

Recombination rates vary across the genome and across populations in a way that depends on PRDM9 and chromatin structure.
Abstract
Fine-scale rates of meiotic recombination vary by orders of magnitude across the genome and differ between species and even populations. Studying cross-population differences has been stymied by the confounding effects of demographic history. To address this problem, we developed a demography-aware method to infer fine-scale recombination rates and applied it to 26 diverse human populations, inferring population-specific recombination maps. These maps recapitulate many aspects of the history of these populations including signatures of the trans-Atlantic slave trade and the Iberian colonization of the Americas. We also investigated modulators of the local recombination rate, finding further evidence that Polycomb group proteins and the trimethylation of H3K27 elevate recombination rates. Further differences in the recombination landscape across the genome and between populations are driven by variation in the gene that encodes the DNA binding protein PRDM9, and we quantify the weak effect of meiotic drive acting to remove its binding sites.
INTRODUCTION
Meiotic recombination is a fundamental genetic process and a critical evolutionary force, which generates haplotypic diversity in sexually reproducing species. In many species, including humans, a zinc finger–containing protein, PRDM9, directs recombination, resulting in hotspots of recombination at its binding sites (1). Yet, PRDM9 binds ubiquitously throughout the genome, including at promoters, and only a subset of these corresponds to recombination hotspots, suggesting that PRDM9 binding may be necessary but not sufficient (2). PRDM9 is capable of trimethylating H3K4 and H3K36 (3), and in species that lack a functional copy of PRDM9, recombination is concentrated at promoters (4), indicating that chromatin structure plays a role in recombination (5).
PRDM9-directed recombination has fundamental consequences: recombination hotspots partition the genome into blocks with low interblock linkage but high intrablock linkage, shaping patterns of linked selection (6). In addition, an excess of sites where PRDM9 binds one chromosome but not its homolog can lead to male sterility (7, 8). Such asymmetric binding sites are common in interspecies hybrids, providing a mechanism for the long-known phenomenon of PRDM9 acting as a speciation gene (9). Furthermore, asymmetric binding followed by the introduction of a double-strand break and subsequent homology-directed repair results in meiotic drive against the PRDM9 binding allele, which is equivalent to genic selection at the population level (10). Over evolutionary time scales, this meiotic drive erodes the binding sites of PRDM9, generating strong positive selection on PRDM9 mutants with new binding sites (11, 12), explaining why PRDM9 is one of the fastest evolving genes (13). These evolutionary dynamics have been studied theoretically (10, 14) and between species (12), but previous empirical investigations have been primarily qualitative rather than quantitative.
We developed a new method, called pyrho, to infer fine-scale recombination rates while taking population demography into account and applied it to 26 diverse human populations from phase 3 of the 1000 Genomes Project (1KG) (15). We then used the resulting accurate, high-resolution maps to investigate the determinants, impacts, and dynamics of recombination rate variation. Software implementing our method and the inferred recombination maps are available at https://github.com/popgenmethods/pyrho.
RESULTS
Fast, accurate inference of fine-scale recombination rates
Our method uses polymorphism data from unrelated individuals to infer fine-scale recombination maps and can be applied to either phased or unphased data. We make use of a composite-likelihood approach (16–18) that has been shown to have favorable statistical properties (19), but unlike previous methods, we avoid computationally expensive Markov chain Monte Carlo (MCMC) by using a penalized likelihood framework and gradient-based optimization (20, 21). Increasing computational efficiency by moving from a Bayesian formulation to a frequentist formulation is a common approach [e.g., (22)]. Our approach is between 10 and 450 times faster than LDhat (17), a popular MCMC-based method, while improving accuracy (Materials and Methods; Fig. 1A, fig. S1, and table S1). We also make use of our recent work on computing two-locus likelihoods (23): This allows us to scale to hundreds of individuals, whereas LDhat can accommodate at most 100 diploid individuals, and, importantly, enables us to account for nonequilibrium demographic histories. Failing to account for past fluctuations in population size has been shown to substantially affect the accuracy of inferred fine-scale recombination rates (23–25). The details of our method are presented in Materials and Methods.
Fig. 1. Accuracy of inference on simulated and real data.
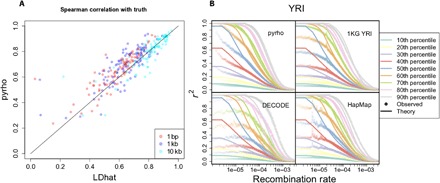
(A) Spearman correlation between inferred and true maps for 100 simulations, each 1 Mb long, for both pyrho and LDhat, with our method showing improved performance especially at finer scales. (B) Our inferred recombination maps provide a better fit to observed patterns of linkage disequilibrium as measured by r2. For a pair of SNPs, r2 is a random quantity and depends on the rate of recombination between the SNPs. Solid lines show theoretical deciles of this distribution for pairs of sites separated by different recombination distances with MAF >0.1 at both sites as calculated under the population size for YRI in Fig. 2A. Shaded points are the deciles of the empirical distribution obtained by considering pairs of sites with MAF >0.1 binned by the recombination rate separating them according to the different recombination maps. 1KG YRI is the population-specific recombination map for YRI in (15); DECODE is the sex-averaged recombination map in (28); and HapMap is the recombination map in (34).
Using samples of unrelated individuals, we are able to produce more accurate, higher-resolution maps from tens to hundreds of individuals than admixture-based (26, 27) or trio-based methods (28), which require data from thousands or tens of thousands of individuals, making our method applicable to a broader set of species and populations, including unadmixed populations and populations with few sequenced individuals. Many recent studies have used approaches similar to ours in a number of nonmodel organisms [e.g., flycatchers (29), monkey flowers (30), house mice (31), and sticklebacks (32)] and would benefit from properly accounting for the demographic histories of these species.
A major difference between our method and trio- or admixture-based methods is the sex and temporal resolution of our inferred recombination rates. Trio-based study designs are capable of inferring sex-specific fine-scale recombination rates and measure the present-day recombination rate. Our method and admixture-based methods infer time- and sex-averaged recombination maps because they implicitly average over many generations. In the case of admixture-based maps, the inferred recombination maps are an average since the time of admixture, whereas in our case the averaging is over much longer time scales in a way that depends on sample size but, in humans, is on the order of hundreds of thousands of years. Larger sample sizes will cause the inferred maps to depend more on recent recombination rates, but the exact temporal dependence of such methods depending on sample size is an open theoretical question.
To explore variation in fine-scale recombination rates across human populations, we inferred population size histories for each of the 26 populations in 1KG (15) using smc++ (Fig. 2A) (33) and used these size histories to infer population-specific fine-scale recombination maps. Our maps provide a significantly better fit of the observed r2, a commonly used measure of linkage disequilibrium, especially at finer scales [mean square error between empirical and theoretical quantiles: P < 1 × 10−5 for each population considered—CEU (Utah residents with northern and western European ancestry), CHB (Han Chinese in Beijing, China), and YRI (Yoruba in Ibadan, Nigeria)—for all comparisons between our maps and those inferred in (15, 26, 28, 34); two-sided permutation test; Materials and Methods, Fig. 1B, and fig. S2]. This improvement is particularly pronounced in non-European populations, such as YRI, and could be due to unrealistic assumptions of equilibrium demography made by other methods, a mismatch between the populations used to compute the other maps [e.g., the recombination maps from DECODE (28) are inferred using Icelanders] or to previous methods having hyperparameters tuned to European-like demographies.
Fig. 2. Interplay of demographic history and fine-scale recombination rates.
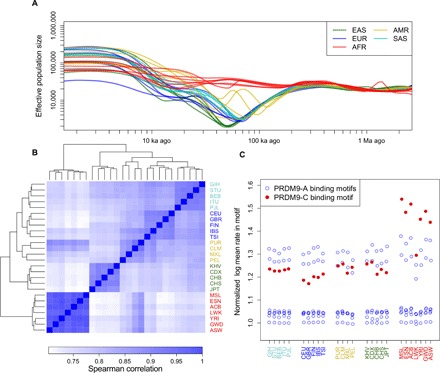
(A) Population sizes as inferred by smc++. All non-African populations show an out-of-Africa bottleneck, which is deepest in East Asian populations. (B) Heatmap of the Spearman correlation between the inferred recombination maps at single–base pair resolution. All maps show a high degree of correlation, yet the relative correlations agree with continental levels of population differentiation. (C) Recombination rates at different PRDM9 binding motifs in each population, normalized by the log average recombination rate in a shuffled version of that motif. PRDM9-A binding motifs show consistent recombination rates across all populations, while PRDM9-C binding motifs show particularly elevated rates in African populations. Three-letter population codes are defined in Table 1.
Recombination maps reflect demographic history
Our inferred recombination maps are largely concordant between populations, with high correlation between all maps, even at the single–base pair resolution (Spearman’s ρ > 0.70 for all pairs), but some differences remain. As seen in Fig. 2B, the correlation between recombination maps largely recapitulates known demographic history, clustering continental-level super-populations, and at a finer resolution separating northern and southern European populations, and to a lesser extent separating the eastern African Luhya in Webuye, Kenya (LWK) from west African and primarily west African–descended populations. Admixed American populations show similarity to both African and European populations, particularly the Iberian population in Spain, especially in Puerto Ricans, providing evidence that the trans-Atlantic slave trade and European colonization, respectively, may have affected the recombination rates of present-day admixed American populations. In fig. S3, we show a direct comparison of the correlation of fine-scale recombination maps to the correlation of fine-scale nucleotide diversity across populations, showing that across all scales we considered (1 kb, 10 kb, 100 kb, and 1 Mb), populations with more similar patterns of diversity have more similar recombination maps. Overall, the correlation of inferred recombination rates is greater than the correlation of nucleotide diversity, which may suggest that fine-scale recombination rates are more stable than local mutation rates. Yet, for the reasons discussed below as well as the fact that measures of nucleotide diversity are noisy estimates of the mutation rate, we note that this may be a result of regression attenuation and leave a more thorough comparison of the evolution of local mutation and recombination rates as a subject for future study.
While such correlations in fine-scale recombination rates across populations could be due to increased sharing of recombinations in the genealogy of individuals from more closely related populations, they could reflect population-level differences in the determinants of fine-scale recombination rate, such as differences in local chromatin structure, PRDM9 binding site locations, or PRDM9 alleles. There are multiple PRDM9 alleles that bind different motifs in humans (35), and while the PRDM9-A allele predominates in all non-African populations, both the PRDM9-A and PRDM9-C alleles are common in African populations, suggesting that African populations may have additional recombination hotspots. This is borne out in our inferred maps, with computationally predicted PRDM9-A binding motifs showing elevated recombination rates in all populations but computationally predicted PRDM9-C binding motifs showing exceptionally elevated rates in African populations (Fig. 2C) relative to matched shuffled versions of the motifs. A number of PRDM9-A motifs have only very slightly elevated recombination rates relative to their shuffled controls, indicating that these motifs likely have low specificity. While computational prediction of binding motifs for PRDM9 is difficult (36), imperfect predictions should not result in the population-specific elevation of recombination rates within predicted PRDM9-C binding motifs.
Rate of erosion of PRDM9 binding sites
An important consequence of PRDM9-driven recombination is meiotic drive against PRDM9 binding alleles, resulting from homology-directed repair of double-strand breaks initiated at the binding motif. While this process has been examined using the divergence between humans and closely related species (12, 37), the magnitude of the effect has not been quantified. As meiotic drive is equivalent to genic selection on evolutionary time scales (10), we may summarize its strength in terms of an effective selection coefficient, s, acting against PRDM9 binding alleles. This selection must be strong enough to explain the substantial divergence between humans and closely related species at PRDM9 binding sites (12, 37) but not so strong as to drive population-level differences within humans: Male hybrids from species of mice with substantial differences in the locations of PRDM9 binding sites are infertile (7, 8), whereas such incompatibilities obviously do not exist in humans.
To estimate the selection coefficient s, we computationally predicted genomic regions that bind PRDM9-A across the autosomes for each haplotype in 1KG and constructed a diallelic sample frequency spectrum (SFS) for each population by treating sequences that can putatively bind PRDM9-A as one allele and sequences that cannot as the alternative allele (Materials and Methods). Because PRDM9 is predicted to bind ubiquitously and not all PRDM9 binding sites are recombination hotspots, we subdivided each SFS by local recombination rate. We then used each SFS to infer s while controlling for background selection and misspecification of the demography (Materials and Methods). For low to moderate recombination rates, we inferred selection coefficients close to zero, consistent with these PRDM9 binding sites not being “true” recombination hotspots, while for the highest recombination rates, we inferred weak but nonzero selection against the PRDM9 binding allele (s ≈ 5 × 10−5 to 20 × 10−5; Fig. 3).
Fig. 3. Gene conversion acts like weak selection to remove PRDM9 binding sites.
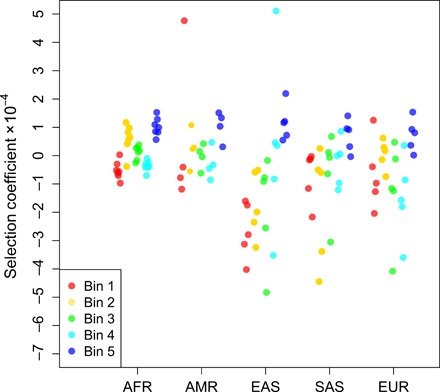
Strength of selection acting against PRDM9-A binding alleles for different populations in different bins of recombination rate. Bin 1 is for per-generation rates, r ∈ [0,1.45 × 10−9); bin 2 is r ∈ [1.45 × 10−9,2.78 × 10−9); bin 3 is r ∈ [2,78 × 10−9,5.25 × 10−9); bin 4 is r ∈ [5.25 × 10−9,1.19 × 10−8); and bin 5 is r ∈ [1.19 × 10−8, ∞). Bins were chosen such that approximately the same number of polymorphic PRDM9 binding sites falls within each bin. Selection is stronger at bins with higher recombination rates.
The above analysis implicitly assumes that the strength of selection has been temporally constant, which is certainly violated as the motif-determining zinc finger array of PRDM9 evolves extremely rapidly (e.g., archaic hominins likely do not have the PRDM9-A allele) (38). We therefore caution that we may be underestimating s.
Overall, this indicates that the meiotic drive acting against PRDM9 binding sites is equivalent to selection on the order of the inverse of the effective population size, meaning that it is a fairly weak evolutionary force. This is in contradiction to previous assumptions that PRDM9 rapidly erodes its own binding sites (14) and calls into question the hypothesis that this erosion causes the rapid evolution of PRDM9. A more plausible explanation for the rapid evolution of PRDM9 is that a small number of frequently used hotspots are crucial for the proper segregation of chromosomes during meiosis and that the strength of meiotic drive at these hotspots is much stronger. PRDM9 binding sites on short chromosomes—especially in the pseudo-autosomal region in males—are promising candidates because recombination is necessary for proper chromosomal segregation and there are likely only a handful of potential PRDM9 binding sites in such small regions (10, 39). This would be consistent with our findings since we infer only an average strength of meiotic drive on autosomes, which does not preclude that a small number of sites on the autosomes or sites on the sex chromosomes might be experiencing extremely strong meiotic drive.
Chromatin affects fine-scale recombination rates
Because fine-scale recombination rates vary substantially even outside of PRDM9-driven hotspots, we also searched for modulators of fine-scale recombination rates beyond PRDM9, finding a role for chromosome length, distance to the telomere, and chromatin state. Specifically, there is a nearly linear relationship between total physical and total genetic length across chromosomes, with a significantly positive slope and intercept (slope, P = 7.76 × 10−13; intercept, P = 1.30 × 10−7; fig. S4A). The positive intercept confirms that chromosomes require some minimum number of crossovers during meiosis, while the positive slope indicates that longer chromosomes can and do have more crossovers. Furthermore, recombination rates are elevated in subtelomeric regions (fig. S4B), likely due to the geometry of the chromosomes during meiosis (40).
We also found a significant role for chromatin structure in shaping fine-scale recombination rates. We used annotations from chromHMM (41) called on 127 ENCODE epigenomes (42); because this dataset does not contain calls in gametic cells, we used the most common chromatin state across the 127 cell types as the label for each locus. As a result, our chromatin state labels are at best a proxy for the true chromatin state in premeiotic cells, and there may be substantive differences between such cells and the 127 cell types in the ENCODE dataset (43). Furthermore, as mentioned above, our recombination estimates represent a historical average, whereas the ENCODE dataset measures modern chromatin structure. As both recombination rate and chromatin structure are likely changing over time, there is a mismatch of time scales. With these caveats in mind, we found that recombination rates vary significantly across chromatin states [P < 2.2 × 10−16, analysis of variance (ANOVA); Fig. 4] and that this effect is not driven by differences in background selection (fig. S5 and Materials and Methods). Repetitive regions of the genome have the lowest recombination rates, consistent with a previous finding that a motif present in THE1B repeats is associated with lower recombination rates (2), and suggesting that recombination suppression in repetitive regions is a broader phenomenon. We also found lower recombination rates in transcribed regions, providing support for the hypothesis that PRDM9 evolved to direct recombination away from functionally important regions (44). Furthermore, recombination rates are low in “closed” heterochromatic or quiescent regions perhaps because these regions preclude access to the recombination machinery.
Fig. 4. PRDM9 and chromatin structure shape fine-scale recombination rates.
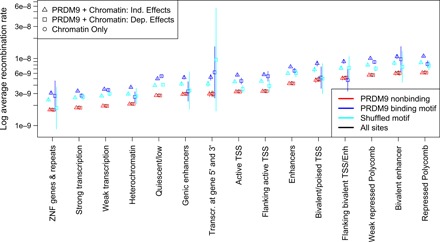
Different chromatin states have substantially different average recombination rates as determined by fitting a model using only chromatin state (Chromatin only), a model with independent chromatin state and PRDM9 binding effects (PRDM9 + Chromatin: Ind. Effects), and a model where PRDM9 binding may have a different effect in different chromatin states (PRDM9 + Chromatin: Dep. Effects). Sites characterized by H3K27me3 marks (bivalent states and regions repressed by Polycomb) have the highest recombination rates, while repetitive regions, transcribed regions, and heterochromatic or quiescent regions all have depressed recombination rates. ZNF, zinc finger genes; TSS, transcription start site.
We found that chromatin states partially characterized by H3K27me3, especially those called as being repressed by Polycomb group proteins (PcGPs), have the highest recombination rates, suggesting a role for H3K27me3 and PcGPs in meiotic recombination. This connection has been noted before, with PcGPs being recruited to double-strand breaks (45) and disruption of the PcGP repression pathway leading to improper chromosomal segregation (46). This improper segregation in PcGP mutants may be due to a reduced number of successful crossover events in the absence of the H3K27me3 marks deposited by PcGPs. We also note that the substantial impact of chromatin on local recombination rates, along with differences between chromatin structure in male and female gametic progenitor cells, could explain previously observed sex-specific differences in fine-scale recombination rates (47). While this manuscript was in preparation, a pedigree-based analysis of crossover recombinations in a large number of Icelandic parent-offspring pairs also found that H3K27me3 and PcGPs are associated with higher local recombination rates (48), corroborating our finding based on population genetic analysis.
The distribution of PRDM9 binding sites across chromatin states is nonuniform (P < 2.2 × 10−16, χ2 test; fig. S4D), and putative PRDM9 binding is associated with a 26% increase in recombination rate over the shuffled control (P < 2.2 × 10−16, t test; fig. S4C), but the variation in recombination rate across chromatin state cannot be explained by differences in PRDM9 binding (P < 2.2 × 10−16 when controlling for PRDM9 binding status). Differences in PRDM9 binding across chromatin state can be partially explained by sequence identity by comparing to a shuffled version of the motif, but the distribution of motifs across the chromatin states is not the same for the shuffled and true motifs (P < 1 × 10−5, χ2 test).
To investigate the interplay of PRDM9 and chromatin state, we compared a model where PRDM9 affects recombination rate in a chromatin-independent fashion (independent effects model) with a model where PRDM9 can have different effects in different chromatin contexts (dependent effects model), and found that the dependent effects model fits better (P < 2.2 × 10−16, F test). In spite of favoring the dependent effects model, we found that in most chromatin states, the predicted mean recombination rate is similar to that in the independent effects model (Fig. 4), indicating that PRDM9 and chromatin state usually act independently. A notable exception is at transcription start sites, where PRDM9 binding is found to have an attenuated effect on recombination rate. Yet, this effect is largely recapitulated even by shuffled PRDM9 binding motifs, indicating a PRDM9-independent mechanism for preventing sequence-based increases in recombination rate.
DISCUSSION
Despite its role as one of the primary evolutionary forces in sexually reproducing populations, much remains unknown about the modulators of the fine-scale recombination rate and the mechanisms by which they act. While PRDM9 has been known to direct the positioning of crossovers in many species (1), our results and a number of recent works (2, 48) suggest that this is far from the complete picture: The fact that many distinct biological processes are associated with differences in local recombination rates suggests that these fine-scale recombination rates are highly polygenic traits. This polygenic view of recombination may explain our observation that the correlation of fine-scale recombination maps between populations largely recapitulates demographic history. While such population-scale differences are due, in part, to differences in the frequency of the various PRDM9 alleles as well as differences in the frequencies of PRDM9 binding sites, these cannot explain differences between closely related populations because the PRDM9-A allele predominates in non-African populations and it only erodes its binding sites very weakly. Thus, other forces must explain the observed correlation between fine-scale recombination rates and demography.
To gain a better understanding of these determinants of recombination, fine-scale rates of recombination should be inferred and compared across the tree of life, especially in closely related species where one contains a functional copy of PRDM9 and one does not. By explicitly accounting for differences in demographic history between different populations and species, and working with fewer samples than required by trio- or admixture-based approaches, our method will facilitate such comparisons, hopefully illuminating the mechanisms underlying the fundamental process of recombination.
MATERIALS AND METHODS
Gradient-based estimation of fine-scale recombination rates
Penalized composite-likelihood method for phased data
To infer a fine-scale recombination map using n haplotypes with L single-nucleotide polymorphisms (SNPs), an obvious first approach would be to attempt to either maximize the likelihood
or obtain a posterior
where ρ1, …, ρL − 1 are the recombination rates between each pair of adjacent SNPs, and hi,ℓ is the allele of haplotype i at position ℓ. Unfortunately, the full likelihood of the data is intractable. Many methods then make the following approximation (16) [but see (49–52), which use machine learning or regression approaches to infer recombination rates based on simulations, and (53, 54), which use hidden Markov models]
with some window size w, which works well in practice and has attractive theoretical properties (19). This also has a justification from the composite-likelihood literature (55). This pairwise likelihood only depends on the total recombination rate separating the two points, which suggests that one could precompute these likelihoods for each possible two-locus haplotype configuration at a grid of recombination rates. Recent work (23) has enabled the computation of these likelihoods for sample sizes in the hundreds.
A drawback of this composite-likelihood approach is that it tends to produce extremely variable estimates. To reduce the variance in the estimate, past approaches have included a prior over-recombination maps that explicitly enforce smoothness and have used MCMC to obtain samples from the composite posterior over-recombination maps (17, 18, 56). Note that these samples are from a “composite posterior” and not a true posterior because we have replaced the true likelihood with the composite likelihood. Thus, although these methods provide some sense of the uncertainty in the estimated recombination map, the estimated uncertainty is likely to be inaccurate (56). A downside of MCMC is that it is slow due to the need to repeatedly evaluate the composite likelihood.
We circumvent MCMC by performing penalized composite-likelihood inference. To enforce that recombination maps are smooth, but to allow some large jumps (e.g., at hotspots), we added an ℓ1 penalty to the difference of the log of adjacent recombination rates, which is referred to in other settings as the fused-LASSO (20). Specifically, we seek to solve the following optimization problem
Note that this is a high-dimensional optimization problem, making derivative-free optimization methods prohibitively slow. We therefore seek to compute gradients of the likelihood with respect to (ρ1, …, ρL − 1), which is problematic because we have replaced exact evaluation of the pairwise log-likelihoods by looking up entries in a precomputed table. To sidestep this issue, we linearly interpolate between the precomputed log-likelihoods, which makes computing gradients an elementary exercise in linear algebra. Note that, due to using linear interpolation, there are nondifferentiable points of the likelihood function, but we circumvent this issue by arbitrarily using the slope of the line infinitesimally to the right of any nondifferentiable point. We find that this does not markedly affect the results. Furthermore, for values of the recombination rate that lie outside of the ranges precomputed in the lookup table, we use the closest entry in the lookup table (either the maximum or minimum recombination rate in the table) and treat the derivative as zero. We use these gradients in a proximal gradient descent method for fused-LASSO problems (21). We found that this optimization scheme usually converges within tens of evaluations of the objective function, making it highly efficient.
One further subtlety is that this optimization problem is nonconvex, implying that there may be local optima in which our optimization scheme could get stuck. One could initialize the optimization at a number of random points and then take the best result, but we take an alternate approach. We first perform a univariate minimization, treating the region as having a single, constant recombination rate. We then use this estimate as our initialization, which should further regularize the optima we find toward being “close” to the constant recombination map.
To further speed up inference, we divide the genome into windows that contain 4001 SNPs that overlap by 100 SNPs and optimize each window independently. We then trim the inferred recombination rates corresponding to the first and last 50 SNPs from each window, and combine the resulting estimates to obtain a recombination map. This process of windowing the genome allows us to run many optimizations in parallel.
Our method is implemented in python and makes extensive use of numba (57), a just-in-time LLVM compiler for python, to optimize numerical routines. We also make use of cyvcf2 (58) to enable the rapid parsing of VCF, bgzipped VCF, and BCF file formats.
Handling unphased data
Our method can also handle unphased data for genotypes from diploid organisms. In principle, one would want to maximize
where g is the observed unphased data, and “h consistent with g” would be the set of phased haplotypes that are equivalent to g when unphased. We could then apply our composite-likelihood approximation to obtain
but unfortunately, the outer sum is intractable as it requires phasing all sites simultaneously. Furthermore, it would be difficult to compute gradients under this formulation due to the product. Instead, we make a further approximation by swapping the sum and product to obtain
While this approximation is admittedly dubious, we may arrive at the same result by first using the composite-likelihood approximation and then summing over consistent haplotypes as follows
In either case, we now only need to phase two loci at a time, and having the product on the outside allows us to take the log and obtain a linear expression
Furthermore, we may precompute the values inside of the log by using our lookup table of haploid likelihoods at a grid of recombination rates. Summing over the consistent haplotypes may be performed efficiently using equations 10 to 12 in (16). We may then use these new precomputed lookup tables as a drop-in replacement when running our optimization scheme.
Benchmarking
Timing
To obtain timings for our method and LDhat for a realistic use case, we computed the time it took to infer a recombination map for chromosome-scale data. Both methods make use of the same precomputed lookup table of two-locus likelihoods, and so we did not benchmark the creation of those tables, which has been done previously (23). Thus, we compare only the amount of time to infer a recombination map. Using msprime (59), we simulated 10 replicates of data matching the length of chromosome 1 with the HapMap recombination map (34) and under the demography inferred for CEU, for a sample size of n = 196 haploids. Because LDhat does not allow parallelization, we wrote a python script to separate the data into the same overlapping windows used in our method (windows of 4001 SNPs overlapping by 100 SNPs). We ran our method pyrho using 32 cores and also used 32 cores to parallelize LDhat runs. For the LDhat runs, we then used a python script to combine the output of the runs. Because our scripts for splitting and combining the data for LDhat are not optimized, we only timed the total runtime of LDhat and compared that to the total time pyrho required, which is slightly advantageous for LDhat. We used the “optimal” hyperparameters for pyrho as discussed below and used the default parameters for LDhat, which were tuned to a human-like setting. The timings are presented in fig. S6, showing that in our simulations pyrho was, on average, at least 10 times faster than LDhat. Yet, when generating the 1KG maps, LDhat was run on windows of 2000 SNPs, and the MCMC was run for 22.5 million iterations per window, whereas we used only 1 million iterations per window in our timing benchmark. Computing the recombination maps for chromosome 1 for 1KG thus likely took between 22.5 and 45 times longer than the results reported here, suggesting that our method is closer to between 225 and 450 times faster.
Accuracy on simulated data
To assess the accuracy of our method, we used msprime (59) to simulate 100 sequences of 1 Mb with recombination maps randomly drawn from the HapMap recombination map (34) under the demography inferred for CEU. We then used the lookup table generated for CEU, which takes demography into account, for pyrho, while using a constant-demography lookup table for LDhat, as is the default for that program. For each simulation, we took the middle 500 kb and computed the correlation between the true recombination map and the inferred recombination map. We computed the Pearson correlation in both natural and log scale, and also the Spearman correlation. To avoid issues with autocorrelation, we look at windows centered at every 10,000th position. To assess the correlation at different spatial scales, we considered windows of different sizes [1 base pair (bp), 1 kb, and 10 kb].
We also performed simulations as above but with smaller sample sizes, n ∈ {20,40,60,80,100,120} and per-generation mutation rates a factor of 10 times lower or higher than humans, μ ∈ {1.25 × 10−9,1.25 × 10−8,1.25 × 10−7}, to show the applicability of pyrho to other species and sample sizes. In all cases, we used the demography for CEU and scaled the fine-scale recombination rates such that the ratio of the mutation and recombination rates remained fixed across different mutation rates. We performed hyperparameter optimization as described below for each sample size and mutation rate combination and then ran pyrho. The results are presented in fig. S1C. Overall, we find that the method performs better for species with higher levels of diversity (i.e., those with a larger mutation rate) and for larger sample sizes, although with some diminishing returns.
We also investigated whether the differences between pyrho and LDhat are due to the optimization scheme (i.e., fused-LASSO versus MCMC) or due to the effect of taking demographic history into account. Using the same simulations as described above, we reran LDhat using the demography-aware lookup table used by pyrho and computed the same measures of correlation between these inferred maps and the true maps. We found that at fine scales pyrho outperforms LDhat by any measure regardless of whether LDhat used a constant-demography lookup table or the demography-aware lookup table. At broader scales, pyrho outperformed LDhat if LDhat used a constant-demography lookup table but performed comparably to LDhat using the demography-aware lookup table. Meanwhile, the demography-aware version of LDhat outperformed the version of LDhat that assumed a constant-demography at all scales. The results are summarized in table S1.
Comparison of r2 on the 1000 Genomes Project dataset
To get a sense of accuracy on real data, we computed a measure of linkage disequilibrium, r2, between pairs of nearby SNPs. We used vcftools (60) with the --hap-r2, −-ld-window 15, −-thin 2000,--maf 0.1, and --max-missing 1 flags. Briefly, this removes all missing data, removes SNPs until they are all separated by at least 2 kb, and removes SNPs with a minor allele frequency (MAF) less than 10% and then computes the r2 for all SNPs within 15 SNPs of each other. For each pair of SNPs, we then computed the recombination rate between them as determined by a given fine-scale recombination map. We sorted the pairs of SNPs by the recombination rate between them, grouped them into bins of 1000 pairs of SNPs, and reported the empirical deciles from that bin. We compared these against the theoretical deciles of the distribution of r2 for sample sizes matched to the observed sample sizes for SNPs with MAF greater than 10%, which we computed from the lookup tables we generated as discussed below. The results for YRI are presented in Fig. 1B, and the results for CEU and CHB are presented in fig. S2. See Table 1 for a list of three-letter population codes.
Table 1. Populations in the 1KG dataset (15).
The super-populations are African (AFR), admixed American (AMR) East Asian (EAS), European (EUR), and South Asian (SAS).
|
Population code |
Population |
Super- population code |
| ACB | African Caribbeans in Barbados | AFR |
| ASW | Americans of African Ancestry in SW USA |
AFR |
| BEB | Bengali from Bangladesh | SAS |
| CDX | Chinese Dai in Xishuangbanna, China | EAS |
| CEU | Utah residents (CEPH) with Northern and Western European ancestry |
EUR |
| CHB | Han Chinese in Beijing, China | EAS |
| CHS | Southern Han Chinese | EAS |
| CLM | Colombians from Medllin, Colombia | AMR |
| ESN | Esan in Nigeria | AFR |
| FIN | Finnish in Finland | EUR |
| GBR | British in England and Scotland | EUR |
| GIH | Gujarati Indian from Houston, Texas | SAS |
| GWD | Gambian in Western Divisions in the Gambia |
AFR |
| IBS | Iberian population in Spain | EUR |
| ITU | Indian Telugu from the United Kingdom |
SAS |
| JPT | Japanese in Tokyo, Japan | EAS |
| KHV | Kinh in Ho Chi Minh City, Vietnam | EAS |
| LWK | Luhya in Webuye, Kenya | AFR |
| MSL | Mende in Sierra Leone | AFR |
| MXL | Mexican ancestry from Los Angeles, USA |
AMR |
| PEL | Peruvians from Lima, Peru | AMR |
| PJL | Punjabi from Lahore, Pakistan | SAS |
| PUR | Puerto Ricans from Puerto Rico | AMR |
| STU | Sri Lankan Tamil from the United Kingdom |
SAS |
| TSI | Toscani in Italia | EUR |
| YRI | Yoruba in Ibadan, Nigeria | AFR |
To determine whether the differences between maps are statistically significant, we computed the mean square error between the empirical and theoretical deciles, averaged across all of the bins of pairs of SNPs for different maps. To compare two maps, we used the difference in their mean square error as a test statistic and obtained a null distribution by performing 1 million permutations of the bins (i.e., randomly assigning each bin to one map or the other, making sure that each map has the correct number of bins). We compared the maps that we inferred to the linkage disequilibrium-based maps, HapMap (34) and 1KG (15); a trio-based map, DECODE (28); and an admixture-based map (26). We performed this comparison for CEU, CHB, and YRI using the appropriate population for our population-specific maps and the population-specific maps of 1KG. That is, overall, we performed 12 comparisons (comparing our maps against four others in three different populations). For each comparison, we found that our recombination maps had a lower mean square error between the empirical and theoretical deciles with no permutations providing an equal or greater improvement, conservatively implying P < 1 × 10−5 for each comparison.
Inference of population size histories
We applied smc++ (v1.11.1) (33) to infer population size histories using a previous build of the genome (hg19). All individuals for a given population were included in the analysis, with the first five individuals (alphabetically by sample name) being used as “distinguished” individuals in the composite likelihood. We assumed a mutation rate of 1.25 × 10−8 per base per generation and masked out sites according to Stephan Schiffels’ mappability mask available at https://oc.gnz.mpg.de/owncloud/index.php/s/RNQAkHcNiXZz2fd. Otherwise, all default parameter settings of smc++ were used, and a generation time of 29 years (61) was used to convert generations to years.
Lookup table generation
When using the population sizes inferred in the previous section to build lookup tables for pyrho, we made some approximations to reduce the computational cost. The population size functions returned by smc++ using the plot command are piecewise constant, with many pieces. To reduce the number of pieces, we started at present and combined adjacent pieces by taking the harmonic mean of the population sizes for those pieces (weighted by their lengths) if all of the pieces that were combined had population sizes within 10% of the resulting harmonic mean. Furthermore, computing the initial stationary distribution of two-locus configurations, which depends on the most ancient population size, is computationally expensive, and so after reducing the number of pieces, the most ancient size was set to 19,067 for all populations. Computing the exact two-locus likelihoods requires O(n6) time, where n is the sample size, and is too computationally prohibitive for sample sizes in the hundreds for 26 populations. In previous work (23), we showed that downsampling approximate two-locus likelihoods for a larger sample size, N, results in little loss in accuracy, and these approximate likelihoods may be computed in O(N3) time and downsampled in O(N3 × (N − n)) time as well. As such, we used this approximation, with N = 256 for each population, downsampling to the observed sample size, which ranged from n = 122 to n = 226 haploids.
Hyperparameter optimization
Our method has two important hyperparameters, namely, the window size w, which determines how far apart pairs of SNPs must be before we ignore them, and the ℓ1 regularization penalty, λ, that determines the smoothness of resulting map. Because our method is extremely fast, we were able to optimize these parameters for each population to account for differences in sample size and demography. For each population, we used msprime (59) to simulate 100 regions, each of 1 Mb in length, with a recombination map randomly drawn from the HapMap recombination map (34) and with sample size matching the observed sample size. On this dataset, we then ran our method with all possible combinations of (w, λ) ∈ {30,40,50,60,70,80,90} × {15,20,25,30,35,40,45,50}. Our method does not estimate a recombination rate before the first SNP or after the last SNP, so we took the estimated recombination rate in the region between the first and last SNP for each simulation and concatenated them together into a single vector, and did the same with the true recombination maps under which we had simulated. We then computed the Pearson correlation of these vectors in both natural and log scale, and also the Spearman correlation; we also computed these correlations at broader scales by taking our estimates and dividing them into nonoverlapping windows of length 10 or 100 kb and concatenating the average recombination rate within each window and doing the same to the true recombination maps. We also computed the squared ℓ2 norm between the inferred recombination maps and the true recombination maps in both natural and log scale. We computed all of these quantities for each setting of the hyperparameters. To choose the “best” hyperparameters, we looked at each measure of quality and ranked the hyperparameter settings for that measure (e.g., the hyperparameter setting that produced the smallest square ℓ2 norm in natural scale between the estimates and the truth would be ranked 1 for that measure). We then chose the hyperparameter setting that minimized the sum of these ranks over all of the measures we considered. Non-African populations tended to have higher values of λ and lower values of w than African populations, likely due to the lower SNP density in non-African populations resulting from the out-of-Africa bottleneck.
Comparison of recombination maps and nucleotide diversity
To compare changes in nucleotide diversity and recombination rate, we divided the genome into nonoverlapping windows of size 1 kb. For each window, we computed the average recombination rate and the average proportion of pairwise nucleotide differences, πℓ ∝ fℓ(1 − fℓ), where fℓ is the frequency of the derived allele at locus ℓ. We averaged only over sites that were not masked out as described above, and treated windows with greater than half of their positions masked out as missing. For each pair of populations, we then computed the Spearman correlation of the recombination rates across the windows, as well as the Spearman correlation of the average value of π across windows. We repeated this analysis for windows of size 10 kb, 100 kb, and 1 Mb, and the results are plotted in fig. S3.
Prediction of PRDM9 binding sites and SFS construction
To predict PRDM9-A binding sites, we obtained empirical position weight matrices (PWMs) from (2). In (2), a number of different motifs are presented, but following that paper, we only used their motifs Human1, …, Human7 as true PRDM9-A binding motifs. For PRDM9-C binding sites, we obtained the PWMs from (62). These PWM matrices describe the probability pX(ℓ) of observing a nucleotide X ∈ {A, C, G, T} for each position ℓ in the motif. To determine a cutoff for whether to call a particular sequence as matching a particular binding motif or not, we generated 10,000,000 random nucleotide sequences by sampling each position independently, and drawing A or T with probability 0.3 and C or G with probability 0.2, which approximately matches the marginal distribution of nucleotides in the human genome. We then computed the log-likelihood, of each sequence by
where is the nucleotide at position ℓ in simulation i and M is the length of the motif. We chose the 9,999,990th largest log-likelihood as the cutoff for calling a motif. This is equivalent to an approximate P value of 1 × 10−6.
We then called PRDM9-A alleles in each haploid sequence in the 1KG dataset on the hg38 genome build as follows. We considered only diallelic SNPs where all individuals have reported genotypes. Sites with more than two alleles or structural variants were treated as missing. Individuals were treated as having the reference allele at all other positions. Then, starting at the first base in the genome, we computed the log-likelihood, as above, for each motif (or its reverse complement) starting at that position, reporting log-likelihoods that are greater than the empirical cutoff for that motif, and then moving to the next base and repeating. We skipped any starting points where any motif overlapped a missing position. Instead of performing this for each haploid individually, we instead constructed all of the unique haplotypes in the dataset that spanned the region from the starting position to the end of the longest motif and only computed the log-likelihood of each motif on these unique haplotypes.
To construct the PRDM9-A binding site SFS, we took these calls and looked for starting positions where some individuals were called as matching one of the PRDM9-A binding motifs, and other individuals were not predicted to bind any PRDM9-A motif. We then treated binding and nonbinding as the two alleles and constructed a standard diallelic SFS. We also constructed SFSs for each population by restricting to only sites with a recombination rate inferred in that population within some range. To insure that our results were due to PRDM9 binding and not due to other factors such as GC content in the motifs, we also repeated the above procedure with shuffled PWMs obtained by randomly permuting the positions of each PWM.
Inference of selection coefficients
While a number of software packages exist to fit a selection coefficient to an SFS [e.g., (63, 64)], there were a number of peculiarities about the PRDM9 binding SFS that prevented us from using these previous methods; we expected selection to act against PRDM9 binding alleles regardless of whether they are ancestral or derived, and hence, we wanted to “polarize” our SFS by considering the frequency of PRDM9 binding alleles, instead of the frequency of the derived allele or the frequency of the minor allele as is usual. Yet, mutations may act to introduce new PRDM9 binding sites or to disrupt PRDM9 binding, meaning that new mutants may arise at either end of the SFS. To account for this issue, we derived and implemented a method to fit selection coefficients for this particular setting.
Let be the observed PRDM9 binding SFS. That is, is the number of segregating sites, where k individuals have a haplotype that binds PRDM9 and n − k individuals have a haplotype that does not bind PRDM9. As in previous methods (63, 64), we fit a selection coefficient by maximizing a multinomial log-likelihood
| (1) |
where ξn, k(s, θbind, θnonbind) is the probability that a segregating site has k binding alleles given a selection coefficient of s, a rate θbind of new PRDM9 binding sites appearing via mutation, and a rate θnonbind of all nonsegregating PRDM9 binding sites generating a new nonbinding PRDM9 allele. As has been shown previously (65), we have
where the approximation is exact in the limit of small mutation rates, and the final equality follows from the fact that absolute scaling of the mutation rates only determines the total number of segregating sites and not their relative proportions, causing a multiplicative factor to cancel in the numerator and denominator. Therefore, we only need to be able to compute , where ϕ = θnonbind/θbind. Assuming a panmictic population, this expectation depends on the unscaled effective population size history, η(t), as well as s and ϕ.
We have thus far suppressed the dependence of this expectation on η for notational convenience, but now define to be for the population size history . That is, we truncate the population size history at some point t and treat the resulting function as a new population size history to compute the expectation. Furthermore, define . The idea behind our method is to set up and solve a system of differential equations of the form
to obtain , which is our desired expectation. In the case where s = 0, this system of equations turns out to be equivalent to the Moran model (66) with a continuous injection of new mutants into classes at the boundary, a result that follows from (67) and is further explored in (68, 69). That is
where the minus signs arise from our convention of having time run backward, ei is the ith basis vector, and Mn(t, s) ∈ Rn −1 × n −1 is the well-known generator of the Moran process scaled by the population size
In the case where there is selection (s ≠ 0), there is no closed system of differential equations exactly describing the evolution of this vector (64, 67). Yet, it is known that the Moran model with selection converges to the Wright-Fisher diffusion with selection in the limit of large n (70). We therefore approximate the dynamics with selection by the Moran process with selection, and we compute these expectations for a larger sample size and then downsample to our observed sample size. With this approximation, we obtain the following system of equations
where
Now that we have set up the system of differential equations, we show how to efficiently solve it. We assume that η is piecewise constant, with sizes η1, …, ηT + 1 and breakpoints t1, …, tT, setting t0 ≔ 0 for ease of notation. For convenience, denote the lengths of the pieces as Δ1, Δ2, …, ΔT where Δk = tk − tk − 1 for k > 1, and also let for any t in the kth epoch. We begin at the most ancient interval, which runs from tT to ∞ and has size ηT + 1. Since this epoch is infinitely long, we can compute by finding the stationary distribution of this process. That is, we solve
for , using a sparse linear solver implemented in SciPy (71).
Now, assume that we have computed . We may compute by separately considering what happens to mass already in the system and what happens to mass that is injected during this epoch. Mass already in the system simply evolves according to , so the contribution of existing mass is , which can be efficiently computed using expm_multiply implemented in SciPy (72). For newly arising mass, we further condition on when the mass arose, resulting in
which can be computed efficiently, again with expm_multiply and a sparse linear solver to avoid needing to invert a matrix. Combining, this results in
and iterating this computation, we arrive at as desired. Last, to find selection coefficients, we can numerically maximize Eq. 1 using Powell’s direction set method (73) as implemented in SciPy.
To minimize the effect of using the Moran process with selection to approximate the Wright-Fisher process, we used a larger sample size of 256 and then downsampled to the desired sample size. We performed this for each population for each window of recombination rates, using the decimated smc++ inferred population sizes.
We were concerned about potential biases arising from either misspecification of the population sizes or differences in background selection due to differences in the recombination rate. To alleviate this bias, we also computed selection coefficients using the shuffled PRDM9 binding motifs as a putatively neutral control. We constructed these as described above for the original PRDM9 PWMs. Our reported debiased estimate for a given recombination bin and population are then the selection coefficients inferred for the PRDM9 binding SFS minus the selection coefficient inferred for the shuffled motif SFS. Because of the results shown in Fig. 2C, we restricted our selection analyses and the analyses in subsequent sections to the two motifs that showed substantially elevated recombination rates relative to their shuffled versions: Human2 and Human6.
Data processing and analysis for determinants of recombination rate variation
For the analyses presented in Fig. 4 and fig. S4, we preprocessed the data as follows. We first restricted our analyses to only sites satisfying the previously mentioned mappability mask for which we inferred recombination rates. Then, to partially alleviate issues of spatial dependency, we subsetted these data by taking every 1000th element. Throughout, we converted the population-scaled recombination rates inferred by pyrho to per-generation rates by multiplying by μ/θ, where μ is the per-generation mutation rate (assumed to be 1.25 × 10−8) and θ is the population-scaled mutation rate (chosen to be 5 × 10−4). We calculated the expected number of recombinations per chromosome by averaging the subsetted data within each chromosome and then multiplying by the chromosome length. For analyzing the subtelomeres, we averaged all entries within the first 10 Mb of each chromosome to obtain an average for the “left subtelomere” and the last 10 Mb for the “right subtelomere.” We ignored the missing subtelomeric regions in the acrocentric chromosomes 13, 14, 15, 21, and 22 and only presented results for the right subtelomere for these chromosomes.
For PRDM9 binding, we would ideally use actual, measured PRDM9-A binding sites (e.g., determined by ChIP-seq), but no such dataset exists. Binding locations of the PRDM9-B allele were determined by ChIP-seq in (2), and binding locations of a PRDM9 variant were inferred in the mouse genome using affinity-seq (74). Pratto and colleagues determined putative PRDM9-A binding sites by performing ChIP-seq on DMC1, a protein recruited to double-strand breaks, in individuals with different PRDM9 alleles (62). This approach is problematic for our purposes because inferring PRDM9-A binding positions by their induced double-strand breaks effectively conditions on those binding sites having elevated recombination rates. We were interested in finding genomic features that modulate the effect of PRDM9 binding on recombination rate, which would be impossible if we only included PRDM9 binding sites with high recombination rates. Ultimately, we labeled each position as affected by PRDM9 binding if it is within 100 bp of a computationally predicted PRDM9-A allele binding motif. Note that we focused on PRDM9-A because that is the predominant allele in humans and is primarily responsible for the historical recombinations we implicitly used in our inference of the recombination maps.
When analyzing the effect of putative PRDM9 binding or chromatin status, we performed all our analyses in log space. In our benchmarking, we found that pyrho produces errors that are approximately normally distributed in log space, making the use of t tests, ANOVA, and linear models more appropriate in log space. All statistical tests were performed in R (75).
Comparison with previous recombination maps
We used LiftOver (76) to remap previously inferred recombination maps to the current genome build (hg38). We compared our maps with maps released with the 1KG project for CEU, CHB, and YRI (15); the sex-averaged DECODE recombination map (28); the HapMap recombination map (34); and the admixture-based maps reported by Hinch et al. (26) and Wegmann et al. (27). We then computed correlation (Pearson in natural and log scales, and Spearman, at various spatial resolutions, as described above) between all pairs of maps. The results are presented in fig. S8.
Effect of genome build
We inferred recombination maps on both the current genome build (hg38) and the previous genome build (hg19) to explore the effect of using LiftOver (76) to move recombination maps from one coordinate system to another. This is common in practice, with, for example, the DECODE map being originally called on hg18 (28) but commonly used on hg19 following LiftOver. There appears to be only a modest overall effect: even at the single–base pair resolution, the Spearman correlation between maps inferred on hg38 and those inferred on hg19 and lifted to hg38 ranged from ρ = 0.986 to ρ = 0.998 across all populations. Similarly, the Pearson correlation in log space varied from r = 0.984 to r = 0.998. The Pearson correlation in natural scale was somewhat less reliable, however, ranging from r = 0.474 to r = 0.987, likely due to the extreme leverage of hotspots in natural scale. The results are summarized in table S2.
Effect of background selection on inferred recombination rates
To investigate the effect of background selection on our inferred recombination rates, we downloaded a genome-wide measure of background selection (B-statistics) from (77). B-statistics range in value from 0 to 1000 and reflect the relative loss in genetic diversity as a result of background selection, with 0 being a total loss in diversity and 1000 representing the truly neutral level of genetic diversity. The available B-statistics are reported in terms of coordinates on hg18, so we used LiftOver (76) to remap the coordinates to hg38. We took the data processed for analyzing determinants of recombination rate variation and further restricted to sites with reported B-statistics. We repeated the analyses of the effect of putative PRDM9 binding and chromatin state while controlling for background selection by including the B-statistics as linear covariates. The results are presented in fig. S5. While we observe a high correlation between the inferred recombination rate and B-statistics (Spearman’s ρ = 0.375, P < 2.2 × 10−16), the overall impact of chromatin state and PRDM9 binding remains comparable whether or not we control for B-statistics.
Note that B-statistics were originally computed by fitting distributions of selection coefficients for exonic and nonexonic regions to observed patterns of diversity (77). The impact of these distributions on the diversity at linked neutral sites depends on the genetic distance between the selected site and the neutral site, and hence requires knowledge of the fine-scale recombination map. Due to this circularity, it is difficult to determine whether the observed correlation between B-statistics and our inferred recombination rates is due to lower recombination rates directly causing higher levels of background selection (and hence lower inferred recombination rates being associated with lower B-statistics), or if higher levels of background selection result in a lower apparent effective population size, resulting in underestimated recombination rates. It may be possible to disentangle background selection from changes in local recombination rate by jointly inferring B-statistics and fine-scale recombination rates, but we leave such an undertaking for future work.
Results on unphased data
To test the performance of our method on unphased data, we performed the hyperparameter optimization described above for each population with genotype data from diploid individuals. We then tested our method on the same benchmarking data mentioned earlier using the optimal hyperparameters for CEU. The results are presented in fig. S8, which also shows a scatterplot of the inferred recombination rates compared to the true recombination rates for both phased and unphased data. Both settings are fairly unbiased for all but the smallest recombination rates. For the most part, inference using unphased individuals results in performance indistinguishable from that on perfectly phased data. As such, we recommend using genotype calls when phasing may be inaccurate.
Supplementary Material
Acknowledgments
We would like to thank E. Jewett for testing our software and J. Yu for assistance in downloading and preparing the ENCODE data. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Funding: This research was supported, in part, by NIH grant R01-GM108805 and a Packard Fellowship for Science and Engineering. Y.S.S. is a Chan Zuckerberg Biohub investigator. Author contributions: J.P.S. implemented the software and performed the analysis of the data. J.P.S. and Y.S.S. conceived and designed the work and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Software implementation of pyrho and inferred recombination maps: https://github.com/popgenmethods/pyrho; 1KG data, hg19: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/; 1KG data, hg38: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/; smc++: https://github.com/popgenmethods/smcpp; LDhat: http://ldhat.sourceforge.net; DECODE recombination maps: https://www.decode.com/addendum/; HapMap recombination maps: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20110106_recombination_hotspots/; Wegmann et al. recombination maps: https://www.eeb.ucla.edu/Faculty/Novembre/software/AfricanAmerican_AfricanCaribbean_recombination_maps.zip; Hinch et al. recombination maps: https://www.well.ox.ac.uk/~anjali/AAmap/; 1KG recombination maps: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20130507_omni_recombination_rates/; PRDM9 binding motif PWMs from Altemose et al.: https://doi.org/10.7554/eLife.28383.009; VCFtools: https://vcftools.github.io; LiftOver: http://hgdownload.soe.ucsc.edu/admin/exe/; ENCODE chromHMM calls: https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html; b-statistics: http://www.phrap.org/othersoftware.html; genomic masks: https://oc.gnz.mpg.de/owncloud/index.php/s/RNQAkHcNiXZz2fd. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw9206/DC1
Fig. S1. Additional measures of accuracy on simulated data.
Fig. S2. Goodness-of-fit of inferred recombination maps.
Fig. S3. Recombination rates are more similar across populations than a measure of diversity.
Fig. S4. Modulators of fine-scale recombination rates.
Fig. S5. Interplay of background selection and inferred recombination rates.
Fig. S6. Comparison of runtime for pyrho and LDhat.
Fig. S7. Correlation between maps inferred by pyrho and maps inferred by previous methods.
Fig. S8. Accuracy on simulations of pyrho using phased and unphased data.
Table S1. Correlation on simulated data at different spatial resolutions.
Table S2. Effect of genome build.
REFERENCES AND NOTES
- 1.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B., PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altemose N., Noor N., Bitoun E., Tumian A., Imbeault M., Chapman J. R., Aricescu A. R., Myers S. R., A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. eLife 6, e28383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powers N. R., Parvanov E. D., Baker C. L., Walker M., Petkov P. M., Paigen K., The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLOS Genet. 12, e1006146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singhal S., Leffler E. M., Sannareddy K., Turner I., Venn O., Hooper D. M., Strand A. I., Li Q., Raney B., Balakrishnan C. N., Griffith S. C., McVean G., Przeworski M., Stable recombination hotspots in birds. Science 350, 928–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lange J., Yamada S., Tischfield S. E., Pan J., Kim S., Zhu X., Socci N. D., Jasin M., Keeney S., The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167, 695–708.e16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schumer M., Xu C., Powell D. L., Durvasula A., Skov L., Holland C., Blazier J. C., Sankararaman S., Andolfatto P., Rosenthal G. G., Przeworski M., Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360, 656–660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies B., Hatton E., Altemose N., Hussin J. G., Pratto F., Zhang G., Hinch A. G., Moralli D., Biggs D., Diaz R., Preece C., Li R., Bitoun E., Brick K., Green C. M., Camerini-Otero R. D., Myers S. R., Donnelly P., Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregorova S., Gergelits V., Chvatalova I., Bhattacharyya T., Valiskova B., Fotopulosova V., Jansa P., Wiatrowska D., Forejt J., Modulation of Prdm9-controlled meiotic chromosome asynapsis overrides hybrid sterility in mice. eLife 7, e34282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forejt J., Iványi P., Genetic studies on male sterility of hybrids between laboratory and wild mice (Mus musculus L.). Genet. Res. 24, 189–206 (1974). [DOI] [PubMed] [Google Scholar]
- 10.Coop G., Myers S. R., Live hot, die young: Transmission distortion in recombination hotspots. PLOS Genet. 3, e35 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Úbeda F., Wilkins J. F., The Red Queen theory of recombination hotspots. J. Evol. Biol. 24, 541–553 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Myers S., Bowden R., Tumian A., Bontrop R. E., Freeman C., MacFie T. S., McVean G., Donnelly P., Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 3, 876–879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker Z., Schumer M., Haba Y., Bashkirova L., Holland C., Rosenthal G. G., Przeworski M., Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. eLife 6, e24133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latrille T., Duret L., Lartillot N., The Red Queen model of recombination hot-spot evolution: A theoretical investigation. Philos. Trans. R. Soc. B 372, 20160463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The 1000 Genomes Project Consortium , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudson R., Two-locus sampling distributions and their application. Genetics 159, 1805–1817 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McVean G., Awadalla P., Fearnhead P., A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160, 1231–1241 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan A. H., Jenkins P. A., Song Y. S., Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLOS Genet. 8, e1003090 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fearnhead P., Consistency of estimators of the population-scaled recombination rate. Theor. Popul. Biol. 64, 67–79 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Tibshirani R., Saunders M., Rosset S., Zhu J., Knight K., Sparsity and smoothness via the fused lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 67, 91–108 (2005). [Google Scholar]
- 21.J. Liu, L. Yuan, J. Ye, An efficient algorithm for a class of fused lasso problems, in KDD '10 Proceedings of the 16th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (ACM, 2010), pp. 323–332. [Google Scholar]
- 22.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamm J. A., Spence J. P., Chan J., Song Y. S., Two-locus likelihoods under variable population size and fine-scale recombination rate estimation. Genetics 203, 1381–1399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston H. R., Cutler D. J., Population demographic history can cause the appearance of recombination hotspots. Am. J. Hum. Genet. 90, 774–783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dapper A. L., Payseur B. A., Effects of demographic history on the detection of recombination hotspots from linkage disequilibrium. Mol. Biol. Evol. 35, 335–353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinch A. G., Tandon A., Patterson N., Song Y., Rohland N., Palmer C. D., Chen G. K., Wang K., Buxbaum S. G., Akylbekova E. L., Aldrich M. C., Ambrosone C. B., Amos C., Bandera E. V., Berndt S. I., Bernstein L., Blot W. J., Bock C. H., Boerwinkle E., Cai Q., Caporaso N., Casey G., Cupples L. A., Deming S. L., Diver W. R., Divers J., Fornage M., Gillanders E. M., Glessner J., Harris C. C., Hu J. J., Ingles S. A., Isaacs W., John E. M., Kao W. H. L., Keating B., Kittles R. A., Kolonel L. N., Larkin E., Marchand L. L., McNeill L. H., Millikan R. C., Murphy, Musani S., Neslund-Dudas C., Nyante S., Papanicolaou G. J., Press M. F., Psaty B. M., Reiner A. P., Rich S. S., Rodriguez-Gil J. L., Rotter J. I., Rybicki B. A., Schwartz A. G., Signorello L. B., Spitz M., Strom S. S., Thun M. J., Tucker M. A., Wang Z., Wiencke J. K., Witte J. S., Wrensch M., Wu X., Yamamura Y., Zanetti K. A., Zheng W., Ziegler R. G., Zhu X., Redline S., Hirschhorn J. N., Henderson B. E., Taylor H. A. Jr., Price A. L., Hakonarson H., Chanock S. J., Haiman C. A., Wilson J. G., Reich D., Myers S. R., The landscape of recombination in African Americans. Nature 476, 170–175 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wegmann D., Kessner D. E., Veeramah K. R., Mathias R. A., Nicolae D. L., Yanek L. R., Sun Y. V., Torgerson D. G., Rafaels N., Mosley T., Becker L. C., Ruczinski I., Beaty T. H., Kardia S. L. R., Meyers D. A., Barnes K. C., Becker D. M., Freimer N. B., Novembre J., Recombination rates in admixed individuals identified by ancestry-based inference. Nat. Genet. 43, 847–853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong A., Thorleifsson G., Gudbjartsson D. F., Masson G., Sigurdsson A., Jonasdottir A., Walters G. B., Jonasdottir A., Gylfason A., Kristinsson K. T., Gudjonsson S. A., Frigge M. L., Helgason A., Thorsteinsdottir U., Stefansson K., Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Kawakami T., Mugal C. F., Suh A., Nater A., Burri R., Smeds L., Ellegren H., Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol. Ecol. 26, 4158–4172 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Puzey J. R., Willis J. H., Kelly J. K., Population structure and local selection yield high genomic variation in Mimulus guttatus. Mol. Ecol. 26, 519–535 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Booker T. R., Ness R. W., Keightley P. D., The recombination landscape in wild house mice inferred using population genomic data. Genetics 207, 297–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shanfelter A. F., Archambeault S. L., White M. A., Divergent fine-scale recombination landscapes between a freshwater and marine population of threespine stickleback fish. Genome Biol. Evol. 11, 1552–1572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terhorst J., Kamm J. A., Song Y. S., Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers S., Bottolo L., Freeman C., McVean G., Donnelly P., A fine-scale map of recombination rates and hotspots across the human genome. Science 310, 321–324 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Berg I. L., Neumann R., Lam K.-W. G., Sarbajna S., Odenthal-Hesse L., May C. A., Jeffreys A. J., PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 42, 859–863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ségurel L., The complex binding of PRDM9. Genome Biol. 14, 112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesecque Y., Glémin S., Lartillot N., Mouchiroud D., Duret L., PLOS Genet. 10, e1004790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz J. J., Roach D. J., Thomas J. H., Shendure J., Primate evolution of the recombination regulator PRDM9. Nat. Commun. 5, 4370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinch A. G., Altemose N., Noor N., Donnelly P., Myers S. R., Recombination in the human pseudoautosomal region PAR1. PLOS Genet. 10, e1004503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batté A., Brocas C., Bordelet H., Hocher A., Ruault M., Adjiri A., Taddei A., Dubrana K., Recombination at subtelomeres is regulated by physical distance, double-strand break resection and chromatin status. EMBO J. 36, 2609–2625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ernst J., Kellis M., ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The roadmap epigenomics consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meiss A., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y., Wang H., Zhang Y., Du Z., Si W., Fan S., Qin D., Wang M., Duan Y., Li L., Jiao Y., Li Y., Wang Q., Shi Q., Wu X., Xie W., Reprogramming of meiotic chromatin architecture during spermatogenesis. Mol. Cell 73, 547–561.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Auton A., Li Y. R., Kidd J., Oliveira K., Nadel J., Holloway J. K., Hayward J. J., Cohen P. E., Greally J. M., Wang J., Bustamante C. D., Boyko A. R., Genetic recombination is targeted towards gene promoter regions in dogs. PLOS Genet. 9, e1003984 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou D. M., Adamson B., Dephoure N. E., Tan X., Nottke A. C., Hurov K. E., Gygi S. P., Colaiácovo M. P., Elledge S. J., A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 107, 18475–18480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu W., Starmer J., Fedoriw A. M., Yee D., Magnuson T., Repression of the soma-specific transcriptome by Polycomb-repressive complex 2 promotes male germ cell development. Genes Dev. 28, 2056–2069 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhérer C., Campbell C. L., Auton A., Dimorphism in human meiotic recombination at multiple scales. Nat. Commun. 8, 14994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halldorsson B. V., Palsson G., Stefansson O. A., Jonsson H., Hardarson M. T., Eggertsson H. P., Gunnarsson B., Oddsson A., Halldorsson G. H., Zink F., Gudjonsson S. A., Frigge M. L., Thorleifsson G., Sigurdsson A., Stacey S. N., Sulem P., Masson G., Helgason A., Gudbjartsson D. F., Thorsteinsdottir U., Stefansson K., Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, eaau1043 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Lin K., Futschik A., Li H., A fast estimate for the population recombination rate based on regression. Genetics 194, 473–484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao F., Ming C., Hu W., Li H., New software for the fast estimation of population recombination rates (FastEPRR) in the genomic era. G3 6, 1563–1571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flagel L., Brandvain Y., Schrider D. R., The unreasonable effectiveness of convolutional neural networks in population genetic inference. Mol. Biol. Evol. 36, 220–238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hermann P., Heissl A., Tiemann-Boege I., Futschik A., LDJump: Estimating variable recombination rates from population genetic data. Mol. Ecol. Resour. 19, 623–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li N., Stephens M., Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165, 2213–2233 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.G. Valadares Barroso, N. Puzovic, J. Dutheil, Inference of recombination maps from a single pair of genomes and its application to archaic samples. bioRxiv 452268 [Preprint]. 25 October 2018. 10.1101/452268. [DOI]
- 55.Varin C., Reid N., Firth D., An overview of composite likelihood methods. Stat. Sin. 21, 5–42 (2011). [Google Scholar]
- 56.Auton A., McVean G., Recombination rate estimation in the presence of hotspots. Genome Res. 17, 1219–1227 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.S. K. Lam, A. Pitrou, S. Seibert, in Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC (LLVM '15) (ACM, 2015), pp. 7:1–7:6. [Google Scholar]
- 58.Pedersen B. S., Quinlan A. R., cyvcf2: Fast, flexible variant analysis with Python. Bioinformatics 33, 1867–1869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelleher J., Etheridge A. M., McVean G., Efficient coalescent simulation and genealogical analysis for large sample sizes. PLOS Comput. Biol. 12, e1004842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fenner J. N., Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am. J. Phys. Anthropol. 128, 415–423 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Pratto F., Brick K., Khil P., Smagulova F., Petukhova G. V., Camerini-Otero R. D., Recombination initiation maps of individual human genomes. Science 346, 1256442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutenkunst R. N., Hernandez R. D., Williamson S. H., Bustamante C. D., Inferring the joint demographic history of multiple populations from multidimensional snp frequency data. PLOS Genet. 5, e1000695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jouganous J., Long W., Ragsdale A. P., Gravel S., Inferring the joint demographic history of multiple populations: Beyond the diffusion approximation. Genetics 206, 1549–1567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffiths R., Tavaré S., The age of a mutation in a general coalescent tree. Commun. Stat. 14, 273–295 (1998). [Google Scholar]
- 66.Moran P. A. P., Random processes in genetics. Math. Proc. Camb. Philos. Soc. 54, 60–71 (1958). [Google Scholar]
- 67.Evans S. N., Shvets Y., Slatkin M., Non-equilibrium theory of the allele frequency spectrum. Theor. Popul. Biol. 71, 109–119 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Kamm J. A., Terhorst J., Song Y. S., Efficient computation of the joint sample frequency spectra for multiple populations. J. Comput. Graph. Stat. 26, 182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamm J. A., Terhorst J., Durbin R., Song Y. S., Efficiently inferring the demographic history of many populations with allele count data. J. Am. Stat. Assoc. 114, 10.1080/01621459.2019.1635482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krone S. M., Neuhauser C., Ancestral processes with selection. Theor. Popul. Biol. 51, 210–237 (1997). [DOI] [PubMed] [Google Scholar]
- 71.E. Jones, T. Oliphant, P. Peterson, SciPy: Open source scientific tools for Python (2001).
- 72.Al-Mohy A. H., Higham N. J., Computing the action of the matrix exponential, with an application to exponential integrators. SIAM J. Sci. Comput. 33, 488–511 (2011). [Google Scholar]
- 73.Powell M. J. D., An efficient method for finding the minimum of a function of several variables without calculating derivatives. Comput. J. 7, 155–162 (1964). [Google Scholar]
- 74.Walker M., Billings T., Baker C. L., Powers N., Tian H., Saxl R. L., Choi K., Hibbs M. A., Carter G. W., Handel M. A., Paigen K., Petkov P. M., Affinity-seq detects genome-wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin 8, 31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.R Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2017).
- 76.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McVicker G., Gordon D., Davis C., Green P., Widespread genomic signatures of natural selection in hominid evolution. PLOS Genet. 5, e1000471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw9206/DC1
Fig. S1. Additional measures of accuracy on simulated data.
Fig. S2. Goodness-of-fit of inferred recombination maps.
Fig. S3. Recombination rates are more similar across populations than a measure of diversity.
Fig. S4. Modulators of fine-scale recombination rates.
Fig. S5. Interplay of background selection and inferred recombination rates.
Fig. S6. Comparison of runtime for pyrho and LDhat.
Fig. S7. Correlation between maps inferred by pyrho and maps inferred by previous methods.
Fig. S8. Accuracy on simulations of pyrho using phased and unphased data.
Table S1. Correlation on simulated data at different spatial resolutions.
Table S2. Effect of genome build.