

Marine refractory dissolved organic carbon is emitted to the atmosphere in primary marine aerosol.
Abstract
Breaking waves produce bubble plumes that burst at the sea surface, injecting primary marine aerosol (PMA) highly enriched with marine organic carbon (OC) into the atmosphere. It is widely assumed that this OC is modern, produced by present-day biological activity, even though nearly all marine OC is thousands of years old, produced by biological activity long ago. We used natural abundance radiocarbon (14C) measurements to show that 19 to 40% of the OC associated with freshly produced PMA was refractory dissolved OC (RDOC). Globally, this process removes 2 to 20 Tg of RDOC from the oceans annually, comparable to other RDOC losses. This process represents a major removal pathway for old OC from the sea, with important implications for oceanic and atmospheric biogeochemistry, the global carbon cycle, and climate.
INTRODUCTION
Most dissolved organic carbon (DOC) in the sea is remarkably old [averaging up to 6500 radiocarbon (14C) years] (1–3), persisting much longer than the time scales for mixing by the global system of ocean currents (250 to 1700 14C years) (4). Combined with the small range of DOC concentrations in the deep sea (~34 to 45 μM), these 14C ages suggest that most marine DOC is refractory (RDOC) with respect to its biogeochemical degradation and is present everywhere in the ocean as mixtures with lesser amounts of recently produced DOC (2, 3, 5, 6). Since marine RDOC has neither accumulated to high concentrations (ca. ≤45 μM, globally) nor attained immeasurable 14C ages, it must be removed somewhere. However, this apparent longevity implies slow loss mechanisms that are difficult to identify and constrain (7). Despite large uncertainties (2), several removal mechanisms for RDOC have been proposed, including biological losses in the water column (7, 8), photochemical oxidation in sunlit surface waters (9), interactions with particulate OC (POC) at depth (8, 10), and degradation in hydrothermal vent systems (11–13). Recently, an additional removal pathway for RDOC was proposed on the basis of observed organic enrichments of primary marine aerosol (PMA) produced by bursting bubble plumes at the sea surface from the North Atlantic surface and deep seawater (14). PMA flux calculations further suggested that the injection of RDOC into the atmosphere in association with PMA and its subsequent photochemical oxidation was a potentially important and hitherto unrecognized RDOC loss mechanism (14). The proposed atmospheric loss of marine RDOC is of particular interest owing to the associated impacts on nutrient cycling, tropospheric chemistry, and Earth’s radiation balance (15). Despite its potential importance, the fraction of RDOC that was injected into the atmosphere associated with newly formed PMA was not determined (14).
Here, we directly determined the fraction of RDOC associated with nascent PMA using natural abundance 14C signatures (Δ14C values) as robust tracers of PMA organic matter (OM) sources. We measured the Δ14C values of model PMA (mPMA) OM produced from natural, near-surface (5 m) seawater pumped at 4 liters min−1 into the base of a high-capacity PMA generator (16) modified by replacing the glass frit with a Venturi nozzle to generate bubbles (see Materials and Methods). The seawater in the generator was taken from two biologically productive and two oligotrophic hydrographic stations in the Northwest Atlantic Ocean during a research cruise from September to October 2016 aboard the R/V Endeavor (Fig. 1). Basic physical and chemical properties were also measured for each station to provide context when interpreting Δ14C values; ancillary measurements included seawater temperature, wind speed, salinity, chlorophyll a (Chl a), and DOC concentrations (Table 1).
Fig. 1. Map of hydrographic stations (BI, GB, SSW, and SSN; circles), cruise track (red line), and Chl a concentrations (color field) during research cruise EN-589 aboard the R/V Endeavor.
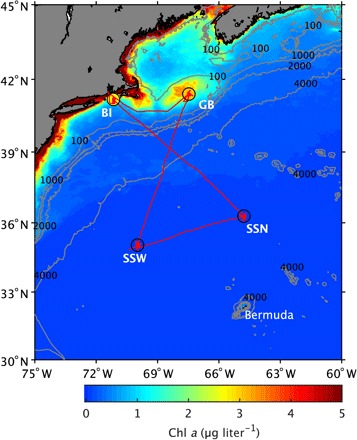
The average Chl a concentrations during occupation of each hydrographic station are indicated by the colors of their corresponding markers. The background color field indicates the spatial distribution of monthly averaged surface Chl a concentrations retrieved by the Moderate Resolution Imaging Spectroradiometer (MODIS) sensor on the Aqua satellite from September for 2013 through 2017. Satellite-derived Chl a data were downloaded from NASA (https://disc.gsfc.nasa.gov/). Isobaths in meters are depicted by gray contour lines. Bathymetry data were downloaded from GEBCO (www.gebco.net/). Additional descriptions of each station are listed in Table 1. BI, Block Island; GB, Georges Bank; SSW, Sargasso Sea West; SSN, Sargasso Sea North.
Table 1. Hydrographic stations occupied in 2016 during research cruise EN-589 aboard the R/V Endeavor.
Sea surface temperature (SST), sea surface salinity, wind speed, and Chl a are reported as the mean ± 1 SD.
| Property | Hydrographic stations | |||
| GB | SSW | SSN | BI | |
| Station name | Georges Bank | Sargasso West | Sargasso North | Coastal Rhode Island |
| Occupation dates | 18 to 22 September | 24 to 30 September | 1 to 7 October | 10 to 14 October |
| Latitude (°N), longitude (°W) | 41.40, 67.47 | 35.04, 69.98 | 36.26, 64.78 | 41.18, 71.16 |
| Bottom depth (m) | ~ 41 | ~ 5190 | ~ 4960 | ~ 42 |
| SST (°C)* | 18.6 ± 0.3 | 27.5 ± 0.5 | 26.3 ± 0.3 | 17.3 ± 0.2 |
| Salinity (ppt)* | 31.8 ± 0.1 | 35.3 ± 0.1 | 35.6 ± 0.1 | 31.7 ± 0.7 |
| Wind speed (m s−1)* | 9.0 ± 3.3 | 13.5 ± 5.5 | 13.5 ± 5.2 | 15.2 ± 10.7† |
| Chl a at 5 m (μg liter−1)‡ | 3.05 ± 0.59 | 0.04 ± 0.01 | 0.05 ± 0.01 | 2.49 ± 0.75 |
| DOC at 5 m (μM)§ | 89.4 ± 5.4 | 74.0 ± 1.0 | 71.2 ± 1.8 | 91.9 ± 1.0 |
*Mean ± 1 SD calculated from measurements recorded every 10 min using R/V Endeavor’s array of underway sensors and continuous in-line seawater system, which sampled seawater from ~5 m below the sea surface. Measurements for calculating means and SDs spanned 6.06 days at GB (n = 873), 4.97 days at SSW (n = 717), 4.99 days at SSN (n = 720), and 2.69 days at BI (n = 389).
†Wind speeds ranged from 2 to 44 m s−1 while occupying BI, averaging 26.19 ± 8.23 m s−1 on 10 October.
‡Mean Chl a concentrations and SDs (n = 30 to 62).
§Mean ± 1 SD reported for DOC concentrations from SSW (n = 2) and SSN (n = 3). Single SDs calculated from replicates at BI (±0.7 μM, n = 2) or propagated from manometry (±0.6 μM, n = 1) were less than the typical reproducibility of replicated standards (±1 SD = ±1.0 μM) and therefore reported as ±1.0 μM.
RESULTS
Since all Δ14C values are corrected for isotopic fractionation (see Materials and Methods) and the 14C half-life (5730 ± 40 years) is much longer than the time scale of aerosol formation, the Δ14C values of mPMA OM constituents must have been equal to the Δ14C values of their sources in the ocean (1, 17). Therefore, we compared Δ14C mPMA OM values to Δ14C signatures of possible source materials, including RDOC (DOC samples collected from a depth of 2500 m), recently produced OM [i.e., the Δ14C value of near-surface dissolved inorganic carbon (DIC)], and DOC in surface seawater at these stations.
Except for the coastal Rhode Island station near Block Island, which was occupied during the passage of a gale with winds gusting to 44 m s−1 (Fig. 1 and Table 1), all mPMA OM Δ14C values (−188 to −149‰) were statistically indistinguishable, averaging −164 ± 20‰ (1370 ± 210 14C years; Table 2 and table S1). The range of Δ14C values was dominated by measurement uncertainty (table S1), constraining the natural variability of Δ14C values of mPMA OM sources across these stations to less than ±20‰. These mPMA OM Δ14C values were higher than the Δ14C values of bulk DOC from near-surface seawater at each station (−291 to −234‰; Table 2) and lower than the “modern” DIC Δ14C values in near-surface seawater (+19 to +43‰; Table 2 and Fig. 2). Therefore, the aerosol generation process must have selectively scavenged an isotopically distinct reservoir of surface-active organic constituents that were not exclusively generated from recently produced OM.
Table 2. Radiocarbon signatures (Δ14C values) and proportions of RDOC in mPMA OM.
Proportions assume mPMA OM result from mixtures of OM recently produced in the near-surface ocean (Δ14C of DIC at 5 m) and RDOC, with RDOC Δ14C values constrained between those of fossil molecules (−1000‰) and DOC collected from 2500 m (averaging −457 ± 8‰, n = 3; table S6). Near-surface DOC Δ14C values are shown for comparison. All mPMA Δ14C values were measured from individual samples generated over the course of 24 hours, except where noted, and reported with single SDs propagated from blank-correction calculations. All DIC and DOC Δ14C values are reported as the average and single SD of duplicate measurements at each station, except where noted. All Δ14C identification numbers are reported in tables S1 and S4 to S7.
| Station | Δ14C (‰) | Proportion of RDOC in mPMA | |||
| DIC | DOC | mPMA | Fossil RDOC | 2500-m DOC | |
|
GB
|
19 ± 2 | −248 ± 14 | −140 ± 23* | 0.16 ± 0.02 | 0.33 ± 0.05 |
| 19 ± 2 | −248 ± 14 | −177 ± 56† | 0.19 ± 0.06 | 0.41 ± 0.12 | |
| 19 ± 2 | −248 ± 14 | −188 ± 47 | 0.20 ± 0.05 | 0.43 ± 0.10 | |
| SSW | 43 ± 2‡ | −291 ± 4§ | −166 ± 20 | 0.20 ± 0.02 | 0.42 ± 0.04 |
| SSN | 43 ± 2‡ | −234 ± 7|| | −149 ± 69 | 0.18 ± 0.07 | 0.38 ± 0.14 |
|
BI
|
23 ± 3 | −227 ± 4 | −50 ± 11 | 0.07 ± 0.01 | 0.15 ± 0.02 |
| 23 ± 3 | −227 ± 4 | −6 ± 29 | 0.03 ± 0.03 | 0.06 ± 0.06 | |
| Averages excluding coastal Rhode Island | −164 ± 20 | 0.19 ± 0.02 | 0.40 ± 0.04 | ||
*mPMA generated during daytime (~12 hours between sunrise and sunset).
†mPMA generated during nighttime (~12 hours between sunset and sunrise).
‡Individual measurements at SSW and SSN were not significantly different and therefore averaged (n = 2).
§Individual measurement with uncertainty reported as single SD propagated from blank-correction calculations.
||Average and SD of triplicate measurements (n = 3).
Fig. 2. Radiocarbon constraints on the proportion of RDOC in mPMA from the Sargasso Sea and Georges Bank.
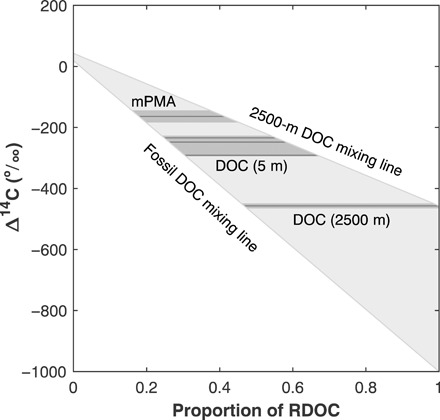
Possible proportions of RDOC in mPMA, DOC from 5-m-deep seawater, and DOC from 2500-m-deep seawater are constrained by their measured Δ14C values (horizontal solid lines), Δ14C uncertainties (±1 SD; dark gray shading), and conservation of mass to lie within the gray wedge. The fraction of RDOC in each reservoir (i.e., mPMA, 5 m of DOC, and 2500 m of DOC) has (i) a lower value that is bounded by the conservative mixing line between Δ14C measurements of DOC from 2500 m (−457‰) and DIC from 5 m (+19 to +43‰, y-axis intercept), denoted as the 2500-m DOC mixing line, and (ii) an upper value that is bounded by the mixing line between fossil DOC (assumed −1000‰) and DIC from 5 m, denoted as the fossil DOC mixing line.
It is possible that the mPMA formation process selectively incorporated surface-active OM with a single age of 1370 ± 210 14C years. However, this is unlikely because it implies that only a small percentage of OM within a relatively narrow age range (0.0002 to 0.0011% of total DOC at a depth of 5 m) was aerosolized to the exclusion of surfactants associated with both RDOC (≥~50% of surface DOC) and recently produced OM that had a Δ14C value equal to that of DIC in the surface water (2). It is also possible but unlikely that, at the other extreme, only fossil RDOC (Δ14C = −1000‰) and modern OM were emitted into the atmosphere in association with mPMA. If this were the case, then 19 ± 2% of mPMA OM would have been generated from fossil RDOC in near-surface waters. However, surface-active RDOC competes for space on rising bubble surfaces with other fractions of surface-active OM present in seawater. Therefore, it is more likely that the mPMA generated from near-surface seawater at Georges Bank and in the Sargasso Sea was derived from blends of surfactant OM (including both RDOC and modern OC) in near-surface seawater that span the full range of possible RDOC Δ14C signatures. Assuming that bulk DOC from the deep ocean (2500 m) is representative of RDOC in the surface ocean, then mPMA RDOC Δ14C values cannot be greater than the lowest regionally observed deep-water DOC Δ14C value (i.e., −457 ± 8‰, n = 3) (2, 3, 5, 18). These constraints dictate that the maximum proportion of RDOC in mPMA OM generated from coastally productive and oligotrophic seawater (stations: Georges Bank, Sargasso Sea West, and Sargasso Sea North) ranges from 19 ± 2 to 40 ± 4% on average (Table 2 and Fig. 2). This calculated range is insensitive to whether mPMA OM originated exclusively from DOC or, at the other extreme, exclusively from POC and RDOC, assuming that the Δ14C signatures of POC, DIC, and recently produced DOC are approximately equal (see Materials and Methods). Accordingly, these Δ14C observations place strong constraints on the proportion of RDOC in mPMA, but they cannot constrain whether the surface-active constituents of RDOC and modern DOC are incorporated into mPMA in the same proportions that they are found in near-surface seawater.
The mPMA OM generated from coastal Rhode Island seawater had Δ14C values that were significantly higher (−50 and −6‰) than near-surface DOC (−227‰) but comparable to near-surface DIC (+23‰) (Table 2). This indicates that the relative contributions of RDOC to mPMA OM at the coastal Rhode Island station ranged from undetectable to statistically significant (7 to 15%) (Table 2), which were considerably less than that observed at Georges Bank and the oligotrophic Sargasso Sea (19 to 40% RDOC). These results suggest that the OM source for mPMA may be more variable in coastal regions worldwide because of influences such as surface runoff, estuaries, submarine groundwater discharge, wind-driven coastal upwelling, or storm-driven sediment resuspension. Therefore, the mPMA OM constituents at this station could have been almost entirely modern and marine in origin or, by conservation of mass, mixtures containing various proportions of RDOC, modern marine OM, and terrestrially derived OM enriched with bomb 14C (1, 18, 19). For example, on the basis of conservation of mass, one possible scenario is that the highest observed mPMA OM Δ14C value (−6‰) could have resulted from a mixture of 19 to 40% RDOC (−1000 to −457‰) and as much as 60 to 81% of riverine DOC (+242 to +295%) that was modestly enriched with bomb 14C. The small mPMA sample sizes in this study precluded additional measurements, such as δ13C, that could have been used to constrain the influence of terrestrial OM, if any, on the observed Δ14C values at this station.
Variability in the proportion of mPMA-associated RDOC was not attributed to differences in Chl a or the proportion of recently produced DOC among these stations because Georges Bank and coastal Rhode Island had different mPMA Δ14C values despite similar algal biomass and OC concentrations (2.49 ± 0.75 and 3.05 ± 0.59 μg liter−1 Chl a; ~90 μM DOC), while Georges Bank and the Sargasso Sea had indistinguishable mPMA Δ14C values even though the Sargasso Sea stations had considerably less Chl a and DOC (~0.04 ± 0.01 μg liter−1 Chl a; ~72.5 μM DOC) (Table 1). Since the Sargasso Sea is representative of ~90% of the ocean surface and the proportion of mPMA-associated OM is not positively correlated to Chl a (20), we assume that 19 to 40% represents a reasonable estimate of the global annually averaged proportion of RDOC in PMA OM (table S2).
DISCUSSION
Bursting bubbles generated from breaking waves at the sea surface deliver between about 8 and 50 Tg C year−1 of OM from the ocean to the atmosphere in association with PMA (21–24). Since between 19 and 40% of this material is RDOC, PMA production will remove between 2 and 20 Tg RDOC year−1 from the global ocean (Fig. 3 and table S2). Once in the atmosphere, the RDOC associated with PMA may be degraded photochemically (as discussed below), transported landward, and/or redeposited at the sea surface (Fig. 3). The production rate of PMA RDOC represents the maximum loss rate of RDOC from the ocean assuming that 100% is mineralized, degraded into more biologically labile DOC (14, 15), and/or sequestered inland.
Fig. 3. Major production and loss pathways for marine RDOC.
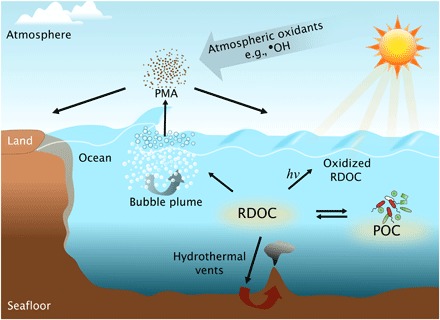
These include (i) photochemical degradation via sunlight (hν) in seawater to form CO2 and other oxidized products, some of which are biologically labile or chemically reactive; (ii) biological, chemical, and physical formation and loss processes associated with living and nonliving POC; (iii) degradation during transit through hydrothermal vent systems; and (iv) the association of RDOC with rising bubble plumes and subsequent emission into the atmosphere as a component of PMA. The RDOC associated with PMA may then be photochemically degraded (see Discussion) and/or (v) transported inland or (vi) returned to the sea.
The PMA-mediated efflux of RDOC to the atmosphere (2 to 20 Tg C year−1) is a significant component of the global RDOC budget, comparable to rates of degradation in hydrothermal systems (1 to 1.4 Tg C year−1) (11–13), biological loss in the water column (≤43 Tg C year−1) (2), and incorporation into POC in the deep ocean (25 to 50 Tg C year−1) (8, 10) but significantly less than published photochemical loss rates (300 to 1300 Tg C year−1) (table S3) (9, 25). The photochemical degradation of RDOC is larger than all other losses by nearly two orders of magnitude with uncertainties that are larger than all other processes combined. Even after considering these uncertainties, the total RDOC loss rate (326 to 1394 Tg C year−1) remains significantly larger than the estimated production rate (43 Tg C year−1) (8) by 283 to 1351 Tg year−1, with the PMA-mediated RDOC efflux exacerbating the imbalance to 285 to 1371 Tg C year−1. Since all rates in this budget are based on limited observations, this imbalance is likely due to large uncertainties associated with all sources and losses. A better understanding of RDOC cycling in the ocean requires additional high-precision observations of natural variability and modeling of all of these processes.
Marine-derived OC is highly enriched in submicrometer mPMA (particle diameter, <1 μm) relative to seawater (14, 26) with enrichments as high as 104 to 106 for particle diameters ca. ≤0.1 μm (14). Inorganic constituents of mPMA produced from open ocean seawater are not significantly enriched relative to seawater composition (26). In addition, the mass of inorganic sea salt constituents is associated primarily with the relatively short-lived supermicrometer-size fractions, whereas approximately 70% of the mass of the OM in freshly produced marine aerosol is associated with submicrometer-size fractions (14, 26) that exhibit atmospheric lifetimes against deposition of several days to a week or more and, thus, may be transported long distances from production sites. Given this lengthy residence time in the atmosphere and the expected rapid (hours) chemical evolution of marine-derived PMA under highly acidic and oxidizing conditions (15), it is reasonable to conclude that most RDOC associated with PMA will chemically evolve through primary and secondary photochemical reactions (Fig. 3). As PMA ages, these processes could generate biologically labile organic compounds including aldehydes, ketones, and organic acids, as well as mineralized products (e.g., carbon dioxide, carbon monoxide, nitrate, ammonia, and phosphate). Subsequent deposition of these marine-derived aerosols to the ocean or land will deliver substrates and nutrients to fuel aquatic food webs. In addition to the production of biological substrates and photomineralized products, the photochemical aging of PMA RDOC will also result in the photochemical formation of reactive volatile products (e.g., CO, glyoxal, acetaldehydye, and halogens) and radical transients (OH, peroxides, etc.) (15, 27) that will affect NOx cycling, ozone formation, and secondary organic aerosol production, especially in marine regions remote from anthropogenic influences.
Our estimated total removal flux of RDOC via PMA production represents upper limits for the combined losses of transport to land and oxidation in the atmosphere to biologically labile or volatile forms of DOC since not all products of atmospheric oxidation will be biologically labile or volatile, as polymerization reactions may also occur. Additional research is needed to further constrain the fluxes. Notwithstanding these limitations, we expect that the production of PMA represents a significant removal pathway for RDOC from the oceans not only in terms of its impact on the cycling of carbon in the oceans but also with respect to its expected impact in the atmosphere. These impacts will be sensitive to increases in surface ocean turbulence predicted to accompany climate change (28), which are expected to increase fluxes of PMA RDOC to the atmosphere. Overall, the results of this study have wide-ranging implications for the interrelated disciplines of chemical and biological oceanography, atmosphere-ocean interactions, atmospheric chemistry, the global carbon cycle, and climate.
MATERIALS AND METHODS
Study sites
A research cruise (EN-589) was conducted from mid-September to mid-October 2016 aboard the R/V Endeavor to study chemical and physical properties of mPMA at four hydrographic stations in the northwestern Atlantic Ocean. Two stations were in biologically productive waters at Georges Bank and off Block Island in coastal Rhode Island, and two stations were in the oligotrophic Sargasso Sea (SSW and SSN) (Fig. 1). Associated physical and chemical characteristics of each hydrographic station are presented in Table 1.
mPMA generator
The design and operation of the mPMA generator are described in detail elsewhere (16), with the following modifications. The lower portion of the model ocean in the generator was reconfigured to accommodate two machined Teflon, force-air Venturi nozzles that produced adjustable bubble-size distributions bracketing those generated by breaking waves on the surface of the open ocean (29–32). The Venturi nozzles replaced the banks of sintered glass frits, the pipe section housing, the air manifold, and associated plumbing used in the design by Long et al. (16). In the reconfigured design, two ports were added to mount the Venturi nozzles to the lower portion of the model ocean at depths of 35 and 74 cm below the air-water interface. All results reported here corresponded to mPMA produced using the 74-cm-deep Venturi nozzle.
Unfiltered seawater from a depth of ~5 m was delivered by the ship’s clean seawater line to the base of the generator at 4 liters min−1. Seawater exited the generator by draining evenly over an annular rim at the model air-water interface, which continuously removed the seawater surface and minimized formation of standing bubble rafts that suppress mPMA production (26, 33). Ultrapure sweep air hydrated to a relative humidity of 80 ± 2% was flowed through the headspace above the seawater reservoir at 70 liters min−1 to entrain mPMA produced by bubbles that rose to and burst at the air-water interface. Particles and reactive trace gases in the feed air were removed upstream of the generator by pumping high-efficiency particulate air (HEPA) filtered air through an in-line 47-mm-diameter quartz-fiber filter (Pall Tissuquartz, no. 2500 QAT-UP), two columns containing activated charcoal positioned in tandem, a second quartz fiber filter, and three mist chamber assemblies containing high-purity deionized, low-OC water (26). The generator was a closed system maintained at slight positive pressure with respect to the ambient atmosphere, which precluded contamination from the ambient atmosphere during PMA generation, collection, and characterization of physical properties. Blank tests revealed no detectable particles (<2 cm−3) in the headspace air.
Properties of bubble plumes produced by the Venturi within the generator (including plume depth, void fraction, bubble size distribution, Hinze scale, and bubble residence time in the water column) overlapped the reported ranges of those produced in the surface ocean by ambient wind waves [e.g., see (29–32)]. These results indicated that bubble plumes within the generator and, by extension, the corresponding mPMA production via bubble bursting were reasonably representative of ambient conditions over the open ocean.
The sweep air transferred particles produced by bubble bursting in the generator to the sizing instruments and sampling devices and was not meant to mimic winds over the sea surface. The relative humidity of sweep air was set to ~80% so that particles dehydrated to conditions representative of those in the marine boundary layer before characterization. However, the total flow rate of sweep air was prescribed operationally on the basis of the combined flow rates required by sizing instruments and sampling devices.
Seawater characterization
Sea surface temperature (SST) and salinity were determined at each station with Sea-Bird Electronic SBE conductivity, temperature, and pressure (CTD) sensors attached to a rosette used to collect seawater. For Chl a, unfiltered seawater was collected from Niskin bottles into 1-liter brown high-density polyethylene bottles or 5-liter polyethylene Cubitainers, and then 40 to 2000 ml of each sample was filtered through a glass microfiber filter (Grade GF/F). Filters were folded and placed into 10-ml borosilicate test tubes that were tightly capped and stored at −20°C until analysis was performed on the ship. Chl a was extracted from the filters by adding 4 ml of 90% acetone in Milli-Q water and allowing the samples to extract overnight at −20°C. Extracted samples were analyzed for Chl a according to standard methods (34). SST, salinity, and Chl a were also measured with R/V Endeavor’s array of underway sensors and continuous in-line seawater system, which collected seawater from ~5 m below the sea surface.
Seawater DOC and DIC analyses
Seawater for analysis of DOC was collected following established procedures (35). Briefly, seawater samples were gravity-filtered from 30-liter Niskin bottles through prebaked 47-mm-diameter Whatman GF/F filters (no. 1825-047) mounted in precleaned stainless steel filter holders (Pall Life Sciences, no. 2220) directly into clean 1-liter amber Boston round bottles (Chemglass, no. CG-827-06). Each bottle was rinsed three times with the sample before filling with ~750 ml of seawater. Each filled bottle was placed in a polyethylene zip-sealed bag and stored frozen (−20°C). Before use, sample bottles were precleaned with Dr. Weigert neodisher LaboClean UW detergent, 10% HCl, Milli-Q water (18.2 megohm∙cm), combusted at 550°C for 2 hours, sealed with acid-rinsed (10% HCl) Teflon-lined caps (Chemglass, no. CG-895-09), and stored in polyethylene zip-sealed bags. Whatman filters were precombusted at 500°C for 2 hours inside separate, previously cleaned aluminum foil envelopes [rinsed with dilute detergent, deionized water, 10% HCl, and Milli-Q water, and then combusted at 500°C for 2 hours]. Each baked filter was held in its aluminum foil envelope and stored inside a polyethylene zip-sealed bag. Filter holders were disassembled, precleaned by soaking in 10% HCl, rinsed with Milli-Q water, reassembled, wrapped in precleaned aluminum foil, and stored in polyethylene zip-sealed bags.
DOC was extracted from the seawater samples by ultraviolet (UV) oxidation to CO2 (35, 36) at the University of California, Irvine (UCI). Samples were diluted with Milli-Q water to ~1 liter, acidified with 1 ml of 85% high-performance liquid chromatography–grade phosphoric acid, purged of residual DIC with ultrahigh purity (UHP) He, and oxidized under UV radiation for 4 hours. The resulting CO2 was stripped from the seawater with UHP He, cryogenically purified, manometrically quantified, and split into separate aliquots for δ13C and Δ14C measurements by isotope ratio mass spectrometry (IRMS) and accelerator mass spectrometry (AMS), respectively (see below). DOC concentrations were calculated from the volumes of seawater oxidized, and corresponding blank-corrected manometric yields of CO2.
DIC samples were collected directly from the Niskin bottles through silicone tubing into custom 500-ml borosilicate bottles and poisoned with 100 μl of saturated aqueous solution of HgCl2 according to convention (37). Sample bottles were sealed with Apiezon N Grease and solid ground glass stoppers and then stored in polyethylene zip-sealed bags at room temperature. Before use, bottles and glass stoppers were precleaned and stored using procedures identical to those described above for DOC Boston round bottles. DIC was extracted from seawater as CO2 at UCI by the headspace extraction method and split into separate aliquots for δ13C and Δ14C measurements (see below) (38).
Sampling and analysis of mPMA for OC
Paired mPMA samples were collected in parallel at 30 liters min−1 on precombusted 47-mm-diameter quartz fiber filters (Pall Tissuquartz, no. 2500 QAT-UP) mounted in precleaned stainless steel in-line holders (Pall Life Sciences, no. 2220). One set of samples was processed and analyzed for OC concentrations and 14C at UCI, and the second paired set was analyzed for major ions by high-performance ion chromatography at the University of Virginia. Major ion data were not evaluated in our study. They are available through our project website (www.bco-dmo.org/project/708310). Sampling times ranged from 12 (discrete daytime or nighttime) to 24 hours. The initial pretreatment procedures for filters and holders were the same as those used for seawater sampling. In addition, pairs of filters were rebaked and held warm (500°C) in a small kiln (Skutt Firebox 8×6) for at least 2 hours immediately before use. The filter housing was loaded and unloaded in a Class 100 clean bench. Exposed filters for analysis of OC and 14C were folded in half, transferred back into their original aluminum foil envelopes and polyethylene bags, and stored frozen.
In addition to mPMA samples, dynamic handling blanks were also evaluated for all analytes. These blanks were prepared by loading filters into housings, mounting on the sampling port, and removing after several seconds with no air flow. Blanks were subsequently processed using the same procedures as samples.
At UCI, the quartz filters were fumigated with HCl to remove carbonates (below). Briefly, each filter was transferred into a precleaned, prebaked, 60-mm-diameter borosilicate petri dish and cored with a custom 38-mm ID cork borer to remove the edge and reduce the blank. Batches of six filters in their petri dishes were fumigated inside an all-glass desiccator for 2 hours with 20 ml of concentrated HCl that was held in a separate 60-mm-diameter petri dish. The desiccator lid was sealed to the kettle with 85% phosphoric acid to reduce contamination from the decomposition or degassing of O-rings or greases.
The mPMA OM was converted to CO2 for 14C analysis via closed double-tube combustion. Briefly, the fumigated filter centers were rolled with precleaned tweezers and inserted into 9-mm outer diameter (OD) × 6.5-cm-long quartz test tubes that were held inside 12-mm OD × 20 cm-long-quartz test tubes. The shorter inner tubes were preloaded with ~60 mg of cupric oxide (CuO, an oxidant) and two grains of silver wire, precombusted at 850°C for 2 hours, and preloaded into the precombusted 12-mm tubes before inserting the filters. The tubes were evacuated to ~21 mtorr, flame-sealed, combusted at 850°C for 2 hours, and allowed to cool overnight. Five of the 28 filters collected at sea were lost to quartz tube devitrification and rupture during combustion. The CO2 produced from each of the remaining 23 filters was cryogenically purified and manometrically quantified in a vacuum line. The small mass of each mPMA sample (5 to 40 μg C) precluded complementary IRMS δ13C measurements, and therefore, the samples were reserved entirely for 14C measurements.
Acid fumigation of mPMA filters
The fumigation and double–closed tube combustion procedures were developed by exposing a series of blank filters loaded with powdered calcite (0.1 to 9 mg CaCO3) to HCl vapors in a desiccator. Following fumigation, residual carbonates were isolated as CO2 by acidification with 85% H3PO4 in 5-ml Monoject vacutainer vials and manometrically quantified on a vacuum line. Residual carbonate was undetectable after 36 min of fumigation on filters that originally supported up to 38 μg C and after 2 hours of fumigation on filters that originally supported up to 134 μg C. This was sufficiently long to remove the maximum anticipated mass of bicarbonate on the filters (≤22 μg C filter−1), estimated from the maximum concentration of mPMA Na+ observed during our EN-589 cruise (8579 nmol Na+ m−3), a typical ratio of HCO3− to Na+ in seawater (~0.005 mol mol−1), the average air flow rate (0.03 m3 min−1 filter−1), and the maximum duration of mPMA sampling (24 hours) (26, 39). Fumigation for 2 hours was deemed sufficiently long to also remove any additional mass from carbonate ions (<80 μg C filter−1) in the unlikely event that all Ca2+ in mPMA was exclusively CaCO3 at concentrations equal to the maximum observed during EN-589 (153 nmol Ca2+ m−3).
Isotopic analyses of DOC, DIC, and mPMA OC
All isotopic measurements were performed by the Keck Carbon Cycle Accelerator Mass Spectrometry (KCCAMS) laboratory at UCI. Aliquots of CO2 extracted from DOC, DIC, and mPMA OM were reduced to graphite using the closed-tube, zinc method (40, 41) before 14C analyses by accelerator mass spectrometry (AMS). The δ13C values of DOC and DIC were measured on separate aliquots of CO2 using a Thermo Electron DELTA Plus IRMS equipped with a Gas Bench II. All isotope results were corrected for graphitization and machine backgrounds by KCCAMS. All 14C abundances were also corrected for isotopic fractionation by KCCAMS, via simultaneous measurements of 12C, 13C, and 14C abundances on the accelerator mass spectrometer. All 14C results were reported according to accepted conventions (17). Briefly, Δ14C values were corrected to the year of sample collection (i.e., 2016), any 14C ages less than 200 14C years were termed “modern,” and 14C ages corresponding to dates after 1950 were termed “> modern” (tables S1 and S4 to S7).
Sample-handling blank filters (table S4) were used to correct raw mPMA OM masses and 14C abundances (table S5) for OC that may have accumulated on the quartz filters during standard operating procedures. One blank (UCI AMS no. 192366) yielded an anomalously high mass of C (504 μg C) compared to all others (2 to 9 μg C) and was rejected on the basis of Chauvenet’s criterion (table S4). Specifically, the Z score for UCID 192366’s mass exceeded a two-tailed critical value of 1.96 = 1 − 1/4n for normally distributed data (n = 10) and therefore was excluded from further analyses (42). Neither the masses nor 14C abundances of the remaining blanks exhibited statistically significant differences between stations or trends over time. Therefore, all mPMA samples were corrected using the mean mass and fraction modern (Fm) of the remaining nine sample-handling blanks based on conservation of mass (Eqs. 1 and 2). Corrected conventional 14C ages and Δ14C values were then calculated from the corrected Fm values (17)
| (1) |
| (2) |
The mPMA blank masses and 14C abundances had SDs that were ~10× greater than the uncertainty of the mean propagated from individual manometric (pressure, temperature, and volume) and AMS measurement uncertainties, suggesting that the observed blank variability was largely due to filter handling rather than instrument precision. Weighted errors of the mean ( were also examined because the uncertainties of individual 14C abundances varied considerably (e.g., Δ14C with ±7 to ±103‰; table S4) (43) but were found to be even smaller than the propagated SDs of the means and not considered further. SEs, which represent likelihoods of the mean blank properties rather than the likelihood of an additional blank measurement, were intermediate between SDs and propagated SDs of the mean, and therefore used as the best estimates for propagating uncertainties through the blank corrections (tables S1, S4, and S5). Except for mPMA samples from the coastal Rhode Island station, all blank-corrected mPMA OM samples generated from 5 m of seawater had an average Δ14C value of −164 ± 20‰ (n = 5; Table 2 and table S1). This uncertainty was equal to the single SD propagated from individual measurement uncertainties (±21‰), which suggested that the range of observed mPMA OM Δ14C values at these sites was explained by methodological precision rather than natural variability.
mPMA RDOC isotopic source apportionment
The maximum proportions of RDOC (XRDOC/PMA) in each mPMA OM sample (Table 2) were estimated from conservation of mass and the 14C signatures of organic source materials (Eq. 3). The proportions were calculated with Fm, but equivalent values would have been obtained from Δ14C values because radioactive decay corrections for sample storage periods were identical for all samples studied here
| (3) |
The proportions of RDOC in PMA OM calculated by this method were insensitive to whether any or all the modern PMA OM originated from DOC or POC, assuming that DIC, POC, and recently produced DOC in the near-surface mixed layer had nearly the same Δ14C values (e.g., Δ14CDIC ≈ Δ14CPOC). For example, if rising bubbles scavenged all DOC constituents nonselectively, then the Δ14C value bulk DOC in seawater would have been transferred with fidelity with the DOC to the PMA. In these cases, the maximum proportions of RDOC in mPMA OM would have been equal to the proportions of DOC in mPMA (XDOC/PMA) multiplied by the proportion of RDOC in DOC found in seawater (XRDOC/DOC). As before, these two proportions were estimated by 14C-based expressions for conservation of mass (Eq. 4). Under these assumptions, substituting the Δ14C value of near-surface DIC for the Δ14C values of POC reduced Eq. 4 to Eq. 3, which was the overall proportion of RDOC in PMA
| (4) |
Accordingly, 14C-based conservation of mass arguments yielded the same proportions of RDOC in PMA OM regardless of whether it was assumed that PMA OM originated exclusively from DOC or, at the other extreme, exclusively from POC and RDOC.
Global mPMA RDOC production rate assumptions
Coastal Rhode Island mPMA Δ14C values indicated that the flux of RDOC to the atmosphere with PMA was possibly smaller and more variable in coastal environments than in the open ocean. Since the Sargasso Sea environment is representative of ~90% of the ocean surface (i.e., oligotrophic, “blue” water), we assumed that the average proportion of RDOC in mPMA observed at Georges Bank and the Sargasso Sea represented a reasonable estimate for the annual average proportion found throughout the world oceans. Accordingly, the range of globally averaged annual rates of RDOC transferred from the ocean to the atmosphere via PMA formation (2 to 20 Tg C year−1) was constrained by the products of the ranges of published globally averaged annual PMA OM production rates (8 to 50 Tg C year−1) (21–24) and the ranges of 14C-based proportions of RDOC in mPMA OM (19 to 40%) (table S2).
Estimating the concentration of aerosolizable DOC and DO14C in seawater
The concentration of DOC aerosolized in near-surface seawater was estimated for each mPMA sample from the total volume of seawater that passed through the generator (e.g., V = 4 liters min−1 integrated over 12 or 24 hours), the branching ratio of sweep air (r = 30 liters min−1/70 liters min−1) that delivered aerosol to the quartz filters, the blank-corrected mass of aerosol OC (ma) collected per filter (table S1), and the molar mass of carbon (mC = 12.011 g mol−1) (Eq. 5)
| (5) |
Accordingly, aerosolized DOC concentrations ranged from 0.48 to 1.25 nM at the coastal Rhode Island station and from 0.17 nM (Sargasso Sea North) to 1.06 nM (Georges Bank) at all other stations. Dividing these concentrations by near-surface DOC concentrations at each station yielded the percentage of aerosolized DOC, which ranged from 0.0005 to 0.0014% at the coastal Rhode Island station and from 0.0002 (Sargasso Sea North) to 0.0011% (Georges Bank) at all other stations.
The concentration of 14C atoms associated with aerosolized DOC in seawater (Eq. 6) was estimated from the molar concentration of aerosolized DOC (Eq. 5), the blank-corrected fraction modern value (table S1), and the 14C/12C ratio of the radiocarbon community’s absolute international standard activity (AISA = 1.176 × 10−12), assuming mPMA δ13C ≈ −25‰ to simplify the calculation
| (6) |
On the basis of this calculation, the concentration of 14C atoms associated with aerosolized DOC in seawater ranged from 340 to 850 atoms liter−1 (0.6 to 1.4 zM) at the coastal Rhode Island station and from 100 (Sargasso North) to 650 (Georges Bank) 14C atoms liter−1 at all other stations (0.2 to 1.1 zM). Assuming an AMS 14C efficiency of 5%, these concentrations correspond to having counted approximately 2 to 20 aerosolized DOC 14C atoms liter−1 of near-surface seawater per quartz filter.
Supplementary Material
Acknowledgments
We thank the captain and crew of the R/V Endeavor for logistical support; E. Druffel, S. Griffin, B. Walker, J. Walker, X. Xu, J. Southon, and G. dos Santos at UCI for laboratory support and 14C measurements; M. Kurz and A. McNichol at Woods Hole Oceanographic Institution for laboratory support; and four anonymous reviewers for constructive comments on the original manuscript. Funding: Financial support was provided by the U.S. National Science Foundation through awards to the State University of New York, College of Environmental Science and Forestry (OCE-1536605 to D.J.K.); University of Virginia (OCE-1536674 to W.C.K.); Stony Brook University (OCE-1536597 to S.R.B.); and Harvard University (OCE-1536608 to M.S.L.). Additional support was provided by the University of Georgia Investment in Sciences initiative and Office of Research. Author contributions: D.J.K., W.C.K., S.R.B., and M.S.L. designed the research. S.R.B. was responsible for all 14C analyses; D.J.K. and J.D.K. for chemical characterization of seawater; W.C.K., A.A.F., P.D., and R.Y.-W.C. for physical and chemical characterization of mPMA; and W.C.K., J.R.M., and M.S.L. for generating mPMA. S.R.B. and D.J.K. evaluated the 14C data and wrote the main drafts of the manuscript, which were reviewed and edited by all authors. All authors participated in field sampling and contributed to the generation and interpretation of results reported herein. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Data for this paper and our research cruise are also archived at the National Science Foundation, Division of Ocean Sciences’ Biological and Chemical Oceanography Data Management Office (BCO-DMO) at the following project website: www.bco-dmo.org/project/708310.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6535/DC1
Table S1. Blank-corrected mPMA sample masses and 14C abundances.
Table S2. Globally averaged rates of RDOC transfer from the ocean to the atmosphere via PMA formation.
Table S3. Published sources and losses of marine RDOC.
Table S4. mPMA handling blank masses and 14C abundances.
Table S5. Uncorrected mPMA sample masses and 14C abundances.
Table S6. Seawater DOC concentrations, δ13C values, and 14C abundances.
Table S7. Seawater DIC concentrations, δ13C values, and 14C abundances.
REFERENCES AND NOTES
- 1.S. R. Beaupré, in Biogeochemistry of Marine Dissolved Organic Matter, 2nd Edition. D. A. Hansell, C. A. Carlson, Eds. (Academic Press, 2015), pp. 335–368. [Google Scholar]
- 2.Hansell D. A., Recalcitrant dissolved organic carbon fractions. Ann. Rev. Mar. Sci. 5, 421–445 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Williams P. M., Druffel E. R. M., Radiocarbon in dissolved organic matter in the central North Pacific Ocean. Nature 330, 246–248 (1987). [Google Scholar]
- 4.Stuiver M., Quay P. D., Ostlund H. G., Abyssal water carbon-14 distribution and the age of the world oceans. Science 219, 849–851 (1983). [DOI] [PubMed] [Google Scholar]
- 5.Beaupré S. R., Aluwihare L., Constraining the 2-component model of marine dissolved organic radiocarbon. Deep-Sea Res. II Top. Stud. Oceanogr. 57, 1494–1503 (2010). [Google Scholar]
- 6.Beaupré S. R., Druffel E. R. M., Constraining the propagation of bomb-radiocarbon through the dissolved organic carbon (DOC) pool in the northeast Pacific Ocean. Deep-Sea Res. I Oceanogr. Res. Pap. 56, 1717–1726 (2009). [Google Scholar]
- 7.C. A. Carlson, D. A. Hansell, in Biogeochemistry of Marine Dissolved Organic Matter, D. A. Hansell, C. A. Carlson, Eds. (Academic Press, 2015), pp. 65–126. [Google Scholar]
- 8.Hansell D., Carlson C., Repeta D., Schlitzer R., Dissolved organic matter in the ocean: A controversy stimulates new insights. Oceanography 22, 202–211 (2009). [Google Scholar]
- 9.Mopper K., Zhou X., Kieber R. J., Kieber D. J., Sikorski R. J., Jones R. D., Photochemical degradation of dissolved organic carbon and its impact on the oceanic carbon cycle. Nature 353, 60–62 (1991). [Google Scholar]
- 10.Druffel E. R. M., Williams P. M., Identification of a deep marine source of particulate organic carbon using bomb 14C. Nature 347, 172–174 (1990). [Google Scholar]
- 11.Hawkes J. A., Rossel P. E., Stubbins A., Butterfield D., Connelly D. P., Achterberg E. P., Koschinsky A., Chavagnac V., Hansen C. T., Bach W., Dittmar T., Efficient removal of recalcitrant deep-ocean dissolved organic matter during hydrothermal circulation. Nat. Geosci. 8, 856–860 (2015). [Google Scholar]
- 12.Lang S. Q., Butterfield D. A., Lilley M. D., Paul Johnson H., Hedges J. I., Dissolved organic carbon in ridge-axis and ridge-flank hydrothermal systems. Geochim. Cosmochim. Acta 70, 3830–3842 (2006). [Google Scholar]
- 13.Shah Walter S. R., Jaekel U., Osterholz H., Fisher A. T., Huber J. A., Pearson A., Dittmar T., Girguis P. R., Microbial decomposition of marine dissolved organic matter in cool oceanic crust. Nat. Geosci. 11, 334–339 (2018). [Google Scholar]
- 14.Kieber D. J., Keene W. C., Frossard A. A., Long M. S., Maben J. R., Russell L. M., Kinsey J. D., Tyssebotn I. M. B., Quinn P. K., Bates T. S., Coupled ocean-atmosphere loss of marine refractory dissolved organic carbon. Geophys. Res. Lett. 43, 2765–2772 (2016). [Google Scholar]
- 15.Zhou X., Davis A. J., Kieber D. J., Keene W. C., Maben J. R., Maring H., Dahl E. E., Izaguirre M. A., Sander R., Smoydzyn L., Photochemical production of hydroxyl radical and hydroperoxides in water extracts of nascent marine aerosols produced by bursting bubbles from Sargasso seawater. Geophys. Res. Lett. 35, (2008). [Google Scholar]
- 16.Long M. S., Keene W. C., Kieber D. J., Frossard A. A., Russell L. M., Maben J. R., Kinsey J. D., Quinn P. K., Bates T. S., Light-enhanced primary marine aerosol production from biologically productive seawater. Geophys. Res. Lett. 41, 2661–2670 (2014). [Google Scholar]
- 17.Stuiver M., Polach H. A., Discussion reporting of 14C data. Radiocarbon 19, 355–363 (1977). [Google Scholar]
- 18.Mortazavi B., Chanton J. P., Use of Keeling plots to determine sources of dissolved organic carbon in nearshore and open ocean systems. Limnol. Oceanogr. 49, 102–108 (2004). [Google Scholar]
- 19.Raymond P. A., Bauer J. E., Riverine export of aged terrestrial organic matter to the North Atlantic Ocean. Nature 409, 497–500 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Quinn P. K., Bates T. S., Schulz K. S., Coffman D. J., Frossard A. A., Russell L. M., Keene W. C., Kieber D. J., Contribution of sea surface carbon pool to organic matter enrichment in sea spray aerosol. Nat. Geosci. 7, 228–232 (2014). [Google Scholar]
- 21.Gantt B., Meskhidze N., Kamykowski D., A new physically-based quantification of marine isoprene and primary organic aerosol emissions. Atmos. Chem. Phys. 9, 4915–4927 (2009). [Google Scholar]
- 22.Long M. S., Keene W. C., Kieber D. J., Erickson D. J., Maring H., A sea-state based source function for size- and composition-resolved marine aerosol production. Atmos. Chem. Phys. 11, 1203–1216 (2011). [Google Scholar]
- 23.Roelofs G. J., A GCM study of organic matter in marine aerosol and its potential contribution to cloud drop activation. Atmos. Chem. Phys. 8, 709–719 (2008). [Google Scholar]
- 24.Spracklen D. V., Arnold S. R., Sciare J., Carslaw K. S., Pio C., Globally significant oceanic source of organic carbon aerosol. Geophys. Res. Lett. 35, 10.1029/2008GL033359, (2008). [Google Scholar]
- 25.Anderson T. R., Williams P. J. l. B., A one-dimensional model of dissolved organic carbon cycling in the water column incorporating combined biological-photochemical decomposition. Global Biogeochem. Cycles 13, 337–349 (1999). [Google Scholar]
- 26.Keene W. C., Long M. S., Reid J. S., Frossard A. A., Kieber D. J., Maben J. R., Russell L. M., Kinsey J. D., Quinn P. K., Bates T. S., Factors that modulate properties of primary marine aerosol generated from ambient seawater on ships at sea. J. Geophys. Res. Atmos. 122, 11,961–911,990 (2017). [Google Scholar]
- 27.Kasibhatla P., Sherwen T., Evans M. J., Carpenter L. J., Reed C., Alexander B., Chen Q. J., Sulprizio M. P., Lee J. D., Read K. A., Bloss W., Crilley L. R., Keene W. C., Pszenny A. A. P., Hodzic A., Global impact of nitrate photolysis in sea-salt aerosol on NOx, OH, and O3 in the marine boundary layer. Atmos. Chem. Phys. 18, 11185–11203 (2018). [Google Scholar]
- 28.IPCC, in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, P. M. Midgley, Eds. (Cambridge Univ. Press, 2013), pp. 1535. [Google Scholar]
- 29.Deane G. B., Stokes M. D., Scale dependence of bubble creation mechanisms in breaking waves. Nature 418, 839–844 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Hinze J. O., Fundamentals of the hydrodynamic mechanism of splitting in dispersion processes. AIChE J. 1, 289–295 (1955). [Google Scholar]
- 31.Baylar A., Ozkan F., Unsal M., On the use of venturi tubes in aeration. Clean 35, 183–185 (2007). [Google Scholar]
- 32.Sundararaj S., Selladurai V., Numerical and experimental study on jet trajectories and mixing behavior of venturi-jet mixer. J. Fluids Engr. 132, 101104 (2010). [Google Scholar]
- 33.Keene W. C., Maring H., Maben J. R., Kieber D. J., Pszenny A. A. P., Dahl E. E., Izaguirre M. A., Davis A. J., Long M. S., Zhou X., Smoydzin L., Sander R., Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface. J. Geophys. Res. 112, (2007). [Google Scholar]
- 34.Welschmeyer N. A., Fluorometric analysis of chlorophyll-a in the presence of chlorophyll-b and pheopigments. Limnol. Oceanogr. 39, 1985–1992 (1994). [Google Scholar]
- 35.Beaupré S. R., Druffel E. R. M., Griffin S., A low-blank photochemical extraction system for concentration and isotopic analyses of marine dissolved organic carbon. Limnol. Oceanogr. Methods 5, 174–184 (2007). [Google Scholar]
- 36.Griffin S., Beaupré S. R., Druffel E. R. M., An alternate method of diluting dissolved organic carbon seawater samples for 14C analysis. Radiocarbon 52, 1224–1229 (2010). [Google Scholar]
- 37.A. P. McNichol, P. D. Quay, A. R. Gagnon, J. R. Burton, in The GO-SHIP Repeat Hydrography Manual: A Collection of Expert Reports and Guidelines. Version 1, E. M. Hood, C. L. Sabine, B. M. Sloyan, Eds. (Technical Report, IOCCP Report Number 14, ICPO Publication Series Number 134, 2010), pp. 1–12.
- 38.Gao P., Xu X., Zhou L., Pack M. A., Griffin S., Santos G. M., Southon J. R., Liu K., Rapid sample preparation of dissolved inorganic carbon in natural waters using a headspace-extraction approach for radiocarbon analysis by accelerator mass spectrometry. Limnol. Oceanogr. Methods 12, 174–190 (2014). [Google Scholar]
- 39.Keene W. C., Pszenny A. A. P., Galloway J. N., Hawley M. E., Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. Atmos. 91, 6647–6658 (1986). [Google Scholar]
- 40.Walker B. D., Xu X. M., An improved method for the sealed-tube zinc graphitization of microgram carbon samples and 14C AMS measurement. Nucl. Instrum. Methods Phys. Sec. B 438, 58–65 (2019). [Google Scholar]
- 41.Xu X. M., Trumbore S. E., Zheng S. H., Southon J. R., McDuffee K. E., Luttgen M., Liu J. C., Modifying a sealed tube zinc reduction method for preparation of AMS graphite targets: Reducing background and attaining high precision. Nucl. Instrum. Methods Phys. Sec. B 259, 320–329 (2007). [Google Scholar]
- 42.D. M. Glover, W. J. Jenkins, S. C. Doney, Modeling Methods for Marine Science (Cambridge Univ. Press, 2011). [Google Scholar]
- 43.P. R. Bevington, D. K. Robinson, Data Reduction and Error Analysis for the Physical Sciences (McGraw-Hill, ed. 3, 2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6535/DC1
Table S1. Blank-corrected mPMA sample masses and 14C abundances.
Table S2. Globally averaged rates of RDOC transfer from the ocean to the atmosphere via PMA formation.
Table S3. Published sources and losses of marine RDOC.
Table S4. mPMA handling blank masses and 14C abundances.
Table S5. Uncorrected mPMA sample masses and 14C abundances.
Table S6. Seawater DOC concentrations, δ13C values, and 14C abundances.
Table S7. Seawater DIC concentrations, δ13C values, and 14C abundances.