

Both the endothelial transporter Mfsd2a and docosahexaenoic acid uptake are disrupted in Zika virus–induced microcephaly models.
Abstract
Zika virus (ZIKV) infection during pregnancy increases the risk of postnatal microcephaly. Neurovascular function provides a homeostatic environment for proper brain development. The major facilitator superfamily domain-containing protein 2 (Mfsd2a) is selectively expressed in human brain microvascular endothelial cells (hBMECs) and is the major transporter mediating the brain uptake of docosahexaenoic acid (DHA). We have discovered a pivotal role for Mfsd2a in the pathogenesis of ZIKV. ZIKV disrupted Mfsd2a both in cultured primary hBMECs and in the neonatal mouse brain. ZIKV envelope (E) protein specifically interacted with Mfsd2a and promoted Mfsd2a polyubiquitination for proteasome-dependent degradation. Infection with ZIKV or ectopic expression of ZIKV E impaired Mfsd2a-mediated DHA uptake. Lipidomic analysis revealed obvious differences in DHA-containing lipids after ZIKV infection. Supplementation with DHA rescued ZIKV-caused growth restriction and microcephaly. Our findings suggest endothelial Mfsd2a as an important pathogenic mediator and supplementation with DHA as a potential therapeutic option for ZIKV infection.
INTRODUCTION
Zika virus (ZIKV) is an emerging mosquito-borne virus in the genus Flavivirus and the family Flaviviridae (1). Although ZIKV was first identified in Uganda in 1947 in rhesus monkeys, a widespread epidemic of ZIKV infection during 2015–2016 in South and Central America raised major concerns because ZIKV infection during pregnancy increases the risk of congenital microcephaly and severe fetal brain defects (2). In congenital infected fetal brain samples, ZIKV particles and viral RNA were found and associated with agyria, hydrocephalus, and multifocal dystrophic calcifications (3). In mice and cultured cells, ZIKV frequently leads to increased death of neural progenitor cells (NPCs) (4). Despite all these findings, the mechanism of ZIKV-associated fetal brain defects has not been elucidated to a large extent, especially with respect to vascular brain microenvironment changes.
Embryonic brain development depends on the transfer of energy, nutrition, and signals from the bloodstream (5). The blood-brain barrier (BBB), composed of brain microvascular endothelial cells (BMECs), provides a constant homeostatic brain environment by preventing vascular leakage and promoting specific transport (5). Docosahexaenoic acid (DHA) is an essential ω-3 fatty acid that is highly enriched in brain phospholipids and critical for normal brain growth and cognitive function (6). However, DHA cannot be synthesized de novo by mammals to maintain a normal DHA composition (7). The major route of DHA uptake into the brain is transport by a sodium-dependent lysophosphatidylcholine (LPC) symporter, namely, major facilitator superfamily domain-containing 2a (Mfsd2a), which is selectively expressed in BMECs (8). Inactivating mutations in human Mfsd2a causes a lethal microcephaly syndrome (9). In addition, genetic ablation of Mfsd2a in mice results in a leaky BBB, reduced uptake of LPC DHA into the brain, cognitive deficits, and, importantly, microcephaly (10).
Here, we report an important role of Mfsd2a in ZIKV infection and neuropathogenicity. We found that ZIKV infection of human BMECs (hBMECs) substantially reduced Mfsd2a levels by viral envelope protein through a proteasome-dependent pathway. ZIKV infection blocked LPC uptake and altered DHA-containing lipids. Administration of DHA could rescue ZIKV infection–induced abnormal brain development in mice. Our results reveal a previously unidentified pathogenic mechanism and potential interventions for ZIKV-related symptoms.
RESULTS
ZIKV infection decreases Mfsd2a at the protein level
ZIKV infection leads to an abnormal brain vasculature morphology, BBB leakage (11), and microcephaly (3), which strongly suggests a disruption of the substantial function of Mfsd2a (9). To investigate whether the Mfsd2a expression level changes during ZIKV infection, we established a cell culture model of ZIKV-infected primary hBMECs. Endothelial marker CD31 was well expressed in the junction of adjacent cells (fig. S1, A and B). In addition, the commercial antibody against Mfsd2a was characterized using brain tissues isolated from wild type, heterozygotes, or mfsd2a knockout mice (fig. S1C). Chinese ZIKV isolate SZ01 (12) and South American ZIKV isolate PRVABC59 (13) readily infected primary hBMECs without reducing cell viability (fig. S2). In primary hBMECs, endogenous Mfsd2a protein was notably down-regulated after ZIKV infection in both a time course– and a multiplicity of infection (MOI)–dependent manner (Fig. 1, A and B). The same pattern of Mfsd2a down-regulation by ZIKV was confirmed in human placenta choriocarcinoma JEG-3 cells that expressed high levels of Mfsd2a (fig. S4). However, Mfsd2a mRNA levels were unaltered following infection (Fig. 1C), suggesting that posttranscriptional modification occurred. In addition to the endogenous Mfsd2a, we made a stable A549 cell line ectopically expressing a green fluorescent protein (GFP)–tagged Mfsd2a and demonstrated a similar suppression of Mfsd2a levels by ZIKV infection using immunofluorescence staining (Fig. 1D). Western blotting bands for Mfsd2a and ZIKV envelope (E) in Fig. 1 were quantified with ImageJ software, and fold change was shown in fig. S3.
Fig. 1. ZIKV infection decreases Mfsd2a at the protein level both in vitro and in vivo.
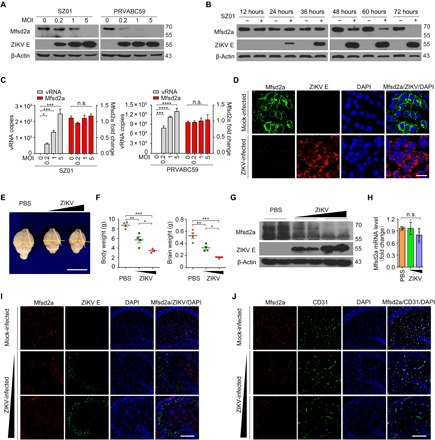
(A and B) hBMECs were challenged with ZIKV SZ01 or PRVABC59 at the indicated MOI for 48 hours (A) or were infected with ZIKV SZ01 at an MOI of 1 for various time courses (B). A representative Western blot analysis (of n = 3 independent experiments) is demonstrated. The expression of Mfsd2a, ZIKV E, and β-actin was assessed. (C) hBMECs were challenged with SZ01 or PRVABC59, and viral RNA (vRNA) and Mfsd2a mRNA were determined by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR). Data are presented as means ± SD of n = 3 experiments run with duplicate samples. n.s., not significant. (D) A549 stable cells expressing Mfsd2a-GFP were infected with ZIKV strains. Immunofluorescent (IF) staining with ZIKV E antibody was performed using the indicated antibodies. Scale bar, 20 μm. (E to G) The neonatal BALB/c mice were infected with ZIKV via intracerebral injection with 10 or 100 plaque-forming units (PFU). The mice were euthanized at 11 days post-infection (dpi) to isolate the brain tissues. The brain morphology (E) (scale bar, 1 cm), body weights and brain weights (F), protein levels of Mfsd2a and ZIKV E in the brain (G), and Mfsd2a mRNA level (H) were measured by weighing, qRT-PCR, or Western blotting. PBS, phosphate-buffered saline. (I and J) Representative IF images (of n = 4 mice per treatment) of mouse brain hippocampus dentate gyrus serial pathological section by staining for ZIKV E, Mfsd2a, the endothelial cell marker CD31, and the nuclei with DAPI (4′,6-diamidino-2-phenylindole) in the infected or control brains. Scale bars, 200 μm. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, by one-way analysis of variance (ANOVA). Photo credit: Jia Zhou, Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College.
To further explore the regulation of Mfsd2a by ZIKV in vivo, using the intracerebral inoculation of ZIKV into neonatal mice, we found that ZIKV infection leads to postnatal growth restriction, including microcephaly (Fig. 1, E to H, and fig. S5). With an increased inoculation dose, ZIKV inhibited brain Mfsd2a protein levels in mice (Fig. 1G) without influencing Mfsd2a mRNA levels (Fig. 1H). Brain regions near the hippocampus were serially sectioned and double-stained for Mfsd2a/ZIKV E or Mfsd2a/CD31, respectively. As shown in Fig. 1 (I and J), the morphologies of stained Mfsd2a and CD31 are tubular shape with length-cutting and dot shape with cross-cutting in the uninfected brain and are mostly colocalized. With increased virus dosage, Mfsd2a-positive cells were markedly decreased, whereas CD31-positive cells remained unchanged (Fig. 1J and fig. S5C). In addition to intracerebral inoculation, systematic infection with ZIKV also down-regulated Mfsd2a protein in the brains of A129 immunodeficient mice (fig. S5D) and the embryonic brains of pregnant Institute of Cancer Research (ICR) mice (fig. S5E). Together, these results confirm that Mfsd2a is a specific target inhibited by ZIKV in hBMECs.
ZIKV E protein targets Mfsd2a for degradation
To further dissect the mechanism of Mfsd2a inhibition, we cotransfected Mfsd2a along with individual ZIKV-encoded proteins into human embryonic kidney (HEK) 293T cells. ZIKV E protein, but not other viral proteins, disrupted Mfsd2a expression (Fig. 2A), which is in line with immunofluorescence results indicating the loss of stable transfected Mfsd2a expression in ZIKV E–positive cells (Fig. 2B). Moreover, overexpressed ZIKV E decreased endogenous Mfsd2a protein in primary hBMECs and JEG-3 cells in a dose-dependent manner (Fig. 2C). To test the specificity, we evaluated envelope proteins from selected members of the family Flaviviridae, including West Nile virus (WNV) and hepatitis C virus (HCV), and only ZIKV E showed effectiveness (Fig. 2D). We next investigated the physical interaction of the two proteins. In a pull-down assay, recombinant His-tagged ZIKV E protein was mixed with HEK293T cell lysates containing transfected Myc-tagged Mfsd2a and captured by Ni-NTA (nitrilotriacetic acid) agarose. The results indicated a direct interaction between Mfsd2a and ZIKV E (Fig. 3A). These observations support the idea that ZIKV E is the major factor involved in Mfsd2a dysregulation during infection.
Fig. 2. ZIKV E protein disrupts Mfsd2a expression.
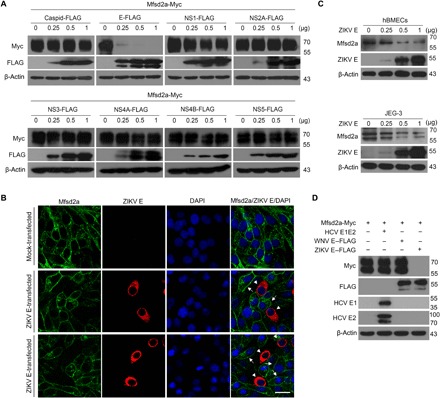
(A) HEK293T cells were cotransfected with plasmids encoding Mfsd2a-Myc and either ZIKV structural C/E or nonstructural NS1/2A/3/4A/4B/5 FLAG fusion proteins. The expression of Mfsd2a-Myc and individual ZIKV proteins was analyzed by Western blotting at 48 hours after transfection. (B) A549 stable cells expressing Mfsd2a-GFP were transfected with a ZIKV E–FLAG plasmid or an empty vector control for 48 hours. Representative GFP and ZIKV E IF confocal images (n = 3 per treatment) were captured. DAPI staining indicates the nuclei. Scale bar, 40 μm. (C) hBMECs or JEG-3 cells were transfected with various amounts of the ZIKV E–Flag plasmid or empty vector control for 48 hours, and the expression of endogenous Mfsd2a was measured by Western blot. (D) An Mfsd2a-Myc plasmid was cotransfected with one of the plasmids encoding ZIKV E–Flag, WNV E–Flag, or HCV E1E2 into HEK293T cells. The expression of Mfsd2a-Myc and individual viral envelope proteins was determined by Western blotting at 48 hours after transfection.
Fig. 3. ZIKV E protein targets Mfsd2a for degradation.
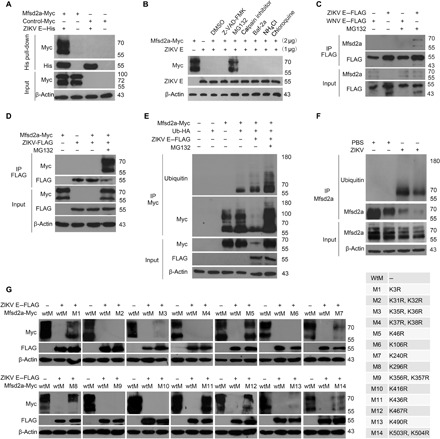
(A) Pull-down assay using recombinant His-tagged ZIKV E with the cell lysates containing transiently expressed Mfsd2a-Myc. (B) HEK293T cells were cotransfected with Mfsd2a-Myc and ZIKV E–Flag plasmids and then treated with one of the indicated pathway inhibitors (10 μM). DMSO, dimethyl sulfoxide. (C) Immunoprecipitation (IP) and immunoblot analysis of endogenous Mfsd2a with transfected ZIKV E–Flag or WNV E–Flag in HUVECs. (D) IP and immunoblot analysis of Mfsd2a-Myc with ZIKV E–Flag in HEK293T cells. (E) Coimmunoprecipitation analysis of Mfsd2a-Myc ubiquitination in HEK293T cells cotransfected with ZIKV E–Flag and ubiquitin-HA (hemagglutinin) in the presence or absence of MG132 (10 μM). (F) In vivo ubiquitination assay of endogenous Mfsd2a in mouse brain after ZIKV infection. (G) ZIKV E–mediated degradation analysis of wild-type Mfsd2a-Myc (wtM) and mutant Mfsd2a-Myc with a single point or combinational lysine mutation in HEK293T cells. Data are representative of three independent experiments with similar results.
To identify specific pathways involved in Mfsd2a degradation, Mfsd2a was cotransfected with ZIKV E in the presence of distinct pathway inhibitors. MG132 (N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal) was the only tested inhibitor preventing ZIKV E from degrading Mfsd2a (Fig. 3B and fig. S7), suggesting that the proteasome degradation pathway plays a critical role during this process. We next performed coimmunoprecipitation to investigate the interaction between endogenous Mfsd2a and ZIKV E in the presence of 10 μM MG132. When transfected into human umbilical vein endothelial cells (HUVECs), Flag-tagged ZIKV E, but not WNV E, coimmunoprecipitated with the endogenous Mfsd2a after the proteasome pathway was blocked by MG132 (Fig. 3C). Furthermore, Myc-tagged Mfsd2a was coimmunoprecipitated with Flag-tagged ZIKV E in HEK293T cells in the presence of MG132 (Fig. 3D). These findings confirm that ZIKV E targets Mfsd2a for proteasome-dependent degradation.
The degradation mechanism of Mfsd2a has never been reported. To investigate whether ZIKV E triggers Mfsd2a polyubiquitination, we cotransfected HEK293T cells with Myc-Mfsd2a and hemagglutinin (HA)–ubiquitin, followed by transfection of Flag–ZIKV E in the absence or presence of MG132. Coimmunoprecipitation experiments showed that Mfsd2a ubiquitination was markedly increased by ZIKV E (Fig. 3E). Notably, the Mfsd2a polyubiquitination level was further enhanced after MG132 treatment (Fig. 3E). In ZIKV-infected mouse brain, ubiquitinated Mfsd2a was detected by coimmunoprecipitation-coupled immunoblot analysis (Fig. 3F). Next, we made a series of Mfsd2a mutants in which individual or adjacent lysine residues were replaced with arginine. Cotransfections of ZIKV E together with each Mfsd2a mutant were performed in HEK293T cells; we found that K3R, K46R, K296R, and K436R mutants showed notable resistance against ZIKV E–mediated degradation (Fig. 3G). These data indicate that ZIKV E mediates the site-specific arginine-linked degradation of Mfsd2a.
ZIKV infection impairs DHA uptake and lipid homeostasis
The neurovascular unit is responsible for brain microenvironment maintenance and a continuous supply of nutrients, in which an influx of DHA is essential for normal brain development and cognitive function. Brain uptake of DHA is mediated by Mfsd2a only in the form of esters with LPCs, but not as free unesterified fatty acids (8). We next sought to evaluate the LPC uptake efficiency in primary hBMECs or A549 cells stably expressing Mfsd2a in the contexts of either ZIKV infection or envelope protein transfection. First, ZIKV isolates SZ01 and PRVABC59 were used, and as the infection dose increased, the hBMEC uptake of TopFluor-LPC was remarkably reduced by up to 95% at a high MOI (Fig. 4, A and B). In a separate approach to confirm Mfsd2a participation in the process of ZIKV-mediated LPC transport blockade, we generated stable Mfsd2a-overexpressing A549 cells. Compared with the negative results in parental A549 cells, Mfsd2a-overexpressing cells showed a strong uptake of TopFluor-LPC (fig. S7). Infection of ZIKV isolates or transfection of ZIKV E almost abolished LPC uptake in Mfsd2a-overexpressing A549 cells (Fig. 4, C to E). In addition, because we screened a degradation-resistant mutant of Mfsd2a (K46R, identified in Fig. 3G), we evaluated the ZIKV E–regulated LPC uptake efficiency in 293T cells overexpressing either wild-type or K46R mutant Mfsd2a. Uptake of TopFluor-LPC in wild-type Mfsd2a-transfected 293T cells was gradually inhibited by ZIKV E in a dose-dependent manner. However, the efficiency of LPC uptake was not altered in K46R mutant Mfsd2a-transfected cells (Fig. 4F). These functional observations were consistent with our previous finding that ZIKV induced Mfsd2a degradation, suggesting a dysregulation of the major mechanism by which DHA enters the brain after ZIKV infection.
Fig. 4. Uptake of TopFluor LysoPC is reduced in ZIKV-infected hBMECs.
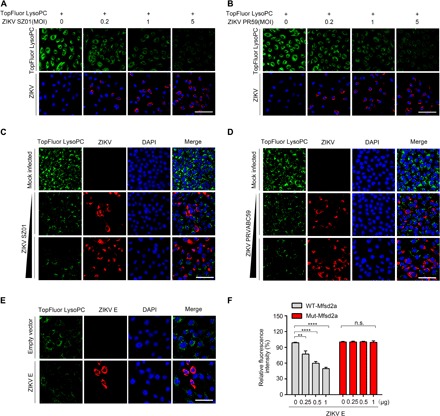
(A and B) TopFluor LysoPC (green) uptake assay in hBMECs that were infected with the ZIKV SZ01 strain (A) or PRVABC59 (PR59) strain (B) at the indicated MOI for 48 hours. IF staining of ZIKV E is shown in red. DAPI staining indicates the nuclei. Scale bars, 100 μm. (C and D) TopFluor LysoPC (green) uptake assay in A549 stable cells expressing Mfsd2a-Flag. Cells were infected with the ZIKV SZ01 strain (C) or PRVABC59 strain (D) at an MOI of 1 or 5 for 48 hours. IF staining of ZIKV E is shown in red. DAPI staining indicates the nuclei. Scale bars, 50 μm. (E) TopFluor LysoPC (green) uptake assay in A549 Mfsd2a stable cells that were transfected with either the ZIKV E plasmid or empty vector as the negative control (NC). Scale bar, 50 μm. (F) HEK293T cells were cotransfected with ZIKV E and either wild-type (WT) Mfsd2a or mutant Mfsd2a (K46R) plasmids for 48 hours. The cells were incubated with TopFluor LysoPC, and the fluorescence intensity was quantified.
Intrigued by the impaired DHA uptake, we further focused on profiling and comparing the lipid alterations in primary hBMECs with or without ZIKV infection. Using a lipidomic analysis, we found that the total level of DHA was reduced in ZIKV-infected cells compared to that in the mock infection group (Fig. 5, A and B, and table S1). Most of the DHA-containing phospholipid species in phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), and phosphatidylserine (PS) were notably decreased in the infection group (Fig. 5B). On the other hand, decreases in several non–DHA-containing phospholipids were also observed in ZIKV-infected cells (Fig. 5A). In addition, the lipidomic changes in ZIKV-infected neonatal mouse brain were also profiled (Fig. 5, C and D, and table S2). Comparable lipidomic changes were observed between in vitro and in vivo infections. These data suggest that differences in the lipid signature of ZIKV-infected cells may contribute to the inhibition of Mfsd2a expression by ZIKV infection.
Fig. 5. ZIKV infection impairs DHA uptake and lipid homeostasis.

(A) Heat map representation of the percentage of individual phospholipid species in hBMECs. The hBMECs were infected with the ZIKV SZ01 strain at an MOI of 1 for 48 hours. Mock-infected hBMECs were used as the NC. Cells were harvested for lipidomic analysis. DHA-containing phospholipid species are highlighted in red. Each group includes three experimental repeats. Ctrl, control. (B) Lipidomic analysis of the total DHA levels and DHA-containing phospholipid species in mock- or ZIKV-infected hBMECs. The results are representative of three independent experiments. The value for each species is normalized against the total peak areas. (C and D) In vivo lipidomic analysis with ZIKV-infected neonatal mouse brains. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, by one-way analysis of variance (ANOVA).
Supplementation with DHA rescues ZIKV-caused microcephaly phenotype
DHA has wide-ranging functions in the brain as components of phospholipids. Intake of ω-3 polyunsaturated fatty acids, particularly DHA, has been recommended for its potential benefits during pregnancy and for several developmental disorders, such as autistic spectrum disorders (14). In view of our in vitro findings that ZIKV impairs the uptake of LPC in hBMECs, we then attempted to investigate whether supplementation with DHA could influence the abnormal brain development in newborn mice after ZIKV infection. Intracerebral inoculation of neonatal mice represents a useful tool for modeling ZIKV-caused postnatal microcephaly and growth restriction (4). To examine the beneficial effects of DHA in vivo, we injected ZIKV SZ01 or phosphate-buffered saline (PBS) (mock) into the λ point of the mouse brain at day 0 after birth. DHA or the same amount of DMSO as a control was supplemented twice by intraperitoneal injection at postnatal day 0 (D0) and D3, respectively. At D5 and D7, the mice were weighed and were administered inhaled carbon dioxide for euthanasia. ZIKV-infected pups had a significantly smaller body and brain (Fig. 6A) than the controls. Notably, significant restoration of the ZIKV-induced growth restriction by supplementation with DHA could be observed by measuring the brain size, brain weight, and body weight (Fig. 6, B to E). Unexpectedly, the administration of DHA increased the brain Mfsd2a level and suppressed ZIKV RNA copies in the brain at D5 (Fig. 6, F to H) and D7 (Fig. 6, G and I). Together, these results suggest a beneficial role of DHA supplementation for alleviating ZIKV-caused postnatal microcephaly and growth restriction by up-regulating Mfsd2a.
Fig. 6. Supplementation with DHA rescues ZIKV-induced microcephaly phenotype.
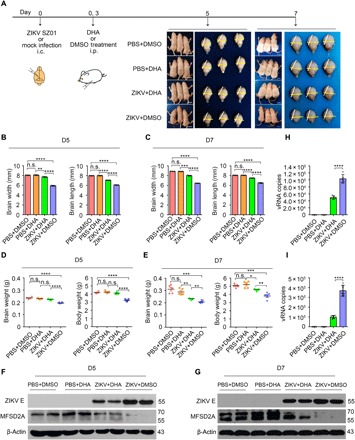
(A) Experimental scheme of the DHA protective effect in a neonatal mouse model of ZIKV infection. The ZIKV SZ01 strain was intracerebrally (i.c.) injected into neonatal BALB/c mice at day 0 after birth. Supplementation with DHA was achieved twice via intraperitoneal (i.p.) injection at day 0 and day 3, respectively. The infected brains were inspected at day 5 and day 7. Yellow and white bars represent the brain width and brain length, respectively. Scale bars, 1 cm. (B to E) Brain width and length (B and C) and brain weight and body weight (D and E) were measured. (F to I) Brain tissues were collected at 5 or 7 dpi, and Mfsd2a and ZIKV E protein levels were analyzed by Western blotting (F and G). Viral RNA copies were analyzed by qRT-PCR (H and I). The data from three independent experiments are shown as means ± SD. Significance was calculated using a one-way ANOVA statistical test with a Dunnett’s multiple comparisons test or a two-sample Student t test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Photo credit: Jia Zhou, Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College.
DISCUSSION
The current evidence from the recent outbreak of ZIKV in the Americas supports the existence of a causal relationship between prenatal ZIKV infection and microcephaly as well as other serious brain anomalies. There are several mechanisms by which ZIKV-induced microcephaly occurs. Sequence evolution of the prM protein of ZIKV and NPC disruption are considered to be the major inducing factors for microcephaly (15). However, it remains unclear whether other cell types or mechanisms contribute to ZIKV infection–related brain abnormalities.
Here, we demonstrated that endothelial cells (including hBMECs) are critical target cells for ZIKV infection. ZIKV E glycoprotein E specifically interacts with the orphaned transporter Mfsd2a and promotes the ubiquitination of Mfsd2a for proteasome-dependent degradation, in both cultured primary hBMECs and the mouse brain. A number of lysine residues within Mfsd2a are responsible for the ZIKV-mediated degradation of Mfsd2a. Our results also showed that ZIKV infection or overexpressed virus E protein impairs Mfsd2a-mediated LPC uptake into cells. Lipidomic analysis indicated that ZIKV infection causes a decrease in DHA-related lipid abundance. Supplementation with DHA can positively up-regulate the expression of its transport receptor Mfsd2a and partially rescue ZIKV-induced growth restriction and microcephaly.
The major facilitator superfamily is a superfamily of membrane transport proteins that facilitate the movement of small solutes across cell membranes (16). Mfsd2a, also known as sodium-dependent LPC symporter 1, is a type of membrane transport protein predominantly expressed in the endothelium of the BBB. Mfsd2a was previously reported to play a dual role in both establishment of BBB integrity and uptake of the fatty acid DHA. Mfsd2a knockout mice are born with a phenotype of microcephaly and cognitive disorders (8). Highly consistent with the dysfunction of Mfsd2a, ZIKV infection has been associated with BBB leakage (11). Our results revealed that the protein level, rather than mRNA abundance, of Mfsd2a was disrupted during ZIKV infection and that the down-regulation of Mfsd2a protein is fairly sensitive to ZIKV E but not to the HCV or WNV envelopes. Our findings establish a direct mechanistic link between ZIKV infection and neurovascular abnormalities through Mfsd2a. Recently, Andreone et al. (17) reported that blocking the ω-3 fatty acid transporter Mfsd2a could open the BBB for drug delivery to treat a range of disorders such as Alzheimer’s disease, stroke, and brain cancer. However, a way to safely and quickly manipulate the BBB through the regulation of Mfsd2a is still lacking. Our findings provide ZIKV E protein this potential.
Although dietary DHA treatment failed to rescue the abnormal brain phenotypes in Mfsd2a knockout mice (8), supplementation with DHA alleviated the ZIKV-induced postnatal microcephaly and growth restriction in our experiments. This result may be attributed to ZIKV inhibition by DHA. Our data suggest that the administration of ω-3 polyunsaturated fatty acids, especially DHA, has potential benefits for ZIKV-infected individuals to reduce harm to the brain. For the potential mechanisms of this DHA protection against ZIKV-induced microcephaly, we observed two corresponding phenomena. One is ZIKV inhibition, and the other is protection of Mfsd2a degradation to a certain extent. It is hard to distinguish which happened first, but we believe that ZIKV inhibition is helpful for slowing Mfsd2a degradation. Together, DHA protection effects, when supplemented as early as possible, seem to be a comprehensive result. We assume that supplementation of ω-3 fatty acids may alter the fatty acid composition of cells, which plays critical roles in regulating nutrient intake of nerve cells and even antiviral response and the like.
In summary, we report a previously unknown mechanism whereby ZIKV affects the neurovascular microenvironment in terms of disrupting Mfsd2a and reducing DHA uptake. Given the beneficial role of DHA in ZIKV-induced brain developmental restriction, our data might provide some potential therapeutic clues for ZIKV infections in pregnant women.
MATERIALS AND METHODS
Antibodies and reagents
Antibodies were obtained from Abcam (Cambridge, MA, USA) for Mfsd2a (ab105399) and CD31 (ab28364); BioFront Technologies (Tallahassee, FL, USA) for ZIKV E (1176-56) and ZIKV non-structural protein 1 (NS1) (1225-36); Sigma-Aldrich (St. Louis, MO, USA) for Flag-tag (F3165) and β-actin (A5441); Abmart (Shanghai, China) for GFP (M20004M); Santa Cruz Biotechnology (Santa Cruz, CA, USA) for ubiquitin (sc-8017); J. McKeating for HCV E2; H. Greenberg for HCV E1; and Thermo Fisher Scientific (Rockford, IL, USA) for His-tag (372900), Myc-tag (MA1-21316), and HA-tag (326700). Fluorescence (111-545-003) or horseradish peroxidase (HRP)–conjugated secondary antibodies (111-035-003, 115-035-003, and 705-035-003) were obtained from Jackson ImmunoResearch (West Grove, PA, USA). HRP Goat Anti-Mouse immunoglobulin G (IgG) Light Chain (A25012) and HRP AffiniPure Mouse Anti-Rabbit IgG Light Chain (A25022) were purchased form Amyjet Scientific (Wuhan, China). The IRDye 800CW–labeled anti-mouse IgG (926-32210) antibody was obtained from LI-COR Biotechnology (Lincoln, Nebraska, USA). DAPI (4′,6-diamidino-2-phenylindole) was purchased from Pierce (Rockford, IL, USA), and other chemicals including chloroquine, Baf-2a, calpain inhibitor, MG132, and Z-VAD-FMK were purchased from Selleck (Houston, TX, USA). Transfection reagents Lipofectamine RNAiMAX and Lipofectamine 3000 and Mem-PER Plus Membrane Protein Extraction Kit were obtained from Invitrogen (Carlsbad, CA, USA), and polyethylenimine was purchased from Polysciences (Warrington, PA, USA). cis-4,7,10,13,16,19-DHA (53171) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cells and cell culture
Primary hBMECs (passage 1) isolated from human brain were purchased from ScienCell Research Laboratories (Santiago, CA, USA). HUVECs (passage 3) were purchased from the American Tissue Culture Collection (ATCC) (Manassas, VA, USA). Both hBMECs and HUVECs were cultured in endothelial cell basal medium supplemented with endothelial cell growth supplement, penicillin-streptomycin solution, and 5% fetal bovine serum (FBS) (Invitrogen) at 37°C in a 5% CO2 atmosphere. The human placenta choriocarcinoma cell line JEG-3, human lung adenocarcinoma cancer cell line A549, African green monkey kidney cell line Vero, and HEK293T cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin (100 U/ml), streptomycin (100 mg/ml), 2 mM glutamine, and 10% FBS and maintained at 37°C in 5% CO2.
Viruses
Two Asian lineage ZIKV strains, SZ01 (GenBank: KU866423) and PRVABC59 (GenBank: KU501215), were used in this study. ZIKV strain SZ01 was isolated from an imported Chinese patient returning from Samoa in 2016 (12). ZIKV strain PRVABC59 is a contemporary strain that was isolated from the serum sample of a ZIKV-infected patient who traveled to Puerto Rico in 2015 (13). ZIKV stocks were propagated in Vero cells, and virus-containing cultural supernatants were harvested at the fourth and sixth day post-infection (dpi). Both strains had undergone fewer than five passages. Viral titers were determined by a cytopathogenic effect–based plaque assay on Vero cells. ZIKV stocks were serially diluted and incubated with Vero cell monolayers for 2 hours to initiate viral attachment to the cells. The inoculum was then removed, and the cells were overlaid with fresh MEM containing 1.2% methylcellulose and 2% FBS. After 5 days of continuous incubation at 37°C, the cells were stained with crystal violet for plaque counting. Viral stocks were stored at −80°C until use. Studies with infectious ZIKV were conducted under biosafety level 2 facilities at the Institute of Pathogenic Biology, Chinese Academy of Medical Sciences.
ZIKV infection of hBMECs, HUVECs, and JEG-3 cells
Cells were seeded into 12-well plates 24 hours before virus addition. Virus stocks were thawed and diluted to the desired MOI (0.2, 1, or 5) according to different experimental purposes and added into the culture medium of the cells for 2 hours at 37°C with 5% CO2. The inoculum was then removed; the cells were washed once with PBS to remove unbound viruses, and fresh culture medium was added to each well. The cells were harvested at the indicated time points. For mock-infected controls, the cells were incubated with the equivalent volume of noninfected culture supernatant.
ZIKV infection of mice
The animal operation procedure was reviewed and approved by the institutional committees for animal ethics and biosafety. A neonatal mouse model of ZIKV infection was established. Briefly, 20 μl of DMEM containing 10 or 100 plaque-forming units (PFU) of ZIKV strain SZ01 or PBS was injected into the λ point of BALB/c mouse brains at D0, and the infected brains were inspected at different dpi. Body weights and brain weights were measured. For DHA administration, neonatal BALB/c mice received a DHA injection [10 mg/kg in DMSO, intraperitoneally (i.p.)] after the injection of ZIKV (10 PFU, intracerebrally) at day 0 after birth, followed by an additional dose of DHA at day 3 after infection. Mice received a DMSO injection (1 ml/kg, i.p.) as sham controls. The infected brains were inspected at 5 or 7 dpi. Mouse body weights and brain weights were measured. Brain sizes (brain width and cerebral cortex length) were measured with ImageJ software (version 1.48). The embryo infection assays were performed as described previously (4); DMEM (1 μl) containing 10 or 100 PFU of ZIKV strain SZ01 or PBS was injected into the lateral ventricle (LV) of embryonic littermate brains of the embryonic day 13.5 (E13.5) ICR mice. The mice were euthanized at E18.5 to isolate the brain tissues. The interferon-α/β receptor–deficient adult A129 mouse model was applied using 3- to 4-week-old A129 mice. For the ZIKV infection, mice were given an intraperitoneal injection with 10,000 PFU ZIKV SZ01, and the mice were euthanized at 7 dpi to isolate the brain tissues. Each experiment was performed with four to five mice per group.
Mfsd2a knockout mice
The Mfsd2a knockout was generated on the C57BL/6N using CRISPR-Cas9 technique by Cyagen Biosciences Inc. The Mfsd2a gene was targeted with Cas9 protein using a guide microRNA (miRNA) in zygote cells. The targeting structure introduced two pairs of guide miRNA matching sites in the transcript flanking exons 1 to 14 and 2 to 13, respectively. Microinjection of homologous embryonic stem cell (ESC) clones with Cas9-recombinase resulted in the loss of the sequences between the two-matching guide miRNA sites, leading to deletion of exons 1 to 14 or exons 2 to 13 of the Mfsd2a gene. When Mfsd2a mutant ESCs divided into blastocysts, embryo transplantation was performed in C57BL/6N mice. Three genotypes were produced from these mice: Mfsd2a mutant heterozygote mice (+/−), Mfsd2a mutant homozygous mice (−/−), and wild-type Mfsd2a mice (+/+). The efficiency of knockout was detected by polymerase chain reaction (PCR) amplification.
Plasmids and stable cell lines
A human Mfsd2a open reading frame was purchased from OriGene (Rockville, MD, USA; RC204264) and subcloned into pMIG(pac) (18)–derived lentiviral plasmids pGFP-TEV-CBP-MIG, pFLAG-TEV-CBP-MIG, or pCMV-cMyc to generate GFP-, FLAG-, or Myc-fused Mfsd2a. Mfsd2a point mutants were generated by using KOD Plus Mutagenesis Kits (Toyobo, Osaka, Japan; SMK-101) with wild-type Mfsd2a as the template. All mutants were confirmed by DNA sequencing. Genes of ZIKV capsid (6-122), envelope (302-749), NS1 (795-1148), NS2A (1158-1307), NS2B (1377-1502), NS3 (1521-1975), NS4A (2125-2269), NS4B (2270-2512), and NS5 (2774-3412) were amplified from the SZ01 strain (GenBank: KU866423.2) and inserted into the eukaryotic expression vector pcDNA3.1 with Flag-tag fused at the C terminus. A eukaryotic expression plasmid for WNV E protein was purchased from Sino Biological (VG40345; Beijing, China).
To generate retroviruses, HEK293T cells were cotransfected with pMIR-Mfsd2a-GFP or pMIR-Mfsd2a-FLAG together with gag/pol and vesicular stomatitis virus glycoprotein. The medium was changed 6 hours after transfection. Retrovirus-containing culture supernatants were collected at 36 and 48 hours after transfection and filtered through a 0.45-μm syringe filter. To establish stable cells, A549 cells were spin-infected with lentiviruses in the presence of polybrene (2 μg/ml). After 48 hours of incubation, cells were selected by 10% FBS-DMEM containing puromycin (2 μg/ml) for 2 weeks to acquire stable cell lines.
Western blotting
Monolayer cells or brain tissues were lysed with lysis buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 50 mM NaF, 1 mM Na3VO4, 5 mM β-glycerophosphate, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride] supplemented with a protease inhibitor cocktail (Sigma-Aldrich) on ice. Lysates were cleared by centrifugation at 14,000g for 20 min. The samples were boiled with 2× SDS loading buffer and loaded onto a 10 to 12% polyacrylamide gel. After electrophoresis, the separated proteins were transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The resulting blots were blocked with 10% milk for 1 hour and then incubated with a primary monoclonal antibody overnight at 4°C. After three washes of the blots, an HRP-linked secondary antibody was applied at a 1:2000 dilution. The ECL reagent (Amersham Biosciences, Piscataway, NJ, USA) was used as the substrate for detection. The intensity of individual bands was quantified using ImageJ software.
Pull down
HEK293T cells were seeded onto 100-mm plates (Corning, Acton, MA, USA) and then transfected with 15 μg of either the Mfsd2a-Myc or control-Myc plasmid for exogenous protein expression. At 24 hours after transfection, the cells were lysed on ice with radioimmunoprecipitation assay buffer supplemented with a protease inhibitor cocktail. The lysates were cleared by centrifugation at 14,000g for 20 min at 4°C. Simultaneously, 50 μl of pre-equilibrated Ni-NTA Sepharose beads (GE Healthcare, Madison, WI, USA) were incubated with 10 μg of His-tagged ZIKV E protein (Sino Biological). Subsequently, 500 μl of cleared cell lysates was added into Ni-NTA Sepharose beads and incubated for another 4 hours at 4°C, thereby allowing the capture of His-tagged ZIKV E protein. Ni-NTA Sepharose beads were washed eight times with TNE buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA, and protease inhibitor], resuspended with 50 μl of protein loading buffer, and loaded onto a 15% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel. Mfsd2a protein was detected by Western blotting using an anti-Myc antibody.
Coimmunoprecipitation
For immunoprecipitation of ectopically expressed Mfsd2a and ZIKV E, HEK293T cells in 100-mm plates were transfected with Mfsd2a-Myc plasmids, followed by Flag-tagged ZIKV E transfection 12 hours later. At 24 hours after transfection, MG132 was added, and a further 6-hour incubation was allowed before lysing the cells. For immunoprecipitation of the ZIKV E protein with endogenous Mfsd2a, Flag-tagged ZIKV E or Flag-tagged WNV E plasmids were transiently transfected into HUVECs in the presence or absence of MG132. The lysate was cleared by centrifugation at 14,000g for 20 min at 4°C and incubated for 1 hours at 4°C with 4 μg of M2 anti-Flag antibody (Sigma-Aldrich). Then, the immune complex was coupled to Protein A/G agarose beads (Sigma-Aldrich) overnight at 4°C. The beads were washed with lysis buffer five times, and samples were boiled in 2× SDS loading buffer, resolved by 10% SDS-PAGE, and then probed with monoclonal antibodies against c-Myc, Mfsd2a, or Flag, respectively. Five percent of the inputs were detected with corresponding antibodies to ensure the expression of transfected genes.
Ubiquitination assay
HEK293T cells were transfected with Mfsd2a-Myc in combination with HA-ubiquitin or ZIKV E in the presence or absence of MG132. At 24 hours after transfection, the cells were lysed, and the cell lysates were immunoprecipitated with anti-Myc antibody and then analyzed by Western blotting with anti-Myc antibody and anti-HA antibody. Neonatal BALB/c mouse brains that were infected by 100 PFU of ZIKV strain SZ01 or PBS were inspected at 3 dpi. The membrane proteins were extracted from the brains using the Mem-PER Plus Membrane Protein Extraction Kit and were immunoprecipitated with anti-Mfsd2a antibody. The precipitated proteins were separated on SDS-PAGE and detected by Western blotting using anti-ubiquitin antibody.
Real-time reverse transcription PCR
Quantifications of ZIKV RNA and Mfsd2a mRNA transcript levels were performed by real-time reverse transcription PCR (RT-PCR). Total RNAs were isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The absolute quantification of ZIKV RNA levels was performed by comparison to a standard curve, an RNA transcript copy of the ZIKV sequence with known concentrations was used for drawing the standard curve, and viral RNA copies were expressed as ZIKV copies per milliliter. ZIKV RNA was measured with the primer set 5′-GGTCAGCGTCCTCTCTAATAAACG-3′ and 5′-GCACCCTAGTGTCCACTTTTTCC-3′. The relative expression of Mfsd2a mRNA was measured with the primer set 5′-CTCCTGGCCATCATGCTCTC-3′ and 5′-GGCCACCAAGATGAGAAA-3′. The expression of glyceraldehyde-3-phosphate dehydrogenase served as the endogenous control, and the following primer set was used: 5′-CCAACCGCGAGAAGATGA-3′ and 5′-CCAGAGGCGTACAGGGATAG-3′. Amplification was performed using a QuantiFast SYBR Green RT-PCR Kit (QIAGEN, Düsseldorf, Germany), and the following real-time PCR conditions were applied: 50°C for 10 min and 95°C for 5 min followed by 40 cycles of 95°C for 10 s and 60°C for 30 s on an Applied Biosystems Prism 7500 Real-Time PCR System (Foster City, CA, USA). Each sample was assayed with three repeats.
Fluorescent LPC transport assay
Transport assays were performed as described previously (8). Briefly, TopFluor LPC (Avanti Polar Lipids) was dissolved in 150 mM NaCl containing 12% bovine serum albumin. A549 cells stably expressing Mfsd2a or NC cells (empty lentivirus transduced) were used for an uptake assay of fluorescent LPC. After 36 hours of infection with ZIKV or transfection with the ZIKV E plasmid, the cells were incubated with 30 μM TopFluor LPC for 30 min at 37°C. For the Mfsd2a wild-type and mutant-mediated LPC uptake assay, HEK293T cells were transfected with Mfsd2a-mutant or Mfsd2a wild-type plasmid in combination with ZIKV E. At 48 hours after transfection, the cells were incubated with 30 μM TopFluor LPC for 30 min at 37°C. The fluorescence intensity was quantified and expressed as the fluorescence per pixel.
Immunofluorescence microscopy
For the immunofluorescence assay, cultured cells on coverslips or brain slices were fixed using 4% paraformaldehyde for 15 min at room temperature and then permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in PBS for 10 min. The cells were then incubated with the following primary antibodies overnight at 4°C: anti-Mfsd2a (1:200), anti–ZIKV E (1:200), anti–ZIKV NS1 (1:200), anti-GFP (1:200), and anti-FLAG (1:400). After three washes with PBS, the cells were incubated with Alexa Fluor594–conjugated goat anti-mouse IgG (Invitrogen) for 1 hour at room temperature. The nuclei were stained with DAPI (1:10,000) diluted in PBS for 5 min and mounted with an antifade reagent (Invitrogen). Images were acquired with a Leica TCS SP5 confocal microscope system. Data were analyzed and processed using LAS AF Lite, ImageJ, and Photoshop software.
Sample preparation for lipidomic analysis
Cell lipids were extracted from mock- or SZ01-infected hBMECs using a liquid-liquid extraction with the tert-butyl methyl ether/methanol/H2O protocol (19). Briefly, cell samples were resuspended in 200 μl of prechilled ultrapure water and 80 μl of ice-cold aqueous methanol (v/v), followed by 7 cycles of 1-min ultrasonication with a 1-min interval in an ice-water bath. After the addition of 400 μl of methyl tert-butyl ether, samples were centrifuged at 16,000 rpm for 15 min at 4°C to remove debris, dried under a gentle nitrogen stream, and then redissolved in 100 μl of dichloromethane/methanol (1:1, v/v) before ultrahigh-performance liquid chromatography–quadrupole time-of-flight mass spectrometry (UHPLC-QTOF/MS) analysis. For in vivo lipidomic analysis, 20 μl of DMEM containing 100 PFU of ZIKV strain SZ01 or PBS was injected into the λ point of neonatal BALB/c mouse brains, and the mice were euthanized at 7 dpi to isolate the brain tissues. The lipids were extracted with methyl tert-butyl ether MTBE as previously described (20).
Nontargeted lipidomics analysis by UHPLC-QTOF/MS platform
Phospholipid species were measured by the Shanghai Profleader Biotech Company (Shanghai, China). Lipidome was analyzed by a 1290 UHPLC (Agilent 1290 Infinity II LC Systems) coupled to a TripleTOF 6600 QTOF mass analyzer equipped with a DuoSpray ion source in positive ion mode [electrospray ionization positive (ESI+)] and negative ion mode (ESI−), respectively (SCIEX, Comcord, Ontario, Canada). The chromatographic separation was performed using a Kinetex C18 column (2.1 mm by 100 mm, 1.7 μm; Phenomenex) at a flow rate of 300 μl/min and a temperature at 55°C. Quality control (QC) sample was obtained by pooling all prepared samples and served to equilibrate chromatographic system and correct variations during lipidomics instrumental analysis. The mobile phases comprised (A) acetonitrile:water (60:40, v/v) with 10 mM ammonium formate and (B) isopropanol:acetonitrile (90:10, v/v) with 10 mM ammonium formate. A linear gradient elution was performed with the following procedure: 0 min, 40% B; 12 min, 100% B; 13.5 min, 100% B; 13.7 min, 40% B and held to 18 min. Eight runs of QC sample was conducted to balance chromatographic system before formal determination, and one run of QC sample was inserted when three samples were analyzed.
The main parameters of MS were as follows. Ion source temperature was 600°C, and ion spray voltage floating (ISVF) was set to 5000 V (ESI+) or 4500 V (ESI−). The pressures of nebulizer gas, heater gas, and curtain gas were set to 60, 60, and 30 psi, respectively. A typical information-dependent acquisition including TOF MS scan and then tandem MS (MS/MS) experiment was performed in the analysis. The TOF MS scan was performed under high-resolution settings with a range of 200 to 2000 mass/charge ratio (m/z) and an accumulation time of 200 ms. The declustering potential and collision energy (CE) were set at 100 V and 10 eV for ESI+ or at −100 V and −10 eV for ESI−, respectively. In the second experiment, top 10 candidate precursors per scan cycle were fragmented in collision-induced dissociation by a CE setting at 45 eV (ESI+) or −45 eV (ESI−) both with CEs of 25 eV, and the data were collected at a range of 100 to 2000 m/z with 50-ms accumulation time for the products of each precursor. The software for controlling instrument and acquiring data was Analyst TF 1.7.1 (SCIEX, Comcord, Ontario, Canada).
Data preprocessing of raw mass spectral data
The raw data of UHPLC-QTOF-MS were firstly transformed to mzXML format using ProteoWizard and then processed by XCMS and CAMERA packages in R software platform. In the XCMS package, the peak picking [method = centWave, ppm = 15, peakwidth = c (5,20), snthresh = 10], alignment (bw = 6 and 3 for the first and second grouping, respectively), and retention time correction (method = obiwarp) were conducted. In the CAMERA package, the annotations of isotope peak, adducts, and fragments were performed with default parameters. The final data were exported as a peak table file, including observations (sample name), variables (rt_mz), and peak areas. After correction by the data of QC samples, the integrated peak areas were then normalized against total peak areas before statistical testing. Average normalized peak intensities were used for comparisons between groups.
Structural identification of lipids
The accurate m/z of precursors and product ions were matched against in silico MS/MS database of LipidBlast (21). The threshold of matching similarity is larger than 80%. Manual curation strategy was also performed to improve the quality of lipid identification.
Statistical analysis
Data were analyzed using GraphPad Prism 6.01 (GraphPad Software, San Diego, CA, USA). The values shown in the graphs are presented as means ± SD of at least three independent experiments. Statistical differences between groups were analyzed using a one-way analysis of variance (ANOVA) statistical test with Dunnett’s multiple comparisons tests or two-tailed unpaired t tests; P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by CAMS Initiative for Innovative Medicine Grant 2016-I2M-3-020, National Basic Research Program of China Grant 2015CB554301, National Natural Science Foundation of China Grants 81871667 and 81672030, and National Science and Technology Major Project 2017ZX10304402-001-013. Author contributions: W.Y., J.Z., and X.C. designed experiments and interpreted the data. J.Z., X.C., M.C., J.F., H.X., T.L., X.L., and X.H. performed experiments and analyzed the data. C.Q. and L.S. discussed the data. W.Y. conceived the study, supervised the work, and wrote the paper. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax7142/DC1
Fig. S1. The expression of the endothelial cell marker CD31 in hBMECs.
Fig. S2. Infection with both SZ01 and PRVABC59 only induces a slightly cytopathic effect at a high dose.
Fig. S3. The relative protein densities of Western blot in Fig. 1.
Fig. S4. ZIKV infection down-regulates Mfsd2a expression in JEG-3 cells.
Fig. S5. ZIKV infection down-regulates Mfsd2a expression in vivo.
Fig. S6. The degradation of Mfsd2a by ZIKV E can be inhibited by MG132.
Fig. S7. Mfsd2a-overexpressing cells show a strong uptake of TopFluor-LPC.
Table S1. Phospholipid species in mock- or ZIKV-infected hBMECs, related to Fig. 5A.
Table S2. Phospholipid species in mock- or ZIKV-infected neonatal mouse brains, related to Fig. 5C.
REFERENCES AND NOTES
- 1.Dick G. W., Kitchen S. F., Haddow A. J., Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 46, 509–520 (1952). [DOI] [PubMed] [Google Scholar]
- 2.Heymann D. L., Hodgson A., Sall A. A., Freedman D. O., Staples J. E., Althabe F., Baruah K., Mahmud G., Kandun N., Vasconcelos P. F., Bino S., Menon K. U., Zika virus and microcephaly: Why is this situation a PHEIC? Lancet 387, 719–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mlakar J., Korva M., Tul N., Popovic M., Poljšak-Prijatelj M., Mraz J., Kolenc M., Resman Rus K., Vesnaver Vipotnik T., Fabjan Vodušek V., Vizjak A., Pižem J., Petrovec M., Avšič Županc T., Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Li C., Xu D., Ye Q., Hong S., Jiang Y., Liu X., Zhang N., Shi L., Qin C.-F., Xu Z., Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Obermeier B., Daneman R., Ransohoff R. M., Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S.-H., Chang C.-D., Chen P.-H., Su J.-R., Chen C.-C., Chaung H.-C., Docosahexaenoic acid and phosphatidylserine supplementations improve antioxidant activities and cognitive functions of the developing brain on pentylenetetrazol-induced seizure model. Brain Res. 1451, 19–26 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Scott B. L., Bazan N. G., Membrane docosahexaenoate is supplied to the developing brain and retina by the liver. Proc. Natl. Acad. Sci. U.S.A. 86, 2903–2907 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen L. N., Ma D., Shui G., Wong P., Cazenave-Gassiot A., Zhang X., Wenk M. R., Goh E. L., Silver D. L., Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Guemez-Gamboa A., Nguyen L. N., Yang H., Zaki M. S., Kara M., Ben-Omran T., Akizu N., Rosti R. O., Rosti B., Scott E., Schroth J., Copeland B., Vaux K. K., Cazenave-Gassiot A., Quek D. Q., Wong B. H., Tan B. C., Wenk M. R., Gunel M., Gabriel S., Chi N. C., Silver D. L., Gleeson J. G., Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 47, 809–813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ben-Zvi A., Lacoste B., Kur E., Andreone B. J., Mayshar Y., Yan H., Gu C., Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 509, 507–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao Q., Herrlinger S., Yang S.-L., Lai F., Moore J. M., Brindley M. A., Chen J.-F., Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 143, 4127–4136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng Y.-Q., Zhao H., Li X.-F., Zhang N.-N., Liu Z.-Y., Jiang T., Gu D.-Y., Shi L., He J.-A., Wang H.-J., Sun Z.-Z., Ye Q., Xie D.-Y., Cao W.-C., Qin C.-F., Isolation, identification and genomic characterization of the Asian lineage Zika virus imported to China. Sci. China Life Sci. 59, 428–430 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ellington S. R., Devine O., Bertolli J., Martinez Quiñones A., Shapiro-Mendoza C. K., Perez-Padilla J., Rivera-Garcia B., Simeone R. M., Jamieson D. J., Valencia-Prado M., Gilboa S. M., Honein M. A., Johansson M. A., Estimating the number of pregnant women infected with zika virus and expected infants with microcephaly following the zika virus outbreak in Puerto Rico, 2016. JAMA Pediatr. 170, 940–945 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Parletta N., Niyonsenga T., Duff J., Omega-3 and omega-6 polyunsaturated fatty acid levels and correlations with symptoms in children with attention deficit hyperactivity disorder, autistic spectrum disorder and typically developing controls. PLOS ONE 11, e0156432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan L., Huang X.-Y., Liu Z.-Y., Zhang F., Zhu X.-L., Yu J.-Y., Ji X., Xu Y.-P., Li G., Li C., Wang H.-J., Deng Y.-Q., Wu M., Cheng M.-L., Ye Q., Xie D.-Y., Li X.-F., Wang X., Shi W., Hu B., Shi P.-Y., Xu Z., Qin C.-F., A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358, 933–936 (2017). [DOI] [PubMed] [Google Scholar]
- 16.yan N., Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Andreone B. J., Chow B. W., Tata A., Lacoste B., Ben-Zvi A., Bullock K., Deik A. A., Ginty D. D., Clish C. B., Gu C., Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94, 581–594.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Parijs L., Refaeli Y., Lord J. D., Nelson B. H., Abbas A. K., Baltimore D., RETRACTED: Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11, 281–288 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Xuan Q., Hu C., Yu D., Wang L., Zhou Y., Zhao X., Li Q., Hou X., Xu G., Development of a high coverage pseudotargeted lipidomics method based on ultra-high performance liquid chromatography-mass spectrometry. Anal. Chem. 90, 7608–7616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bozek K., Wei Y., Yan Z., Liu X., Xiong J., Sugimoto M., Tomita M., Pääbo S., Sherwood C. C., Hof P. R., Ely J. J., Li Y., Steinhauser D., Willmitzer L., Giavalisco P., Khaitovich P., Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron 85, 695–702 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Kind T., Liu K.-H., Lee D. Y., DeFelice B., Meissen J. K., Fiehn O., LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 10, 755–758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax7142/DC1
Fig. S1. The expression of the endothelial cell marker CD31 in hBMECs.
Fig. S2. Infection with both SZ01 and PRVABC59 only induces a slightly cytopathic effect at a high dose.
Fig. S3. The relative protein densities of Western blot in Fig. 1.
Fig. S4. ZIKV infection down-regulates Mfsd2a expression in JEG-3 cells.
Fig. S5. ZIKV infection down-regulates Mfsd2a expression in vivo.
Fig. S6. The degradation of Mfsd2a by ZIKV E can be inhibited by MG132.
Fig. S7. Mfsd2a-overexpressing cells show a strong uptake of TopFluor-LPC.
Table S1. Phospholipid species in mock- or ZIKV-infected hBMECs, related to Fig. 5A.
Table S2. Phospholipid species in mock- or ZIKV-infected neonatal mouse brains, related to Fig. 5C.