could restore MAP across the range of infarct sizes simulated, while much larger increases in contractility of the surviving myocardium were required to compensate for a given size of infarct (Fig. 6(a)). We then compared the effects of these different compensation patterns on predicted left ventricular dilation (change in end-diastolic volume at 6 weeks). Venoconstriction (increased stressed blood volume) induced the greatest degree of dilation for any given infarct size while increases in heart rate and contractility resulted in the lowest degree of predicted dilation (Fig. 6(b)). We observed the same trends for all sham animals.
Fig. 6.
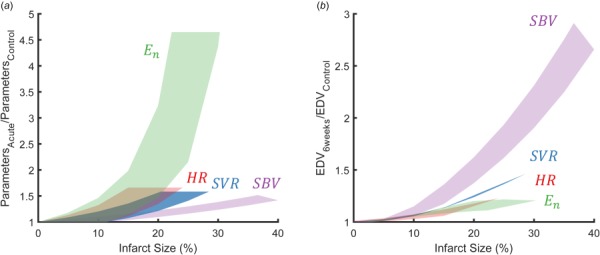
(a) The change in each parameter (heart rate, HR, the end-systolic elastance of the surviving myocardium, En, systemic vascular resistance, SVR, stressed blood volume, SBV) necessary to restore mean arterial pressure to within 90% of its control value. (b) The predicted change in end-diastolic volume (EDV) at 6 weeks resulting when mean arterial pressure was restored to within 90% of its control value using a single compensatory mechanism.
Discussion
The goal of this study was to reinterpret available published hemodynamic measurements to better understand the contributions and variability of the individual compensatory mechanisms following MI and to explore how that variability might translate to variability in ventricular hypertrophy. There are three novel aspects of this study relative to prior work. First, we developed a method to identify an individual's post-MI compensatory mechanism balance based on routinely accessible hemodynamic data. In particular we were able to estimate the contractility of the surviving myocardium and venoconstriction, responses that cannot be measured directly. Second, we demonstrated that variability in postinfarction hemodynamic compensation can plausibly generate variability in predicted LV dilation post-MI. Third, we used the model to determine the effects of a therapeutic intervention on hemodynamic compensation and were able to correctly predict how that intervention altered LV dimensions over 6 weeks post-MI. Together, the methods and results presented here suggest that the idea of customizing postinfarction therapies to manage hemodynamic responses and LV dilation merits further study.
Fitting Compensatory Responses.
We used a computational model to determine the compensatory balance in dogs postinfarction by reinterpreting published hemodynamic measurements. While two of the parameters quantifying the post-MI compensatory response—heart rate and systemic vascular resistance—can be directly measured or easily estimated for individuals, the other two—stressed blood volume and surviving myocardial elastance—are very difficult to measure and therefore must be estimated from other measured data using an appropriate model. In this study, we created an optimization routine to fit these model parameters by minimizing the sum squared error in reported LV end-diastolic pressure, the maximum derivative of LV pressure, and mean arterial pressure. Our optimization procedure produced parameters with high levels of accuracy (the average error was below 9% in all cases). We found the error landscape for this optimization to rise fairly steeply from a single minimum, indicating that the optimized values were both precise and insensitive to initial guesses (Fig. 3).
Variability in Compensation.
When we fitted data from individual dogs measured after myocardial infarction, we found a wide range of compensatory reflex profiles. To test that this variability was due to natural differences in reflex compensation rather than other causes, we also fit the compensatory responses for dogs in which propranolol and atropine were administered to block sympathetic and parasympathetic reflex responses, and confirmed that there was much less variability in the compensatory responses of the blocked dogs.
While no previous study has comprehensively characterized all four compensatory reflex responses, several pieces of published data support our finding of variable compensation post-MI. In some cases, we found that heart rate, systemic vascular resistance, or myocardial elastance were actually lower than the sham ranges following infarction (Table 1). Such reductions in a parameter that might normally be expected to increase post-MI were observed experimentally by Costantino et al. [33], who made serial measurements in dogs before and after occluding the left anterior descending artery. Though they observed increases in heart rate and systemic vascular resistance in most dogs, they reported decreases in at least three dogs. A second consistent piece of evidence is that the coefficients of variation for heart rate and systemic vascular resistance increased following infarction in the data reported by Hood et al. [17], Endo et al. [34], and Yamaguchi et al. [35], indicating greater variability post-MI.
The Effects of Infarct Size on Predicted Dilation.
Infarct size is one important determinant of cardiac function and dilation post-MI: larger infarcts are associated with decreased LV ejection fraction and increased LV end-diastolic volume index [7,8,36–39]. However, other factors clearly play a critical role, since different patients with the same size infarct display different levels of dilation [7,8,38–41]. Our analysis may explain why patients with moderately sized infarcts have the most variable outcomes. For small infarcts, the amount of predicted growth appears small regardless of what mixture of compensatory mechanisms are employed; when we simulated compensation for an infarct size of 5% by increasing each compensatory mechanism alone the predicted increase in end-diastolic volume at 6 weeks post-MI ranged from 0% to 3% (Fig. 6(b)). Conversely, for very large infarcts, our simulations suggest that venoconstriction likely plays a dominant role, since it is the only mechanism that can fully restore MAP (Fig. 6(a)); at the same time, it is also the mechanism most likely to drive LV dilation (Fig. 6(b)). For moderate infarcts, many different mixtures of compensatory mechanisms can restore MAP (Fig. 6(a)), and those mechanisms induce very different levels of LV dilation (Fig. 6(b)). Thus, moderate infarcts trigger a variable mix of compensation mechanisms that result in a variable degree of LV dilation.
Treatment Implications.
Following MI, pharmacologic agents such as β-blockers, angiotensin-converting-enzyme inhibitors, and angiotensin receptor blockers are often prescribed to prevent adverse remodeling and reduce chronic adrenergic stimulation. These agents and others such as diuretics or vasodilators that may be used in patients with hypertension alter hemodynamic loading on the heart, to a degree that likely depends upon an individual patient's initial compensatory response following MI. For example, a vasodilator would be expected to reduce arterial resistance much more in a patient with a large increase in arterial resistance post-MI than in one with no increase. Our simulations suggest that accounting for individual variations in reflex compensation and hemodynamic responses to therapy might facilitate patient-specific prediction of postinfarction hypertrophy, or even enable customization of postinfarction pharmacotherapy to reduce LV remodeling. However, some aspects of our results also raise questions that must be addressed in future studies before employing growth models in a clinical setting. For example, our model predicts less LV dilation when heart rate and/or myocardial contractility are increased following MI, despite the fact that β-blockers, which reduce heart rate and LV contractility, have been shown to reduce long-term mortality following infarction [42–45]. One possible explanation for this apparent contradiction is that other effects of β-blockers outweigh the dilation-promoting effects of lowering heart rate and contractility. In terms of hemodynamics, β-blockers also reduce mean arterial pressure, which in our model does reduce predicted postinfarction dilation. In terms of the underlying biology, β-adrenergic signaling pathways directly modulate hypertrophy, so that realistically predicting the effects of β-blockers on growth may ultimately require modeling their effects on those signaling pathways [46,47], in addition to their effects on hemodynamics and myocardial mechanics.
Limitations.
As we reported previously [16], we found that we were only able to obtain unique fits for parameters reflecting myocardial contractility () and venoconstriction (SBV) from commonly reported hemodynamic variables if we constrained the optimization using literature-based data on the plausible range of end-diastolic stretch. The value of stretch used here was based on experiments in dogs, and would need to be adapted when fitting data from patients or other animal models. More broadly, in our growth simulations we assumed that hemodynamic compensations remained constant over time, when in reality these reflexes frequently adapt over longer time scales; modeling this adaptation using approaches such as the one employed by Beard et al. [48] might allow our growth models to better predict the impact of hemodynamic compensation on LV hypertrophy. When predicting growth, we employed a computational model that was previously validated to predict experimental hypertrophy following pressure overload, volume overload, and myocardial infarction in dogs, all with a single set of parameters [16]. Despite the success of this model in retrospectively matching multiple experimental datasets, it has not yet been validated for prospectively predicting the effects of altering hemodynamics on postinfarction remodeling. Because the model predicts hypertrophy based on myocardial stretch, we would expect interventions or diseases that alter the relationship between tissue stretch and myocyte hypertrophy—including some drugs (see above), myocardial fibrosis, or even prior hypertrophy—to adversely affect the predictive power of our phenomenologic growth model.
Conclusion
The model simulations presented here suggest that reflex compensation could be both a source of individual variability in the extent of LV hypertrophy and a target for therapies aimed at reducing that remodeling following myocardial infarction. We found a strikingly wide range of compensatory reflex profiles in response to MI. This variability in compensation translated to variability in the degree of predicted LV dilation consistent with variability reported in the literature. Treatment with a vasodilator had the expected result of shifting the compensatory response away from arterial and venous vasoconstriction and toward increased heart rate and myocardial contractility. Importantly, this shift reduced predicted LV dilation, a prediction that matched prior experimental studies well. The next steps toward using predictive growth models to customize postinfarction therapy include incorporating models of long-term changes in compensatory reflexes and direct effects of drugs on hypertrophy, as well as prospective validation of the ability of an integrated model to predict the modulation of growth responses by drug treatment in individual animals or patients.
Acknowledgment
This study was funded by the Seraph Foundation. The authors wish to thank Wade Zhang for his contributions to early versions of this model.
In addition, JWH would like to acknowledge Dr. Fung's role as a mentor, role model, and inspiration to pursue a career in biomechanics. Although I only spent a summer in his laboratory as a rotating MD/Ph.D. student at UC San Diego, Dr. Fung did more than any other mentor to teach me about the power and importance of teaching and mentoring. I first learned solid mechanics from his Foundations of Solid Mechanics textbook, was introduced to biomechanics through his Biomechanics book series, and had the opportunity to host lab tours for many of his laboratory's alumni during the 1990 World Congress of Biomechanics. Through these experiences, I realized that even given the tremendous impact of his groundbreaking research, Dr. Fung has likely exerted an even greater influence on our field through the thousands of students his books have inspired, and the hundreds of students his students have trained. Thank you Dr. Fung and best wishes on your 100th birthday.
Contributor Information
Colleen M. Witzenburg, Biomedical Engineering, , University of Wisconsin, , Madison, WI 53706; Mechanical Engineering, , University of Wisconsin, , Madison, WI 53706; Cardiovascular Research Center, University of Wisconsin, Madison, WI 53706
Jeffrey W. Holmes, Biomedical Engineering, , Robert M. Berne Cardiovascular , Research Center, , University of Virginia, , Charlottesville, VA 22908; , Medicine, , Robert M. Berne Cardiovascular , Research Center, , University of Virginia, , Charlottesville, VA 22908; , Center for Engineering in Medicine, , University of Virginia, , Charlottesville, VA 22908
Funding Data
National Institutes of Health (U01 HL127654; Funder ID: 10.13039/100000002).
American Heart Association (17 POST 33660943; Funder ID: 10.13039/100000968).
References
- [1]. Benjamin, E. J. , Virani, S. S. , Callaway, C. W. , Chamberlain, A. M. , Chang, A. R. , Cheng, S. , Chiuve, S. E. , Cushman, M. , Delling, F. N. , Deo, R. , de Ferranti, S. D. , Ferguson, J. F. , Fornage, M. , Gillespie, C. , Isasi, C. R. , Jiménez, M. C. , Jordan, L. C. , Judd, S. E. , Lackland, D. , Lichtman, J. H. , Lisabeth, L. , Liu, S. , Longenecker, C. T. , Lutsey, P. L. , Mackey, J. S. , Matchar, D. B. , Matsushita, K. , Mussolino, M. E. , Nasir, K. , O'Flaherty, M. , Palaniappan, L. P. , Pandey, A. , Pandey, D. K. , Reeves, M. J. , Ritchey, M. D. , Rodriguez, C. J. , Roth, G. A. , Rosamond, W. D. , Sampson, U. K. A. , Satou, G. M. , Shah, S. H. , Spartano, N. L. , Tirschwell, D. L. , Tsao, C. W. , Voeks, J. H. , Willey, J. Z. , Wilkins, J. T. , Wu, J. H. Y. , Alger, H. M. , Wong, S. S. , and Muntner, P. , 2018, “ Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association,” Circulation, 137(12), pp. E67–E492. 10.1161/CIR.0000000000000558 [DOI] [PubMed] [Google Scholar]
- [2]. Bahit, M. C. , Kochar, A. , and Granger, C. B. , 2018, “ Post-Myocardial Infarction Heart Failure,” JACC Heart Failure, 6(3), pp. 201–208. 10.1016/j.jchf.2017.09.015 [DOI] [PubMed] [Google Scholar]
- [3]. Chen, J. , Hsieh, A. F. C. , Dharmarajan, K. , Masoudi, F. A. , and Krumholz, H. M. , 2013, “ National Trends in Heart Failure Hospitalization After Acute Myocardial Infarction for Medicare Beneficiaries 1998-2010,” Circulation, 128(24), pp. 2577–2584. 10.1161/CIRCULATIONAHA.113.003668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Hartupee, J. , and Mann, D. L. , 2017, “ Neurohormonal Activation in Heart Failure With Reduced Ejection Fraction,” Nat. Rev. Cardiol., 14(1), pp. 30–38. 10.1038/nrcardio.2016.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Yancy, C. W. , Jessup, M. , Bozkurt, B. , Butler, J. , Casey, D. E. , Colvin, M. M. , Drazner, M. H. , Filippatos, G. S. , Fonarow, G. C. , Givertz, M. M. , Hollenberg, S. M. , Lindenfeld, J. A. , Masoudi, F. A. , McBride, P. E. , Peterson, P. N. , Stevenson, L. W. , and Westlake, C. , 2017, “ 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of Amer,” J. Am. Coll. Cardiol., 70(6), pp. 776–803. 10.1016/j.jacc.2017.04.025 [DOI] [PubMed] [Google Scholar]
- [6]. Gerdes, A. M. , and Capasso, J. M. , 1995, “ Structural Remodeling and Mechanical Dysfunction of Cardiac Myocytes in Heart Failure,” J. Mol. Cell. Cardiol., 27(3), pp. 849–856. 10.1016/0022-2828(95)90000-4 [DOI] [PubMed] [Google Scholar]
- [7]. Wu, E. , Ortiz, J. T. , Tejedor, P. , Lee, D. C. , Bucciarelli-Ducci, C. , Kansal, P. , Carr, J. C. , Holly, T. A. , Lloyd-Jones, D. , Klocke, F. J. , and Bonow, R. O. , 2008, “ Infarct Size by Contrast Enhanced Cardiac Magnetic Resonance Is a Stronger Predictor of Outcomes Than Left Ventricular Ejection Fraction or End-Systolic Volume Index: Prospective Cohort Study,” Heart, 94(6), pp. 730–736. 10.1136/hrt.2007.122622 [DOI] [PubMed] [Google Scholar]
- [8]. Bulluck, H. , Go, Y. Y. , Crimi, G. , Ludman, A. J. , Rosmini, S. , Abdel-Gadir, A. , Bhuva, A. N. , Treibel, T. A. , Fontana, M. , Pica, S. , Raineri, C. , Sirker, A. , Herrey, A. S. , Manisty, C. , Groves, A. , Moon, J. C. , and Hausenloy, D. J. , 2017, “ Defining Left Ventricular Remodeling Following Acute ST-Segment Elevation Myocardial Infarction Using Cardiovascular Magnetic Resonance,” J. Cardiovasc. Magn. Reson., 19(1), p. 26. 10.1186/s12968-017-0343-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Lin, I. E. , and Taber, L. A. , 1995, “ A Model for Stress-Induced Growth in the Developing Heart,” ASME J. Biomech. Eng., 117(3), pp. 343–349. 10.1115/1.2794190 [DOI] [PubMed] [Google Scholar]
- [10]. Taber, L. A. , 1998, “ Biomechanical Growth Laws for Muscle Tissue,” J. Theor. Biol., 193(2), pp. 201–213. 10.1006/jtbi.1997.0618 [DOI] [PubMed] [Google Scholar]
- [11]. Kroon, W. , Delhaas, T. , Arts, T. , and Bovendeerd, P. , 2009, “ Computational Modeling of Volumetric Soft Tissue Growth: Application to the Cardiac Left Ventricle,” Biomech. Model. Mechanobiol., 8(4), pp. 301–309. 10.1007/s10237-008-0136-z [DOI] [PubMed] [Google Scholar]
- [12]. Göktepe, S. , Abilez, O. J. , Parker, K. K. , and Kuhl, E. , 2010, “ A Multiscale Model for Eccentric and Concentric Cardiac Growth Through Sarcomerogenesis,” J. Theor. Biol., 265(3), pp. 433–442. 10.1016/j.jtbi.2010.04.023 [DOI] [PubMed] [Google Scholar]
- [13]. Arts, T. , Delhaas, T. , Bovendeerd, P. , Verbeek, X. , and Prinzen, F. W. , 2005, “ Adaptation to Mechanical Load Determines Shape and Properties of Heart and Circulation: The CircAdapt Model,” Am. J. Physiol. Heart Circ. Physiol., 288(4), pp. H1943–H1954. 10.1152/ajpheart.00444.2004 [DOI] [PubMed] [Google Scholar]
- [14]. Kerckhoffs, R. C. P. , Omens, J. H. , and McCulloch, A. D. , 2012, “ A Single Strain-Based Growth Law Predicts Concentric and Eccentric Cardiac Growth During Pressure and Volume Overload,” Mech. Res. Commun., 42, pp. 40–50. 10.1016/j.mechrescom.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Witzenburg, C. M. , and Holmes, J. W. , 2017, “ A Comparison of Phenomenologic Growth Laws for Myocardial Hypertrophy,” J. Elasticity, 129(1–2), pp. 257–281. 10.1007/s10659-017-9631-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Witzenburg, C. M. , and Holmes, J. W. , 2018, “ Predicting the Time Course of Ventricular Dilation and Thickening Using a Rapid Compartmental Model,” J. Cardiovasc. Transl. Res., 11(2), pp. 109–122. 10.1007/s12265-018-9793-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Hood, W. B. , McCarthy, B. , and Lown, B. , 1967, “ Myocardial Infarction Following Coronary Ligation in Dogs. Hemodynamic Effects of Isoproterenol and Acetylstrophanthidin,” Circ. Res., 21(2), pp. 191–199. 10.1161/01.RES.21.2.191 [DOI] [PubMed] [Google Scholar]
- [18]. Fomovsky, G. M. , Clark, S. A. , Parker, K. M. , Ailawadi, G. , and Holmes, J. W. , 2012, “ Anisotropic Reinforcement of Acute Anteroapical Infarcts Improves Pump Function,” Circ.: Heart Failure, 5(4), pp. 515–522. 10.1161/CIRCHEARTFAILURE.111.965731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Theroux, P. , Ross, J. , Franklin, D. , Kemper, W. S. , and Sasyama, S. , 1976, “ Regional Myocardial Function in the Conscious Dog During Acute Coronary Occlusion and Responses to Morphine, Propranolol, Nitroglycerin, and Lidocaine,” Circulation, 53(2), pp. 302–314. 10.1161/01.CIR.53.2.302 [DOI] [PubMed] [Google Scholar]
- [20]. Jugdutt, B. I. , and Khan, M. I. , 1994, “ Effect of Prolonged Nitrate Therapy on Left Ventricular Remodeling After Canine Acute Myocardial Infarction,” Circulation, 89(5), pp. 2297–2307. 10.1161/01.CIR.89.5.2297 [DOI] [PubMed] [Google Scholar]
- [21]. Burkhoff, D. , and Tyberg, J. V. , 1993, “ Why Does Pulmonary Venous Pressure Rise After Onset of LV Dysfunction: A Theoretical Analysis,” Am. J. Physiol., 265(5 Pt 2), pp. H1819–H1828. 10.1152/ajpheart.1993.265.5.H1819 [DOI] [PubMed] [Google Scholar]
- [22]. Sunagawa, K. , Maughan, W. L. , and Sagawa, K. , 1983, “ Effect of Regional Ischemia on the Left Ventricular End-Systolic Pressure-Volume Relationship of Isolated Canine Hearts,” Circ. Res., 52(2), pp. 170–178. 10.1161/01.RES.52.2.170 [DOI] [PubMed] [Google Scholar]
- [23]. Jugdutt, B. I. , Humen, D. P. , Khan, M. I. , and Schwarz-Michorowski, B. L. , 1992, “ Effect of Left Ventricular Unloading With Captopril on Remodelling and Function During Healing of Anterior Transmural Myocardial Infarction in the Dog,” Can. J. Cardiol., 8(2), pp. 151–163.https://www.ncbi.nlm.nih.gov/pubmed/1559191 [PubMed] [Google Scholar]
- [24]. Jugdutt, B. I. , Schwarz-Michorowski, B. L. , and Khan, M. I. , 1992, “ Effect of Long-Term Captopril Therapy on Left Ventricular Remodeling and Function During Healing of Canine Myocardial Infarction,” J. Am. Coll. Cardiol., 19(3), pp. 713–721. 10.1016/S0735-1097(10)80298-0 [DOI] [PubMed] [Google Scholar]
- [25]. Jugdutt, B. I. , Khan, M. I. , Jugdutt, S. J. , and Blinston, G. E. , 1995, “ Effect of Enalapril on Ventricular Remodeling and Function During Healing After Anterior Myocardial Infarction in the Dog,” Circulation, 91(3), pp. 802–812. 10.1161/01.CIR.91.3.802 [DOI] [PubMed] [Google Scholar]
- [26]. Hood, W. B. , Covelli, V. H. , Abelmann, W. H. , and Norman, J. C. , 1969, “ Persistence of Contractile Behaviour in Acutely Ischaemic Myocardium,” Cardiovasc. Res., 3(3), pp. 249–260. 10.1093/cvr/3.3.249 [DOI] [PubMed] [Google Scholar]
- [27]. Suga, H. , Sagawa, K. , and Shoukas, A. A. , 1973, “ Load Independence of the Instantaneous Pressure-Volume Ratio of the Canine Left Ventricle and Effects of Epinephrine and Heart Rate on the Ratio,” Circ. Res., 32(3), pp. 314–322. 10.1161/01.RES.32.3.314 [DOI] [PubMed] [Google Scholar]
- [28]. Grossman, W. , Braunwald, E. , Mann, T. , McLaurin, L. P. , and Green, L. H. , 1977, “ Contractile State of the Left Ventricle in Man as Evaluated From End-Systolic Pressure-Volume Relations,” Circulation, 56(5), pp. 845–852. 10.1161/01.CIR.56.5.845 [DOI] [PubMed] [Google Scholar]
- [29]. Magder, S. , and De Varennes, B. , 1998, “ Clinical Death and the Measurement of Stressed Vascular Volume,” Crit. Care Med., 26(6), pp. 1061–1064. 10.1097/00003246-199806000-00028 [DOI] [PubMed] [Google Scholar]
- [30]. Gelman, S. , 2008, “ Venous Function and Central Venous Pressure: A Physiologic Story,” Anesthesiology, 108(4), pp. 735–748. 10.1097/ALN.0b013e3181672607 [DOI] [PubMed] [Google Scholar]
- [31]. Santamore, W. P. , and Burkhoff, D. , 1991, “ Hemodynamic Consequences of Ventricular Interaction as Assessed by Model Analysis,” Am. J. Physiol., 260(1 Pt 2), pp. H146–57. 10.1152/ajpheart.1991.260.1.H146 [DOI] [PubMed] [Google Scholar]
- [32]. Theroux, P. , Ross, J. , Franklin, D. , Kemper, W. S. , and Sasayama, S. , 1976, “ Coronary Arterial Reperfusion—Part III: Early and Late Effects on Regional Myocardial Function and Dimensions in Conscious Dogs,” Am. J. Cardiol., 38(5), pp. 599–606. 10.1016/S0002-9149(76)80009-4 [DOI] [PubMed] [Google Scholar]
- [33]. Costantino, C. , Corday, E. , Lang, T. W. , Meerbaum, S. , Brasch, J. , Kaplan, L. , Rubins, S. , Gold, H. , and Osher, J. , 1975, “ Revascularization After 3 Hours of Coronary Arterial Occlusion: Effects on Regional Cardiac Metabolic Function and Infarct Size,” Am. J. Cardiol., 36(3), pp. 368–384. 10.1016/0002-9149(75)90492-0 [DOI] [PubMed] [Google Scholar]
- [34]. Endo, T. , Nejima, J. , Kiuchi, K. , Fujita, S. , Kikuchi, K. , Hayakawa, H. , and Okumura, H. , 1988, “ Reduction of Size of Myocardial Infarction With Nicorandil, a New Antianginal Drug, After Coronary Artery Occlusion in Dogs,” J. Cardiovasc. Pharmacol., 12(5), pp. 587–592. 10.1097/00005344-198811000-00013 [DOI] [PubMed] [Google Scholar]
- [35]. Yamaguchi, K. , Suzuki, K. , Niho, T. , Sato, M. , Ito, C. , and Ohnishi, H. , 1983, “ Reduction of Myocardial Infarct Size by Trapidil in Anesthetized Dogs,” J. Cardiovasc. Pharmacol., 5(3), pp. 499–505. 10.1097/00005344-198305000-00023 [DOI] [PubMed] [Google Scholar]
- [36]. Bello, D. , Einhorn, A. , Kaushal, R. , Kenchaiah, S. , Raney, A. , Fieno, D. , Narula, J. , Goldberger, J. , Shivkumar, K. , Subacius, H. , and Kadish, A. , 2011, “ Cardiac Magnetic Resonance Imaging: Infarct Size is an Independent Predictor of Mortality in Patients With Coronary Artery Disease,” Magn. Reson. Imaging, 29(1), pp. 50–56. 10.1016/j.mri.2010.03.031 [DOI] [PubMed] [Google Scholar]
- [37]. Stone, G. W. , Selker, H. P. , Thiele, H. , Patel, M. R. , Udelson, J. E. , Ohman, E. M. , Maehara, A. , Eitel, I. , Granger, C. B. , Jenkins, P. L. , Nichols, M. , and Ben-Yehuda, O. , 2016, “ Relationship Between Infarct Size and Outcomes Following Primary PCI Patient-Level Analysis From 10 Randomized Trials,” J. Am. Coll. Cardiol., 67(14), pp. 1674–1683. 10.1016/j.jacc.2016.01.069 [DOI] [PubMed] [Google Scholar]
- [38]. Tarantini, G. , Razzolini, R. , Cacciavillani, L. , Bilato, C. , Sarais, C. , Corbetti, F. , Perazzolo Marra, M. , Napodano, M. , Ramondo, A. , and Iliceto, S. , 2006, “ Influence of Transmurality, Infarct Size, and Severe Microvascular Obstruction on Left Ventricular Remodeling and Function After Primary Coronary Angioplasty,” Am. J. Cardiol., 98(8), pp. 1033–1040. 10.1016/j.amjcard.2006.05.022 [DOI] [PubMed] [Google Scholar]
- [39]. Chareonthaitawee, P. , Christian, T. F. , Hirose, K. , Gibbons, R. J. , and Rumberger, J. A. , 1995, “ Relation to Initial Infarct Size to Extent of Left Ventricular Remodeling in the Year After Acute Myocardial Infarction,” J. Am. Coll. Cardiol., 25(3), pp. 567–573. 10.1016/0735-1097(94)00431-O [DOI] [PubMed] [Google Scholar]
- [40]. Westman, P. C. , Lipinski, M. J. , Luger, D. , Waksman, R. , Bonow, R. O. , Wu, E. , and Epstein, S. E. , 2016, “ Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction,” J. Am. Coll. Cardiol., 67(17), pp. 2050–2060. 10.1016/j.jacc.2016.01.073 [DOI] [PubMed] [Google Scholar]
- [41]. Schachinger, V. , Erbs, S. , Elsasser, A. , Haberbosch, W. , Hambrecht, R. , Holschermann, H. , Yu, J. , Corti, R. , Mathey, D. G. , Hamm, C. W. , Suselbeck, T. , Assmus, B. , Tonn, T. , Dimmeler, S. , and Zeiher, A. M. , 2006, “ Intracoronary Bone Marrow-Derived Progenitor Cells in Acute Myocardial Infarction,” N. Engl. J. Med., 355(12), pp. 1210–1221. 10.1056/NEJMoa060186 [DOI] [PubMed] [Google Scholar]
- [42]. Fox, K. , Borer, J. S. , Camm, A. J. , Danchin, N. , Ferrari, R. , Lopez Sendon, J. L. , Steg, P. G. , Tardif, J. C. , Tavazzi, L. , and Tendera, M. , 2007, “ Resting Heart Rate in Cardiovascular Disease,” J. Am. Coll. Cardiol., 50(9), pp. 823–830. 10.1016/j.jacc.2007.04.079 [DOI] [PubMed] [Google Scholar]
- [43]. Kjekshus, J. K. , 1986, “ Importance of Heart Rate in Determining Beta-Blocker Efficacy in Acute and Long-Term Acute Myocardial Infarction Intervention Trials,” Am. J. Cardiol., 57(12), pp. 43F–49F. 10.1016/0002-9149(86)90888-X [DOI] [PubMed] [Google Scholar]
- [44]. Freemantle, N. , Cleland, J. , Young, P. , Mason, J. , and Harrison, J. , 1999, “ Beta Blockade After Myocardial Infarction: Systematic Review and Meta Regression Analysis,” BMJ, 318(7200), pp. 1730–1737. 10.1136/bmj.318.7200.1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45]. López-Sendón, J. , Swedberg, K. , McMurray, J. , Tamargo, J. , Maggioni, A. P. , Dargie, H. , Tendera, M. , Waagstein, F. , Kjekshus, J. , Lechat, P. , Torp-Pedersen, C. , and Task Force On Beta-Blockers of the European Society of Cardiology, 2004, “ Expert Consensus Document on Beta-Adrenergic Receptor Blockers,” Eur. Heart J., 25(15), pp. 1341–1362. 10.1016/j.ehj.2004.06.002 [DOI] [PubMed] [Google Scholar]
- [46]. Ryall, K. A. , Holland, D. O. , Delaney, K. A. , Kraeutler, M. J. , Parker, A. J. , and Saucerman, J. J. , 2012, “ Network Reconstruction and Systems Analysis of Cardiac Myocyte Hypertrophy Signaling,” J. Biol. Chem., 287(50), pp. 42259–42268. 10.1074/jbc.M112.382937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Frank, D. U. , Sutcliffe, M. D. , and Saucerman, J. J. , 2018, “ Network-Based Predictions of In Vivo Cardiac Hypertrophy,” J. Mol. Cell. Cardiol., 121, pp. 180–189. 10.1016/j.yjmcc.2018.07.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48]. Beard, D. A. , Pettersen, K. H. , Carlson, B. E. , Omholt, S. W. , and Bugenhagen, S. M. , 2013, “ A Computational Analysis of the Long-Term Regulation of Arterial Pressure,” F1000Research, 2, p. 208. 10.12688/f1000research.2-208.v2 [DOI] [PMC free article] [PubMed] [Google Scholar]