

Abstract
Lynch syndrome (LS) predisposes patients to cancer and is caused by germline mutations in the DNA mismatch repair (MMR) genes. Identifying the deleterious mutation, such as a frameshift or nonsense mutation, is important for confirming an LS diagnosis. However, discovery of a missense variant is often inconclusive. The effects of these variants of uncertain significance (VUS) on disease pathogenesis are unclear, though understanding their impact on protein function can help determine their significance. Laboratory functional studies performed to date have been limited by their artificial nature. We report here an in cellulo functional assay in which we engineered site-specific MSH2 VUS using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 gene editing in human embryonic stem cells. This approach introduces the variant into the endogenous MSH2 loci, while simultaneously eliminating the wild-type gene. We characterized the impact of the variants on cellular MMR functions including DNA damage response signaling and the repair of DNA microsatellites. We classified the MMR functional capability of eight of ten VUS providing valuable information for determining their likelihood of being bona fide pathogenic LS variants. This human cell-based assay system for functional testing of MMR gene VUS will facilitate the identification of high risk LS patients.
Keywords: Lynch syndrome, DNA mismatch repair, variants of uncertain significance, human embryonic stem cells, MSH2, CRISPR-Cas9
1. INTRODUCTION
DNA mismatch repair (MMR) performs multiple important cellular functions to protect genomic integrity. The primary function is to repair DNA polymerase errors. Recognition of a misincorporated base is carried out by the heterodimer composed of Mutator S homologs (MSH) MSH2-MSH6 while small insertion or deletion loops (indels) are recognized by a heterodimer of MSH2-MSH3. Upon detection of a mismatch, the MSH complexes recruit the Mutator L homolog (MLH) heterodimer MLH1-PMS2 which together regulate the activity of the DNA exonuclease Exo1 to excise the newly synthesized DNA strand prior to DNA re-synthesis (Jiricny, 2013; Kunkel & Erie, 2015). MMR also attempts to repair mismatches introduced by polymerase mispairing of modified bases due to exposure to certain DNA damaging agents such as DNA alkylating agents. Futile repair processing of these lesions ultimately results in cell cycle arrest and apoptosis which prevents subsequent accumulation of mutation (Kaina, Ziouta, Ochs, & Coquerelle, 1997; Li, Pearlman, & Hsieh, 2016; Stojic, Brun, & Jiricny, 2004). In the absence of functional MMR, cells have an increased rate of spontaneous mutation generation and heightened resistance to the cytotoxic effects mediated by certain DNA damaging agents (Heinen, 2016). Germline mutations in the MMR genes are associated with Lynch syndrome (LS; MIM # 120435), a hereditary cancer predisposition condition. LS is inherited in an autosomal dominant fashion with patients inheriting a mutation in one allele of a MMR gene. Somatic loss of the functioning wild type (WT) allele leads to a MMR deficient cell and the establishment of a well-characterized mutator phenotype. The increased mutation rate increases the likelihood of acquiring mutations in tumor suppressors or oncogenes ultimately leading to development of cancer (Lynch, Snyder, Shaw, Heinen, & Hitchins, 2015).
Among the four canonical MMR genes, germline mutations in MSH2 (MIM# 609309) account for 33% of LS cases (Plazzer et al., 2013). Apart from the clearly deleterious changes such as nonsense or frameshift mutations that result in the loss of the MSH2 protein, a significant portion (~ 27%) of LS-associated MSH2 variants are missense variants that result in the change of a single amino acid (Plazzer et al., 2013). For a large subset of missense variants, it has been difficult to unambiguously ascertain their impact on MSH2 function and therefore their significance to disease pathogenesis. Thus, they are aptly termed variants of uncertain significance (VUS). To help clinicians definitively diagnose suspected LS patients carrying germline VUS, a group of scientists and clinicians have created a 5-point classification scheme for MMR variants (Thompson et al., 2014). The classification scheme attempts to determine the extent of pathogenic significance of a given variant based on co-segregation with disease, occurrence in multiple LS families, molecular characteristics of the tumor, and other features. In addition, determining whether the variant affects the function of the predicted protein product in a laboratory assay is often a crucial piece of information in determining its pathogenic likeliness (Thompson et al., 2014). Similarly, the American College of Medical Genetics and Genomics (ACMG) also recommended guidelines for interpreting sequence variants (Richards et al., 2015). The classification of variants as pathogenic, likely pathogenic, VUS, likely benign or benign is based on various weighted criteria ranging from very strong to supporting. Evidence that a variant damages the effect of the gene product in a well-established laboratory assay or shows no damaging effect on protein function is considered strong evidence for pathogenicity or benign impact, respectively. A recent analysis indicated that the existence of strong functional data may allow for reclassification of 97% of non-conflicting missense VUS evidence combinations demonstrating the importance of developing lab-based functional assays for disease variants (Brnich, Rivera-Munoz, & Berg, 2018).
The impact of MSH2 VUS on protein function has been examined by multiple assays (Heinen & Rasmussen, 2012). In vitro reconstitution of the MMR reaction with cellular extracts or recombinant proteins, ectopic expression of the variant protein in MMR-deficient cancer cells, or modeling mutations in conserved yeast or mice Msh2 residues have been utilized (Belvederesi et al., 2008; Brieger, Trojan, Raedle, Plotz, & Zeuzem, 2002; Christensen et al., 2009; Drost et al., 2018; Drost et al., 2012; Gammie et al., 2007; Geng et al., 2012; Houlleberghs et al., 2016; Lutzen, de Wind, Georgijevic, Nielsen, & Rasmussen, 2008; Mastrocola & Heinen, 2010; Ollila, Dermadi Bebek, Greenblatt, & Nystrom, 2008; Ollila, Dermadi Bebek, Jiricny, & Nystrom, 2008; Wielders, Dekker, Holt, Morris, & te Riele, 2011). However, possible caveats in these studies such as lack of a cellular environment, non-physiological level of mutant protein expression, or species-specific differences reduce confidence in their outcome. To this end, we have employed CRISPR-Cas9 as a tool to model a panel of LS-associated MSH2 VUS in human embryonic stem cells (hESCs). As a non-transformed cell system, hESCs provide several advantages. Unlike commonly used cancer cell lines, hESCs are an immortalized yet genetically stable, isogenic population. We created a panel of cell lines each harboring a specific MSH2 variant in both endogenous alleles and tested their ability to perform MMR cellular functions including repair and damage response signaling. These proof-of-principle experiments establish genetically engineered hESCs as a novel and valid cellular model to study the functional significance of LS-associated VUS in order to improve their clinical interpretation and better identify at-risk LS patients.
2. METHODS
2.1. Cell Line and Culture Conditions
hESCs (H1) were obtained from the University of Connecticut Stem Cell Core and scored to have a normal karyotype. H1s were cultured on growth factor reduced Matrigel coated plates (Corning) and fed daily with PeproGrow hESC medium (Peprotech). Upon reaching ~ 80% confluency, the cells were passaged either by microdissection or by using StemPro Accutase Cell Dissociation Reagent (ThermoFisher Scientific).
2.2. Generation of MSH2 Variant Expressing Cell Lines
For each MSH2 (GenBank NG_007110.2) variant, a suitable guide RNA (gRNA) sequence upstream of a 3’ protospacer adjacent motif (PAM; NGG/NGA for SpCas9 used in this study) in close proximity to the site of mutation (less than ten nt) was chosen using an in silico program (crispr.mit.edu and CRISPOR) based on high on-target specificity and low off-target probability (Supp. Table S1). DNA oligos containing the gRNA target sequence were cloned into the Px459V2.0 vector (Addgene, plasmid# 62988). For each transfection, one million H1 hESCs were pre-treated with ROCK inhibitor (Selleckchem) for two h and then transfected using Amaxa Stem Cell Nucleofector Kit 2 (Lonza) following the protocol recommended by the manufacturer using an Amaxa Nucleofector II machine. Two µg of the specific gRNA and SpCas9 expressing plasmid (Px459v2.0 backbone) were transfected along with two µL of 100 µM single strand deoxyoligonucleotide (ssODN) (90 nt with 5’ PO4 modification) (IDT) carrying the mutation to be incorporated and silent mutations (if any) for screening purposes (Supp. Table S1). To increase the rate of homology directed repair, DNA ligase IV inhibitor, SCR-7 (Selleckchem), and Rad51 activator, L755507 (Selleckchem), were used for 48 h starting from the day of transfection (at one µM and five µM final concentration, respectively). Transfected cells were transiently selected by treating with one µg/mL Puromycin (Sigma) starting 24 h after transfection for a period of 48 h. At the end of the selection, surviving cells were treated with CloneR (STEMCELL Technologies) as recommended. Subsequently, daily medium change was carried out until the surviving single cells grew to become a visible colony (7–10 days). Surviving colonies were picked in a PCR hood; a fraction of the cells were used for preparing genomic hotshot DNA for screening by PCR while the remaining cells were transferred to a Matrigel coated 96-well or 24-well plate for further passaging.
2.3. Off-target Analysis
Probable top five off-target binding sites for each guide RNA used in this study was determined using the crispr.mit.edu or crispor.tefor.net website. Each site was PCR amplified using high fidelity Phusion DNA polymerase (NEB) and sequenced using Sanger sequencing. The integrity of each site was verified by doing pairwise sequence alignment with the reference genome along with manual examination of the DNA chromatogram. Whenever, a homozygous/heterozygous single nucleotide polymorphism (SNP) was observed, Sanger sequencing results were cross-checked with the PCR amplicon from the corresponding site in parental H1 genomic DNA.
2.4. Screening Strategy
For each targeting event, hotshot DNA collected from single-cell derived clones was subjected to PCR amplification using primers flanking the target site (Supp. Table S2). For a subset of generated lines, the ssODN used in the targeting process carried silent mutations to facilitate screening by inserting or removing a restriction enzyme site. In such instances, homozygous clones were identified by restriction enzyme (RE) digestion of the PCR amplicons. Otherwise, direct Sanger sequencing of the PCR amplicons was used to identify the homozygous clones. Each identified positive clone was further expanded, genomic DNA was obtained from the cell population using Qiagen DNA Mini Kit (Qiagen), and sequenced (Genewiz) again to re-confirm the homozygous presentation of the given mutation in the population.
2.5. Immunoblotting
Approximately one million cells for each line under study were lysed in 200 µL RIPA buffer in presence of protease inhibitors. 25 µg of each cell lysate was run on an 8% SDS-PAGE gel and probed with anti-MSH2 (BD Pharmingen, 1:1000), anti-MSH6 (Bethyl laboratories, 1:1000), and anti-beta-actin (Sigma-Aldrich, 1:2000) antibodies. Percentage estimation of MSH2 and MSH6 levels in variant expressing lines were carried out in comparison to WT H1 MSH2 or MSH6 levels after being normalized for protein loading using beta-actin levels via ImageJ software.
2.6. Exon inclusion assay
The assay was carried out as described previously (Tricarico et al., 2017). Briefly, indicated cell lines were treated with 10 μg/mL cycloheximide (Sigma-Aldrich) for four h to suppress nonsense-mediated decay and allow accumulation of potential unstable transcripts. Subsequently, total RNA was extracted and cDNA was synthesized by reverse transcription using the iScript cDNA synthesis kit (Bio-Rad). Each variant-containing exon was detected by PCR amplification using primers in the flanking exons (Supp. Table S3) and the PCR products were analyzed on a 1.5% agarose gel.
2.7. MNNG drug toxicity assay
Approximately 8,000 cells of each cell line were seeded into individual wells of a Matrigel coated 24-well plate along with 10 µM ROCK inhibitor. The medium was replaced with hESC culture medium without the ROCK inhibitor the next day and cells were allowed to grow overnight. Approximately 36 h after seeding, cells were initially treated with 25 µM O6-Benzylguanine (O6-BG) (Sigma) for 2 h to inhibit action of the O6-methylguanine-DNA methyltransferase (MGMT) which normally functions to remove aberrant alkyl groups from the O6 position of guanine. Subsequently, cells were treated with different concentrations of N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) (obtained from the National Cancer Institute Chemical Carcinogen Reference Standard Repository) as indicated for 24 h in the presence of O6-BG and surviving cell population was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (ThermoFisher Scientific) following the manufacturer’s instruction.
2.8. Statistical analyses
In order to identify how variant-containing cell lines respond to different doses of MNNG, we applied clustering methods. In this unsupervised problem, three homogenous clusters were created based on similarity to an abrogated cell-line control (MSH2 KO), a proficient wild-type control (WT) or neither. K-means clustering was applied which can partition a data set into K distinct, non-overlapping clusters (Hastie, Tibshirani, & Friedman, 2009). We specified the desired number of clusters as three and each variant was assigned to one of the clusters. Statistical algorithms in R software (kmeanCluster) was used to identify the clusters. To examine the effects of the I561S reversion mutant on MNNG sensitivity, the mean change scores at the 2 μM MNNG concentration were compared using a two-group t-test with a two-sided alpha level of significance of 0.05 to assess statistical significance.
2.9. Microsatellite instability (MSI) analysis
Dilution cloning was employed to isolate and grow multiple single cell clones for each cell line for 7–10 days. Approximately 32 single cell clones were picked for each line and genomic DNA was isolated. All the cell lines used for MSI analysis were between passage number p15-p20. The MSI loci BAT-26 and NR-27 were amplified using high fidelity Phusion DNA polymerase (NEB) with 6-FAM labeled fluorescent primers and the PCR products were sent for fragment analysis (Genewiz). An additional analysis of 12 p.H639R clones was performed utilizing the Promega MSI Analysis System, Version 1.2 assay, that uses a multiplex PCR to amplify five mononucleotide repeat makers (BAT-26, NR-21, BAT-25, MONO-27, NR-24) and two pentanucleotide repeat markers (Penta D and Penta E). These PCR products were separated on the ABI 3730 DNA Analyzer using capillary electrophoresis. Clones showing changes in fragment length compared to wild-type H1 hESC genomic DNA were scored as MSI-positive and the percentage of positive clones were calculated for each line. Peak Scanner 2 and Geneious software programs were used to analyze the data.
2.10. Next Generation Sequencing
Next Generation Sequencing (NGS) was performed using a 501 gene panel to examine mutation levels in nine cell lines. We isolated genomic DNA from the indicated cell lines grown for 15–20 passages after editing and sequenced it using a targeted-enrichment hybrid-capture of 501 cancer related genes for which all coding exons and flanking intronic regions were sequenced (Agilent Technologies). Illumina NextSeq sequencers generated 149 bp paired-end sequence reads with a mean coverage of greater than or equal to 500X. Sequence analysis was performed using the ActionSeq™ Genome Analytics 2.0 (AGA2) pipeline, developed at The Jackson Laboratory, linking an in-house QC Toolkit, BWA, Picard, BreaKmer and the GATK4 MuTect2 tool kit for QC, alignment, and variant discovery. Variants were called against human genome build GRCh38 and a limit of detection (LOD) was set at 2% variant allele frequency cutoff for downstream analysis. The WT cell line was used to determine germline polymorphisms which were excluded from analysis. Mutations in mononucleotide repeat tracks were identified using the MSIseq R package. Total mononucleotide repeat mutations were divided by the size of the panel (2.3 Mb) to determine the number of frameshifts/Mb.
3. RESULTS
3.1. Generation of MSH2 VUS Expressing Cell Lines.
We selected nine MSH2 VUS as characterized by the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) database to determine their effects on cellular MMR function: c.131C>T, p.Thr44Met, p.T44M; c.1223A>G, p.Tyr408Cys, p.Y408C; c.1321A>C, p.Thr441Pro, p.T441P; c.1547G>T, p.Ser516Ile, p.S516I; c.1808A>T, p.Asp603Val, p.D603V; c.1916A>G, p.His639Arg, p.H639R; c.2141C>T, p.Ala714Val, p.A714V; c.2168C>T, p.Ser723Phe, p.S723F; c.2242G>T, p.Asp748Tyr, p.D748Y (Thompson et al., 2014) (Figure 1A, Supp. Table S1). We selected a tenth VUS (c.2021 G>C, p.Gly674Ala, p.G674A) that caused cancer in mice, yet appeared to separate MSH2 DNA repair and damage response functions (Lin et al., 2004). We also selected five variants previously determined to be Class 1 benign variants (c.23C>T, p.Thr8Met, p.T8M; c.381_382delinsTC, p.Asn127Ser, p.N127S; c.499G>C, p.Asp167His, p.D167H; c.965G>A, p.Gly322Asp, p.G322D; c.1168C>T, p.Leu390Phe, p.L390F) and five others previously deemed Class 5 pathogenic variants (c.595T>C, p.Cys199Arg, p.C199R; c.1046C>G, p.Pro349Arg, p.P349R; c.1865C>T, p.Pro622Leu, p.P622L; c.2089T>C, p.Cys697Arg, p.C697R; c.2251G>A, p.Gly751Arg, p.G751R) as positive and negative controls, respectively. We also used the parental H1 hESCs and an H1 hESC clone in which we used CRISPR-Cas9 to disrupt the MSH2 loci in a homozygous fashion as described previously (Gupta, Lin, Cowan, & Heinen, 2018) as additional positive and negative controls, respectively. To assess the functional effects of MSH2 VUS on MMR function in a human cell culture model, we used CRISPR-Cas9 gene editing to introduce each variant directly into the endogenous MSH2 locus of H1 hESCs. A plasmid vector-based expression of both guide RNA (gRNA) and Streptococcus pyogenes Cas9 (SpCas9) was used to introduce a genomic DNA double-strand break (DSB) at the appropriate location in MSH2. We then selected clones which successfully utilized homology directed repair (HDR) using a single strand deoxyoligonucleotide (ssODN) as the external template to site-specifically introduce the mutations in a homozygous fashion (Supp. Figure S1, Supp. Table S1). To enhance the efficient generation of single-cell derived hESC clones carrying the desired mutation, we employed several strategies. First, silent mutations were incorporated in the ssODN template along with the target amino acid-altering mutations to prevent Cas9 from re-cleaving after HDR of the break site. In some cases, these silent mutations also provided us with a way to screen for the mutant clone using restriction enzyme (RE) digestion of the PCR amplicon (Supp. Figure S2). Second, SCR-7 (DNA ligase IV inhibitor) and L-755507 (Rad51 activity stimulator) were used during the targeting process to reduce non-homologous end joining repair of the DSB and increase the HDR efficiency (Maruyama et al., 2015; Yu et al., 2015). Third, ssODNs with canonical 5’-PO4 and 3’-OH ends were used for increasing the incorporation efficiency into genomic DNA (Radecke, Radecke, Peter, & Schwarz, 2006). The results of our gene editing experiments are summarized in Supp. Table S4 and Supp. Figure S3 which show successful generation of 20 different MSH2 homozygous variant-expressing cell lines.
Figure 1.
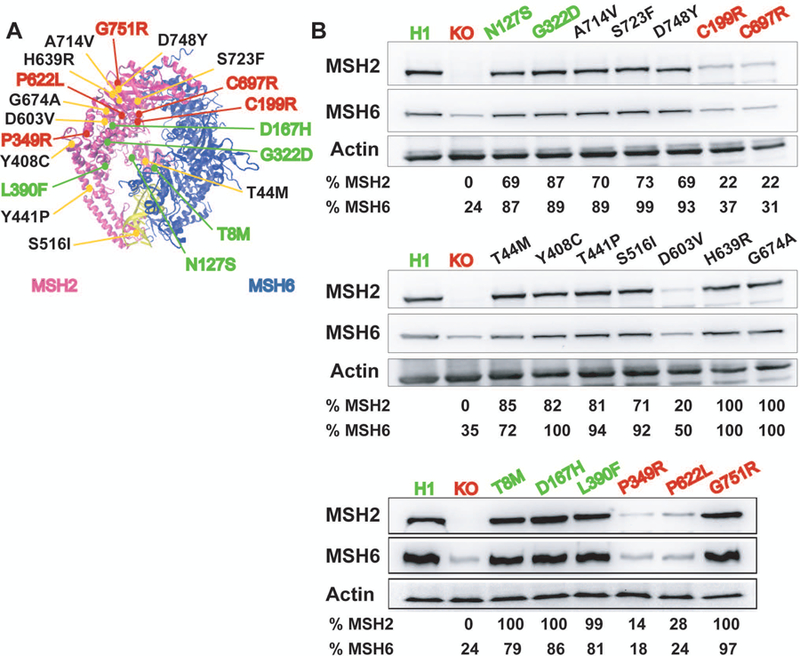
MSH2 variants tested and quantification of MSH2 and MSH6 steady-state protein levels. A: The MSH2 missense variants examined in this study as mapped to the DNA bound MSH2-MSH6 crystal structure (Warren et al., 2007). DNA is shown in yellow. B: Steady-state level of MSH2 and MSH6 proteins in each of the engineered MSH2 variant expressing lines is shown. Wild type H1 and MSH2 Class 1 benign variants are depicted in green. MSH2 KO and Class 5 pathogenic variants are shown in red. Actin was used as a loading control. Relative MSH2 and MSH6 levels were calculated with respect to the level in H1 WT cells after normalization for total protein loading.
As shown in Supp. Table S4, we identified homozygous clones with efficiencies ranging from 5–27% as determined by restriction enzyme (RE) digestion screening, when applicable. To further confirm the incorporation of the target variant in a homozygous fashion, we employed Sanger sequencing. In certain instances, sequencing revealed that the positive result in the RE digestion screen only indicated presence of the silent mutation and not the target variant of interest. As an example, we identified two potentially targeted clones expressing the p.D748Y variant using the RE digestion assay. However, upon sequencing, we noted that the amino acid-altering mutation was only incorporated in a heterozygous fashion (D748Yhet). Fortuitously, homozygous incorporation of the silent mutations provided a new PAM and gRNA binding sequence closer to the target codon. Thus, the D748Yhet cell line was further re-targeted using a re-designed gRNA that bound the modified protospacer region with the already incorporated silent mutations to generate a homozygous p.D748Y expressing line (Supp. Figure S4).
For a subset of variants where the PAM site was in close enough proximity to the target codon, we were able to introduce the target variant with relatively high efficiency without introducing additional silent mutations. If the target variant occurs in the PAM proximal regions (< ten nt), then it should be sufficient to prevent re-cutting while simultaneously allowing for efficient targeting as Cas9 is intolerant to mismatches in this region (Hsu et al., 2013; Kuscu, Arslan, Singh, Thorpe, & Adli, 2014; Wang, Wei, Sabatini, & Lander, 2014; Wu et al., 2014). As shown in Supp. Table S4, we generated homozygous clones confirmed by Sanger sequencing at a modestly improved efficiency (compared to the lines in which additional silent mutations were included) for all such variants except p.C199R. Of note, the efficiency of single nucleotide insertion was higher for the variants within 1–3 nt of 5’ end of the PAM sequence (12–27%). However, an increase in distance, as in the p.C199R line (seven nt away from PAM), significantly decreased the targeting efficiency (6%).
Availability of a canonical PAM sequence for Cas9 in the genomic region of interest is a known limiting factor for a desired targeting event. In our study, we generally observed a relationship between the efficiency of targeting and the distance of the target codon from the PAM site, in agreement with previously reported observations (Hsu et al., 2013; Yang et al., 2013). Not surprisingly, the variants most difficult to target were the aforementioned p.D748Y which had a distance of 14 nt between target codon and its closest PAM site and the p.D603V variant which was 31 nt away from an available PAM. For p.D603V, we did not obtain a single homozygous clone initially as judged from the RE digestion assay (Supp. Table S4). Thus, we redesigned a gRNA using an NGA sequence which has been reported previously to be an alternate PAM site for SpCas9 (Hu et al., 2018; Zhang et al., 2014). We were able to successfully generate a single heterozygous clone which was subsequently re-targeted to obtain multiple p.D603V expressing homozygous clones (Supp. Figure S5).
An important concern during CRISPR-Cas9 gene editing is off-target binding of the gRNA and the possibility of introducing a DSB by Cas9 mediated cleavage at unintended loci. To check for evidence of off-target cleavage in all 20 single-cell derived lines, we identified the top five predicted off-target sites by in silico analysis based on the probability of binding of a given gRNA to other locations in the genome. For each cell line, genomic DNA was extracted and the putative off-target regions were amplified by PCR. Purified PCR amplicons were subjected to Sanger sequencing and matched with the H1 hESC genomic reference sequence. We did not detect any sequence alterations at these sites in the genome suggesting an absence of off-target cleavage (Supp. Table S5) in line with similar observations from previous studies (Smith et al., 2014; Veres et al., 2014). Taken together, these results demonstrate the practicality of using CRISPR-Cas9 as a versatile tool to engineer multiple, site-specific LS-associated MSH2 VUS in an isogenic cell system to further analyze their functional impact on MMR function.
3.2. Variant Effects on MSH2 Protein Level.
Many pathogenic variants of MSH2 impair MMR by conferring a defect in the stability of MSH2 protein (Heinen, 2016; Heinen & Rasmussen, 2012). To check the steady-state level of MSH2 in the different variant expressing lines, we measured MSH2 protein levels in comparison to the levels in WT H1 hESCs via Western blot. The known pathogenic variants p.C199R, p.P349R, p.P622L and p.C697R showed an approximately 70–85% reduction in MSH2 protein levels compared to the WT control. However, with the exception of p.D603V (20% of WT levels), we did not observe any appreciable changes in the MSH2 level of any other variant expressing lines (Figure 1B). To determine if any of the variants interfered with the ability of the MSH2 protein to form a heterodimer with its partner MMR protein MSH6, we also examined MSH6 protein levels. MSH6 is an obligate heterodimer partner of MSH2 and so the inability of the two to form a heterodimer should lead to reduced stability of MSH6 (Marra et al., 1998). We found that the MSH6 protein levels were stable for all variant expressing lines except for those in which the MSH2 protein was also reduced suggesting that none of these variants likely interfere with heterodimer formation.
To test whether the reduced protein levels for p.D603V were due to a defect in RNA splicing, we performed an exon inclusion assay from cDNA (Tricarico et al., 2017). Using primers placed in exons 11 and 13 which flanked the c.1808A>T (p.D603V) variant in exon 12, we detected only the full-length product containing exon 12. We did not detect any novel splice forms excluding exon 12 nor did we see a reduction in the levels of RT-PCR product suggesting the variant did not have an effect on mRNA splicing (Figure 2). We similarly examined the known pathogenic control variants p.C199R and p.C697R for splicing defects as they also showed reduced protein levels. Similar to p.D603V, we did not detect any changes in the mRNA in these two variant lines either indicating all three alterations likely affect protein stability (Figure 2).
Figure 2.
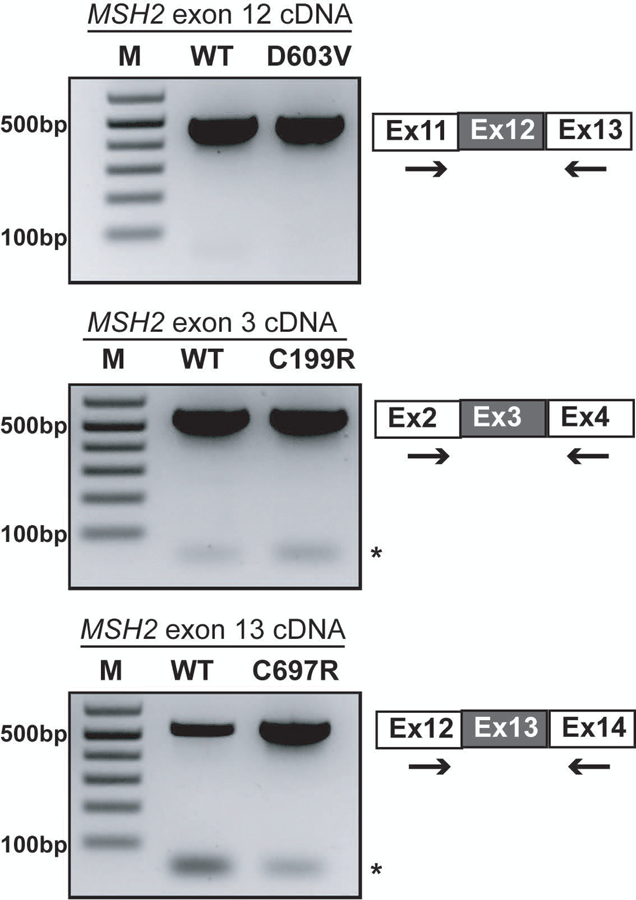
The c.1808A>T (p.D603V) variant does not impact splicing of exon 12. Ethidium stained agarose gels of the cDNA PCR products from the WT H1 and indicated variant cell lines. For each, exons depicted as grey boxes represent the exon carrying the genomic variant being tested. →, indicates location of the primers used for PCR amplification of the cDNA for each cell line. *, indicates primer dimers.
3.3. DNA Damage Response Signaling.
To test the functional effects of the altered MSH2 proteins in hESCs, we next examined the ability of these cells to elicit a DNA damage response to the alkylating agent MNNG. Human cancer cells undergo a permanent cell cycle arrest in response to various DNA damaging agents including SN1 alkylating agents like MNNG, however, this response is dependent on a functional MMR pathway (Heinen, 2016; Li et al., 2016). More recently, we have demonstrated that human pluripotent stem cells instigate a rapid apoptotic response to MNNG, also in a MMR-dependent manner (Gupta et al., 2018; Lin, Gupta, & Heinen, 2014). To assess the ability of the MSH2 VUS lines to induce cell death in response to MNNG, we used an MTT assay to measure cell survival following treatment. A 24 h challenge with MNNG resulted in a significant survival advantage for MSH2 knock-out hESCs compared to WT cells. Approximately 85% of the knock-out cells survived treatment at the highest concentration of MNNG tested (two μM) compared to only 35% survival for WT cells (Figure 3A). As expected, the response of hESC lines expressing known MSH2 benign variants (p. T8M, p.N127S, p.D167H, p.G322D and p.L390F) and MSH2 pathogenic variants (p.C199R, p.P349R, p.P622L, p.C697R and p.G751R) matched closely to the proficient WT and abrogated MSH2 knock-out control cell lines, respectively (Figure 3A). For the ten MSH2 VUS expressing lines under study, we used a statistical clustering approach to identify those variants whose survival curves either overlapped or formed a separate subgroup from the abrogated or proficient controls. We created three, non-overlapping homogenous clusters with one based on data from the abrogated standards, a second created from the proficient standards, and a third intermediate cluster that was not similar to either of these groups. Using this statistical method, we were able to segregate the ten variant lines into the three clearly distinct phenotypic groups. p.T44M, p.Y408C, p.T441P, and p.A714V expressing lines exhibited a similar response to that of the proficient controls. The p.D603V, p.G674A, p.S723F, and p.D748Y cell lines were resistant to MNNG and clustered with the abrogated controls. However, the p.S516I and p.H639R lines showed an intermediate phenotype upon MNNG challenge (66% and 67% survival, respectively at two μM MNNG) that formed an independent non-overlapping intermediate cluster (Figure 3A). We reasoned that the impaired damage signaling in the p.S516I variant line should be rescued by reverting the mutant isoleucine residue back to serine. As we did not destroy the Cas9 target site during the initial targeting event, we were able to use CRISPR-Cas9 to correct the introduced single nucleotide variant and generate a p.I516S line. As shown in Figure 3B, reversion of the mutant residue back to the original sequence restored MNNG induced apoptotic signaling to WT levels. Taken together, these results show the ability of our cell based assay system to clearly distinguish phenotypic variation between different MSH2 VUS lines.
Figure 3.
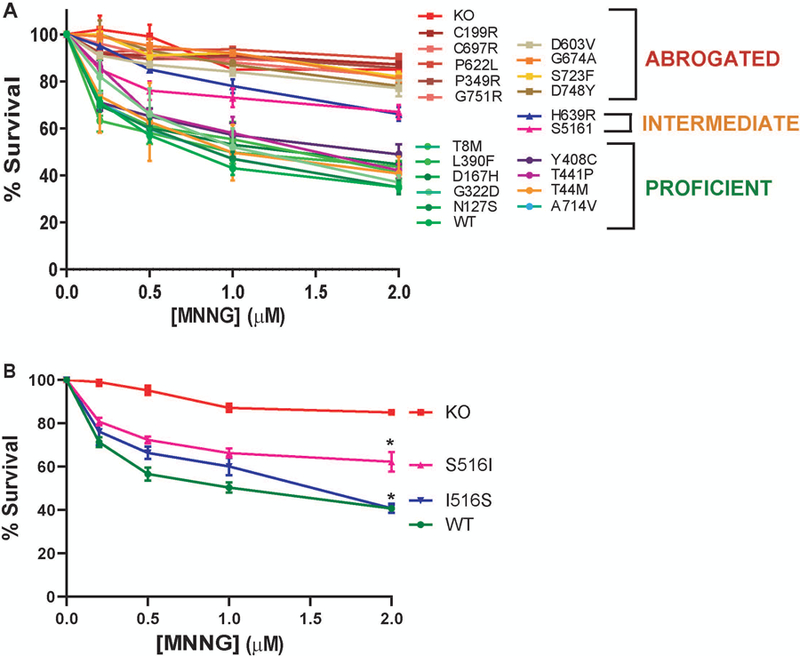
The MMR dependent damage response in variant cell lines. Cell survival as measured by MTT assay showing percentage survival after 24 h treatment with increasing doses of the DNA alkylating agent MNNG in the indicated cell lines. The values are represented as the mean ± S.E.M. N = 3–5. A: Survival curves for 10 VUS lines along with 10 control variants, and WT and MSH2 knockout (KO) controls. A statistical clustering analysis was performed which grouped the variants into three populations based on their survival. One cluster resembled the Abrogated controls, one cluster resembled the Proficient controls and a third resembled an Intermediate cluster. B: Survival curves for S516I line and I516S reversion mutant line. *, p < 0.05
3.4. Mismatch Repair Ability.
The canonical function of MMR is to detect and repair mismatches generated during DNA replication due to DNA polymerase errors (Kunkel & Erie, 2015). The MMR pathway also repairs small indels created by polymerase errors at simple repeat sequences. To examine the ability of the MSH2 variants to repair endogenous chromosomal sequences, we tested for the presence of instability at two DNA microsatellites. Lack of functional MMR manifests as instability in genomic microsatellite regions which is the hallmark of bona fide LS tumors (Boland & Goel, 2010; Liu et al., 1996). We tested for evidence of microsatellite instability (MSI) in the VUS expressing lines at two mononucleotide repeat loci, BAT-26 and NR-27, which are well-established predictive biomarkers for impaired MMR function (Goel, Nagasaka, Hamelin, & Boland, 2010). Single-cell derived clones were grown from control and VUS cell populations using dilution cloning. The MSI status at the BAT-26 and NR-27 loci was assessed employing fragment analysis for multiple clones for each cell line. Table 1 shows the percentage of clones showing a variable allele length compared to the reference H1 genomic sequence suggesting an alteration in number of repeat units (indicative of MSI) for both loci. As expected, no MSI was detected in any of the subclones for H1 WT hESCs at either marker. In contrast, every clone obtained from MSH2 KO cells exhibited an altered repeat length. Inter-clonal variability with regards to repeat length was also observed among the MSH2 KO clones (data not shown). Among the missense variants, no MSI was detected for the p.T44M, p.Y408C, p.T441P, and p.A714V lines along with the Class 1 proficient controls indicative of retained normal MSH2 repair function. However, a varying degree of MSI was detected for the p.D603V, p.G674A, p.S723F, and p.D748Y lines along with the Class 5 abrogated controls suggesting different degrees of functional impairment in these lines. Interestingly, the H639R clones also did not exhibit any defect in MSI length whereas S516I displayed a very low percentage of clones with MSI despite their apparent functional defects in the alkylation damage response assay.
Table 1.
Microsatellite Instability in Variant Cell Lines
| Cell Line | BAT-26a | NR-27a | Clones Tested |
|---|---|---|---|
| H1 (WT) | 0 | 0 | 24 |
| MSH2-KO | 100 | 100 | 24 |
| Class 1 | |||
| T8M | 0 | 0 | 32 |
| N127S | 0 | 0 | 27 |
| D167H | 0 | 0 | 32 |
| G322D | 0 | 0 | 32 |
| L390F | 0 | 0 | 32 |
| Class 3 | |||
| T44M | 0 | 0 | 32 |
| Y408C | 0 | 0 | 32 |
| T441P | 0 | 0 | 32 |
| S516I | 19 | 6 | 32 |
| D603V | 71 | 60 | 31 |
| H639R | 0 | 0 | 32 |
| G674A | 52 | 26 | 31 |
| A714V | 0 | 0 | 32 |
| S723F | 41 | 25 | 32 |
| D748Y | 85 | 88 | 26 |
| Class 5 | |||
| C199R | 34 | 53 | 32 |
| P349R | 47 | 44 | 32 |
| P622L | 38 | 47 | 32 |
| C697R | 100 | 100 | 20 |
| G751R | 44 | 84 | 32 |
Percentage of clones with altered alleles compared to WT sequence
To further explore the effect of the variants on DNA repair function, we measured mononucleotide repeat instability levels in a subset of variants via NGS. Genomic DNA from nine cell lines including the WT and KO controls were sequenced using a targeted-enrichment hybrid-capture panel of 501 cancer related genes for which all exons and flanking introns were evaluated. We examined the rate of indels per megabase (Mb) within mononucleotide repeats as another measurement of MSI. Over 13,000 mononucleotide repeats were assessed within the gene panel. Consistent with our clonal PCR-based MSI analyses described above, we found high levels of indels for the KO and two abrogated control variants (p.C199R and p.C697R) and no difference in the two proficient control variants (p.N127S and p.G322D) compared to the WT genome (Figure 4). We also tested p.S723F, a VUS with abrogated function in the alkylation damage response (Figure 3A) and clonal PCR-based MSI studies (Table 1), albeit at a slightly decreased level than the KO and Class 5 controls. While this variant showed fewer indels in simple repeats than the abrogated controls, the level of instability was > 0.395 indels in simple repeats/Mb, a threshold consistent with MSI-high (MSI-H) tumors (Ni Huang et al., 2015) (Figure 4). Interestingly, both the p.S516I and p.H639R variants displayed some indels compared to WT, though neither met the threshold for MSI-H (Figure 4). These results are consistent with the clonal PCR-based MSI studies for p.S516I, but not for p.H639R (Table 1). In light of this result, we reanalyzed 12 p.H639R clones for MSI by PCR using a panel of five mononucleotide repeat markers and found instability at one marker for a single clone suggesting a possible MSI-low (MSI-L) phenotype (Supp. Table S6). Thus, while a simpler two-marker test is likely sufficient for most variants, we suggest that any variant displaying an intermediate DNA damage response function, but no MSI with the two-marker test, be examined further using the five-marker panel.
Figure 4.
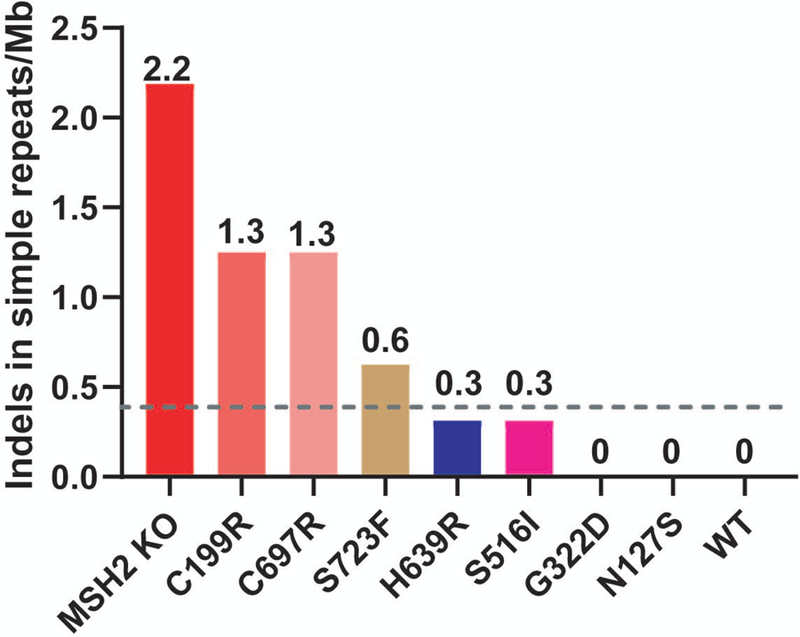
Next-Generation sequencing of simple repeats from a subset of variant cell lines. Indels in simple repeat sequences were measured from genomic DNA by next-generation sequencing (NGS) for the p.H639R, p.S516I and p.S723F VUS lines as well as two abrogated (p.C199R and p.C697R) and two proficient controls (p.N127S and p.G322D) along with the WT and MSH2 knockout (KO) controls. Values indicate the number of indels in simple repeat sequences per megabase (Mb). The dashed gray line represents the threshold value for MSI-H (0.395 indels in simple repeats/Mb) as determined by NGS analysis of human tumors (Ni Huang et al., 2015).
4. DISCUSSION
Limited genetic and functional information on a MMR gene missense variant can be a bottleneck in the definitive diagnosis of LS. In absence of a robust family pedigree, variant specific functional information becomes crucial in determining the likely pathogenic significance of the variant (Starita et al., 2017). Thus, a suitable model system to study the functional impact of a gene variant is necessary. The advent of CRISPR-Cas9 gene editing provides the ability to engineer specific genetic variants at their endogenous chromosomal locus. Thus, any impact of a given variant on protein function can be examined in the context of the gene’s cell-intrinsic regulatory mechanisms that govern its expression and function. This approach resolves concerns that have arisen from previous functional studies of MMR VUS (Heinen & Rasmussen, 2012). In vitro reconstitution of repair of a single base pair mismatch has been a valuable tool to identify variants that clearly disrupt biochemical activity, however, this approach likely does not reveal all variants that could interfere with MMR activity in vivo. Additionally, cell based assays relying on exogenous expression of variant-containing transgenes provides cellular context, but may be prone to non-specific effects due to improper regulation of transgene expression. More so, the availability of an established variant expressing mutant line opens up possibilities for modeling other potentially interesting aspects of the disease. For example, putative epistatic or synthetic lethal interactions can now be studied in the variant background. In addition, although outside the scope of this study, our use of hESCs allows for future studies to examine the ramifications of the variant in specific cell types. Pluripotency confers the ability to differentiate these cells into any tissue type in the body and hence provides potential avenues to carry out functional testing in a three-dimensional model system and, if need be, do drug testing as a proxy for the tissue of interest. Through the course of the current study, we have developed a robust CRISPR-Cas9 targeting methodology for use in human pluripotent stem cells which are generally not very amenable to genetic manipulations using HDR (Lin, Staahl, Alla, & Doudna, 2014; Mali et al., 2013; Yang et al., 2013).
A key cellular function of MMR that we assessed was the ability of MSH2 to instigate apoptosis in response to DNA damage. This function is an important gate-keeping mechanism that prevents cells from going forward with unrepaired DNA lesions (Heinen, 2016; Li et al., 2016; Stojic et al., 2004). Absence of this protective mechanism due to MMR deficiency may be an important indicator of the transformative potential of a cell (Heinen, Schmutte, & Fishel, 2002; Wojciechowicz et al., 2014). Based on the response of the variant lines to MNNG, we were able to cluster eight MSH2 VUS into two distinct groups indicating presence or absence of a functional DNA damage signaling pathway (Figure 3A). Of those proficient lines (p.T44M, p.Y408C, p.T441P, and p.A714V), the steady state levels of MSH2 protein are comparable to WT cells. In the abrogated line p.D603V, the MSH2 protein is present at about 20% of the WT level which may partially underlie the defective damage response. As the c.1808A>T variant that encodes for p.D603V was knocked-into the endogenous MSH2 allele, we were also able to determine that it did not disrupt splicing of exon 12 (Figure 2). The ability to simultaneously test effects at both the protein and mRNA level is another advantage of our gene editing approach. In the other three abrogated lines (p.G674A, p.S723F and p.D748Y), while the protein levels appear stable, the variants affect residues in the ATPase domain of MSH2 spanning amino acids 620–855, which is crucial to MSH2 function. Thus, the impairment of MSH2 dependent signaling may be due to defective adenosine nucleotide processing (Heinen, Wilson, et al., 2002). Interestingly, the resistance observed in the p.G674A line contrasts with the original study describing this variant in which p.G674A-expressing mouse embryonic fibroblasts retain sensitivity to damaging agents (Lin et al., 2004). Our result was more consistent with a latter study using mouse ESCs to screen for Msh2 variants that provided resistance to the guanine analog 6-thioguanine (Houlleberghs et al., 2016). In that study, p.G674A-expressing cells also displayed an abrogated DNA damage response. The p.S516I variant displays an intermediate response in the MNNG toxicity assay while displaying no observable difference in the MSH2 protein level compared to WT. S516 is present in the clamp domain of the MSH2 protein very near residues K512 and R524 which make significant, non-specific contacts with the DNA backbone upon mismatch binding (Warren et al., 2007). The change of a polar amino acid (serine) to a hydrophobic residue (isoleucine) at position 516 may affect these non-specific interactions to partially impair mismatch binding affinity. As shown in Figure 3B, we were able to rescue the sensitivity of this variant to WT levels upon correcting the mutation in the variant line to re-express p.S516 indicating the specific contribution of the isoleucine substitution to the observed MMR response before correction. The p.H639R variant also displays an intermediate damage signaling function. This variant lies within the ATPase domain of MSH2 as well as within a domain between amino acids 601–671 that has previously been shown to be important for Exo1 binding in a peptide-binding assay (Schmutte, Sadoff, Shim, Acharya, & Fishel, 2001). Further biochemical studies will be required to ascertain the specific mechanistic defect in these mutant proteins.
We also assessed the MSI status of all the generated lines in this study. No MSI was detected for the p.T44M, p.Y408C, p.T441P, and p.A714V lines, which when combined with their normal damage signaling functions suggests that these four MSH2 VUS do not alter MSH2 function providing strong evidence of a benign impact from these variants (BS3, Table 2) (Richards et al., 2015). In contrast, the p.D603V, p.G674A, p.S723F, and p.D748Y lines display both an MSI and damage signaling phenotype closely matching that of the MSH2 knock out and known pathogenic variants providing strong functional evidence of pathogenicity (PS3, Table 2) (Richards et al., 2015). Curiously, the p.S516I and p.H639R variants show either only a minor MSI phenotype or no defect at all by PCR analysis of the two markers. A much more extensive analysis of mononucleotide repeat instability by NGS revealed that both lines displayed low levels of indels, consistent with an MSI-L phenotype (Ni Huang et al., 2015). In general, the use of NGS to analyze the variant cell lines is likely unnecessary as the results mostly recapitulated the simpler, less expensive assays. However, for those variants with intermediate effects in the MSI and damage response assays, the use of NGS may provide important insights such as the determination of an MSI-L phenotype for the p.S516I and p.H639R lines. Given their modest abrogation of function, these variants may be weak disease alleles, though additional testing is clearly warranted to more accurately gauge their impact on disease.
Table 2.
Summary of Functional Studies and Prior Evidence
| Variant | MSI Dataa | Previous Functional Studiesb | PolyPhen-2 Predictionc | Mutation Assessor Predictiond | MAPP-MMR Scorese | Protein Stabilityf | Damage Response Signalingf | DNA Repair Functionf | ACMG Evidence Codeg |
|---|---|---|---|---|---|---|---|---|---|
| Class 1 | |||||||||
| T8M | MSS1, 2 | Interm19 | Possibly Damaging¥ | Low | − | + | + | + | BS3 |
| N127S | Prof20 | Possibly Damaging | High | 3.26± | + | + | + | BS3 | |
| D167H | MSS3 | Prof21,22, Interm23, Mixed24 | Probably Damaging | Medium | 9.62 | + | + | + | BS3 |
| G322D | MSS4-8 | Prof20,23,25 | Benign | Medium | 3.24± | + | + | + | BS3 |
| L390F | MSS2, 9 | Prof23 | Benign | Medium | 5.71 | + | + | + | BS3 |
| Class 3 | |||||||||
| T44M | Interm23, Prof26 | Probably Damaging | High | 3.92± | + | + | + | BS3 | |
| Y408C | Probably Damaging | High | 3.94± | + | + | + | BS3 | ||
| T441P | MSI-L10 | Benign | Low | 5.90 | + | + | + | BS3 | |
| S516I | Benign | Low | 3.11± | + | +/− | +/− | Inc | ||
| D603V | Probably Damaging | High | 22.25 | − | − | − | PS3 | ||
| H639R | MSI-H11,h | Interm23, Mixed24 | Probably Damaging | High | 31.55 | + | +/− | +/− | Inc |
| G674A | Abrog27,28, Mixed29 | Probably Damaging | High | 22.05 | + | − | − | PS3 | |
| A714V | Probably Damaging | High | 30.41 | + | + | + | BS3 | ||
| S723F | MSI-H12 | Abrog23,28 | Probably Damaging | High | 21.57 | + | − | − | PS3 |
| D748Y | Probably Damaging | High | 19.62 | + | − | − | PS3 | ||
| Class 5 | |||||||||
| C199R | MSI-H13, 14 | Abrog19 | Probably Damaging | Medium | 31.22 | − | − | − | PS3 |
| P349R | MSI-H15, 16 | Abrog28 | Probably Damaging | High | 41.48 | − | − | − | PS3 |
| P622L | MSI-H17 | Abrog21,22,23,24,28 | Probably Damaging | High | 26.92 | − | − | − | PS3 |
| C697R | MSI-H16 | Abrog30 | Probably Damaging | High | 37.64 | − | − | − | PS3 |
| G751R | MSI-H18h | Probably Damaging | High | 36.52 | + | − | − | PS3 |
MSI analysis in human tumors from patients carrying variant as reported in literature when available.
Effects of variant on function in previous studies reported in literature when available. Abrog, abrogated; Prof, proficient; Intermed, intermediate; Mixed, conflicting results in different assays from same study.
PolyPhen-2 scores were calculated from http://genetics.bwh.harvard.edu/pph2/ (Adzhubei et al., 2010). Scores range between 0–1. Scores close to 0 are predicted to be benign while scores close to 1 indicate damaging impact on protein function. Predictions shown concur between the HumDiv and HumVar model.
indicates conflict in prediction between the two models.
Mutation Assessor Release 3 functional impact scores were calculated from http://mutationassessor.org/r3/ (Reva et al., 2011). A higher score predicts a greater functional impact by the given mutation.
MAPP-MMR prediction scores obtained from http://mappmmr.blueankh.com/ (Chao et al., 2008). Higher score indicates greater predicted deleterious impact on protein function. Scores > 3.5 are considered either VUS or deleterious in nature.
indicates predicted borderline deleterious nature of the variant. There is no available MAPP-MMR score for the p.T8M variant.
From this study.
Expected ACMG evidence code resulting from our data; BS3, strong evidence of benign impact; PS3, strong evidence of pathogenicity; Inc, inconclusive.
p.H639R co-occurred in germline with a MLH1 p.Gly* variant in patient with MSI-H tumor.
Scartozzi et al.,2002
Capozzi et al., 1990
Traditionally, functional evaluation of any gene variant is carried out in a post hoc fashion which is a time consuming process. Hence, to provide quick guidance as to whether a new-found variant is deleterious or neutral, a number of in silico variant prediction platforms have been developed (Adzhubei et al., 2010; Chao et al., 2008; Reva, Antipin, & Sander, 2011). These platforms mainly predict the effect of a gene variant on protein function based on evolutionary conservation and amino acid charge. However, often times different prediction algorithms provide conflicting information. For example, p.N127S is a well-established benign variant of MSH2 which is also confirmed in this study to be MMR-proficient (Ollila, Dermadi Bebek, Greenblatt, et al., 2008; Tricarico et al., 2017). p.N127S along with p.G322D are also represented in the ExAC database (Lek et al., 2016) as known polymorphisms prevalent in the normal population. However, multiple variant predictive platforms identify p.N127S as deleterious in nature (Table 2). Similar observations were also made for p.T44M and p.A714V which were likewise shown to be MMR-proficient in our assays. These results demonstrate the inaccuracy that still exists with such in silico approaches, and highlights the need for a robust dataset of functionally characterized variants for training the in silico platforms.
In summary, we demonstrate the utility of using CRISPR-Cas9 genome-engineered hESCs to obtain relevant functional information for better clinical interpretation of LS associated, patient-derived MSH2 VUS. Recently, a CRISPR-based approach was used to perform a large-scale screen of single nucleotide variants in the BRCA1 (MIM # 113705) gene (Findlay et al., 2018). Combining a CRISPR-based editing approach with NGS allowed for a relatively rapid assessment of thousands of variants. The use of such a strategy to characterize MMR gene variants should also be possible. However, such an approach does have limitations. First, the variants are categorized based on their ability to survive selection for gain or loss of some function. In the case of MMR variants, selection for loss of alkylation damage sensitivity is one obvious tactic. Any variants that affect DNA repair function without significantly altering the damage response would be missed in such a study. Second, no individual cell lines carrying distinct variants are created by this approach, eliminating the possibility for follow-up studies such as on variants with intermediate effects. Third, it is unclear how expert panels that currently classify the pathogenic significance of variants will utilize the data from these large-scale screens. Likely, significant validation will be required. Thus, a combination of large-scale screens with a more methodical one-at-a-time approach will likely be necessary for the immediate future. In this current study, we were able to collect data on multiple MMR functions, allowing us to determine the functional capability of eight of ten VUS (Table 2). This functional characterization provides important evidence for both the InSiGHT variant classification scheme (Thompson et al., 2014) and the ACMG guidelines for determining pathogenic potential (Richards et al., 2015).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge technical support provided by Ms. Qingfen Yang, Ms. Kaussar Rahman, Mr. John Glynn and Mr. Chris Stoddard in preparing the reagents and providing helpful suggestions.
Funding Information: National Institutes of Health (CA115783 and CA222477); State of Connecticut Regenerative Medicine Fund (13SCB-UCHC-06).
Footnotes
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnetson R, Cartwright N, van Vliet A, Haq N, Drew K, Farrington S, … Dunlop M (2008). Classification of ambiguous mutations in DNA mismatch repair genes identified in a population-based study of colorectal cancer. Human Mutation, 29(3), 367–374. [DOI] [PubMed] [Google Scholar]
- Belvederesi L, Bianchi F, Galizia E, Loretelli C, Bracci R, Catalani R, … Cellerino R (2008). MSH2 missense mutations and HNPCC syndrome: pathogenicity assessment in a human expression system. Human Mutation, 29, E296–E309. [DOI] [PubMed] [Google Scholar]
- Boland CR, & Goel A (2010). Microsatellite Instability in Colorectal Cancer. Gastroenterology, 138(6), 2073–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieger A, Trojan J, Raedle J, Plotz G, & Zeuzem S (2002). Transient mismatch repair gene transfection for functional analysis of genetic hMLH1 and hMSH2 variants. Gut, 51(5), 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich SE, Rivera-Munoz EA, & Berg JS (2018). Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Human Mutation, 39(11), 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi E, Della Puppa L, Fornasarig M, Pedroni M, Boiocchi M, & Viel A (1999). Evaluation of the replication error phenotype in relation to molecular and clinicopathological features in hereditary and early onset colorectal cancer. European Journal of Cancer, 35(2), 289–295. [DOI] [PubMed] [Google Scholar]
- Chan TL, Yuen ST, Chung LP, Ho JWC, Kwan K, Fan YW, … Leung SY (1999). Germline hMSH2 and differential somatic mutations in patients with Turcot’s syndrome. Genes, Chromosomes and Cancer, 25(2), 75–81. [PubMed] [Google Scholar]
- Chao E, Velasquez J, Witherspoon M, Rozek L, Peel D, Ng P, … Lipkin S (2008). Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Human Mutation, 29(6), 852–860. [DOI] [PubMed] [Google Scholar]
- Christensen LL, Kariola R, Korhonen MK, Wikman FP, Sunde L, Gerdes A-M, … Ørntoft TF (2009). Functional characterization of rare missense mutations in MLH1 and MSH2 identified in Danish colorectal cancer patients. Familial Cancer, 8(4), 489. [DOI] [PubMed] [Google Scholar]
- Cravo M, Afonso AJ, Lage P, Albuquerque C, Maia L, Lacerda C, … Nobre-Leitão C (2002). Pathogenicity of missense and splice site mutations in hMSH2 and hMLH1 mismatch repair genes: implications for genetic testing. Gut, 50(3), 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lellis L, Aceto GM, Curia MC, Catalano T, Mammarella S, Veschi S, … Cama A (2013). Integrative analysis of hereditary nonpolyposis colorectal cancer: the contribution of allele-specific expression and other assays to diagnostic algorithms. PLoS One, 8(11), e81194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieumegard B, Grandjouan S, Sabourin JC, Le Bihan ML, Lefrere I, Bellefqih, … Bressac-de Paillerets B (2000). Extensive molecular screening for hereditary non-polyposis colorectal cancer. British Journal of Cancer, 82(4), 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost M, Tiersma Y, Thompson BA, Frederiksen JH, Keijzers G, Glubb D, … Tavtigian SV (2018). A functional assay–based procedure to classify mismatch repair gene variants in Lynch syndrome. Genetics In Medicine [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost M, Zonneveld JBM, van Hees S, Rasmussen LJ, Hofstra RMW, & de Wind N (2012). A rapid and cell-free assay to test the activity of lynch syndrome-associated MSH2 and MSH6 missense variants. Human Mutation, 33(3), 488–494. [DOI] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, … Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature, 562(7726), 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Konishi F, Shitoh K, Kojima M, Nagai H, & Tsukamoto T (2002). Evaluation of screening strategy for detecting hereditary nonpolyposis colorectal carcinoma. Cancer, 94(4), 911–920. [PubMed] [Google Scholar]
- Gammie AE, Erdeniz N, Beaver J, Devlin B, Nanji A, & Rose MD (2007). Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics, 177, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Sakato M, DeRocco V, Yamane K, Du C, Erie DA, … Hsieh P (2012). Biochemical Analysis of the Human Mismatch Repair Proteins hMutSα MSH2G674A-MSH6 and MSH2-MSH6T1219D. Journal of Biological Chemistry, 287(13), 9777–9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Nagasaka T, Hamelin R, & Boland CR (2010). An optimized pentaplex PCR for detecting DNA mismatch repair-deficient colorectal cancers. PLoS One, 5(2), e9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D, Lin B, Cowan A, & Heinen CD (2018). ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proceedings of the National Academy of Sciences, 115(7), 1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, & Friedman J (2009). The elements of statistical learning: data mining, inference, and prediction Springer. [Google Scholar]
- Heinen CD (2016). Mismatch repair defects and Lynch syndrome: The role of the basic scientist in the battle against cancer. DNA Repair, 38, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen CD, & Rasmussen LJ (2012). Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hereditary Cancer in Clinical Practice, 10(1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen CD, Schmutte C, & Fishel R (2002). DNA Repair and Tumorigenesis: Lessons from Hereditary Cancer Syndromes. Cancer Biology & Therapy, 1(5), 477–485. [DOI] [PubMed] [Google Scholar]
- Heinen CD, Wilson T, Mazurek A, Berardini M, Butz C, & Fishel R (2002). HNPCC mutations in hMSH2 result in reduced hMSH2-hMSH6 molecular switch functions. Cancer Cell, 1(5), 469–478. [DOI] [PubMed] [Google Scholar]
- Houlleberghs H, Dekker M, Lantermans H, Kleinendorst R, Dubbink HJ, Hofstra RMW, … te Riele H(2016). Oligonucleotide-directed mutagenesis screen to identify pathogenic Lynch syndrome-associated MSH2 DNA mismatch repair gene variants. Proceedings of the National Academy of Sciences, 113(15), 4128–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, … Zhang F (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology, 31(9), 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, … Liu DR (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature, 556(7699), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J (2013). Postreplicative Mismatch Repair. Cold Spring Harbor Perspectives in Biology, 5(4), a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaina B, Ziouta A, Ochs K, & Coquerelle T (1997). Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: facts and models. Mutation Research, 381(2), 227–241. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, & Erie DA (2015). Eukaryotic Mismatch Repair in Relation to DNA Replication. Annual Review of Genetics, 49(1), 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, Singh R, Thorpe J, & Adli M (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nature Biotechnology, 32(7), 677–683. [DOI] [PubMed] [Google Scholar]
- Lee SC, Guo JY, Lim R, Soo R, Koay E, Salto-Tellez M, … Goh BC (2005). Clinical and molecular characteristics of hereditary non-polyposis colorectal cancer families in Southeast Asia. Clinical Genetics, 68(2), 137–145. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … MacArthur DG (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pearlman AH, & Hsieh P (2016). DNA mismatch repair and the DNA damage response. DNA Repair, 38, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Gupta D, & Heinen CD (2014). Human Pluripotent Stem Cells Have a Novel Mismatch Repair-dependent Damage Response. Journal of Biological Chemistry, 289(35), 24314–24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, … Edelmann W (2004). An Msh2 Point Mutation Uncouples DNA Mismatch Repair and Apoptosis. Cancer Research, 64(2), 517–522. [DOI] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, & Doudna JA (2014). Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife, 3, e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, … Kinzler KW (1996). Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nature Medicine, 2(2), 169–174. [DOI] [PubMed] [Google Scholar]
- Lutzen A, de Wind N, Georgijevic D, Nielsen F, & Rasmussen L (2008). Functional analysis of HNPCC-related missense mutations in MSH2. Mutation Research, 645, 44–55. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, & Hitchins MP (2015). Milestones of Lynch syndrome: 1895–2015. Nature Reviews Cancer, 15(3), 181–194. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, … Church GM (2013). RNA-guided human genome engineering via Cas9. Science, 339(6121), 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, & Jiricny J (1998). Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proceedings of the National Academy of Sciences, 95(15), 8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, & Heinen CD (2010). Lynch syndrome-associated mutations in MSH2 alter DNA repair and checkpoint response functions in vivo. Human Mutation, 31(10), E1699–E1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara M, Yokozaki H, Yasui W, Dohi K, & Tahara E (1997). Identification of concurrent germ-line mutations in hMSH2 and/or hMLH1 in Japanese hereditary nonpolyposis colorectal cancer kindreds. Cancer Epidemiology Biomarkers Prevention, 6(12), 1057–1064. [PubMed] [Google Scholar]
- Ni Huang M, McPherson JR, Cutcutache I, Teh BT, Tan P, & Rozen SG (2015). MSIseq: Software for Assessing Microsatellite Instability from Catalogs of Somatic Mutations. Scientific Reports, 5, 13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila S, Dermadi Bebek D, Greenblatt M, & Nystrom M (2008). Uncertain pathogenicity of MSH2 variants N127S and G322D challenges their classification. International Journal of Cancer, 123(3), 720–724. [DOI] [PubMed] [Google Scholar]
- Ollila S, Dermadi Bebek D, Jiricny J, & Nystrom M (2008). Mechanisms of pathogenicity in human MSH2 missense mutants. Human Mutation, 29(11), 1355–1363. [DOI] [PubMed] [Google Scholar]
- Pastrello C, Pin E, Marroni F, Bedin C, Fornasarig M, Tibiletti M, … Viel A (2011). Integrated analysis of unclassified variants in mismatch repair genes. Genetics In Medicine, 13(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Plazzer JP, Sijmons RH, Woods MO, Peltomaki P, Thompson B, Den Dunnen JT, & Macrae F (2013). The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Familial Cancer, 12(2), 175–180. [DOI] [PubMed] [Google Scholar]
- Radecke S, Radecke F, Peter I, & Schwarz K (2006). Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. Journal of Genetic Medicine, 8(2), 217–228. [DOI] [PubMed] [Google Scholar]
- Reva B, Antipin Y, & Sander C (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research, 39(17), e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics In Medicine, 17, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez de Abajo A, de la Hoya M, van Puijenbroek M, Godino J, Diaz-Rubio E, Morreau H, & Caldes T (2006). Dual role of LOH at MMR loci in hereditary non-polyposis colorectal cancer? Oncogene, 25(14), 2124–2130. [DOI] [PubMed] [Google Scholar]
- Schmutte C, Sadoff MM, Shim K-S, Acharya S, & Fishel R (2001). The Interaction of DNA Mismatch Repair Proteins with Human Exonuclease I. Journal of Biological Chemistry, 276(35), 33011–33018. [DOI] [PubMed] [Google Scholar]
- Sheng JQ, Fu L, Sun ZQ, Huang JS, Han M, Mu H, … Li SR (2008). Mismatch repair gene mutations in Chinese HNPCC patients. Cytogenetics and Genome Research, 122(1), 22–27. [DOI] [PubMed] [Google Scholar]
- Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, … Ye Z (2014). Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell, 15(1), 12–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, … Fowler DM(2017). Variant Interpretation: Functional Assays to the Rescue. American Journal of Human Genetics, 101(3), 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojic L, Brun R, & Jiricny J (2004). Mismatch repair and DNA damage signalling. DNA Repair, 3(8–9), 1091–1101. [DOI] [PubMed] [Google Scholar]
- Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, … Tavtigian SV (2013). Calibration of Multiple In Silico Tools for Predicting Pathogenicity of Mismatch Repair Gene Missense Substitutions. Human Mutation, 34(1), 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BA, Spurdle AB, Plazzer J-P, Greenblatt MS, Akagi K, Al-Mulla F, … on behalf of InSi, G. H. T. (2014). Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nature Genetics, 46(2), 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricarico R, Kasela M, Mareni C, Thompson BA, Drouet A, Staderini L, … Committee IVI (2017). Assessment of the InSiGHT Interpretation Criteria for the Clinical Classification of 24 MLH1 and MSH2 Gene Variants. Human Mutation, 38(1), 64–77. [DOI] [PubMed] [Google Scholar]
- Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, … Musunuru K (2014). Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell, 15(1), 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, & Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science, 343(6166), 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R, Meldrum C, Williams R, Mokany E, Scott R, Turner J, … Spigelman A (2002). Impact of microsatellite testing and mismatch repair protein expression on the clinical interpretation of genetic testing in hereditary non-polyposis colorectal cancer. Journal of Cancer Research and Clinical Oncology, 128(8), 403–411. [DOI] [PubMed] [Google Scholar]
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, & Beese LS (2007). Structure of the Human MutSalpha DNA Lesion Recognition Complex. Molecular Cell, 26(4), 579–592. [DOI] [PubMed] [Google Scholar]
- Wielders EAL, Dekker RJ, Holt I, Morris GE, & te Riele H (2011). Characterization of MSH2 variants by endogenous gene modification in mouse embryonic stem cells. Human Mutation, 32(4), 389–396. [DOI] [PubMed] [Google Scholar]
- Wojciechowicz K, Cantelli E, Van Gerwen B, Plug M, Van Der Wal A, Delzenne-Goette E, … Riele HT (2014). Temozolomide Increases the Number of Mismatch Repair–Deficient Intestinal Crypts and Accelerates Tumorigenesis in a Mouse Model of Lynch Syndrome. Gastroenterology, 147(5), 1064–1072.e1065. [DOI] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, … Sharp PA (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology, 32(7), 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Zhong X, Kanazawa S, Koike J, Tsujita K, & Hemmi H (2003). Oncogenic pathway of sporadic colorectal cancer with novel germline missense mutations in the hMSH2 gene. Oncology Reports, 10(4), 859–866. [PubMed] [Google Scholar]
- Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, … Church G (2013). Optimization of scarless human stem cell genome editing. Nucleic Acids Research, 41(19), 9049–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen ST, Chan TL, Ho JW, Chan AS, Chung LP, Lam PW, … Leung SY (2002). Germline, somatic and epigenetic events underlying mismatch repair deficiency in colorectal and HNPCC-related cancers. Oncogene, 21(49), 7585–7592. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ge X, Yang F, Zhang L, Zheng J, Tan X, … Gu F (2014). Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Scientific Reports, 4, 5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.