

Abstract
Coenzyme A (CoA) and acetyl-coenzyme A (acetyl-CoA) are ubiquitous cellular molecules, which mediate hundreds of anabolic and catabolic reactions including energy metabolism. Highly sensitive methods including absorption spectroscopy and mass spectrometry enable their analysis, albeit with many limitations. To date, however, NMR spectroscopy has not been used to analyze these important molecules. Building on our recent efforts, which enabled simultaneous analysis of a large number of metabolites in tissue and blood including many coenzymes and antioxidants [Anal Chem. 2016, 88:4817–24; ibid 2017, 89:4620–4627], we describe here a new method for identification and quantitation of CoA and acetyl-CoA ex vivo in tissue. Using mouse heart, kidney, liver, brain and skeletal tissue, we show that a simple 1H NMR experiment can simultaneously measure these molecules. Identification of the two species involved a comprehensive analysis of the different tissue types using 1D and 2D NMR, in combination with spectral databases for standards, as well as spiking with authentic compounds. Time dependent studies showed that while the acetyl-CoA levels remain unaltered, CoA levels diminish by more than 50% within 24 h, which indicates that CoA is labile in solution; however, degassing the sample with helium gas halted its oxidation. Further, interestingly, we also identified endogenous coenzyme A glutathione disulfide (CoA-S-S-G) in tissue for the first time by NMR and show that CoA, when oxidized in tissue extract also forms the same disulfide metabolite. The new ability to simultaneously visualize absolute concentrations of CoA, acetyl-CoA and endogenous CoA-S-S-G along with redox coenzymes (NAD+, NADH, NADP+, NADPH), energy coenzymes (ATP, ADP, AMP), antioxidants (GSH, GSSG) and a vast pool of other metabolites using a single 1D NMR spectrum offers a new avenue in the metabolomics field for investigation of cellular function in health and disease.
Keywords: NMR, quantitation, CoA, acetyl-CoA, Coenzyme A glutathione disulfide, mouse, tissue, heart, kidney, brain, liver, skeletal muscle, redox and energy coenzymes, antioxidants
Graphical Abstract
INTRODUCTION
Coenzyme A (CoA) and acetyl-coenzyme A (acetyl-CoA) are ubiquitous cellular molecules, which mediate hundreds of anabolic and catabolic reactions and play critical role in the production of energy required for cellular function.1,2 In cells, the ratio of CoA and acetyl-CoA regulates important pathways including glycolysis and fatty acid oxidation.3,4 In addition, new roles for these coenzymes are increasingly being realized; for example, CoA is known to be directly involved in post-translational modification of proteins and the epigenome,2 and acetyl-CoA is recognized as a key indicator of the metabolic state of cells.5 Aberrations in CoA biosynthesis or acetyl-CoA homoeostasis is associated with various human pathologies.2 Synthesis that leads to conversion of acetate to acetyl-CoA is implicated in the growth of numerous types of cancer including hepatocellular carcinoma, glioblastoma, breast cancer and prostate cancer.6,7 The knowledge of cellular levels of these species is therefore critical for investigation of cellular metabolism.
Since the discovery of CoA more than six decade ago, numerous methods have been developed for analysis of the CoA, acetyl-CoA and other thioester derivatives.8 Of these, enzymatic methods were the earliest to develop, which are still used for their analysis.9–12 A major drawback of this approach, however, is that each compound needs to be measured separately, and sample manipulation, chemically or enzymatically, is required between measurements. In addition, varied enzyme activity in samples due to differences in the chemical environment necessitates internal standards for the reliable analysis of the two molecules. The use of reverse phase high performance liquid chromatography and detection using UV absorption is another approach in use currently.13–19 However, the absorption method used in this approach is associated with significant uncertainty regarding the identity and the specificity, which necessitates additional analysis including chemical or enzymatic treatment for further confirmation of the obtained results. An approach that is increasingly promising is mass spectrometry (MS) combined with chromatographic separation; using this approach, the CoA and acetyl-CoA are subjected to analysis either directly or after derivatization.20–24 However, owing to numerous challenges including poor chromatographic separation, ion suppression and signal losses, further advances in MS based approaches are required for reliable analysis of CoA and acetyl-CoA.25
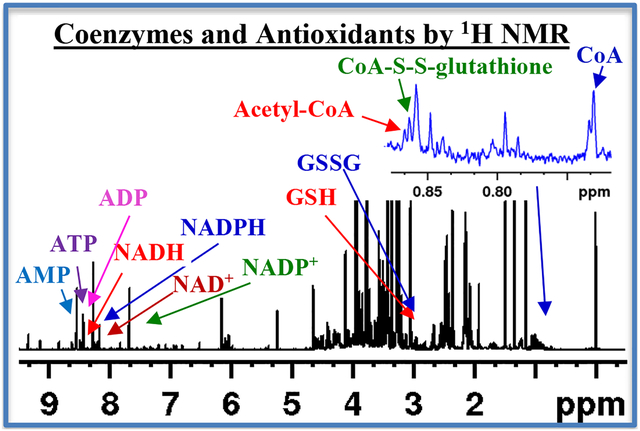
An altogether different approach that enables quantitative analysis of metabolites noninvasively, reproducibly and with no need for separation, is nuclear magnetic resonance (NMR) spectroscopy.26–32 Owing to its unsurpassed characteristics, NMR enables visualization of absolute concentrations of a large number of cellular metabolites in a single step. However, the intrinsically low sensitivity of NMR continues to pose a major challenge, which has restricted the analysis of biological mixtures to relatively high concentration metabolites (>1 μM). Therefore, important molecules such as CoA and acetyl-CoA, which exist near submicromolar levels have been out of reach for analysis using NMR. As a part of alleviating such challenges, recently, our efforts have led to the expansion of quantifiable metabolite pool in biological specimens including tissue and blood;33–35 these methods offer an ability to analyze a vast pool of metabolites including major redox coenzymes, energy coenzymes and antioxidants, many of which were inaccessible to NMR, previously. Building on these studies, we describe here a new method for the identification and quantitation of CoA and acetyl-CoA ex vivo in tissue, routinely. We show that a simple 1H NMR experiment can measure CoA, acetyl-CoA and a newly identified endogenous metabolite, coenzyme A glutathione disulfide, in addition to a large number of other metabolites. The ability to visualize absolute concentrations of ubiquitous and important metabolites such as CoA and acetyl-CoA that are fundamental to cellular functions, in one step and, using a simple 1D NMR experiment, offers new opportunities for the metabolomics field.
MATERIALS AND METHODS
Methanol, chloroform, monosodium phosphate (NaH2PO4), disodium phosphate (Na2HPO4), 3-(trimethylsilyl)propionic acid-2,2,3,3-d4, sodium salt (TSP), sodium azide, potassium hydroxide and perchloric acid were obtained from Sigma-Aldrich (St. Louis, MO). Standard compounds including coenzyme A (CoA), oxidized coenzyme A (CoA-S-S-CoA), acetyl coenzyme A (acetyl-CoA), succinyl coenzyme A, malonyl coenzyme A, reduced glutathione (GSH) and oxidized glutathione (GSSG) used for chemical shift/spectral databases and/or spiking experiments were all obtained from Sigma-Aldrich or Fisher (Waltham, MA). Deuterium oxide (D2O) was obtained from Cambridge Isotope laboratories, Inc. (Andover, MA). Deionized (DI) water was purified using an in-house Synergy Ultrapure Water System from Millipore (Billerica, MA). All chemicals were used without further purification.
Solutions of standard CoA, derivatives of CoA, and oxidized and reduced glutathione:
Stock solutions (1 mM) of standard compounds (Table S1) were prepared in D2O by weighing each compound and dissolving it in D2O. A solution of each compound was mixed with phosphate buffer (0.1 M; pH = 7.4) in D2O containing 25 or 50 μM TSP to obtain final concentration of about 100 μM, which were then transferred to 5 mm NMR tubes for 1D/2D NMR experiments.
Mouse tissue harvesting and metabolites extraction:
The investigations using mouse tissue were performed with the approval of the Institutional Animal Care and Use Committee of the University of Washington. A total of twenty-four wild type (WT) mice aged 3.5 to 6 months were used for method development (Table S2). After each mouse was anesthetized, tissue specimens from heart, kidney, brain, liver and skeletal muscle were separated, quickly rinsed with a solution containing glucose (10 mM) and pyruvate (0.5 mM) and snap frozen in liquid nitrogen. We used a previously optimized protocol for the extraction of tissue metabolites.34 Briefly, tissue specimens (~5 to 100 mg) were mixed with a mixture of cold methanol and chloroform (1 mL; 1:2 v/v; 4 °C) in 2 mL Eppendorf vials. The samples were homogenized, sonicated for 20 s and a mixture of cold chloroform/distilled water (800 μL; 1:1 v/v) was added; the samples were vortexed and set aside for 30 min on ice. The samples were then centrifuged at 2039 rcf and the top aqueous layer was separated, filtered using 1.5 mL 0.2 μm syringe filters and freeze dried. To evaluate the extraction method using recovery experiments, a small set of tissue samples (n=4) were extracted with or without spiking with standard solutions of CoA and acetyl-CoA. The dried extracts were mixed with 200 μL of a cold phosphate buffer (0.1 M; pH = 7.4; 4 °C) in D2O containing 25 μM TSP and the solutions were transferred to 3 mm sample tubes for NMR analysis.
NMR Spectroscopy:
NMR experiments for both standard solutions and tissue extracts were performed at 298 K on a Bruker Avance III 800 MHz spectrometer equipped with a cryogenically cooled probe and z-gradients suitable for inverse detection. One dimensional NOESY pulse sequence with residual water suppression using presaturation, 10204 Hz spectral width, 3 to 16 s recycle delay, 32 or 64 transients (for standard coenzymes) or 128 transients (for tissue extracts) and 32 K time domain points were used for 1H 1D NMR experiments. NMR experiments were performed immediately after preparing the solutions and a second time 24 h after preparation to assess the stability of the CoA species. For a few representative tissue extracts, 1D NMR spectra were also obtained before and after spiking with solutions of the authentic compounds to confirm the identified peaks (Table S1). To measure T1 relaxation times for the peaks used for quantitation, experiments were performed for a typical tissue sample using the inversion recovery pulse sequence. The recovery delay (τ) at which the inverted signal becomes zero (τnull) was used to calculate T1 relaxation time for the coenzymes and TSP reference. Separately, 1D NMR experiments for a few tissue extracts were also performed using degassed NMR solvent (D2O buffer) and sample tubes flushed with helium or nitrogen gas to test whether oxidation could be prevented. The sample tubes were sealed after degassing using parafilm and experiments were performed immediately after preparing the solutions and a second time 24 h after preparation. In addition, to evaluate the oxidation of CoA to CoA-CoA disulfide (CoA-S-S-CoA) or CoA-glutathione disulfide (CoA-S-S-G) or both CoA-S-S-CoA and CoA-S-S-G, 1D NMR experiments were performed for mixtures of standard CoA and reduced glutathione (GSH) at different mole ratios (1:0, 1:0.33 and 1:12) and for three different time points (0, 5 and 13 h) after preparing the mixtures. To aid peak identification, two-dimensional (2D) NMR experiments, which included 1H-1H double quantum filtered correlation spectroscopy (DQF-COSY) and 1H-1H total correlation spectroscopy (TOCSY) experiments, were performed for representative samples from each type of tissue as well as authentic compounds, under similar conditions. The 2D experiments were performed with suppression of the residual water signal by presaturation during the relaxation delay. A sweep width of 9615 Hz was used in both dimensions; 512 or 400 FIDs were obtained with t1 increments for DQF-COSY or TOCSY, respectively, each with 2048 complex data points. The number of transients used was 8 or 16 for DQF-COSY and 8, 16 or 24 for TOCSY. The relaxation delay used was 2.0 or 2.5 s for DQF-COSY and 1.0 or 1.5 s for TOCSY. The resulting 2D data were zero-filled to 1024 points in the t1 dimension and a 90° shifted squared sine-bell window function was applied to both dimensions before Fourier transformation. Chemical shifts were referenced to the internal TSP signal for both the 1H 1D and 2D NMR spectra. Bruker software package TopSpin version 3.5pl6 or 3.5pl7 was used for NMR data acquisition, processing, and analyses.
Peak assignments, coenzyme identification and quantitation:
Initial assignments followed the strategies as used in our earlier studies, which employed databases including the human metabolome database (HMDB),36 the biological magnetic resonance data bank (BMRB),37 and exhaustive 1D/2D NMR spectral assignments of tissue, serum and authentic compounds.33,34 Building on this work, further assignments of tissue NMR spectra focused on CoA and its derivatives were made based on the recent study of whole blood35 and the comprehensive analysis of 1D/2D NMR spectra of standard CoA and its derivatives (Table S1). Chemical shifts, peak multiplicity and J couplings obtained for standard compounds under similar conditions as used for tissue were comprehensively used for identifying the CoA and acetyl-CoA in tissue spectra. Identified CoA species were further confirmed by spiking experiments using authentic compounds. Bruker TopSpin version 3.5pl7 was used for peak integration and quantitation of the coenzymes. Peak integration with reference to the internal TSP signal based on peak deconvolution enabled the determination of absolute concentrations.
RESULTS AND DISCUSSION
Figure 1(a) shows a typical 1D 1H NMR spectrum of a mouse heart tissue extract. Similar to the heart tissue spectrum, spectra of the mouse liver, kidney, brain and skeletal muscle tissue were complex and rich with peaks from a vast pool of metabolites detected by NMR (Figure S1). The NMR spectrum for each tissue type was distinct; however, major features of the spectra were qualitatively similar to each other, which indicates that different tissue types share a large portion of the common metabolome. Comprehensive analysis of a multitude of NMR spectra from the heart, liver, kidney, brain and skeletal muscle tissue enabled unambiguous identification of CoA and Acetyl-CoA as highlighted in Figure 1, apart from other metabolites. The analysis combined results from earlier studies33–35 as well as spectral and chemical shift databases of standard compounds. Figures S2 and S3 and Table S3 show the NMR spectral databases for standard CoA and many of its derivatives obtained under conditions similar to tissue samples and utilized for the identification of CoA and acetyl-CoA in tissue. Results illustrating confirmation of identified coenzyme species in tissue, by spiking experiments, are shown in Figure 2.
Figure 1.

(a) Typical 800 MHz 1H NMR spectrum of a mouse heart tissue extract with labeling of some of the metabolites: BCCA: branched chain amino acids; TSP: reference peak; (b-e) expanded spectral regions highlighting characteristic peaks for (b) coenzyme A (CoA), acetyl coenzyme A (acetyl-CoA) and coenzyme A glutathione disulfide (CoA-S-S-G); (c) CoA, acetyl-CoA, oxidized nicotinamide adenine dinucleotide (NAD+), oxidized nicotinamide adenine dinucleotide phosphate (NADP+), reduced nicotinamide adenine dinucleotide (NADH), reduced nicotinamide adenine dinucleotide phosphate (NADPH), adenosine triphosphate (ATP), adenosine diphosphate (ADP) and adenosine monophosphate (AMP); (d) reduced glutathione (GSH) and oxidized glutathione (GSSG); and (e) creatine (Cr) and phosphocreatine (PCr). Peak labels for coenzymes correspond to the hydrogen atom labeling based on the recently developed ALATIS, which creates a unique and atom-specific InChI labels (Figure S3).38
Figure 2.

Portions of 800 MHz 1H NMR spectra of a mouse heart tissue extract (a) before and (b) after spiking with standard coenzyme A (CoA) solution; (c) portion of the standard CoA spectrum, shown for comparison; portions of spectra of a mouse liver tissue extract (d) before and (e) after spiking with standard acetyl coenzyme A (acetyl-CoA) solution; (f) portion of the standard acetyl-CoA spectrum, shown for comparison. Peak labels correspond to the hydrogen atom labeling based on the recently developed ALATIS, which creates a unique and atom-specific InChI labels (Figure S3).38
The challenges for identification of CoA and acetyl CoA in tissue NMR spectra were manifold: first, their low concentration in tissue translated into very weak peaks in NMR spectra; second, relatively poor spectral resolution led to overlap of all or most of the coenzyme signals with other metabolites in the sample (Figure S1); third, similarity in structures for CoA and its derivatives led to virtually identical NMR spectra resulting in a partial or complete overlap of signals (Table S3 and Figure S2); and fourth, chemical shifts are sensitive to external conditions such as pH, ionic strength, and concentration as well as the composition of biological mixtures, all of which added to the challenge. In this study, we have overcome these challenges and made a one-time establishment of the identities of the coenzymes in different types of tissue. Because of the linear response of NMR signals to 1H nuclei, identification of a single isolated peak for each coenzyme is sufficient for reliable quantitation. As highlighted in bold in Table S3, peaks for protons labeled H49–51, H52–54, H61–62 and H68, for CoA and H55–57, H58–60 and H74 for acetyl-CoA were largely isolated from other signals (Figures 1 and 2, and Figure S4). Note, the atom labeling is based on the recently developed ALATIS, which creates a unique and atom-specific InChI string.38 The CH3 protons labeled H49–51 for CoA (0.7315 ppm) and the CH3 protons labeled H58–60 for acetyl-CoA (0.8674 ppm) (Figure 1) were consistently isolated from other peaks in all tissue types and hence they were found to be suitable for routine quantitation of the coenzymes species. Hence, in this study, we have used these peaks along with the TSP reference peak for quantitation of CoA and acetyl-CoA based on peak deconvolution using TopSpin version 3.5p17 (Figure S5). In view of the weak resonances of CoA/Acetyl-CoA, proper correction of baseline is critical for their reliable quantitation. Figure 3 shows concentrations of CoA and acetyl-CoA thus derived for the mouse heart, kidney, brain and liver tissue. The two coenzyme species were, however, undetectably low for NMR in the skeletal muscle tissue (Figure 4).
Figure 3.

Absolute concentrations of coenzyme A (CoA) and acetyl coenzyme A (acetyl-CoA) in extracts of heart, kidney, brain and liver tissue of wild type mice (n=4; age 3.5 to 5.5 months) obtained using 1H NMR spectroscopy.
Figure 4.

Portions of 800 MHz 1H NMR spectra of a mouse heart, kidney, brain, liver and skeletal muscle tissue extracts with annotation of peaks for the coenzyme A (CoA), acetyl coenzyme A (acetyl-CoA) and coenzyme A glutathione disulfide (CoA-S-S-glutathione). While all three coenzymes were detected in heart, kidney, brain and liver tissue, they were too low to be detected in skeletal muscle tissue. Peak labels correspond to the hydrogen atom labeling based on the recently developed ALATIS, which creates a unique and atom-specific InChI labels (Figure S3).38
Many coenzymes are extremely labile and they can evade detection wholly or partly depending on the procedure used for tissue harvesting/extraction. In an earlier study focused on the development of an NMR method for analysis of redox and energy coenzymes, we evaluated many tissue harvesting and extraction protocols.34 The tissue harvesting protocols investigated included freeze clamping of tissue after 20 min of Langendorff isolated heart perfusion,39 freeze clamping separated hearts after washing with a solution containing glucose (10 mM) and pyruvate (0.5 mM), and freeze clamping separated hearts after washing with a solution of cold PBS. The tissue extraction procedures evaluated included the use of a mixture of methanol and water, perchloric acid (0.6 N), and methanol and chloroform. Based on these results, we have shown that quickly washing the harvested tissue with a solution containing glucose and pyruvate and freeze clamping, followed by extraction using a mixture of methanol and chloroform provided the best results in terms of both the integrity and accuracy of the detected coenzymes.34 Further, using the same solvent combination, we have shown that redox coenzymes, energy coenzymes and antioxidants can be measured quantitatively along with many other metabolites that have also been detected in whole blood.35 Hence, we have used these optimized tissue harvesting and extraction protocols for the analysis of CoA and acetyl-CoA, which also enables analysis of many other metabolites including major redox coenzymes, energy coenzymes, and antioxidants, simultaneously (Figure 1(a–e)). The robustness of the method was also tested based the recovery experiments after the addition of standard CoA and acetyl-CoA (Table S4).
Among the NMR peaks used for quantitation, the peak with the longest T1 relaxation, in our study, was for the TSP reference and the measured value of T1 for TSP was 3.1 s (Figure S6). In principle, a recycle delay of five times the longest T1 peak needs to be used for accurate quantitation or a T1 correction needs to be made. We have used 6.6 s recycle delay for quantitation of CoA and acetyl-CoA (Figure 3). Both CoA (H49–51) and Acetyl-CoA (H58–60) peaks used for quantitation exhibit an order of magnitude lower T1 values (Figure S6) compared to the recycle delay used for data acquisition. Hence, both CoA (H49–51) and Acetyl-CoA (H58–60) peak areas have not been deleteriously affected by T1 relaxation. However, no correction was made for the TSP peak T1 relaxation. Generally 1H NMR spectra are obtained using a recycle delay of 5 s or less for quantitative analysis in the metabolomics field.40 Use of a relaxation delay shorter than five times T1 provides an advantage of high throughput measurement without affecting the inferences of studies, particularly when comparing two or more groups of samples as often encountered in the metabolomics field.
Time dependent analysis showed that while acetyl-CoA is unaltered, CoA oxidizes in solution (Figure 5(a–b), Figures S7 and S8). More than 50% of the CoA was reduced in 24 h after sample preparation. However, degassing of the NMR tube and solvent using helium gas prevented the oxidation (Figure 5(c–d)). Degassing with nitrogen gas, however, did not halt the oxidation of CoA (Figure S9); the reduction in the level of CoA in the degassed samples using nitrogen gas, however, was not as drastic as observed in the absence of degassing (see Figure 5(a–b)). Although the reason for reduction in the level of CoA in the sample degassed with nitrogen is unknown, it is possible that the nitrogen gas may have been contaminated with oxygen. In the non-degassed samples, free thiol group (-SH) from CoA can oxidize with other free thiol group containing molecules such the CoA itself, cysteine or the reduced glutathione (GSH) to form a disulfide compound.41 The propensity of CoA oxidation, however, depends on the concentrations of other thiol group containing compounds relative to the CoA levels. GSH is the most abundant thiol containing molecule in cells and hence, in tissue, the GSH concentration is significantly higher when compared to the levels of both the CoA and cysteine. Our results for the tissue samples were in conformity with these facts as shown in Figure S10, for example, for two liver tissue samples, where GSH levels were higher than the CoA levels by a factor of more than 55; peaks for cysteine were undetectably low for NMR. Therefore, under such conditions, the CoA is anticipated to oxidize predominantly with GSH to form CoA-S-S-G disulfide. Indeed, NMR spectra show oxidation of CoA to form CoA-S-S-G (Figure 5(a–b), Figure S7) and it was observed in all tissue types with NMR detectable CoA (Figure 4). To further confirm this finding, NMR experiments performed using standard mixtures of CoA and GSH at different mole ratios showed that in the absence of GSH, CoA oxidizes with another molecule of CoA to form symmetric CoA-S-S-CoA (Figure 6(a–b)); however, in presence of GSH at 1:0.33 (CoA:GSH) mole ratio, the CoA oxidizes both with itself as well as with GSH to form a nearly 1:2 mixture of symmetric (CoA-S-S-CoA) and mixed (CoA-S-S-G) disulfide (Figure 6(c)); further, in presence of the GSH at 1:12 mole ratio, the CoA oxidizes predominantly with GSH to form CoA-S-S-G (Figure 6(d)). These results substantiate the hypothesis that CoA in tissue extracts oxidizes predominantly with GSH to form mixed disulfide, CoA-S-S-G. The thiol-disulfide exchange observed in this study for tissue extracts is also in conformity with the large body of pioneering works from Rabenstein’s group.42–45 Using NMR spectroscopy, this group has studied a large number of thiol group containing compounds including CoA and GSH, and exhaustively investigated thiol-disulfide chemistry for both symmetric and mixed disulfides.
Figure 5.

Portions of 800 MHz 1H NMR spectra of a typical mouse heart tissue extract without degassing: (a) obtained immediately after sample preparation; and (b) obtained 24 h after preparation of the sample. Note, while the acetyl-CoA level is unaltered, the CoA level reduced drastically with a concomitant increase of the CoA-S-S-glutathione level 24 h after sample preparation. Portions of spectra of a typical mouse heart tissue extract with degassing of both the NMR tube and solvent using helium gas: (c) obtained immediately after sample preparation; and (d) obtained 24 h after preparation. Note, the levels of the CoA, acetyl-CoA, and CoA-S-S-glutathione are unaltered even 24 h after the sample preparation, which indicates no oxidation of CoA. Tissue samples used for (a-b) and (c-d) were from different mouse. Peaks around 0.80 and 0.85 ppm observed in (a-b) are unidentified and need to be investigated in future. Peak labels correspond to the hydrogen atom labeling based on the recently developed ALATIS, which creates a unique and atom-specific InChI labels (Figure S3).38
Figure 6.

Portions of 800 MHz 1H NMR spectra of (a) a 1:0.33 mixture of coenzyme A (CoA; 100 μM) and reduced glutathione (GSH; 33 μM) solution obtained immediately after sample preparation. Note, only CoA peaks are observed indicating no oxidation of CoA; (b) only CoA solution (100 μM) obtained 5 h after sample preparation. Note, a portion of CoA is oxidized to form CoA-S-S-CoA; (c) same sample as in (a) obtained 13 h after sample preparation. Note, a portion of CoA is oxidized to form CoA-S-S-CoA and CoA-S-S-glutathione (CoA-S-S-G); (d) a 1:12 mixture of CoA (43 μM) and GSH (500 μM) obtained 13 h after sample preparation. Note, a portion of CoA is oxidized to CoA-S-S-G; no CoA-S-S-CoA is detected since GSH concentration is significantly higher than CoA. Signals shown in all the spectra originate from the CH3 protons number H49–51 and H52–54 of CoA (Figure 1, Figure S3 and Table S3). Peak labels correspond to the hydrogen atom labeling based on the recently developed ALATIS, which creates a unique and atom-specific InChI labels (Figure S3).38
We have also identified the endogenous CoA-S-S-G metabolite for the first time by NMR in tissue, which is simultaneously quantifiable in addition to the CoA, acetyl-CoA and other metabolites (Figure 1). The CoA-S-S-G as an endogenous compound was first shown in rat liver long ago.46 However, its identification in numerous mammalian tissue including mouse liver,47 rat lung,48 bovine adrenal glands,49 human parathyroid glands,50 and human myocardial tissue and cardiac specific granules51 is relatively recent. The recognition of its cellular function is, therefore, relatively new. To date, it has been attributed to numerous functions including as an endogenous vasoactive substance49–51 and an indicator of intramitochondrial oxidant stresses.48 In view of its important roles in cell metabolism, the ability to detect CoA-S-S-G simultaneously with other metabolites by NMR is significant. It should be noted that in the absence of CoA oxidation the observed CoA-S-S-G peak represents the endogenous concentration (Figure 5(a)); however, if the CoA is allowed to oxidize in solution, then the observed CoA-S-S-G peak represents the sum of endogenous and oxidized forms (Figure 5(b)).
In this study, we have used between 5 and 100 mg of tissue to identify CoA and acetyl-CoA along with CoA-S-S-G. To evaluate the amount of tissue required to analyze the coenzyme species, NMR spectra were obtained for different tissue amounts, under similar conditions. Figure S11 shows portions of typical NMR spectra with highlighting to emphasize the characteristic peaks. Each spectrum was obtained using 128 scans and 6.6 s recycle delay resulting in a 15 min acquisition per sample. As seen in the figure, while measurable peaks for the coenzyme species could be obtained with both 79.0 and 19.1 mg tissue, no measurable peaks were detected with 5.4 mg tissue. In a similar study, phosphorylated redox coenzymes (NADPH and NADP+), were also not detected with 5.6 mg tissue, under similar conditions;34 NADPH and NADP+ are generally low in concentration (a few ng/mg tissue) compared to the non-phosphorylated redox coenzymes (NADH and NAD+) (Figure 1). These results suggest that tissue amounts of 15 mg or more is required for analysis of the coenzymes, although sensitivity enhancement approaches, such as additional signal averaging or utilizing micro-coil probes, can reduce the detection limit further.
In conclusion, in this study, we demonstrate analysis of CoA, acetyl-CoA and CoA-S-S-G in one step. This is in addition to a large number of other metabolites including the major redox coenzymes, energy coenzymes and antioxidants, which we have recently shown can be analyzed simultaneously.34,35 Using a combination of various 1D and 2D NMR experiments on different types of mouse tissue, we were able to unambiguously identify the coenzyme species for the first time using NMR. Distinct peaks for the identified compounds are annotated in the 1H NMR spectra of tissue for their routine identification and quantitation using a single internal reference. The new approach, which provides an ability to simultaneously visualize absolute concentrations of CoA, acetyl-CoA and CoA-S-S-G along with major redox coenzymes (NAD+, NADH, NADP+, NADPH), energy coenzymes (ATP, ADP, AMP), antioxidants (GSH, GSSG) and other metabolites using a single 1D NMR spectrum offers new opportunities in the metabolomics field for investigation of cellular functions in health and disease. Similar to many redox and energy coenzymes,34 CoA is labile and it easily oxidizes in solution. The oxidation of CoA can, however, be prevented by degassing the NMR tube and solvent using a gas such as helium, if samples cannot be analyzed immediately. Moreover, since the GSH level in tissue is significantly higher than the CoA level, CoA predominantly oxidizes with GSH, when it is allowed to oxidize to form a mixed disulfide. Since the CoA-S-S-G is itself an endogenous cellular metabolite, care should be exercised to distinguish between the endogenous and oxidized CoA-S-S-G.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from the NIH R01GM085291, R01GM085291-S02, HL118989, HL129510 and NIH/NIBIB T32EB1650, and Royalty Research Fund, University of Washington. The authors also thank Daniel Raftery for useful input to the manuscript.
Footnotes
The authors declare no competing financial interests.
SUPPORTING INFORMATION: Tables S1 to S4, and Figures S1 to S11.
REFERENCES
- 1.Pietrocola F; Galluzzi L; Bravo-San Pedro JM; Madeo F; Kroemer G Acetyl coenzyme A: a central metabolite and second messenger. CellMetab. 2015, 21(6), 805–21. [DOI] [PubMed] [Google Scholar]
- 2.Theodoulou FL; Sibon OC; Jackowski S; Gout I Coenzyme A and its derivatives: renaissance of a textbook classic. Biochem. Soc. Trans 2014, 42(4), 1025–32. [DOI] [PubMed] [Google Scholar]
- 3.Li Q; Zhang S; Berthiaume JM; Simons B; Zhang GF Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. J. Lipid Res 2014, 55(3), 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abo Alrob O; Lopaschuk GD Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem. Soc. Trans 2014, 42, 1043–1051. [DOI] [PubMed] [Google Scholar]
- 5.Shi L; Tu BP Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 2015, 33, 125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schug ZT; Vande Voorde J; Gottlieb E The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16(11), 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshii Y; Furukawa T; Saga T; Fujibayashi Y Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application. Cancer Lett. 2015, 356(2 Pt A), 211–6. [DOI] [PubMed] [Google Scholar]
- 8.Tsuchiya Y; Phan U; Gout I Methods for measuring CoA and CoA derivatives in biological samples. Biochem. Soc. Trans 2014, 42, 1107–11. [DOI] [PubMed] [Google Scholar]
- 9.Allred JB; Guy DG Determination of coenzyme A and acetyl CoA in tissue extracts. Anal. Biochem 1969, 29, 293–299. [DOI] [PubMed] [Google Scholar]
- 10.Kato T CoA Cycling: An Enzymatic Amplification Method for Determination of CoASH and Acetyl CoA. Anal. Biochem 1975, 66, 373–392. [DOI] [PubMed] [Google Scholar]
- 11.Szutowicz A; Bielarczyk H Elimination of CoASH interference from acetyl-CoA cycling assay by maleic anhydride. Anal. Biochem 1987, 164, 292–296. [DOI] [PubMed] [Google Scholar]
- 12.Takamura Y; Kitayama Y; Arakawa A; Yamanaka S; Tosaki M; Ogawa Y Malonyl-CoA: acetyl-CoA cycling. A new micromethod for determination of acyl-CoAs with malonate decarboxylase. Biochim. Biophys. Acta 1985, 834, 1–7. [DOI] [PubMed] [Google Scholar]
- 13.Shurubor YI; D’Aurelio M; Clark-Matott J; Isakova EP; Deryabina YI; Beal MF; Cooper AJL; Krasnikov BF Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection. Molecules. 2017, 22(9). 10.3390/molecules22091388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata K; Nakai T; Fukuwatari T Simultaneous high-performance liquid chromatography determination of coenzyme A, dephospho-coenzyme A, and acetyl-coenzyme A in normal and pantothenic acid-deficient rats. Anal. Biochem 2012, 430, 151–155. [DOI] [PubMed] [Google Scholar]
- 15.Corkey BE; Hale DE; Glennon MC; Kelley RI; Coates PM; Kilpatrick L; Stanley CA Relationship between unusual hepatic acyl coenzyme A profiles and the pathogenesis of Reye syndrome. J. Clin. Invest 1988, 82, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lysiak W; Lilly K; DiLisa F; Toth P; Bieber LL Quantitation of the effect of L-carnitine on the levels of acid-soluble short-chain acyl-CoA and CoASH in rat heart and liver mitochondria. J. Biol. Chem 1988, 263, 1151–1156. [PubMed] [Google Scholar]
- 17.King MT; Reiss PD Separation and measurement of short-chain coenzyme-A compounds in rat liver by reversed-phase high-performance liquid chromatography. Anal. Biochem 1985, 146, 173–179. [DOI] [PubMed] [Google Scholar]
- 18.Corkey BE; Brand M; Williams RJ; Williamson JR Assay of short-chain acyl coenzyme A intermediates in tissue extracts by high-pressure liquid chromatography. Anal. Biochem 1981, 118, 30–41. [DOI] [PubMed] [Google Scholar]
- 19.Demoz A; Garras A; Asiedu DK; Netteland B; Berge RK Rapid method for the separation and detection of tissue short-chain coenzyme A esters by reversed-phase high-performance liquid chromatography. J. Chromatogr. B 1995, 667, 148–152. [DOI] [PubMed] [Google Scholar]
- 20.Li Q; Zhang S; Berthiaume JM; Simons B; Zhang GF Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. J Lipid Res. 2014, 55(3), 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamvakopoulos CS; Anderson VE Detection of acyl-coenzyme A thioester intermediates of fatty acid beta-oxidation as the N-acylglycines by negative-ion chemical ionization gas chromatography-mass spectrometry. Anal. Biochem 1992, 200, 381–387. [DOI] [PubMed] [Google Scholar]
- 22.Kopka J; Ohlrogge JB; Jaworski JG Analysis of in vivo levels of acyl-thioesters with gas chromatography/mass spectrometry of the butylamide derivative. Anal. Biochem 1995, 224, 51–60. [DOI] [PubMed] [Google Scholar]
- 23.Gao L; Chiou W; Tang H; Cheng X; Camp HS; Burns DJ Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J. Chromatogr. B 2007, 853, 303–313. [DOI] [PubMed] [Google Scholar]
- 24.MacDonald MJ Synergistic potent insulin release by combinations of weak secretagogues in pancreatic islets and INS-1 cells. J. Biol. Chem 2007, 282, 6043–6052. [DOI] [PubMed] [Google Scholar]
- 25.Abrankó L; Williamson G; Gardner S; Kerimi A Comprehensive quantitative analysis of fatty-acyl-Coenzyme A species in biological samples by ultra-high performance liquid chromatography-tandem mass spectrometry harmonizing hydrophilic interaction and reversed phase chromatography. J Chromatogr. A 2018, 1534, 111–122. [DOI] [PubMed] [Google Scholar]
- 26.Nagana Gowda GA; Zhang S; Gu H; Asiago V; Shanaiah N; Raftery D Metabolomics-based methods for early disease diagnostics. Expert. Rev. Mol. Diagn 2008, 8(5), 617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagana Gowda GA; Raftery D Can NMR solve some significant challenges in metabolomics? J. Magn. Reson 2015, 260, 144–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagana Gowda GA; Raftery D Recent Advances in NMR-Based Metabolomics. Anal Chem. 2017, 89(1), 490–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larive CK; Barding GA Jr; Dinges MM NMR spectroscopy for metabolomics and metabolic profiling. Anal. Chem 2015, 87(1), 133–46. [DOI] [PubMed] [Google Scholar]
- 30.Clendinen CS; Lee-McMullen B; Williams CM; Stupp GS; Vandenborne K; Hahn DA; Walter GA; Edison AS 13C NMR metabolomics: applications at natural abundance. Anal. Chem 2014, 86(18), 9242–9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellinger JJ; Chylla RA; Ulrich EL; Markley JL Databases and Software for NMR-Based Metabolomics. Curr. Metabolomics 2013, 1(1): 10.2174/2213235X11301010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halouska S, Fenton RJ, Barletta RG, Powers R. Predicting the in vivo mechanism of action for drug leads using NMR metabolomics. ACS Chem. Biol 2012, 7(1):166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagana Gowda GA; Gowda YN; Raftery D Expanding the limits of human blood metabolite quantitation using NMR spectroscopy. Anal. Chem 2015, 87(1), 706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagana Gowda GA; Abell L; Lee CF; Tian R; Raftery D Simultaneous Analysis of Major Coenzymes of Cellular Redox Reactions and Energy Using ex Vivo (1)H NMR Spectroscopy. Anal. Chem 2016, 88(9), 4817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagana Gowda GA; Raftery D Whole Blood Metabolomics by 1H NMR Spectroscopy Provides a New Opportunity To Evaluate Coenzymes and Antioxidants. Anal. Chem 2017, 89(8), 4620–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wishart DS; Jewison T; Guo AC; Wilson M; Knox C; Liu Y; Djoumbou Y; Mandal R; Aziat F; Dong E; Bouatra S; Sinelnikov I; Arndt D; Xia J; Liu P; Yallou F; Bjorndahl T; Perez-Pineiro R; Eisner R; Allen F; Neveu V; Greiner R; Scalbert A HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41(Database issue), D801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Kent Wenger R; Yao H; Markley JL BioMagResBank. Nucleic Acids Res. 2008, 36 (Database issue), D402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dashti H; Westler WM; Markley JL; Eghbalnia HR Unique identifiers for small molecules enable rigorous labeling of their atoms. Sci. Data 2017, 4, 170073 DOI: 10.1038/sdata.2017.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolwicz SC Jr.; Tian R Assessment of cardiac function and energetics in isolated mouse hearts using 31P NMR spectroscopy. J. Vis. Exp 2010, 42, 2069, doi: 10.3791/2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravanbakhsh S; Liu P; Bjorndahl TC; Mandal R; Grant JR; Wilson M; Eisner R; Sinelnikov I; Hu X; Luchinat C; Greiner R; Wishart DS Accurate, fully-automated NMR spectral profiling for metabolomics. PLoS One 2015, 10(5), e0124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegel D; Permentier H; Reijngoud DJ; Bischoff R Chemical and technical challenges in the analysis of central carbon metabolites by liquid-chromatography mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2014, 966, 21–33. [DOI] [PubMed] [Google Scholar]
- 42.Keire DA; Strauss E; Guo W; Noszal B; Rabenstein DL Kinetics and equilibria of thiol/disulfide interchange reactions of selected biological thiols and related molecules with oxidized glutathione. J. Org. Chem 1992, 57, 123–127. [Google Scholar]
- 43.Keire DA; Robert JM; Rabenstein DL Microscopic protonation equilibria and solution conformations of coenzyme A and coenzyme A disulfides. J. Org. Chem 1992, 57, 4427–4431. [Google Scholar]
- 44.Rabenstein DL; Theriault Y free access A nuclear magnetic resonance study of the kinetics and equilibria for the oxidation of penicillamine and N-acetylpenicillamine by glutathione disulfideCan. J. Chem 1984, 62, 1672–1680. [Google Scholar]
- 45.Keire DA; Rabenstein DL Nuclear magnetic resonance studies of thiol/disulfide chemistry: I. Kinetics and equilibria of the reduction of captopril disulfide and captopril-glutathione mixed disulfide by glutathione. Bioorg. Chem 1989, 17, 257–267. [Google Scholar]
- 46.Ondarza RN Characterization of a nucleotide-peptide from rat liver. Biochim Biophys Acta. 1965, 107(1), 112–9. [DOI] [PubMed] [Google Scholar]
- 47.Rogers LK; Valentine CJ; Szczpyka M; Smith CV Effects of hepatotoxic doses of acetaminophen and furosemide on tissue concentrations of CoASH and CoASSG in vivo. Chem Res Toxicol. 2000, 13(9), 873–82. [DOI] [PubMed] [Google Scholar]
- 48.O’Donovan DJ; Rogers LK; Kelley DK; Welty SE; Ramsay PL; Smith CV CoASH and CoASSG levels in lungs of hyperoxic rats as potential biomarkers of intramitochondrial oxidant stresses. Pediatr Res. 2002, 51(3), 346–53. [DOI] [PubMed] [Google Scholar]
- 49.Schlüter H; Meissner M; van der Giet M; Tepel M; Bachmann J; Gross I; Nordhoff E; Karas M; Spieker C; Witzel H; Zidek W Coenzyme A glutathione disulfide. A potent vasoconstrictor derived from the adrenal gland. Circ Res. 1995, 76, 675–680. [DOI] [PubMed] [Google Scholar]
- 50.Jankowski J; Schröter A; Tepel M; van der Giet M; Stephan N; Luo J; Zidek W; Schlüter H Isolation and characterization of coenzyme A glutathione disulfide as a parathyroid-derived vasoconstrictive factor. Circulation. 2000, 102(20), 2548–52. [DOI] [PubMed] [Google Scholar]
- 51.Luo J; Jankowski V; Henning L; Schlüter H; Zidek W; Jankowski J Endogenous coenzyme A glutathione disulfide in human myocardial tissue. J Endocrinol Invest. 2006, 29(8), 688–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.