

Abstract
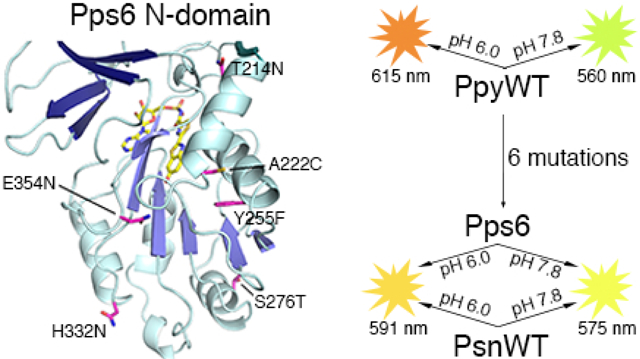
The dazzling yellow-green light emission of the common North American firefly Photinus pyralis and other bioluminescent organisms has provided a wide variety of prominent research applications like reporter gene assays and in vivo imaging methods. While the P. pyralis enzyme has been extensively studied, only recently has a second Photinus luciferase been cloned from the species scintillans. Even though the enzymes share very high sequence identity (89.8%), the color of the light they emit, their specific activity, and their stability to heat, pH, and chemical denaturation are quite different with the scintillans luciferase being generally more resistant. Through the construction and evaluation of the properties of chimeric domain swapped, single point, and various combined variants, we have determined that only 6 amino acid changes are necessary to confer all of the properties of the scintillans enzyme to wild-type P. pyralis luciferase. Altered stability properties were attributed to four of the amino acid changes (T214N/S276T/H332N/E354N) and single mutations each predominantly changed emission color (Y255F) and specific activity (A222C). Results of a crystallographic study of the P. pyralis enzyme containing the 6 changes (Pps6) provide some insight into the structural basis for some of the documented property differences.
Keywords: Bioluminescence, firefly, luciferase, scintillans, thermostability, x-ray crystallography
Graphical Abstract
INTRODUCTION
As typified by the brilliant yellow-green light emission of the common North American firefly Photinus pyralis, beetle bioluminescence (BL) has provided a wide variety of prominent research applications like reporter gene assays1-10 and in vivo imaging methods.11-15 Advances in the development of an impressive diversity of applied procedures have been primarily the result of increased understanding of the basic biochemistry of BL. From the original observation by McElroy16 that ATP is required for light emission, through the first crystal structure17 of a beetle luciferase (Luc), to the present day development of luciferin substrate analogs that produce near-Infrared light,13, 18-22 basic scientific research has been key to the timely development of important novel methodologies.
Among the approximately 30 cloned beetle luciferases, the Photinus pyralis Luc (PpyWT) has been particularly extensively studied. However, until our recent cloning and characterization of Photinus scintillans Luc (PsnWT),23 no other species of Photinus had been substantially studied. While the amino acid sequence of PsnWT is highly similar (89.8%) to the well-characterized PpyWT, light emission from PsnWT is yellow-orange (λmax = 574 nm) compared to the familiar yellow-green glow (λmax = 560 nm) of PpyWT. We made several chimeric proteins from subdomains of the two Lucs and determined that one of these called PpyPsnN2 (previously referred to as PpyN1PsnN2PpyC),23 which is PpyWT containing 19 residues substituted with those found in PsnWT in the region 197-440, produced the same emission color as PsnWT. We then showed that the single residue change Y255F was necessary and sufficient to produce the red-shifted BL. The results of additional mutagenesis and crystallographic studies23 that focused on a H-bond network 23-27 which includes the Y255 residue, supported our general view and that of others25,27-29 that the structural integrity of this network at the active site either through direct or long range effects, e.g. an electrostatic interaction between E311 and R337, is essential to maintaining green BL. We favor the concepts23 that the critical role of the H-bond network is to control the resonance structures that affect charge delocalization of the keto form of emitter oxyluciferin or simply to prevent protonation of the phenolate ion of the emitter. Alternative views of BL color determination including controlling the polarity specifically around the phenolate of the emitter27, 30, 31 and stabilization of the microenvironment at the active site25, 28, 29 have garnered support.
In our prior comparison of the properties of the Photinus Lucs, we noted23 too that PsnWT had several enhanced properties including: (1) 1.55-fold higher specific activity; (2) partial resistance (+18 nm versus +55 nm) to red-shifted emission at low pH (pH sensitivity); (3) a 4.6 °C increased mean aggregation temperature (Tm); and (4) improved (0.5 h versus 0.3 h) thermostability (T1/2) at 37 °C (Table 1). While the Y255F difference was not responsible for any of the other improved properties (Table 1), the PpyPsnN2 chimeric enzyme met or exceeded all of the PsnWT attributes indicating that the residue changes in the 197-440 region were responsible for all of the variations between the two Lucs (Table 1). We also had determined23 that the double mutant PpyR213K/T214N had similarly increased specific activity and a T1/2 at 37 °C of 1 h that exceeded that of PsnWT, but had no additional augmented properties. We decided to take advantage of the opportunity to further explore this interesting example of two highly similar enzymes from the same genus that have distinctly different activity and stability properties. We first directed our investigation at determining the minimum number of amino acid differences in PsnWT that are necessary for its enhanced properties, in part, to make the interpretation of biochemical and structural results more straightforward. We report these results here along with our interpretation of additional mutagenesis and structural studies designed to better understand the basis of the enhanced stability and activity properties.
Table 1.
Identification of a minimal P. pyralis Luc variant with PsnWT properties
| Relative Specific Activity a |
BL Emission (λmax ± 2 nm)b |
||||
|---|---|---|---|---|---|
| Enzyme | Flash Height (± 5%) |
pH 7.8 | pH 6.0 | Tm (± 0.2 °C)c |
T1/2 (± 0.5 h)d |
| PpyWT | 100 | 560 (70) | 615 (61) | 44.0 | 0.3 |
| PsnWT | 155 | 574 (72) | 592 (85) | 48.6 | 0.5 |
| PpyPsnN2 | 156 | 576 (73) | 584 (79) | 50.4 | 2.5 |
| PpyPsnN2A | 153 | 582 (75) | 587 (78) | 50.3 | 3.0 |
| PpyPsnN2B | 80 | 562 (78) | 617 (57) | 43.5 | 0.06 |
| PpyR213K | 86 | 563 (77) | 615 (60) | 43.9 | 0.3 |
| PpyT214N | 95 | 563 (75) | 616 (60) | 44.9 | 2.5 |
| PpyY255F | 93 | 572 (80) | 618 (56) | 45.3 | 0.1 |
| PpyR213K/T214N | 150 | 561 (72) | 616 (65) | 43.9 | 1.0 |
| PpyA | 149 | 573 (73) | 620 (58) | 45.8 | 0.75 |
| PpyB | 136 | 576 (85) | 614 (71) | 47.3 | 4.0 |
| PpyC | 217 | 576 (78) | 610 (78) | 46.0 | 5.5 |
| PpyD | 180 | 574 (71) | 590 (84) | 47.4 | 5.0 |
| PpyE | 228 | 575 (73) | 592 (84) | 48.4 | > 24 |
| PpyF | 185 | 576 (72) | 593 (83) | 48.8 | > 24 |
| PpyG | 144 | 575 (71) | 595 (84) | 48.2 | > 24 |
| Pps6 | 160 | 575 (72) | 591 (81) | 48.9 | >24 |
Specific activities were obtained at pH 7.8 with D-LH2 (0.3 mM) and Mg-ATP (2 mM) and are expressed relative to the PpyWT value, which is defined as 100.
BL emission maxima measured at 22 °C and the indicated pH values. The bandwidth at full-width half-maximum values are shown in parentheses.
Tm, mean aggregation temperature determined by circular dichroism spectroscopy monitored at 222 nm.
Time for the maximum initial activity to decrease 50% at 37 °C.
MATERIALS AND METHODS
Materials and General Methods.
The following materials were obtained from the sources indicated: Mg-ATP (bacterial source) and 5,5′-dithio-bis-(2-nitobenzoic acid) (Ellman’s Reagent) from Sigma-Aldrich (St. Louis, MO); restriction endonucleases from New England Biolabs (Beverly, MA); oligonucleotides from Integrated DNA technologies (Coralville, IA); Glutathione Sepharose 4B media and the pGEX-6P-2 expression vector from GE Healthcare (Piscataway, NJ); and the QuikChange® Lightning Site-Directed Mutagenesis kit from Agilent Technologies (Santa Clara, CA). D-firefly luciferin (D-LH2) was a generous gift from Promega (Madison, WI). Sequences of luciferase genes in the pGEX-6P-2 vector were verified by DNA sequencing at the W. M. Keck Biotechnology Laboratory (Yale University, New Haven, CT). Concentrations of purified proteins were determined with the Bio-Rad Protein Assay system using bovine serum albumin as the standard. Mass spectral analyses were performed by tandem HPLC-electrospray ionization mass spectrometry using a ThermoFinnigan Surveyor HPLC system and a ThermoFinnigan LTQ XL mass spectrometer. Samples were injected via autosampler and UV-Vis data were collected from a photodiode array detector. Water-0.1% trifluoracetic acid (TFA) (solvent A) and acetonitrile-0.1% TFA (solvent B) were used as LC eluents for all LC/MS and HPLC methods. The conditions for protein mass determinations were: column, Phenomenex Aeris 3.6 μm widepore C4 (50 × 2.10 mm); mobile phase, solvent A: solvent B (95:5), gradient after 3 min to 5:95 over 5 min; flow rate, 0.360 mL/min; MS mode, ES+; scan range, m/z = 200-2000. Total mass spectra for protein samples were reconstructed from the ion series using Bioworks Browser 3.2 with BIOMASS deconvolution or using BioPharma Finder software. The found molecular masses (Da) of the luciferase (Luc) proteins containing the N-terminal peptide extension GlyProLeuGlySer- were within the allowable experimental error (0.01%) of the calculated values.
Protein Expression and Purification.
Glutathione S-transferase (GST) fusion constructs of all enzymes in the pGEX-6P-2 bacterial expression vector were expressed in Escherichia coli strain BL21(DE3) pLysS as previously described.32, 33 Cultures (250 mL in Luria-Bertani media with 100 μg/mL ampicillin) were grown in 1-L flasks at 37 °C to mid-log phase (A600 = 0.5–0.7) and then induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated at 22 °C for 18-20 h. Cells were harvested by centrifugation at 4 °C and then frozen at −80 °C for 15 min. Cell pellets were resuspended in 25 mL of phosphate-buffered saline (PBS) containing 0.1 mM phenylmethylsulfonyl fluoride and 0.5 mM dithiothreitol (DTT). After the addition of lysozyme (2.5 mL of 10 mg/mL solution in PBS), the cells were lysed by sonication and treated with DNase (5 μg/mL) and RNase (10 μg/mL) for 10 min on ice. Triton X-100 was added to the lysates (1% final volume), and the whole-cell extracts were isolated by centrifugation at 20,000g for 45 min. Proteins were further purified using Glutathione Sepharose 4B affinity chromatography according to the manufacturer’s instructions. During the purification, luciferases were released from GST by incubation with PreScission protease in 20 mM Tris-HCl (pH 7.4) containing 150 mM NaCl, 1 mM EDTA and 1 mM DTT (CB; cleavage buffer) for 18-20 h at 4 °C with gentle mixing. Proteins were eluted with CB and were either flash frozen in liquid N2 for long-term storage at −80 °C or stored at 4 °C in CB containing 0.8 M ammonium sulfate and 2% glycerol (CBA). Protein yields were determined to be 7-10 mg per 0.25 L protein preparation.
BL Specific Activities.
BL specific activity assays were performed with a custom-built luminometer using equipment previously described.34 Reactions were initiated by the injection of 0.12 mL of 8.8 mM Mg-ATP into 8 × 50 polypropylene tubes containing 0.4 mL of 0.4 mM D-LH2 in 50 mM Tricine buffer (pH 7.8) and 0.5-1 μg enzyme in CBA. The final concentrations of D-LH2 and Mg-ATP were 0.3 and 2.0 mM, respectively, in a final volume of 0.525 mL. Peak maximum intensity was used to determine flash-height based activities. All activity values were corrected for the spectral response of the detector.
BL Emission Spectra.
BL emission spectra were obtained using a Horiba Jobin-Yvon iHR imaging spectrometer equipped with a liquid N2 cooled CCD detector. Data were collected at 22 °C in a 0.8 mL quartz cuvette over the wavelength range 450-750 nm with the emission slit width set to 5 nm and were corrected for the spectral response of the CCD using a correction curve provided by the manufacturer. BL was initiated by adding 3 μL of enzyme in CBA (0.02-0.25 μM final concentration) to a cuvette containing solutions (0.52 mL) of 300 μM D-LH2 and 2 mM Mg-ATP in 50 mM Tricine buffer pH 7.8 or 50 mM MES pH 6.0. The pH values were confirmed before and after spectra were obtained.
Estimated Rates of Half-Reactions.
The estimated rates of the oxidative half-reactions were based on BL activity assays using synthetic D-LH2-AMP as the substrate (Table 3, main text). Assays (0.510 mL) in 50 mM Tricine buffer, pH 7.8, containing aliquots (0.1 mL) of D-LH2-AMP solution (final concentration 2-70 μM) in 10 mM sodium acetate, pH 4.5. The upper concentration limit was imposed by the concentration of the stock substrate solution after purification of synthetic D-LH2-AMP. Light reactions were initiated by injections of 10 μL of luciferase enzymes (0.4-1 μg in CBA). Kinetic constants were determined using a nonlinear least squares method of the Enzyme Kinetics Pro software (SynTex), which fits data from the Michaelis-Menten equation to a rectangular hyperbola. The corresponding kcat values were obtained by dividing the Vmax values by the final amounts (μmol) of each luciferase in the assay mixtures.
Table 3.
Estimated relative reaction rates of the Photinus luciferases and variants
| Relative rates (%)a | Km (μM) b |
Relative ΦBLc D-LH2-AMP |
|||
|---|---|---|---|---|---|
| Enzyme | Overall | Adenylation | Oxidation | ||
| PpyWT | 100 ± 10 | 100 ± 15 | 100 ± 9 | 14 ± 3 | 100 ± 9 |
| PsnWT | 155 ± 3 | 80 ± 10 | 173 ± 10 | 24 ± 8 | 106 ± 3 |
| Pps6 | 160 ± 8 | 97 ± 10 | 182 ± 18 | 11 ± 4 | 120 ± 6 |
| Pps7 | 220 ± 11 | 117 ± 18 | 227 ± 16 | 12 ± 2 | 115 ± 8 |
Overall and oxidation rates (expressed relative to PpyWT) were estimated using kcat values obtained with D-LH2 and Mg-ATP or D-LH2-AMP, respectively. Adenylation rates were estimated from the rates of dehydroluciferyl-adenylate (L-AMP formation).
The Km values were calculated from the oxidation reaction rates.
Determined by measuring the total light emitted from D-LH2-AMP using an excess of enzyme to insure consumption of substrate.
The relative rates of adenylate formation were estimated by fluorescence-based assays of L-AMP formation35 using a Perkin Elmer LS55 luminescence spectrometer operated in the "time-drive" mode. Using an excitation wavelength of 350 nm, the luciferase-catalyzed formation of L-AMP from dehydroluciferin (L), initiated by the addition of Mg-ATP, was assessed by following the decrease in the intensity of the 440 nm fluorescence of the initial enzyme-L complex. The change in fluorescence was used to estimate the rates of L-AMP formation catalyzed by the Lucs. Assays (0.4 mL) in 50 mM Tris buffer, pH 7.4 contained 2.8 μM enzyme and 0.55 μM L. The initial fluorescence at 440 nm was recorded and then the decrease was monitored following the rapid injection of 50 μL solutions of varying concentrations of Mg-ATP in the same buffer. The rates of decrease (slopes) were calculated using the arithmetic function of the UVWinLab software (Perkin Elmer) and used to determine the initial velocities for each Mg-ATP concentration.33 We note that under the assay conditions, dictated, in part, by making the rapid fluorescence measurements, it is possible that L-enzyme formation might have influenced the estimated relative rate determinations.
Relative BL Quantum Yields.
BL quantum yields with limiting D-LH2-AMP were performed in assays (0.5 mL) in 50 mM Tricine buffer, pH 7.8 containing 0.1 mL of 0.5 μM D-LH2-AMP in 10 mM sodium acetate pH 4.5, and were initiated by the injection of 10 μL of enzyme (16-32 μM) in CBA. The final concentrations of enzyme and D-LH2-AMP were 310-630 nM and 100 nM, respectively, in a final volume of 0.51 mL. The light output was monitored at a sampling rate of 100 Hz for 15-30 s until the initial signal intensity decreased by 99%. The equipment used to make these measurements has been previously described.34 An additional aliquot of enzyme was added to the spent mixtures and emission intensity was monitored to ensure that the reactions were completed.33
Thermal Inactivation at 37 °C.
Enzymes (1.2 mg/mL in 20 mM Tris-HCl pH 7.4 at 4 °C containing 150 mM NaCl, 1 mM EDTA and 1 mM DTT) were diluted to 0.12 mg/mL in 0.2 mL of room temperature 25 mM Glycyl-glycine buffer, pH 7.8 and incubated at 37 °C. Aliquots (3 μL) were taken at regular intervals, quenched by diluting the enzyme 133-fold with room temperature 25 mM Glycyl-glycine buffer (pH 7.8) containing 400 μM D-LH2, and assayed for BL activity as stated above. Time for the maximum initial activity to decay to 50% is reported.33
Thermal Inactivation at 45 °C.
Enzymes (1.2 mg/mL in 20 mM Tris-HCl pH 7.4 at 4 °C containing 150 mM NaCl, 1 mM EDTA and 1 mM DTT) were diluted to 0.12 mg/mL in 0.2 mL of room temperature 1X PBS buffer (140 mM NaCl, 13.5 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4), pH 7.4 and incubated at 45 °C. Aliquots (3 μL) were taken at regular intervals, quenched by diluting the enzyme 133-fold with room temperature 50 mM Tricine buffer (pH 7.4) containing 0.4 mM D-LH2, and assayed for BL activity as stated above. Half-lives were calculated using first-order rate constants obtained from log plots of percentage activity remaining versus time at 45 °C, pH 7.4 (Figure S1).
Effect of Guanidinium Chloride (GdmCl) on Luc Activity.
All reactions were performed in 0.1 M sodium phosphate buffer (pH 7.8) with 1 mM EDTA. Loss of BL activity was monitored by incubating 0.8 μM luciferase with varying concentrations of GdmCl (0-1 M) at 25 °C for 10 min. Aliquots (3μL) were removed and added to 8 × 50 polypropylene tubes containing 0.4 mL of 0.4 mM D-LH2 in 50 mM Tricine buffer (pH 7.8). Reactions were initiated by injection of 0.12 mL of 8.8 mM Mg-ATP. Final concentrations of enzyme, D-LH2 and Mg-ATP were 0.5 μM, 0.3 mM and 2.0 mM, respectively in a final volume of 0.525 mL.33 BL activity was measured as described above.
Crystallization and Structure Determination.
The structure of Pps6, the enzyme containing six mutations that confer the stability and activity properties of PsnWT onto PpyWT was determined by X-ray crystallography. Screening of Pps6 protein for crystallization was done with a lab made sparse matrix screen to identify crystallization conditions. An initial hit with fine needle-like crystals grew with a cocktail 50 mM 4-(2-Hydroxyethyl)-1-piperazinepropane sulfonic acid (EPPS) pH 8.0 and 20% PEG 4000. Further optimization with an organic additive screen and micro-seeding led to rod shaped crystals grown with cocktail 50 mM EPPS pH 8.5, 25% PEG 4000 and 2% 2,5 hexanediol. Diffraction data from single crystals were collected remotely at SSRL beam line 9-2. Processing of data was performed with mosflm, XDS, and AIMLESS, as implemented in the SSRL AutoProc pipeline, to a resolution of 2.6 A in space group P21. The protein atoms from the structure of PpyWT luciferase (PDB Code: 4G36) were used as search model for molecular replacement with Phaser. An initial model was identified with three protein chains per asymmetric unit. Electron density for the C-terminal domain was observed only for two chains. The model was iteratively refined using COOT and PHENIX.refine with NCS restrains. Strong positive electron density was observed for ligand 5′-O-[(N-dehydroluciferyl)-sulfamoyl] adenosine in all three chains (Figure S3). Final model building and refinement with ligand was done using PHENIX.refine with TLS parameters. Final diffraction and refinement data are presented in Table S2.
RESULTS AND DISCUSSION
Identification of PpyWT variant Pps6 that contains the minimum number of amino acid changes needed to confer the properties of PsnWT to PpyWT.
A summary of the abbreviations used for the variants and the mutations that they contain is provided in Table S1. Since PpyPsnN2, containing 19 amino acid differences (versus PpyWT) in the region 197-440, displayed all of the PsnWT improved properties (Table 1), we sought to determine the fewest number of changes that were required to make PpyWT behave like PsnWT. We began by simply combining the 3 previously identified mutations23 PpyR213K/T214N and PpyY255F to make the triple mutant PpyR213K/T214N/Y255F (PpyA), which retained the specific activity enhancement and emission maxima increase contributed by R213K/T214N and Y255F, respectively (Table 1). Next, we noted that one of the PpyPsnN2 mutations, E354N, was at a residue well-established to impart thermostability to Lucs. 36-44 Adding the E354N change to PpyA produced PpyR213K/T214N/Y255F/E354N (PpyB), which exhibited an additional +1.5 °C improvement in Tm and an anticipated significant jump in 37 °C thermostability to 4 h compared to PpyA (Table 1).
To narrow down the remaining number of residues for consideration, we used convenient restriction sites in the luc genes to make 2 additional chimeric Lucs PpyPsnN2A and PpyPsnN2B, which contain 13 and 6 amino acid residue differences in the regions 197-394 and 395-440, respectively. Interestingly, the PpyPsnN2A enzyme had stability properties that exceeded the enhancements documented for PsnWT and approximately equivalent specific activity (Table 1). In contrast, the specific activity and stability properties of the PpyPsnN2B enzyme were diminished compared to those of PpyWT. Apparently, the PsnWT determinants were contained in some or all of the following 13 PsnWT residues: R213K, T214N, C216I, A222C, Y255F, V262I, S276T, S320A, H332N, P334S, E354N, D357E, and F368L. Evidently, based on the documented PpyPsnN2A properties (Table 1), the PsnN2B region negatively offset the potentially greater enhancements that PsnWT might achieve. The PsnN2 subdomain results enabled us to at least temporarily eliminate 6 of the 19 PsnWT residues as positive contributors to the enhanced properties of this Luc. Having evaluated 4 of the 13 PsnN2A residues and lacking any other obvious candidates for mutagenesis, we simply proceeded by evaluating the mutations in order starting with the most N-terminal one C216I. This change along with V261I and S320A, failed to improve stability properties. The quintuple variant PpyR213K/T214N/A222C/Y255F/E354N (PpyC), constructed by the addition of A222C to PpyB, surprisingly exhibited a specific activity increase to a level 2.2-fold higher than that of the PpyWT value and 1.4-fold greater than that of PsnWT (Table 1).
We next made PpyR213K/T214N/A222C/Y255F/S276T/E354N (PpyD). This sextuple Luc variant met or exceeded all of the properties of PsnWT except that the Tm, while improved 1.4 °C (to 47.4 °C), fell short of the target 48.6 °C value of PsnWT (Table 1). Interestingly, while PpyC showed a very modest resistance to the pH 6.0 BL emission shift (+34 nm), we observed the full PsnWT protection (+16 nm) after addition of the S276T mutation, an 18 nm total improvement.
The octuple variant PpyR213K/T214N/A222C/Y255F/S276T/H332N/P334S/E354N (PpyE) was then constructed by introducing the H332N and P334S changes together with a single primer. The Tm of PpyE increased 1.0 °C to 48.4 °C, a value equivalent within experimental error to that of PsnWT (Table 1). Additionally, the thermostability was greatly enhanced such that the T1/2 at 37 °C was not reached after 1 day. Moreover, the specific activity of this variant was 2.3-fold higher than PpyWT, exceeding the 1.55-fold enhancement of PsnWT. Thus, after examining 11 of the 13-targeted mutations, we had identified a PpyE octuple variant that met or exceeded the PsnWT properties. We did not evaluate the remaining D357E and F368L changes.
The fact that several activity and stability properties of PpyE exceeded those of PsnWT, PpyPsnN2, and PpyPsnN2A suggested that at least one of the mutations was unnecessary. Since we had introduced both the position 213 and 214 as well as the 332 and 334 changes with single primers, we considered that in each case only one of the mutations might have been responsible for the observed improvements. We first made the individual PpyR213K and PpyT214N Lucs and determined that both had similar properties as PpyWT except that the T214N mutation improved both thermal properties (Table 1); the latter result is consistent with prior reports37, 41 of thermostability improvements. By reverting the position 213 change, we made the septuple variant PpyT214N/A222C/Y255F/S276T/H332N/P334S/E354N (PpyF), an enzyme highly similar to PpyE, except that Tm was improved (+0.4 °C) and the specific activity enhancement of 1.85-fold was closer to the 1.55-fold value observed for PsnWT. To evaluate whether the H332N and P334S mutations were both required, we reverted each one in the PpyE template making 2 new sextuple variants. For PpyG, eliminating the H332N change diminished the specific activity of the PpyE enzyme 1.6-fold. Fortunately, inclusion of H332N alone in PpyT214N/A222C/Y255F/S276T/H332N/E354N, which we termed Pps6, produced a Luc that very closely mimicked all of the properties of PsnWT with the only exception being the substantially greater T1/2 at 37 °C (Table 1). Evidently not only are amino acid variations found in the PpyPsnN2B region offsetting the achievable thermostability, but others among the 36 changes not present in PpyPsnN2 are doing likewise. We noted too that the small differences in the secondary structure of PsnWT, as judged by circular dichroism spectral data (Table S3 and Figure S2), were found in Pps6 as well.
Specific Activity Differences.
While the relative (to PpyWT = 100) flash height or burst specific activity of PsnWT (155) was achieved with the minimal variant Pps6 (160), it was markedly exceeded in mutants PpyC (217) and PpyE (228) (Table 1). Residue change A222C is certainly a strong contributor (140) (Table 2) to this property in these variants, but cannot alone account for the elevated values. As previously reported,23 the specific activity of PpyR213K/T214N (150) is also approximately equal to that of PsnWT. Mutants PpyC and PpyE contain these changes plus A222C, initially hinting that the 3 residues could be acting in an additive fashion. We made PpyR213K/T214N/A222C and noted that the freshly prepared protein had a relative specific activity of 195, but on standing for 1 day the value leveled off to 150 (Table S4). However, the PpyR213K/T214N/A222C/E354N enzyme displayed a stable high activity level (188) (Table S4) as a result of the E354N change. It is likely that an additional small contribution by H332N is sufficient to exceed the PpyWT specific activity value by an impressive 2.2 times as evidenced by data obtained with Pps7 (Pps6 plus R213K) (Table 2).
Table 2.
Properties of Photinus luciferases and variants
| Enzyme | Relative Specific Activity a |
BL Emission λ(max ± 2 nm) b |
||||
|---|---|---|---|---|---|---|
| Flash Height (± 5%) |
pH 7.8 | pH 6.0 | Tmc (± 0.2 °C) |
T1/2d (± 0.5 h) |
T1/2e (± 0.2 min) |
|
| PpyWT | 100 | 560 (70) | 615 (61) | 44.0 | 0.3 | 0.3 |
| PsnWT | 155 | 574 (72) | 592 (85) | 48.6 | 0.5 | 0.6 |
| PpyA222C | 140 | 561 (80) | 615 (61) | 44.0 | 0.4 | 0.3 |
| Pps3 | 105 | 569 (78) | 591 (92) | 48.5 | >24 | 4.5 |
| Pps4 | 110 | 569 (79) | 586 (89) | 49.5 | >24 | 5.5 |
| Pps6 | 160 | 575 (72) | 591 (81) | 48.9 | >24 | 4.4 |
| Pps7 | 220 | 574 (72) | 589 (83) | 48.4 | >24 | 3.0 |
Specific activities were obtained at pH 7.8 with D-LH2 (0.3 mM) and Mg-ATP (2 mM) and are expressed relative to the PpyWT value, which is defined as 100. Mean values were obtained from at least three trials.
BL emission maxima at 22 °C and the indicated pH values. The bandwidths at full-width half-maximum are shown in parentheses.
Tm, mean aggregation temperature determined by CD spectroscopy monitored at 222 nm.
Time for the maximum initial activity to decay to 50% at 37 °C, pH 7.8.
Half-lives at 45 °C, pH 7.4 were calculated using first-order rate constants obtained from log plots of percentage activity remaining versus time (Figure S1).
The Luc flash height-based specific activity measurements reported here relate the maximum achievable overall reaction rates of light emission, a 2-step process in which D-LH2 is first converted into the corresponding adenylate D-LH2-AMP and then oxidized to the eventual light emitter oxyluciferin formed in its electronically excited state. The substantially greater overall rates of light production of PsnWT, Pps6, and Pps7 compared to PpyWT are all attributable to corresponding increases in the relative oxidation half-reaction rates that produce oxyluciferin (Table 3). It is not likely that Km values are a significant determinant because, with the exception of the ~2-fold greater Km of PsnWT, there was little variation in the values of the other enzymes tested (Table 3). BL quantum yields (ΦBL) can be used as measures of the total amount of light emitted by a limiting quantity of D-LH2-AMP. While the relative ΦBL values of the wild-type Lucs and Pps7 are roughly equivalent, the Pps6 value is slightly greater (Table 3). The small variation in the ΦBL values and great differences in oxidation rates (Table 3) suggest that the R213K, T214N and A222C mutations are predominantly responsible for accelerating the process of light emission from D-LH2-AMP. Possibly, the structural changes associated with the three mutations exert their effects by increasing turnover due to decreased competitive inhibition by known inhibitors dehydroluciferin-AMP and oxyluciferin.45
Enhanced stability of PsnWT to heat, acidic pH, and GdmCl.
Among the 7 point mutations in PpyWT that were carefully examined (Table 1, Table 2, and Table S4), all except R213K and A222C produced 0.4 °C to 1.4 °C higher Tm of aggregation values than PpyWT (44 °C). The 4.6 °C increase of PsnWT over PpyWT was achieved by Pps6 (48.9 °C) (Table 1). The 3 mutations T214N, S276T, and E354N found in Pps3 were sufficient to improve the PpyWT Tm value to 48.5 °C; adding the H332N change produced Pps4 with a Tm of 49.5 °C, the highest value of all the Lucs tested. The Tm improvements documented for Pps4 appear to represent incremental increases contributed by each of the 4 mutations.
The thermostability half-life (T1/2) at 37 °C of PsnWT (0.5 h) was greater than that of PpyWT (0.3 h), yet single PpyWT point mutants T214N (2.5 h) and E354N (1.5 h) had even greater T1/2 values than the wild-type Lucs (Table 1 and Table S4). Moreover, Pps3, Pps4, Pps6, and Pps7 all had >24 h half-lives. Similarly, PsnWT and the 4 variants are all more resistant to long wavelength shifts and peak broadening of BL emission at 37 °C compared to PpyWT (Figure S4). We then made measurements at 45 °C (Table 2 and Figure S1) and found that Pps4 (5.5 min) also showed the greatest thermostability, far surpassing that of PsnWT (0.6 min). A comparison of the Tm values and half-lives of Pps6, Pps7, PpyPsnN2A, and PpyPsnN2B reveals similar trends in both of the thermal properties, which are optimal with the 4 point mutations found in Pps4, and that are negatively influenced by amino acid changes outside of the 197-394 region.
The enhanced stability of BL emission, expressed as resistance to red-shifting at acidic pH, of PsnWT compared to PpyWT can be expressed as a +18 ± 2 nm versus + 55 ± 2 nm shift in the BL λmax for measurements at pH 7.8 and 6.0 (Figure 1 and Table 2). For this property, the Pps3 value of +22 ± 2 approaches that of PsnWT, but mutation H332N present in Pps4 is also required to achieve a value of +17 ± 2 nm, which is similar to those of Pps6 and Pps7 (Figure 1 and Table 2). Unlike with the thermal properties, the 4 residues of Pps4 are required to reach the PsnWT λmax value. Interestingly, we note that by mutating residue 354 to Lys in Pps6, a Luc variant is produced with almost complete pH resistance (+ 6 ± 2 nm) and a + 0.7 °C improved Tm compared to Pps6 (Table S4). These results further highlight the importance of residue 354 to improved pH resistance and thermo-aggregation.
Figure 1.

BL emission as a function of pH. Normalized emission spectra of the PpyWT (Photinus pyralis wild type), PsnWT (Photinus scintillans wild type), Pps3 (PpyT214N/S276T/H332N), Pps4 (PpyT214N/S276T/H332N/E354N), Pps6 (PpyT214N/A222C/Y255F/S276T/H332N/E354N), and Pps7 (PpyR213K/T214N/A222C/Y255F/S276T/H332N/E354N) luciferases at pH 7.8 (solid lines) and pH 6.0 (dashed lines).
Additionally, we examined the effect of the chemical denaturant GdmCl on several of the stability enhanced Lucs (Figure 2). The BL activity of the Pps3, Pps4, Pps6, and Pps7 variants was decreased ~50% at a denaturant concentration of ~810 mM about twice the value required for the wild-type Lucs. The similar behavior of PpyWT and PsnWT compared to the much greater resistance of the Luc variants to chemical denaturation is somewhat akin to the thermostability of these enzymes at 45 °C. Apparently, both chemical and thermal denaturation proceed through similar unfolding intermediates.
Figure 2.

Activity loss based on GdmCl denaturation of Photinus variants. Luc variants were incubated with varying concentrations of GdmCl (0 −1.0 M) at 25 °C for 5 min. Aliquots were then assayed for BL activity as described in Materials and Methods. Percent remaining activity was plotted as a function of GdmCl concentration for the following luciferases: PpyWT (Photinus pyralis wild type), blue; PsnWT (Photinus scintillans wild type), red; Pps3 (PpyT214N/S276T/H332N), dark grey; Pps4 (PpyT214N/S276T/H332N/E354N), yellow; Pps6 (PpyT214N/A222C/Y255F/S276T/H332N/E354N), light grey; and Pps7 (PpyR213K/T214N/A222C/ Y255F/S276T/H332N/E354N, purple.
Structural Characterization of the Pps6 Mutant Protein.
Here and in a prior study,23 we have documented significant bioluminescence emission, activity, and stability differences between Lucs from very closely related Photinus species. The PpyWT variant Pps6 contains 6 amino acid changes that are sufficient to impart all of the properties of PsnWT onto the P. pyralis Luc. To better understand the structural basis for the altered properties, we determined the crystal structure of a complex of Pps6 and bound inhibitor 5′-O-[(N-dehydroluciferyl)-sulfamoyl]-adenosine (DLSA),46 a luciferyl-adenylate analog (Figure 3). The structure of Pps6 contains three independent molecules in the asymmetric unit. As has been observed for many members of this enzyme family, the dynamic C-terminal domain is poorly ordered in some protein chains. Only one chain contains density for the complete Pps6 protein. A second contains a poorly ordered C-terminal domain beyond the hinge at residue Lys439, with 24 disordered residues of the 106 residues of the C-terminal domain. The C-terminal domain of the third protein chain is completely disordered beyond the hinge. Despite these differences, the structures of the N-terminal domains are highly conserved, with rms displacements between homologous Cα positions of 0.6-0.9 Å. The most dramatic changes occur in the highly dynamic P-loop and several other surface loops. Chain A, containing residues E2 – G545 was utilized for comparison to prior structures of PpyWT and L. cruciata Luc.
Figure 3.

Structure of Pps6-DLSA complex. A. Ribbon representation of the Pps6 protein (top). The N2 core (residues 192-440) of the protein containing the substitutions between PpyWT and PsnWT is colored blue, while the N1- and C-terminal regions are grey. The molecule of DLSA is bound in the core of the protein. B. Stereorepresentation of the location of the six residues (magenta) in Pps6 that are mutated to the residue present in PsnWT. The protein is rotated slightly compared to the view of panel A.
Near the phenolate moiety of the DLSA ligand, the active site pocket contains a solvent filled cavity that is variably populated with water molecules in different luciferase crystal structures. In the higher resolution structures, this pocket contains as many as six solvent molecules that interact with both protein atoms and DLSA. The proposed basis of the BL emission shift associated with the Y255F change is a disruption of this key H-bonding network resulting from the absence of the phenolic side chain group. In the L. cruciata Luc structure (PDB Code: 2D1S),26 Y255 makes a H-bond to a H2O, which is also bonded to N229 and S284 (PpyWT numbering). This network of hydrogen bonds further connects to E311 and ultimately to the phenolate group of the luciferin analog. In all three chains of the Pps6 protein, there are only two, poorly ordered water molecules in this cavity. While this partly reflects the lower resolution of the Pps6 structure, the highly ordered water network of the L. cruciata protein is incompatible with the Y255F mutation. We also note that the position of F255 in the Pps6 structure is essentially the same as the aromatic side chain of the homologous Y257 in the L. cruciata structure suggesting that the mutation had any other effect than to disrupt the H-bond network (Figure 4). This extended pocket is highly polar as it bordered by both main chain and side chain hetero atoms from the protein. We note that N229 can best be modeled in alternate side chain conformations in the wild-type and Pps6 mutant structures. Although the side chain mostly makes interactions with water molecules, a potential interaction with the side chain of R337 in the mutant structure (3.4 Å) and with E311 in the wild-type structure (3.3 Å) may cause this side chain to flip, and alternately contribute a hydrogen bond donor or an acceptor to the solvent filled cavity, thereby altering the solvent network of the pocket. Interestingly, E311 and R337 have been implicated in maintaining a closed conformation of the active site pocket that stabilizes green emission.28
Figure 4.

Surface representation of the active site pocket. A. The pocket of Pps6 near the phenolate moiety of the DLSA ligand shows a rather polar environment. Mutant residues C222 and F255 are shown, along with the α-helices on which they reside. The protein atoms, as well as the surface they contact, are colored cyan (carbon), red (oxygen), blue (nitrogen), and yellow (sulfur). B. Similar view of the wild-type Luc derived from PDB Code: 4G36. Figures created using PYMOL, with cavity culled option to emphasize internal cavities.
A second finding was that the single amino acid change A222C was sufficient to account for nearly all of the 1.55-fold specific activity increase over that of PpyWT. Based on prior results,23 an alternative way to similarly improve specific activity is through the synergistic effects of the R213K and T214N changes (Table 1). By combining the three changes from these residues located in the same helix (R213-R223), the specific activity is increased 2.2- fold. In both Pps6 containing the A222C change and Pps7 containing all 3 of the mutations, the activity improvements can be mainly attributed to the oxidative half reactions. Since the production of oxyluciferin by PpyWT is already so high (72±7%)23,33, the major effect of the activity enhancing mutations most likely represents an improved microenvironment for efficient formation of the emitter in the excited state and subsequent radiative emission. In Pps6, the major source of the improvement must be the A222C change. In the mutant enzyme, the side chain of C222 adopts a conformation with the thiol directed away from the luciferin binding pocket (Figure 3B). The closest atom of the DLSA ligand is ~8Å from the side chain of C222. Between the C222 side chain and the ligand lies the side chain of R218 and the solvent cavity. R218 adopts a nearly identical conformation as in the PpyWT (PBD code: 4G36) and the higher resolution Japanese luciferase (PBD code: 2D1S). The side chain of C222 is directed towards G254 and I257, which sit in an α-helix that runs from G246 to C258. This helix contains two additional residues, F247 and T251, that pack directly against the dehydroluciferin moiety of DLSA. Thus, the influence of C222 and Y255 on the hydrophobic environment of the ligand may be mediated by this α-helix. Additionally, we considered that the C222 was in a favorable position to C216 to form a disulfide bond that might account for the activity improvement. However, neither the crystal structure nor Ellman titration of enzyme stored for a week produced evidence of disulfide formation (Supplemental Information).
The third finding was an increase in stability to loss of activity by heat or chemical denaturation, aggregation, and acidic pH. The pH and aggregation stability of PsnWT was imparted (and for 45 °C thermostability and GdmCl denaturation exceeded) onto Pps6 by introducing 4 mutations (T214N, S276T, H332N, and E354N). With the single exception that the improved pH sensitivity was slightly less than that of PsnWT, the Pps3 changes T214N, S276T, and E354N were sufficient to achieve all of the stability properties. Adding H332N to make the Pps4 variant provided full PsnWT-type pH stability and somewhat improved thermal properties. Notably, Pps3 and Pps4, which lack the A222C and Y255F substitutions, do not have enhanced specific activity or red-shifted BL emission. It is also interesting that in a prior study47 that identified 3 PpyWT folding domains, the one corresponding to PpyPsnN2 was identified as the most sensitive portion of Luc to GmdCl denaturation.
Since all 4 mutations make partial contributions to the stability improvements, we anticipated that it would be difficult to identify clear structural determinants and this was generally the case. Moreover, with the exception of E354N, the amino acid changes converted PpyWT residues to those found in wild-type L. cruciata Luc, an enzyme that does not possess exceptional stability properties. Additionally, CD measurements did not reveal any significant differences in secondary structure among the wild-type or Pps6 Lucs (Figure S2). Somewhat reassuringly, we observed structural changes that appear to be associated with the E354N mutation that might be related to the enhanced stability properties. This residue is a known major contributor to Luc thermal stability36-41, 43, 44 and, as established in this study, the Ppy E354N point mutant did have greater Tm and T1/2 at 45 °C values than PpyWT.
The side chains of S276T, H332N, and E354N all lie on the surface of the protein (Figure 3), which is perhaps not surprising given their impact on protein stability. We examined the structure for changes that may play a role in the observed stabilization. In the structure of PpyWT, the closest approach of the side chain of E354 to the E311 side chain is 3.7 Å. In contrast, in the Pps6 structure, the side chain of N354 forms a series of interactions with other protein residues, including some that are in close proximity to important active site motifs that are likely responsible for the increased stability. The side chain of N354 is H-bonded to the E311 side chain through an ordered H2O and also to the carbonyl oxygens of P225 and G228 (Figure 5A). Residue E311 is the first residue of a β-strand from 311-314 that, together with the adjacent β-strand formed by residues 337-340, form one wall of the DLSA binding pocket. This latter β-strand then leads directly to the catalytically important motif 2 (residues 340-344) that is present in the ANL superfamily of adenylating enzymes48. In both structures, the E311 side chain interacts with R337, which is 0.7 Å closer to the DLSA in Pps6. In addition to its role in the solvent channel, the R337 residue also plays a structural role at the base of the D-LH2 binding pocket.28
Figure 5.

Protein interactions for stabilizing mutations. Protein environments for the A. E453N, B. S276T, and C. T241N mutations highlighting interactions with neighboring protein atoms that contribute to overall stability of the Pps6 protein.
Although there are no dramatic changes of protein structure near the S276T mutation, the introduced methyl group is directed into a hydrophobic pocket formed by A45, L238, V263, and Y280 (Figure 5B). Also, the side chain of the T214N substitution makes a series of interactions with the side chains of R213, T346, and the main chain carbonyl of I397 (Figure 5C). In sum, these subtle structural changes are likely responsible for the well-documented increased stability of Pps6.
CONCLUSIONS
It is remarkable that only 6 amino acid changes scattered throughout a subdomain of two very similar proteins from very closely related beetle species can so significantly enhance the properties of an already highly evolved enzyme that converts chemical energy into light. Our detailed analysis with multiple mutations in isolation and in combination illustrates our efforts to explore these changes and the impact that they have on both stability and function. The residues of PpyWT that impact both the emission spectra and activity lie deep within the active site pocket that binds to DLSA. Neither residue Y255 nor A222 directly contact the substrate. It appears that the effect of the Y255F mutation is largely mediated through the impact on a water filled pocket at the base of the active site as well as to changes in the hydrophobic environment near the substrate. Comparisons with the highest resolution structures that are available for luciferase enzymes (PDB Codes: 5KYT and 2D1S) show a highly ordered network of solvent molecules. While resolution differences between alternate structures have made it challenging to directly compare these networks, the removal of the hydroxyl of Y255 in the Pps6 mutant will preclude formation of the same solvent network. The stability enhancements contributed by 3 of the mutated residues serve to further highlight the importance of surface residues, particularly that of E354. Additionally, this structure-function investigation illustrates the utility of systematic studies of even closely homologous proteins to better understand how nature has selected small numbers of changes in proteins to alter and improve properties of fundamental importance.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the National Science Foundation MCB-1410390 (BRB), Air Force Office of Scientific Research FA9550-18-1-0017 (BRB), the Hans & Ella McCollum ‘21 Vahlteich Endowment (BRB), as well as a grant from the National Institutes of General Medical Sciences at the NIH, GM116957 (AMG). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the DOE, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
Footnotes
Accession Code
The atomic coordinates and crystallographic structure factors of Pps6 complexed with DLSA have been deposited in the Protein Data Bank as entry 6Q2M.
Supporting Information. The Supporting Information is available free of charge on the ACS Publication website. The following items are included: procedures for steady-state kinetic constant determination; preparation of Pps6-DLSA complex; Circular Dichroism spectroscopy and mean aggregation temperature determination; determination of free thiols. Also included are: a Table listing the abbreviations and descriptions of Luc variants; a Table of crystallographic diffraction and refinement data; 3 Tables of properties of Luc variants; and 4 Figures illustrating thermal inactivation, CD spectra of Lucs, an omit map electron density for the DLSA ligand in all three protein chains, and bioluminescence emission spectra of Lucs recorded at pH 7.8, 37 °C.
The authors declare no competing financial interest.
REFERENCES
- 1.Naylor LH (1999) Reporter gene technology: the future looks bright, Biochem. Pharmacol 58, 749–757. [DOI] [PubMed] [Google Scholar]
- 2.Caysa H, Jacob R, Muther N, Branchini B, Messerle M, and Soling A (2009) A redshifted codon-optimized firefly luciferase is a sensitive reporter for bioluminescence imaging, Photochemical & Photobiological Sciences 8, 52–56. [DOI] [PubMed] [Google Scholar]
- 3.Siebring-van Olst E, Vermeulen C, de Menezes RX, Howell M, Smit EF, and van Beusechem VW (2013) Affordable Luciferase Reporter Assay for Cell-Based High-Throughput Screening, J. Biomol. Screen 18, 453–461. [DOI] [PubMed] [Google Scholar]
- 4.Gabriel GV, and Viviani VR (2014) Macrolampis Firefly Luciferase: A New Dual Reporter Gene for Simultaneous Ratiometric Intracellular pH Sensing and Gene Expression, Luminescence 29, 101–101.24639020 [Google Scholar]
- 5.Ohmiya Y (2015) Simultaneous Multicolor Luciferase Reporter Assays for Monitoring of Multiple Genes Expressions, Combinatorial Chem. High Throughput Screening 18, 937–945. [DOI] [PubMed] [Google Scholar]
- 6.Cevenini L, Calabretta MM, Calabria D, Roda A, and Michelini E (2016) Luciferase Genes as Reporter Reactions: How to Use Them in Molecular Biology?, In Bioluminescence: Fundamentals and Applications in Biotechnology, Vol 3 (Thouand G, and Marks R, Eds.), pp 3–17. [DOI] [PubMed] [Google Scholar]
- 7.Kapitan M, Eichhof I, Lagadec Q, and Ernst JF (2016) Click beetle luciferases as dual reporters of gene expression in Candida albicans, Microbiology-Sgm 162, 1310–1320. [DOI] [PubMed] [Google Scholar]
- 8.Tarnow P, Bross S, Wollenberg L, Nakajima Y, Ohmiya Y, Tralau T, and Luch A (2017) A Novel Dual-Color Luciferase Reporter Assay for Simultaneous Detection of Estrogen and Aryl Hydrocarbon Receptor Activation, Chem. Res. Toxicol 30, 1436–1447. [DOI] [PubMed] [Google Scholar]
- 9.Mezzanotte L, van 't Root M, Karatas H, Goun EA, and Lowik C (2017) In Vivo Molecular Bioluminescence Imaging: New Tools and Applications, Trends Biotechnol. 35, 640–652. [DOI] [PubMed] [Google Scholar]
- 10.Branchini BR, Southworth TL, Fontaine DM, Kohrt D, Florentine CM, and Grossel MJ (2018) A Firefly Luciferase Dual Color Bioluminescence Reporter Assay Using Two Substrates to Simultaneously Monitor Two Gene Expression Events, Scientific Reports 8:5990, doi: 10.1038/s41598-018-24278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans MS, Chaurette JP, Adams ST, Reddy GR, Paley MA, Aronin N, Prescher JA, and Miller SC (2014) A synthetic luciferin improves bioluminescence imaging in live mice, Nat. Methods 11, 393-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu WX, Su J, Tang CC, Bai HX, Ma Z, Zhang TC, Yuan ZL, Li ZZ, Zhou WJ, Zhang HT, Liu ZZ, Wang Y, Zhou YB, Du LP, Gu LC, and Li MY (2017) cybLuc: An Effective Aminoluciferin Derivative for Deep Bioluminescence Imaging, Anal. Chem 89, 4808–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall MP, Woodroofe CC, Wood MG, Que I, Van'T Root M, Ridwan Y, Shi C, Kirkland TA, Encell LP, Wood KV, Löwik C, and Mezzanotte L (2018) Click beetle luciferase mutant and near infrared naphthyl-luciferins for improved bioluminescence imaging, Nat Commun 9, doi: 10.1038/s41467-017-02542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwano S, Sugiyama M, Hama H, Watakabe A, Hasegawa N, Kuchimaru T, Tanaka KZ, Takahashi M, Ishida Y, Hata J, Shimozono S, Namiki K, Fukano T, Kiyama M, Okano H, Kizaka-Kondoh S, McHugh TJ, Yamamori T, Hioki H, Maki S, and Miyawaki A (2018) Single-cell bioluminescence imaging of deep tissue in freely moving animals, Science 359, 935–939. [DOI] [PubMed] [Google Scholar]
- 15.Yao Z, Zhang BS, and Prescher JA (2018) Advances in bioluminescence imaging: new probes from old recipes, Curr. Opin. Chem. Biol 45, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McElroy WD (1947) The Energy Source for Bioluminescence in an Isolated System, Proc Natl Acad Sci U S A 33, 342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti E, Franks NP, and Brick P (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes, Structure 4, 287–298. [DOI] [PubMed] [Google Scholar]
- 18.Iwano S, Obata R, Miura C, Kiyama M, Hama K, Nakamura M, Amano Y, Kojima S, Hirano T, Maki S, and Niwa H (2013) Development of simple firefly luciferin analogs emitting blue, green, red, and near-infrared biological window light, Tetrahedron 69, 3847–3856. [Google Scholar]
- 19.Jathoul AP, Grounds H, Anderson JC, and Pule MA (2014) A Dual-Color Far-Red to Near-Infrared Firefly Luciferin Analogue Designed for Multiparametric Bioluminescence Imaging, Angew Chem Int Edit 53, 13059–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mofford DM, Reddy GR, and Miller SC (2014) Aminoluciferins Extend Firefly Luciferase Bioluminescence into the Near-Infrared and Can Be Preferred Substrates over D-Luciferin, J. Am. Chem. Soc 136, 13277–13282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson JC, Grounds H, Jathoul AP, Murray JAH, Pacman SJ, and Tisi L (2017) Convergent synthesis and optical properties of near-infrared emitting bioluminescent infra-luciferins, Rsc Advances 7, 3975–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson JC, Chang CH, Jathoul AP, and Syed AJ (2018) Synthesis and bioluminescence of electronically modified and rotationally restricted colour-shifting infraluciferin analogues, Tetrahedron. [Google Scholar]
- 23.Branchini BR, Southworth TL, Fontaine DM, Murtiashaw MH, McGurk A, Talukder MH, Qureshi R, Yetil D, Sundlov JA, and Gulick AM (2017) Cloning of the Orange Light-Producing Luciferase from Photinus scintillansA New Proposal on how Bioluminescence Color is Determined, Photochem. Photobiol 93, 479–485. [DOI] [PubMed] [Google Scholar]
- 24.Ugarova NN, and Brovko LY (2002) Protein structure and bioluminescent spectra for firefly bioluminescence, Luminescence 17, 321–330. [DOI] [PubMed] [Google Scholar]
- 25.Viviani VR, Oehlmeyer TL, Arnoldi FGC, and Brochetto-Braga MR (2005) A new firefly luciferase with bimodal spectrum: Identification of structural determinants of spectral pH-sensitivity in firefly luciferases, Photochem. Photobiol 81, 843–848. [DOI] [PubMed] [Google Scholar]
- 26.Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, and Kato H (2006) Structural basis for the spectral difference in luciferase bioluminescence, Nature 440, 372–376. [DOI] [PubMed] [Google Scholar]
- 27.Navizet I, Liu Y-J, Ferre N, Xiao H-Y, Fang W-H, and Lindh R (2010) Color-Tuning Mechanism of Firefly Investigated by Multi-Configurational Perturbation Method, J. Am. Chem. Soc 132, 706–712. [DOI] [PubMed] [Google Scholar]
- 28.Viviani VR, Simoes A, Bevilaqua VR, Gabriel GV, Arnoldi FG, and Hirano T (2016) Glu311 and Arg337 Stabilize a Closed Active-site Conformation and Provide a Critical Catalytic Base and Countercation for Green Bioluminescence in Beetle Luciferases, Biochemistry 55, 4764–4776. [DOI] [PubMed] [Google Scholar]
- 29.Carrasco-López C, Ferreira JC, Lui NM, Schramm S, Berraud-Pache R, Navizet I, Panjikar S, Naumov P, and Rabeh WM (2018) Beetle luciferases with naturally red- and blue-shifted emission, Life Science Alliance 1, doi: 10.26508/lsa.201800072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirano T, Hasumi Y, Ohtsuka K, Maki S, Niwa H, Yamaji M, and Hashizume D (2009) Spectroscopic Studies of the Light-Color Modulation Mechanism of Firefly (Beetle) Bioluminescence, J. Am. Chem. Soc 131, 2385–2396. [DOI] [PubMed] [Google Scholar]
- 31.Naumov P, Ozawa Y, Ohkubo K, and Fukuzumi S (2009) Structure and Spectroscopy of Oxyluciferin, the Light Emitter of the Firefly Bioluminescence, J. Am. Chem. Soc 131, 11590–11605. [DOI] [PubMed] [Google Scholar]
- 32.Branchini BR, Ablamsky DM, Davis AL, Southworth TL, Butler B, Fan F, Jathoul AP, and Pule MA (2010) Red-emitting luciferases for bioluminescence reporter and imaging applications, Anal. Biochem 396, 290–297. [DOI] [PubMed] [Google Scholar]
- 33.Branchini BR, Southworth TL, Fontaine DM, Davis AL, Behney CE, and Murtiashaw MH (2014) A Photinus pyralis and Luciola italica Chimeric Firefly Luciferase Produces Enhanced Bioluminescence, Biochemistry 53, 6287–6289. [DOI] [PubMed] [Google Scholar]
- 34.Branchini BR, Ablamsky DM, Rosenman JM, Uzasci L, Southworth TL, and Zimmer M (2007) Synergistic mutations produce blue-shifted bioluminescence in firefly luciferase, Biochemistry 46, 13847–13855. [DOI] [PubMed] [Google Scholar]
- 35.Branchini BR, Murtiashaw MH, Magyar RA, and Anderson SM (2000) The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase, Biochemistry 39, 5433–5440. [DOI] [PubMed] [Google Scholar]
- 36.White PJ, Squirrell DJ, Arnaud P, Lowe CR, and Murray JA (1996) Improved thermostability of the North American firefly luciferase: saturation mutagenesis at position 354, Biochem. J 319 (Pt 2), 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tisi LC, White PJ, Squirrell DJ, Murphy MJ, Lowe CR, and Murray JAH (2002) Development of a thermostable firefly luciferase, Anal. Chim. Acta 457, 115–123. [Google Scholar]
- 38.Kitayama A, Yoshizaki H, Ohmiya Y, Ueda H, and Nagamune T (2003) Creation of a thermostable firefly luciferase with pH-insensitive luminescent color, Photochem. Photobiol 77, 333–338. [DOI] [PubMed] [Google Scholar]
- 39.Baggett B, Roy R, Momen S, Morgan S, Tisi L, Morse D, and Gillies RJ (2004) Thermostability of firefly luciferases affects efficiency of detection by in vivo bioluminescence, Molecular Imaging 3, 324–332. [DOI] [PubMed] [Google Scholar]
- 40.Law GHE, Gandelman OA, Tisi LC, Lowe CR, and Murray JAH (2006) Mutagenesis of solvent-exposed amino acids in Photinus pyralis luciferase improves thermostability and pH-tolerance, Biochem. J 397, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Branchini BR, Ablamsky DM, Murtiashaw MH, Uzasci L, Fraga H, and Southworth TL (2007) Thermostable red and green light-producing firefly luciferase mutants for bioluminescent reporter applications, Anal. Biochem 361, 253–262. [DOI] [PubMed] [Google Scholar]
- 42.Moradi A, Hosseinkhani S, Naderi-Manesh H, Sadeghizadeh M, and Alipour BS (2009) Effect of Charge Distribution in a Flexible Loop on the Bioluminescence Color of Firefly Luciferases, Biochemistry 48, 575–582. [DOI] [PubMed] [Google Scholar]
- 43.Koksharov MI, and Ugarova NN (2011) Thermostabilization of firefly luciferase by in vivo directed evolution, Protein Engineering Design & Selection 24, 835–844. [DOI] [PubMed] [Google Scholar]
- 44.Mortazavi M, and Hosseinkhani S (2017) Surface charge modification increases firefly luciferase rigidity without alteration in bioluminescence spectra, Enzyme Microb. Technol 96, 47–59. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro C, and da Silva J (2008) Kinetics of inhibition of firefly luciferase by oxyluciferin and dehydroluciferyl-adenylate, Photochemical & Photobiological Sciences 7, 1085–1090. [DOI] [PubMed] [Google Scholar]
- 46.Branchini BR, Murtiashaw MH, Carmody JN, Mygatt EE, and Southworth TL (2005) Synthesis of an N-acyl sulfamate analog of luciferyl-AMP: A stable and potent inhibitor of firefly luciferase, Biorg. Med. Chem. Lett 15, 3860–3864. [DOI] [PubMed] [Google Scholar]
- 47.Frydman J, Erdjument-Bromage H, Tempst P, and Ulrich Hartl F (1999) Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase, Nature Struct. Biol 6, 697–705. [DOI] [PubMed] [Google Scholar]
- 48.Gulick AM (2009) Conformational Dynamics in the Acyl- CoA Synthetases, Adenylation Domains of Non-ribosomal Peptide Synthetases, and Firefly Luciferase, Acs Chemical Biology 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.