

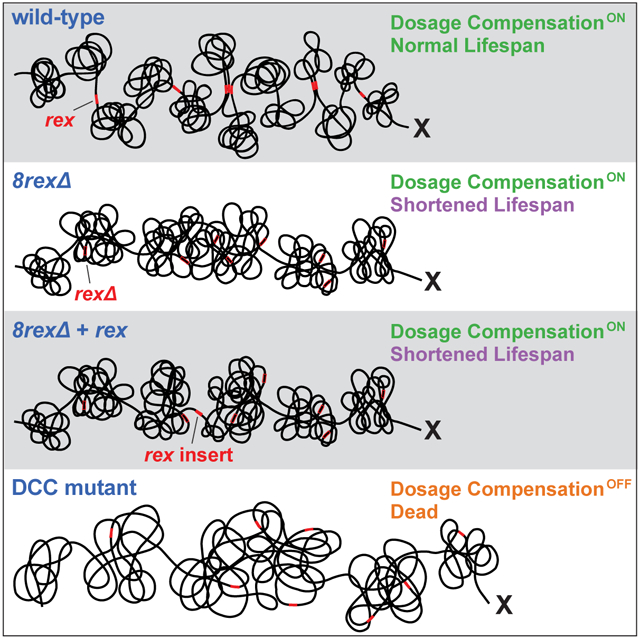
SUMMARY
Mechanisms establishing higher-order chromosome structures and their roles in gene regulation are elusive. We analyzed chromosome architecture during nematode X-chromosome dosage compensation, which represses transcription via a dosage-compensation condensin complex (DCC) that binds hermaphrodite Xs and establishes megabase-size topologically associating domains (TADs). We show that DCC binding at high-occupancy sites (rex sites) defines eight TAD boundaries. Single rex deletions disrupted boundaries, and single insertions created new boundaries, demonstrating a rex site is necessary and sufficient to define DCC-dependent boundary locations. Deleting eight rex sites (8rexΔ) recapitulated TAD structure of DCC mutants, permitting analysis when chromosome-wide domain architecture was disrupted but most DCC binding remained. 8rexΔ animals exhibited no changes in X expression and lacked dosage-compensation mutant phenotypes. Hence, TAD boundaries are neither the cause nor consequence of DCC-mediated gene repression. Abrogating TAD structure did, however, reduce thermotolerance, accelerate aging, and shorten lifespan, implicating chromosome architecture in stress responses and aging.
Graphical Abstract
eTOC
Anderson et al. show that binding of the condensin dosage compensation complex (DCC) to individual high-affinity sites on nematode X chromosomes is necessary and sufficient to establish boundaries for topologically associating domains. Eliminating DCC-dependent domain architecture does not compromise dosage compensation but does reduce thermotolerance, accelerate aging, and shorten lifespan.
INTRODUCTION
Interphase chromosomes are highly organized into a series of structures ranging from kilobase-scale chromatin loops to territories comprising hundreds of megabases (Gibcus and Dekker, 2013; Serizay and Ahringer, 2018; Yu and Ren, 2017). Dissecting the mechanisms underlying the formation of these interrelated structures and unravelling their functions require their precise manipulation, a task made challenging by the paucity of information about DNA sequences and factors required for higher-order chromosome organization.
We focus on topologically associating domains (TADs) of approximately one megabase that are characteristic of metazoan chromosomes. TADs were defined in mammalian cells as self-interacting domains (Dixon et al., 2012; Nora et al., 2012). Loci in one TAD interact predominantly with each other while being insulated from interactions with loci in neighboring TADs. Mechanisms that define TAD boundaries and the biological functions of TADs have been elusive. In mammalian cells, architectural proteins important for establishing TADs, such as the zinc-finger protein CTCF and the SMC complex cohesin, localize at boundaries between TADs (Dixon et al., 2012; Nora et al., 2012). The architectural proteins also play roles in essential cellular processes such as chromosome segregation (Hocquet et al., 2018; Morales and Losada, 2018), making the functional significance of TADs difficult to assess at the organismal level by depleting these proteins to disrupt TADs (Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017). Rather than depleting multi-functional proteins, we eliminate TADs by deleting specific binding sites for an architectural protein complex and assess the effects on gene expression and animal development.
We analyzed chromosome architecture and its impact on gene expression in the context of Caenorhabditis elegans X-chromosome dosage compensation, an essential process in which both gene expression and higher-order chromosome structure are regulated via a specialized condensin complex (Meyer, 2018). This dosage compensation complex (DCC) equalizes X expression between XO males and XX hermaphrodites by binding along both hermaphrodite X chromosomes and reducing transcription by half, while also establishing an X structure composed of one-megabase TADs (Crane et al., 2015). Without DCC binding, eight TAD boundaries are lost, causing X structure to resemble that of autosomes with fewer, less regularly spaced TAD boundaries. These remaining boundaries are DCC-independent. All eight DCC-dependent TAD boundaries contain a high-affinity DCC binding site (rex site) (Crane et al., 2015), which typically includes multiple X-enriched motifs important for DCC binding (Albritton et al., 2017; Ercan et al., 2007; Jans et al., 2009; McDonel et al., 2006; Pferdehirt et al., 2011). The findings that stable DCC binding near a gene is neither necessary nor sufficient for the dosage compensation of the gene and that transgenes integrated at multiple locations along X become compensated indicate that dosage compensation is a chromosome-wide process in which the DCC can act at a distance to repress transcription (Jans et al., 2009; Wheeler et al., 2016). Thus, DCC-dependent TAD structure could plausibly underlie the mechanism of gene repression.
In addition to condensin subunits, the DCC includes proteins important for loading of condensin onto X (SDC-2 and SDC-3) and a histone demethylase (DPY-21) responsible for enriching the histone modification H4K20me1 on X (Bian et al., 2018; Brejc et al., 2017). Loss of H4K20me1 enrichment causes partial disruption of dosage compensation, de-compaction of X, and weakening of DCC-dependent TAD boundaries (Brejc et al., 2017).
Mechanisms used by the DCC to remodel X-chromosome topology and establish TADs are broadly relevant because condensin and other SMC complexes play key roles in shaping chromosome architecture in eukaryotes and prokaryotes. Condensin is essential for mitotic chromosome compaction and segregation in eukaryotes (Gibcus et al., 2018; Hirano, 2016), represses transcription in quiescent yeast (Swygert et al., 2019), and resolves newly replicated sister origins during DNA replication in Bacillis subtilis (Wang et al., 2017). Cohesin is required for the formation of most TAD boundaries in mammalian cells (Rao et al., 2017; Schwarzer et al., 2017).
The functions of TADs in gene regulation have not been well characterized. In principle, loss of TAD boundaries could either be a cause or consequence of transcriptional changes. In mammalian cells, disrupting a TAD boundary leads to merging of adjacent TADs, permitting enhancers to interact with additional genes and activate them ectopically. Developmental defects such as limb malformations can occur as a consequence (Franke et al., 2016; Lupiáñez et al., 2015; Narendra et al., 2015). In other contexts, transcription can control chromosome structure, as in Caulobacter, where transcription drives the formation of boundaries between chromosomal interaction domains (Le and Laub, 2016). Defining the relationship between TAD structure and gene regulation has been challenging because architectural proteins that establish TADs also bind and function at locations other than TAD boundaries, such as promoters, making it unclear whether the transcriptional changes resulting from their depletion are caused exclusively by altered TAD structure or by other roles of the proteins in gene regulation (Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017).
Here we defined the requirements for creating a DCC-dependent boundary by making a series of rex site deletions and insertions on X chromosomes and measuring the resulting chromosome structure. DCC binding at a high-occupancy rex site was necessary and sufficient to define the location of a boundary. By deleting all eight rex sites at the eight DCC-dependent boundaries, we eliminated DCC-dependent TAD structure and found that TADs do not drive dosage compensation. In the absence of TADs, the DCC promotes DNA interactions across X between loci within 0.1–1 Mb, which may underlie transcriptional repression. Although abrogating TAD structure in hermaphrodites by deleting rex sites did not disrupt dosage compensation, it did reduce thermotolerance, accelerate aging, and shorten lifespan, implicating higher-order chromosome structure in regulating stress responses and aging.
RESULTS
Deleting the Single rex Site at Each DCC-Dependent TAD Boundary Eliminated the Boundary
We dissected the mechanisms by which the DCC establishes TAD boundaries by analyzing the potential contributions of multiple factors. First, the presence of a high-occupancy rex site at each DCC-dependent boundary suggested that DCC binding at these strong rex sites might define the locations of boundaries. Second, DCC-mediated interactions between DCC-dependent boundaries are among the strongest long-range interactions on the X chromosome, suggesting that boundary formation might require not only DCC binding at rex sites, but also long-range interactions between rex sites located at DCC-dependent boundaries (Crane et al., 2015). Third, the DCC promotes interactions between rex sites within TADs (Crane et al., 2015), raising the possibility that these intra-TAD interactions might passively create boundaries by bringing loci together.
To assess the role in boundary formation of DCC binding at high-occupancy rex sites, we sequentially deleted the single rex site at each of the eight DCC-dependent TAD boundaries on X (Table S1) and measured the resulting chromosome structure (Figure 1A). Seven rex sites are in intergenic regions, and each rex deletion (400–1100 bp) removed all motifs known to be important for DCC binding. The exception (rex-33) was deleted by removing the entire intron of the essential gene in which it resides. To verify that deletions eliminated DCC binding, we performed ChIP-seq using antibodies for the DCC-specific SMC protein DPY-27 and the non-condensin DCC subunit SDC-3, which helps load all condensin subunits onto X. Binding of both subunits was lost entirely at each deleted site, while DCC binding at dozens of remaining rex sites persisted (Figure S1A).
Figure 1. A Single rex Site Is Necessary for Formation of Each DCC-Dependent TAD Boundary.

(A) Locations on X of the eight rex sites (orange) at DCC-dependent TAD boundaries. All eight sites are deleted on 8rexΔ chromosomes, but only a subset are deleted on rex-47Δ, 3rexΔ (magenta circles), and 6rexΔ (green asterisks) chromosomes.
(B, C, and D) X chromosome heatmaps binned at 50 kb show wild-type Hi-C Z-scores subtracted from rex-47Δ, 3rexΔ, or 6rexΔ Hi-C Z-scores. Red, higher interactions in rex mutant. Blue, higher interactions in wild type. Arrows, positions of deleted rex sites where TAD boundaries are lost. Plots (below) show insulation scores across X in rex deletion mutants (blue) and wild-type embryos (orange) and the insulation score difference between genotypes (red). Blue ticks, positions of deleted rex sites. Orange ticks, DCC-dependent boundaries that persist in the mutant.
(E, F, and G) X heatmaps binned at 20 kb show Hi-C interactions in 8rexΔ, DCC mutant [sdc-2(y93, RNAi)], and wild-type embryos. Arrows mark positions of DCC-dependent boundaries found in wild-type embryos, which are lost in both mutants. Other DCC-independent boundaries remain.
(H and I) Heatmaps binned at 50 kb compare Hi-C Z-scores in 8rexΔ or DCC mutants to those in wild-type embryos. Plots (below) show X chromosome insulation profiles. Black arrows (top) and blue ticks (bottom) show locations of DCC-dependent boundaries.
(J) Heatmaps binned at 10 kb show enlargement of the X region surrounding rex-43 and rex-48 (arrows) in wild-type and mutant embryos and the interaction between the two rex sites in wild-type embryos (circle).
(K) 3D plots show that average Z-scores increase in 8rexΔ versus wild-type embryos for interactions among the 22 non-boundary rex sites with highest SDC-3 binding. Shown are interactions between sites within 4 Mb. DCC-mediated rex interactions occur regardless of the orientation of known X-enriched motifs (Jans et al., 2009) that are important in rex sites for DCC binding (Figure S3A). Insulation scores were calculated by summing interactions in a 500 kb sliding window.
See also Figures S1–S3, Tables S1 and S2.
We examined the effects of rex deletions on chromosome structure by performing in situ Hi-C on wild-type embryos, a series of mutant embryos with increasing numbers of rex deletions, and DCC mutant embryos lacking DCC binding on X [sdc-2(y93, RNAi)] (Table S2). Previous fluorescence in situ hybridization experiments showed that deleting rex-47 greatly diminished the associated TAD boundary (Crane et al., 2015). We confirmed this result by Hi-C. By comparing distance-normalized Hi-C interactions (Z-scores) between rex-47Δ and wild-type embryos, we found an increase in interactions across the deleted rex site (Figures 1B and S2A), verifying boundary loss. To quantify the extent of loss, we created X chromosome insulation profiles (Crane et al., 2015). Briefly, we calculated an insulation score for each locus by summing all interactions within a 500 kb window surrounding that locus. When comparatively few interactions occur across a locus, it has a low insulation score. Such a local minimum in the insulation profile is defined as a TAD boundary. rex-47 is at a local minimum in the wild-type insulation profile, and the valley was eliminated in rex-47Δ, indicating complete loss of the TAD boundary (Figure 1B). Thus, the rex site defined the boundary location.
Next, we examined 3rexΔ and 6rexΔ chromosomes, in which additional rex sites were deleted from the center and right side of X. The boundaries at the deleted rex sites were eliminated, but all other boundaries remained (Figures 1C,1D, S2B and S2C). No new boundaries emerged at the remaining weaker rex sites, indicating that high-occupancy rex sites do not impede lower-occupancy rex sites from forming boundaries.
Deleting all eight rex sites at the DCC-dependent boundaries recapitulated the disrupted TAD structure of X chromosomes in embryonic lethal DCC mutants that lack DCC binding. In both the 8rexΔ and DCC mutants, seven TAD boundaries were eliminated, and the boundary at rex-32 was dramatically weakened (Figures 1E–1J), while DCC-independent TAD boundaries on X and autosomes remained (Figures S2D–S2H). Although the DCC was bound to numerous rex sites across X in 8rexΔ embryos (Figure S1A), removing only the eight rex sites at DCC-dependent TAD boundaries was sufficient to disrupt the TAD structure of X. Thus, a rex site is essential to define the location of each DCC-dependent TAD boundary.
Using 8rexΔ embryos, we tested whether intra-TAD rex-rex interactions might passively create boundaries. We found that DCC-mediated interactions between the 22 highest-affinity non-boundary rex sites persisted in 8rexΔ embryos (Figures 1K, S2I, and S2J), indicating that interactions between rex sites within the same TAD are not sufficient to create TAD boundaries on the native X. Instead, DCC binding to the eight rex sites at DCC-dependent TAD boundaries defines boundary location.
Inserting a Single High-Occupancy rex Site Created a New TAD Boundary on X But Not on an Autosome
Having shown that a rex site is necessary to form a DCC-dependent TAD boundary, we asked whether a single rex site is sufficient to create a boundary. We inserted a high-occupancy rex site (rex-32) that forms a boundary at its endogenous location into a new location in the middle of the 8rexΔ X chromosome (Figure 2A). The inserted rex site bound the DCC at a level comparable to that at its original location (Figure S4A), and it defined a new boundary of similar strength to the boundary at its endogenous site (Figure 2B, 2C, and 2F). Therefore, insertion of a single high-affinity DCC binding site is sufficient to create a TAD boundary on X. This boundary is the sole DCC-dependent boundary on the chromosome, revealing that boundary formation does not require interactions between DCC-dependent boundaries. However, we have not ruled out the possibility that interactions between the ectopic rex site and weaker, non-boundary rex sites may contribute to boundary formation.
Figure 2. A High-Occupancy rex Site is Sufficient to Create a TAD Boundary at a New Location on X.

(A) Locations on X of eight rex deletions and two rex insertions at new locations (arrows). rex-32 was inserted 224 kb from wild-type rex-14; rex-8 was inserted 267 kb from wild-type rex-47.
(B and D) Heatmaps binned at 20 kb show Hi-C interactions on X in embryos with either a rex-32 insertion or rex-32 and rex-8 insertions. Arrows mark locations of inserted rex sites where new TAD boundaries are created. (D, right) Heatmap binned at 10 kb shows enlargement of the X region surrounding the inserted rex sites (arrows) and the interaction between the inserted sites (circle).
(C and E) Z-score subtraction heatmaps binned at 50 kb show increased (red) and decreased (blue) interactions on 8rexΔ chromosomes with rex insertions compared to 8rexΔ chromosomes. Arrows, locations of inserted rex sites.
(F) Plot comparing X insulation scores of 8rexΔ chromosomes (orange) to those of 8rexΔ chromosomes with one (blue) or two (red) inserted rex sites
(G) Diagram shows the location of rex-32 inserted 1.05 Mb from rex-48 on the wild-type X chromosome (arrow). Z-score subtraction heatmap binned at 50 kb shows the difference in Z-scores between rex-32 insertion and wild-type chromosomes.
(H) Plot comparing X chromosome insulation scores for wild-type (orange) and rex-32 insertion (blue) embryos
(I) Boundaries adjacent to deleted boundaries became stronger (p = 0.02, paired t test), while boundaries two away were unchanged (p = 0.60). Using Hi-C data from rex-47Δ, 3rexΔ, 6rexΔ, and 8rexΔ X chromosomes, we calculated the average insulation profile around all boundaries adjacent to a deleted boundary and compared it to the profile at the same boundaries in the strain with the next fewer deletions (see STAR Methods).
See also Figures S3 and S4, Tables S1 and S2.
A second rex site (rex-8) inserted 1.4 Mb downstream of the newly inserted rex-32 site also defined a new TAD boundary (Figure 2D–F). Boundary-to-boundary interactions occurred between the inserted rex-8 and rex-32 sites (Figure 2D, right), and the boundary at rex-32 became slightly weaker (Figure 2F). Therefore, interactions between adjacent DCC-dependent boundaries do not strengthen boundaries.
To determine whether a rex site is sufficient to create a new boundary in the context of wild-type 3D architecture, we inserted rex-32 into a new, non-boundary location on the right side of the wild-type X chromosome. Again, rex-32 bound the DCC at a level similar to that at its endogenous location (Figure S4B) and defined a new boundary (Figures 2G and 2H). Thus, the native X structure does not prevent formation of new TADs but can be further subdivided into new domains by addition of a rex site.
Given that each DCC-dependent boundary contains a high-occupancy rex site and that a single strong rex site is sufficient to create a boundary at a new location on X, we asked whether the high-occupancy rex sites that fail to define boundaries can nonetheless reduce DNA interactions between flanking loci. Five rex sites that have higher SDC-3 binding than the boundary-forming site rex-47 failed to form boundaries but did alter chromosome structure. That is, in DCC mutants, interactions between loci flanking these non-boundary rex sites increased compared to interactions across neighboring loci (Figures S3B and S3C). DCC binding at these rex sites may be insufficient to create strong boundaries due to unknown factors that modulate TAD structure (such as lamin binding at rex-40, rex-45, and Prex-30), or DCC-dependent boundary formation may be overwhelmed by strong DCC-independent boundaries nearby (at rex-23 and rex-16). Regardless of whether a rex site defines a boundary, the level of SDC-3 binding at that site correlates strongly with the DCC-dependent change in insulation score (Figure S3B).
We next sequentially inserted three TAD-boundary rex sites from the center of X into the center of chromosome I in the 3rexΔ strain. Inserted rex sites recruited the DCC, but with only about 20% of the binding of their endogenous X locations (Figure S4C). These inserted rex sites had the same spacing as on X but did not create boundaries on chromosome I (Figure S4D–S4F). Lack of new boundaries is consistent with the observation that rex sites on X with the same low level of DCC binding as at these ectopic sites do not define TAD boundaries, but we cannot rule out the possibility that some other unique characteristic of X chromosomes, such as widespread DCC binding or H4K20me1 enrichment, is necessary for DCC-dependent boundary formation.
Together, our results establish that the DCC forms boundaries via binding to high-affinity rex sites with multiple X-enriched motifs located at boundaries and not through intra-TAD interactions. A single inserted rex site is sufficient to define a boundary at a new location on X, and interactions with a second inserted rex site do not strengthen the boundary, revealing that interactions between rex sites at boundaries are not necessary to form a boundary.
Creating and Destroying Boundaries Affects the Strength of Neighboring and Distant Boundaries
Our deletion series offered a unique setting to investigate how adding and removing boundaries affects the strength of neighboring and distant boundaries. When comparing 8rexΔ versus 6rexΔ X chromosomes, we found that the four DCC-independent TAD boundaries flanking the two additional rex deletions in 8rexΔ became stronger (Figure S4G). Overall, composite profiles showed that when a boundary was eliminated, adjacent boundaries were strengthened (p = 0.02, paired t test; Figure 2I), while boundaries two away from deleted boundaries showed no consistent pattern of change (p = 0.60, Figure 2I).
Conversely, when a new boundary was created, the adjacent boundaries were weakened. For example, when the inserted rex-32 formed a new TAD boundary in the center of the 8rexΔ chromosome, the upstream DCC-independent boundary became weaker, and the local minimum in the 8rexΔ insulation profile downstream of the rex-32 insertion site disappeared (Figure 2F). Changes in DCC-independent boundary strength caused by altering DCC-dependent boundaries reveal that a boundary can influence the strength of an adjacent boundary even when boundaries are established through different mechanisms.
Boundary deletions can also have long-range effects on the strength of distant boundaries. For example, when rex sites were deleted on the right side of X, boundaries were altered over 10 Mb away on the left side of X (Figure S4H). The intricate interplay between distant boundaries highlights the complexity of predicting the long-range effects of altering chromosome structure and suggests the intriguing possibility that perturbation of one site could affect the functions of broad genomic regions.
Most DCC Binding is Preserved on 8rexΔ Chromosomes
Assessing the effect of aberrant TAD structure on gene expression in 8rexΔ embryos required that we understand the degree to which DCC binding along X was affected by rex deletions. Thousands of DCC peaks of varying strengths are detectable on X by ChIP-seq analysis (Albritton et al., 2017; Crane et al., 2015). These peaks represent two categories of DCC binding: autonomous, X-motif-dependent binding to rex sites and non-autonomous binding to secondary sites that lack X-enriched motifs and exhibit DCC binding only when located on X chromosomes (Albritton et al., 2017; Csankovszki et al., 2004; Ercan et al., 2007; Jans et al., 2009; McDonel et al., 2006; Pferdehirt et al., 2011). rex sites confer X-chromosome specificity for DCC binding.
To determine whether loss of strong rex sites affects DCC binding nearby or broadly, we compared SDC-3 and DPY-27 binding on 8rexΔ versus wild-type X chromosomes. In 8rexΔ embryos, DCC binding was preserved at all remaining rex sites (Figure S1A). Furthermore, the change in DCC binding across the entire X chromosome was minimal, but the ratio of DCC binding on 8rexΔ compared to wild-type chromosomes revealed that binding of both DCC components was reduced up to 16-fold immediately adjacent to the rex deletions and gradually returned to wild-type levels at approximately 20 kb on either side of each deleted site (Figures S1B and S1C). Similarly, we compared DCC binding around rex sites inserted into new locations on 8rexΔ X chromosomes to binding on 8rexΔ chromosomes lacking the rex insertion. DCC binding was enriched approximately 16-fold around the inserted sites, and binding gradually decreased to wild-type levels approximately 15 kb from the insertions (Figures S1D and S1E). Two explanations could account for the reduction in DCC binding around deleted sites and increase around inserted sites. First, DCC binding at strong rex sites might facilitate DCC binding nearby, up to about 20 kb. Alternatively, the sequences within 20 kb of the rex site may not be bound directly by the DCC, but instead be in close enough proximity to flanking DNA to become crosslinked, thus giving the appearance of increased DCC binding nearby. Because DCC binding was retained on 8rexΔ X chromosomes beyond 20 kb from each deleted rex site, we could measure the effect of aberrant TAD structure on X gene expression without the complication of a general disruption of DCC binding across X.
TADs Do Not Mediate Transcriptional Repression during Dosage Compensation
To determine whether disruption of TAD structure alters dosage compensation, we first inspected 8rexΔ worms for evidence of canonical dosage compensation defects. Complete disruption of dosage compensation causes XX-specific embryonic lethality, and weak disruption causes dumpy (Dpy) and egg-laying defective (Egl) phenotypes (Dawes et al., 1999). Dosage compensation defects can also be detected by an even more sensitive genetic assay. Males with xol-1 mutations are inviable because the DCC binds inappropriately to the single X and represses X expression. They are rescued by mutations that disrupt dosage compensation (Miller et al., 1988; Rhind et al., 1995). 8rexΔ worms do not display any of these dosage compensation phenotypes. They have a normal brood size, and 100% of animals are viable and not Dpy or Egl (Table S3). Deletions do not rescue xol-1 mutant males (STAR Methods).
To assay dosage compensation more directly and thoroughly, we measured gene expression genome-wide. We performed RNA-seq on mixed-stage embryos of three genotypes: wild-type, a DCC mutant lacking DCC binding on X, and 8rexΔ in which X topology was altered but the majority of DCC binding persisted.
Assaying embryos allowed us to measure elevation in X expression in the severe DCC mutant before the animals died. However, using embryos required that we carefully match the age distribution of embryos in the populations of different genotypes. X chromosome expression is silenced in the germline (Fong et al., 2002; Kelly et al., 2002; Tzur et al., 2018), and as silencing wanes in early embryos, average expression of genes on X, but not autosomes, increases for approximately the first five hours of embryogenesis (Figure S5A). Thus, comparing populations of embryos that are not precisely age matched could introduce X-chromosome-wide artifacts in the analysis of transcript levels (Figure S5B). To avoid such confounding factors, we first showed that embryogenesis proceeds at the same rate in 8rexΔ and wild-type animals (Figure S5C).
For each RNA-seq replicate, we attempted to harvest age-matched embryo populations by growing worms and collecting embryos of all genotypes in parallel. We then computationally compared the age distribution of wild-type and 8rexΔ embryo populations by assessing expression levels of autosomal genes that change during development (STAR Methods, Figure S5D). We selected the five best-matched wild-type and 8rexΔ replicates, excluding other replicates in which the wild-type embryos clearly had an older age distribution than the 8rexΔ population. While median X expression was elevated 1.5-fold in DCC mutant versus wild-type embryos (p = 2 × 10−16, two-sided Wilcoxon rank-sum test comparing changes on X to autosomes), expression of X genes was not elevated in 8rexΔ embryos (median fold change 0.97 compared to wild type, p = 0.1) (Figures 3A and 3B). No individual genes showed significant differential expression between wild-type and 8rexΔ embryos (FDR > 0.06 for all genes).
Figure 3. Expression of X-Linked Genes Is Not Significantly Changed by Loss of TAD Boundaries.

(A and B) Box plots show gene expression changes for each chromosome in 8rexΔ or sdc-2(y93, RNAi) versus wild-type embryos. Numbers of genes per chromosome are listed.
(C) Median gene expression changes in DCC mutant or 8rexΔ versus wild-type embryos in a 400 kb sliding window on X. Blue vertical lines, locations of rex sites deleted in 8rexΔ
To determine whether transcription varied with changes in TAD boundaries, we calculated the average change in gene expression in a 400 kb sliding window along X. No correlation was evident between changes in gene expression and distance to a DCC-dependent boundary for either 8rexΔ or DCC mutant embryos (Figure 3C). The small cohort of genes within 100 kb of an altered boundary did not show dramatic expression differences compared to genes farther from a boundary. These observations reinforce the conclusion that dosage compensation is a chromosome-wide process that represses genes across X regardless of their proximity to TAD boundaries (Crane et al., 2015). In both 8rexΔ and DCC mutant embryos, X chromosome TAD boundaries were lost, but X-linked expression was elevated only in the DCC mutant and not significantly changed in 8rexΔ. Therefore, DCC-dependent TADs are neither a cause nor consequence of transcriptional repression; the changes in chromosome domain architecture and gene expression result from two separate DCC roles.
The DCC Promotes Interactions at the Scale of 0.1–1 Mb Independently of TAD Boundary Formation
Beyond revealing that dosage compensation occurs without TADs, the complete disruption of DCC-dependent TAD boundaries on 8rexΔ chromosomes allowed us to discover DCC-dependent structures that persisted in the absence of TADs and could be important for long-range transcriptional repression. By comparing the X topology of 8rexΔ embryos to DCC mutant embryos, we found that interactions between loci within ~1 Mb were reduced in DCC mutants but persisted across the X chromosome of 8rexΔ embryos (Figures 4A–4C). The X insulation profile in DCC mutants reflects this chromosome-wide reduction in interactions (Figure 4A). In contrast, the pattern of interactions on autosomes was unchanged between wild-type and either 8rexΔ or DCC mutant embryos (Figure 4B).
Figure 4. The DCC Promotes Interactions on X at the Scale of 0.1–1 Mb Independently of TAD Formation.

(A and B) Z-score subtraction heatmaps show increased (red) and decreased (blue) interactions for chromosomes X and I in sdc-2(y93, RNAi) versus 8rexΔ embryos. Arrows mark locations of DCC-dependent TAD boundaries, which are in wild-type embryos but not in either mutant. Plots (below) show changes in insulation profiles of 8rexΔ and DCC mutant versus wild-type embryos. Black ticks, DCC-dependent boundaries on X.
(C) Cumulative plots show Z-score differences between DCC mutant and 8rexΔ chromosomes at different length scales. More interactions within 1 Mb occur on X in 8rexΔ versus DCC mutant embryos.
(D) Scaling plot shows the average interactions between loci at increasing distances (10 kb – 20 Mb) on X and autosomes in wild-type embryos.
(E-G) Scaling plots zoom in on the average interactions between loci within 40 kb to 3.2 Mb on X and autosomes in wild-type and mutant embryos.
(H) Cartoons of X chromosome structure in three genotypes. Red rectangles, rex sites; open rectangles, rex deletions. On 8rexΔ chromosomes, the eight DCC-dependent boundaries found on wild-type chromosomes are lost, while other DCC-mediated interactions and DCC-independent boundaries remain. In DCC mutants, the eight DCC-dependent boundaries are lost as are the DCC-mediated interactions at the 0.1–1 Mb length scale. X volume expands in DCC mutant but not in 8rexΔ embryos.
(I) Boxplots show the fraction of total chromatin (measured by DAPI staining) occupied by X (measured by DPY-27 immunofluorescence) for intestinal nuclei of wild-type, 8rexΔ, and dpy-21(null) adults. X compaction does not require DCC-dependent TAD formation. n, number of nuclei
The differences in interactions on X versus autosomes were apparent from plotting the average interaction frequencies between loci at increasing genomic distances. Interaction frequencies scale with genomic distance but have different slopes at different length scales (Figure 4D). This “scaling factor” was shallowest for loci separated by 0.1–1 Mb, and the X chromosome had more interactions than autosomes at these distances, while X and autosomes had matching scaling factors below 0.1 Mb and over 1 Mb (Figure 4E). The 0.1–1 Mb interactions on X were similarly elevated in 8rexΔ but not DCC mutant embryos, indicating these interactions occur independently of TADs but are mediated by the DCC (Figures 4F and 4G). Therefore, in addition to creating TAD boundaries, the DCC confers a unique structure on X by promoting interactions at the scale of 0.1–1 Mb (Figure 4H).
Besides creating TADs and increasing interactions between loci in the 0.1–1 Mb range, the DCC compacts the overall volume of X (Brejc et al., 2017; Lau et al., 2014). To determine whether this compaction relies on TAD formation, we measured the volume of X in intestines of wild-type, 8rexΔ, and dpy-21(null) adults using antibodies to the DCC condensin subunit DPY-27 to mark X. dpy-21(null) mutations do not block assembly of other DCC subunits onto X but do result in Dpy, viable animals with elevated X chromosome expression (Brejc et al., 2017; Yonker and Meyer, 2003). As shown previously (Brejc et al., 2017; Lau et al., 2014), the volume of X expanded in dpy-21(null) versus wild-type animals. In contrast, X volume was unchanged in 8rexΔ adults (Figure 4I). Therefore, the DCC-mediated compaction of X is unrelated to TAD structure but could reflect the increased chromatin interactions at the 0.1–1 Mb scale and could contribute to dosage compensation. The DCC’s dual functions in establishing TAD boundaries and compacting chromatin at the 0.1–1 Mb scale are consistent with a mechanism in which the DCC remodels chromosome topology through loop extrusion (see Discussion).
Loss of DCC-dependent TAD boundaries via rex deletions also enabled us to determine whether TADs affect the positioning of X chromosomes within the nucleus. X chromosomes of XX hermaphrodites localize to the nuclear periphery somewhat more frequently than those of either XO males or XX DCC mutants (Snyder et al., 2016), both of which lack DCC binding on X and DCC-dependent TADs. Using DCC immunofluorescence we assessed X positioning in 8rexΔ and wild-type embryos (Figure S5E). The radial positioning of X within the nucleus was unchanged in 8rexΔ versus wild-type XX embryos, indicating that DCC-dependent TAD boundaries do not control nuclear positioning of X chromosomes.
Destroying DCC-Dependent TADs Reduces Thermotolerance, Shortens Lifespan, and Accelerates Aging
Although disrupting X structure did not cause statistically significant changes in embryonic gene expression under normal growth conditions, aberrant chromosome architecture did adversely affect the ability of adults to tolerate proteotoxic stress induced by a stressful condition. When worms are exposed to proteotoxic stresses, the presence of unfolded proteins triggers activation of genes needed to refold proteins and restore homeostasis (Higuchi-Sanabria et al., 2018). We assessed the ability of 8rexΔ adults to tolerate three forms of proteotoxic stress. We heat stressed worms at 37°C during day 1 of adulthood and measured their survival. After seven hours, survival of 8rexΔ adults was significantly lower than that of wild-type adults (p = 0.0014, paired t test, Figure 5A and Table S4), indicating reduced thermotolerance. We then induced endoplasmic reticulum (ER) unfolded protein stress using tunicamycin, which causes accumulation of unfolded glycoproteins in the ER, and mitochondrial stress using paraquat, which causes accumulation of reactive oxygen species in mitochondria. 8rexΔ adults were not more sensitive than wild-type adults to either ER or mitochondrial stress (Figures 5B and 5C; Table S5). Thus, removing DCC-dependent TADs specifically impairs thermotolerance but does not generally compromise responses to all forms of proteotoxic stress. Adults with disrupted X structure may be uniquely sensitive to heat because heat treatment is a systemic stress that affects the whole cell rather than individual organelles.
Figure 5. 8rexΔ Hermaphrodites Exhibit Reduced Thermotolerance, Shortened Lifespan, and Accelerated Aging.

(A) Percent survival of 50 wild-type or 8rexΔ day 1 adult worms after 7 hr at 37°C in each of ni ne trials shows reduced thermotolerance in 8rexΔ adults. A gray line links the measurements of the two genotypes in the same trial.
(B) Lifespans were scored for wild-type and 8rexΔ hermaphrodites transferred on day 1 of adulthood to plates with either 1% DMSO (control) or 20 ng/μL tunicamycin to induce ER unfolded protein stress. Replicate experiments and statistics are in Table S5.
(C) Percent survival of day 1 adult wild-type and 8rexΔ hermaphrodites in 0.2 M paraquat to induce reactive oxygen species in mitochondria. Wild-type worms subjected to daf-2 RNAi were used as a control for increased oxidative stress tolerance. For each genotype, the average of at least three replicates is plotted. Error bars, standard error of the mean (SEM).
(D) Comparison of lifespans for wild-type versus 8rexΔ (p < 0.0001, logrank test) and 2rexΔ (p < 0.0001) hermaphrodites shows lifespan shortening in mutants. See replicate experiments and statistics in Table S6.
(E) Comparison of lifespans for wild-type versus 8rexΔ males (p = 0.2485, logrank test).
(F) Average unstimulated speed of wild-type and 8rexΔ hermaphrodites during aging. For each genotype, the speed of 50 worms on each of eight replicate plates was measured throughout adulthood. Measurements included only moving worms. We calculated the mean speed of worms on each plate and plotted the mean ± SEM of all eight plates. Asterisks, significant differences (p < 0.05, t test). mm/s, millimeters per second.
(G and H) Maximal speed and reversal distance during aging of wild-type and 8rexΔ hermaphrodites in response to a mechanical stimulus (plate tap). Mean ± SEM are plotted as in F.
To assess whether rex deletions affect other aspects of adult worm performance, we measured the lifespan of 8rexΔ animals. Median lifespan of 8rexΔ hermaphrodites was 19 days, compared to 23 days for wild-type hermaphrodites (Figure 5D; Table S6; see also Figure 5B). This significant lifespan decrease (p < 0.0001, logrank test) is comparable to that caused by disrupting known regulators (xbp-1, ire-1, and daf-16) of stress responses and lifespan (Calo et al., 2018; Henis-Korenblit et al., 2010; Lin et al., 2001; Taylor and Dillin, 2013).
Because the DCC and rex sites function specifically in hermaphrodites, not males, to control X-chromosome architecture and gene expression, males are not affected by mutations that disrupt these dosage compensation components. Therefore, if the decrease in hermaphrodite lifespan is a bona fide consequence of disrupting sex-specific, DCC-dependent functions of rex sites, the lifespan of males should not be affected by rex deletions. Indeed, lifespans of 8rexΔ and wild-type males were not significantly different (median 14 days, p = 0.25), indicating that the lifespan defect of 8rexΔ hermaphrodites is sex-dependent and DCC-dependent (Figure 5E). The reduction in lifespan is not explained by defects in DCC-independent processes caused by either rex deletions or off-target mutations introduced during deletion of rex sites.
To determine whether 8rexΔ worms die prematurely from accelerated aging rather than general “sickness” throughout development, we measured locomotory behaviors in adult animals using a Multi-Worm Tracker (Swierczek et al., 2011). We first asked whether locomotion declines at the same rate in 8rexΔ mutants and in wild-type animals as a function of age. Hermaphrodite 8rexΔ worms moved normally at day 1 of adulthood, but after a peak in speed comparable to that in wild-type animals, their movement subsequently declined more rapidly than did that of wild-type worms. The decline was evident in both the speed of normal explorative movement in the absence of a stimulus and the speed of movement in response to a controlled mechanical tap stimulus (Figures 5F, 5G, S6A, and S6B). A tap elicits a multifaceted escape response: worms increase their speed, reverse the direction of motion, and then turn. By two metrics, speed and distance traveled backward, the escape response declined more rapidly during adulthood in 8rexΔ mutants than in wild-type animals (Figures 5G and 5H).
We also monitored animal size during adulthood. Wild-type animals increase in size for the first six days of adulthood and then shrink. Shorter-lived wild-type individuals are smaller and stop growing sooner (Hulme et al., 2010; Pincus et al., 2011). Although the 8rexΔ mutants were the same size as wild-type animals at adult day 1 and increased in size somewhat until day 6, they never reached the same size as wild-type animals, and they shrank in size faster (Figure S6C).
The more rapid decline in explorative locomotive speed, maximal escape speed, magnitude of escape response, and body size, despite all of these metrics being similar between young adult 8rexΔ and wild-type worms, indicates that 8rexΔ worms are healthy at the onset of adulthood but have a rapid-aging phenotype. These results suggest that although aberrant chromosome structure does not affect DCC-dependent gene expression early in development, it does cause misregulation of the aging program later in life.
The reduced thermotolerance and accelerated aging could be due to changes in expression of a few genes on X with roles in these processes. Alternatively, the phenotypes could result from the cumulative effect of misregulating many genes across X, as in aneuploid cells in which overexpression of genes from an entire chromosome causes proteotoxic stress regardless of the specific aneuploid chromosome (Oromendia and Amon, 2014). To distinguish between these possibilities, we measured lifespans and stress responses of adults with subsets of the eight rex deletions.
The full reduction in lifespan observed in 8rexΔ was seen when only rex-32 and rex-33 were deleted at the leftmost two boundaries (2rexΔ) (Figure 5D; Table S6). Deleting the six other rex sites had no effect on lifespan (Figure S6D; Table S6). Furthermore, the reduction in lifespan was not suppressed by establishing new DCC-dependent boundaries in the center of X by inserting rex sites (Figure S6E; Table S6). Therefore, the aging defect is likely caused by misregulation of specific genes required for normal lifespan rather than the cumulative consequence of many changes in gene expression across the entire chromosome. Deleting rex-32 or rex-33 individually did not recapitulate the lifespan reduction caused by deleting two sites together (Figure S6F), suggesting that misregulation of multiple genes is responsible for lifespan reduction of 8rexΔ animals. The misregulated genes are likely to be in proximity to rex-32 and rex-33, although other genes on X could also be responsible, given that rex deletions can modulate the strength of distant boundaries.
In contrast to the lifespan results, both 2rexΔ and 6rexΔ adults behaved like 8rexΔ adults in having reduced thermotolerance compared to wild-type adults (p = 0.004 and p = 0.002, respectively, paired t test) (Figure S6G and S6H, Table S4). The thermosensitivity of 2rexΔ, 6rexΔ, and 8rexΔ adults was not increasingly more severe with removal of additional rex sites. Rather, the thermosensitivity was not different among the three genotypes (p > 0.6 for each pairwise comparison, Table S4). Thus, thermosensitivity appears to be similarly influenced by misregulation of genes in different regions of X rather than the cumulative misregulation of many genes all across X. Neither 6rexΔ nor 2rexΔ adults were more sensitive to ER or mitochondrial stress compared to 8rexΔ or wild-type adults (Table S5 and Figure S6I and S6J).
Candidates for the misregulated genes that cause reduced heat tolerance and accelerated aging include genes that are induced in response to heat shock and genes that regulate aging. In a recent genome-wide study, of 815 genes upregulated in wild-type worms after heat shock (Brunquell et al., 2016), 149 genes were on X. In addition, genetic screens identified 22 genes on X that either extend worm lifespan or cause accelerated aging when knocked down, including six genes in the TADs flanking rex-32 and rex-33 (Hamilton et al., 2005; Hansen et al., 2005; Samuelson et al., 2007). Although none of these candidate genes on X exhibited significant differential expression in 8rexΔ embryos, their misregulation in 8rexΔ adults upon heat shock or during aging might contribute to the defective heat shock response and accelerated aging. Reduced thermotolerance, shortened lifespan, and accelerated aging resulting from disrupted chromosome architecture are likely caused by the combined effect of misregulating multiple candidate genes.
DISCUSSION
Metazoan chromosomes are organized into megabase-scale topologically associating domains (TADs), but the mechanisms that establish these domains and their link to gene expression remain unclear. We discovered the requirements for establishing hermaphrodite-specific TAD boundaries during C. elegans dosage compensation. Unlike other studies that abolished TAD structure genome-wide by depleting a protein that is essential for boundary formation and also plays other roles in gene expression and development, our study eliminated DCC-dependent TAD boundaries across X by deleting a series of DCC binding sites. We discovered that removing the single rex site at each of eight DCC-dependent boundaries eliminated the boundary. Moreover, inserting a single rex site into a new location on X created a TAD boundary. Thus, DCC binding at high-occupancy rex sites defines locations of TAD boundaries. By disrupting TAD structure on X while leaving most DCC binding intact, we found that TAD boundaries are neither the cause nor consequence of transcriptional repression during dosage compensation. We also discovered a feature of DCC-mediated chromosome structure that is not reliant on DCC-dependent TAD boundaries and potentially enables long-range gene repression: the DCC promotes DNA interactions at the scale of 0.1–1 Mb. Although rex deletions that abrogate TAD structure in hermaphrodites do not affect dosage compensation, they increase thermosensitivity, accelerate aging, and shorten lifespan, implying a role for chromosome architecture in the regulation of stress responses and aging.
Mechanism of TAD Boundary Formation
Our discovery that a single high-occupancy rex site is necessary and sufficient to define the location of a DCC-dependent TAD boundary distinguishes between models for establishing TAD boundaries. Boundary locations are not defined passively by intra-TAD interactions. Furthermore, although strong interactions are often observed between DCC-dependent boundaries in C. elegans and between adjacent boundaries in mammalian cells, we show that boundary formation does not require boundary-to-boundary interactions.
Instead of favoring a model in which specific DCC-mediated interactions promote TAD boundary formation, our results support the loop extrusion model for boundary formation and chromosome compaction. In this model, a complex extrudes a chromatin loop of increasing size until it reaches a barrier element, which blocks the progression of extrusion (Fudenberg et al., 2016). The loops created by extruders loading across an entire chromosome manifest in Hi-C measurements as increased DNA interactions. Because loops do not cross extrusion barriers, the barriers delineate boundaries between TADs. In mammalian cells, evidence points to the SMC complex cohesin acting as a loop extruder and the DNA-bound protein CTCF acting as a barrier to extrusion (Fudenberg et al., 2018; Sanborn et al., 2015). The related SMC complex condensin can extrude DNA loops in vitro, suggesting its role as a loop extruder in vivo (Ganji et al., 2018).
Our results fulfill all predictions for the DCC, a condensin complex, acting as a loop extruder in vivo and the eight high-occupancy rex sites with multiple binding motifs acting as extrusion barriers. First, eliminating the loop extruder should cause TAD boundary loss and an overall reduction in DNA interactions (Fudenberg et al., 2018). Strong interactions between TAD boundaries that can result from loop extrusion should also be lost in the absence of the loop extruder. Indeed, mutations preventing the DCC from binding to X had all these consequences. Second, eliminating a single extrusion barrier should eliminate the boundary. Each rex deletion did abolish the associated boundary. Third, removing all extrusion barriers should eliminate TADs but leave other DCC-mediated interactions intact. Indeed, TAD boundaries were lost in 8rexΔ embryos, but DCC-dependent 0.1–1 Mb interactions on X were retained. Finally, inserting an extrusion barrier at a new location should delineate a new boundary. Insertion of a rex site at three new locations on X did define a new boundary at each site.
Under the loop extrusion model, different DCC components would function to extrude loops and block extrusion, analogous to the roles played by cohesin and CTCF during boundary formation in mammalian cells. DCC condensin subunits would extrude chromatin loops, and non-condensin DCC subunits such as SDC-2 would bind rex sites and halt extrusion (Figures 6A and 6B). SDC-2, the only DCC subunit with hermaphrodite-specific expression, is essential for robust binding of all DCC components to X and can bind rex sites in the absence of other DCC subunits (Albritton et al., 2017; Dawes et al., 1999; Pferdehirt et al., 2011). Thus, SDC-2 could be responsible both for loading the DCC onto X at all rex sites and rex-dependent secondary sites (as NIPBL loads cohesin) and for binding the highest-affinity rex sites in a manner that blocks loop extrusion (as CTCF apparently blocks cohesin extrusion). In principle, SDC-2 could travel with DCC condensin from loading sites on X to the highest affinity rex sites where SDC-2 binds most stably and blocks extrusion. Alternatively, because SDC-2 can bind rex sites independently of condensin, and condensin can bind secondary sites on X at a low level in the absence of SDC-2 (Albritton et al., 2017; Pferdehirt et al., 2011), condensin alone could extrude a loop until encountering SDC-2 bound at a rex site. A combination of both options might occur.
Figure 6. Model for TAD Formation by the DCC.

(A) DCC condensin (blue) loads onto chromatin with SDC proteins (magenta) and extrudes loops of increasing size until the extrusion is halted by binding to a high-occupancy rex site with multiple X-enriched motifs (pink). Because DCC-mediated loops do not cross high-occupancy rex sites, the rex sites define the locations of TAD boundaries. The SDC loading factors could travel with condensin subunits from loading sites on X to the highest-affinity rex sites where they bind stably and block extrusion. Alternatively, condensin alone could bind at low levels to X and extrude loops until encountering SDC proteins bound independently at a rex site. Both possibilities could occur. Only the boundary rex sites are shown, even though rex sites with a range of DCC binding affinities act as loading sites and confer X specificity.
(B) When high-occupancy rex sites are deleted (orange), TAD boundaries are lost, but other DCC-mediated interactions remain, most notably those at the 0.1–1 Mb length scale. The 8rexΔ X maintains the same level of compaction as the wild-type X.
A loop extrusion model for DCC progression along X chromosomes also provides an explanation for how the DCC subunit DPY-21, a histone H4K20me2 demethylase, enriches H4K20me1 across X. DPY-21 could associate with the condensin complex as it extrudes DNA loops, providing access for DPY-21 to demethylate H4K20me2 at all loci across X regardless of proximity to high-occupancy DCC binding sites.
In mammalian cells, pairs of CTCF binding sites that interact strongly tend to contain CTCF binding motifs oriented in a convergent direction (de Wit et al., 2015; Rao et al., 2014; Sanborn et al., 2015). In contrast, high-occupancy C. elegans rex sites within 2 Mb interact strongly regardless of the orientations of their binding motifs (Figure S3A).
TADs and Transcription
Because the DCC can act over long range to repress transcription on X, DCC-dependent TAD organization was an attractive model for mediating the long-range, chromosome-wide dosage compensation process. However, we found that TAD structure itself does not repress or activate transcription in the context of dosage compensation.
In mammalian cells, loss of a TAD boundary can change transcription by allowing enhancers to ectopically activate genes that were previously in separate TADs (Lupiáñez et al., 2015; Valton and Dekker, 2016), but this mode of regulation by TAD boundaries is likely limited to a few specific genes. Studies that eliminated TADs genome-wide by depleting cohesin, the cohesin loader NIPBL, or CTCF revealed that hundreds of genes had modest increases or decreases in expression but found no widespread ectopic activation (Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017). Similarly, we found no broad activation of genes on the C. elegans X chromosome when TADs were eliminated. Mechanisms involving ectopic activation of a gene by an enhancer in an adjacent TAD after boundary disruption may not be relevant in C. elegans. Although putative enhancers have been identified (Chen et al., 2013; Daugherty et al., 2017; Jänes et al., 2018), no regulatory elements are known to activate gene expression at distances greater than 5 kb.
Our study separates the consequences of eliminating TAD boundaries from other defects caused by removing multifunctional architectural proteins. Interpreting the effects of depleting proteins essential for TAD boundary formation is difficult because the proteins also bind sites outside of TAD boundaries and likely alter gene expression through multiple mechanisms. For example, the majority of genes whose expression in mouse embryonic stem cells was reduced by CTCF depletion had CTCF binding near their transcription start sites, suggesting that CTCF may activate gene expression by binding promoters rather than by establishing TADs (Nora et al., 2017). We found that binding of an architectural protein indeed has distinct functions at different genomic locations. DCC binding to rex sites at TAD boundaries is necessary for the creation of boundaries, but DCC binding at other locations is sufficient for transcriptional repression.
TAD-Independent DCC-Mediated Architecture
By comparing X chromosome structure in 8rexΔ versus DCC mutant embryos, we found that the DCC also promotes chromatin interactions at the 0.1–1 Mb length scale (as expected from a loop extrusion model for condensin action). Loss of these interactions in the DCC mutant is consistent with the effect of removing SMC complexes in other contexts. Loss of cohesin loading in mouse cells and loss of condensin in yeast both result in reduced interactions at distances below 200 kb (Paul et al., 2018; Schwarzer et al., 2017).
The DCC-mediated interactions at the hundred-kb scale are a plausible driver of transcriptional repression because they occur chromosome-wide, consistent with the degree of repression not being correlated with a gene’s distance from a rex site or TAD boundary (Crane et al., 2015; Jans et al., 2009; Wheeler et al., 2016) (Figure 3). Perhaps the increased interactions are indicative of the DCC creating an X chromosome compartment with an environment unfavorable for polymerase recruitment. If the DCC-mediated interactions at the hundred-kb scale drive dosage compensation, then repression is achieved not by loops between specific loci but through a dynamic ensemble of loops that varies between individual cells and requires DCC binding across the chromosome, not only at rex sites.
Chromosome Structure, Lifespan, and Stress Responses
Although aberrant TAD structure does not compromise dosage compensation in embryos, it does reduce lifespan and thermotolerance, suggesting that chromosome structure influences gene regulation in adults. The 8rexΔ phenotypes could be a cumulative effect of subtle changes in many genes across X that collectively disrupt proteostasis, making worms less able to tolerate heat stress and aging. However, our finding that the reduced lifespan is due to deletion of the two left-most boundary rex sites, but not the other six boundary rex sites, favors the idea that a few specific genes in proximity to rex-32 and rex-33 are responsible for accelerated aging. Moreover, the observation that adults with only two or six rex deletions are as thermosensitive as 8rexΔ adults suggests that thermosensitivity likewise results from misregulation of specific genes in discrete regions of X. In addition, 8rexΔ worms were not sensitive to other proteotoxic stresses, including mitochondrial stress and ER unfolded protein stress, further suggesting that rex deletions do not generally misregulate gene expression chromosome wide and thereby disrupt proteostasis. Our discoveries demonstrate the importance of using whole organisms to dissect the function of chromosome structure during different stages of development and offer new directions for understanding the regulation of lifespan and the connection between chromosome organization and gene regulation.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact Barbara J. Meyer (bjmeyer@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans Strains
C. elegans strains were grown at 20°C on either NGM plates with HB101 bacteria, NGM plates with OP50 bacteria, or RNAi plates (NGM agar with 1 mM IPTG and 100 μg/mL carbenicillin) with HT115 bacteria carrying an RNAi plasmid (sdc-2 or pL4440 negative control). Bacteria were grown as in (Crane et al., 2015). To collect embryos, starved L1s were plated on bacteria and grown at 20°C for three days. The gravid adults were then bleached to isolate mixed-stage embryos. When sdc-2(y93) worms (Nusbaum and Meyer, 1989) were grown on RNAi bacteria from the L1 stage, their progeny died as embryos or L1s.
Experiments measuring thermotolerance, sensitivity to mitochondrial stress, or lifespan required the use of staged worms. To stage worms, gravid adults were bleached to release embryos using a solution of 1.8% sodium hypochlorite and 0.375 M KOH. Progeny were grown for 12–16 hr without food in M9 buffer (22 mM KH2PO4 monobasic, 42.3 mM Na2HPO4, 85.6 mM NaCl, 1mM MgSO4) until they developed into arrested L1 larvae.
METHOD DETAILS
rex Deletions and Insertions
rex-32 and rex-47 deletions were previously published (Crane et al., 2015; Lo et al., 2013). All other rex deletions and insertions were made using CRISPR/Cas9. To generate the 6rexΔ strain, the right six rex sites at DCC-dependent boundaries were deleted sequentially, and the resulting strain was outcrossed four times using TY0456 dpy-6(e14) unc-3(e151) X. To generate the rex-32Δ rex-33Δ strain, rex-33 was deleted in TY5648, which carried a rex-32 deletion, and the strain was outcrossed three times using lon-2(e678) X. To build the 8rexΔ strain, 6rexΔ was crossed to the rex-32Δ rex-33Δ strain and recombinant F2s with all eight rex deletions were selected. The rex-32Δ and rex-33Δ strains (TY5910 and TY5911, respectively) used for lifespan experiments were created by crossing rex-32Δ rex-33Δ (TY5826) to wild-type males and selecting recombinant F2s with single deletions. All other strains used for Hi-C experiments were outcrossed at least two times.
For the rex-14, rex-8, rex-43, rex-48, and rex-35 deletions, worms were injected with plasmids encoding Cas9 and the sgRNA along with either a plasmid or oligo repair template as in (Farboud and Meyer, 2015). Repair templates consisted of a 6 bp NcoI restriction site flanked by homology arms (500–1000 bp for plasmids or at least 50 bp for oligos). Each deletion replaced the rex site with a 6 bp NcoI restriction site to facilitate screening for deletions. The rex-33 deletion was made using a Cas9 RNP along with an oligo repair template (Farboud et al., 2019), and all rex site insertions were made as in (Farboud et al., 2019) using the Cas9 RNP, gBlocks (IDT) with ~200 bp homology arms as repair templates, and either dpy-10 or rol-6 co-conversion markers. The locations and sizes of insertions and deletions and guide RNA sequences are listed in Table S1.
Hi-C and Analysis
Wild-type and rex site deletion and insertion worms were grown on NGM plates with HB101 bacteria, and sdc-2(y93) worms were grown on RNAi plates with HT115 bacteria carrying an sdc-2 (RNAi) plasmid. In situ Hi-C was performed on nuclei isolated from mixed stage embryos and data were processed as in (Brejc et al., 2017). Hi-C was performed for two biological replicates of wild type, TY5815 rex-47Δ, TY5824 6rexΔ, TY5827 8rexΔ, and TY0810 DCC mutant and a single replicate for TY5818 3rexΔ, TY5868 8rexΔ plus rex-32, TY5872 8rexΔ plus rex-32 & rex-8, TY5854 rex-32 insert, TY5859 3rexΔ plus rex-47 on I, TY5860 3rexΔ plus rex-47 & rex-8 on I, and TY5867 3rexΔ plus rex-14, rex-47 & rex-8 on I. Z-score subtraction heatmaps and insulation plots were generated by comparing single Hi-C replicates of each genotype. Patterns of boundary loss and gain were completely reproducible between replicates. For strains with two replicates, interaction frequencies after iterative correction from the two replicates were combined. Boundary pileups in Figure 2I, 3D plots, and scaling plots were generated using the combined replicates. Average interaction frequencies at increasing distances in Figures 4D–4G were calculated using fragment-level (unbinned) contacts using the hiclib library for Python (https://bitbucket.org/mirnylab/hiclib) (Imakaev et al., 2012).
To assess how removing a boundary affects the strength of adjacent boundaries (Figure 2I), data from rex-47Δ, 3rexΔ, 6rexΔ, and 8rexΔ X chromosomes were used to calculate the average insulation profile around all boundaries adjacent to a deleted boundary and the average insulation profile at the same boundaries in the strain with the next fewer deletions. For example, the boundary upstream of rex-14 in 3rexΔ was compared to the same boundary in rex-47Δ. Two replicates for each of 12 boundaries were included for 24 total comparisons. Similar analysis was performed for the seven boundaries that are two boundaries away from deleted boundaries for 14 total comparisons. The insulation scores at the boundaries were used to calculate a p value using a paired t test.
ChIP-seq
Wild-type, 8rexΔ, TY5872 8rexΔ plus rex-32 & rex-8, and TY5867 3rexΔ plus rex-14, rex-8 & rex-47 on I strains were grown on RNAi plates with HT115 bacteria carrying the empty plasmid vector pL4440. Approximately 0.5 g of mixed-stage embryos were isolated as above and frozen in liquid nitrogen. Embryos were washed once in 2% formaldehyde in M9 buffer and then fixed for 30 min with gentle rocking in 50 mL 2% formaldehyde in M9 buffer. Embryos were washed in 10mM Tris-HCl (pH 7.5) and then in FA buffer (150 mM NaCl, 50 mM HEPES-KOH pH 7.6, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) and resuspended in FA buffer with protease inhibitors (5 mM DTT, protease inhibitor cocktail, 1 mM PMSF) to a total volume of 1 mL. Embryos were ground with 50 strokes in a 2 mL dounce homogenizer. Sarkosyl was added to a final concentration of 0.1% and chromatin was sheared in an S2 Covaris with duty cycle 20%, intensity 8, and 200 cycles/burst for 30 cycles of 60 sec with 45 sec of rest for a total time of 52 min. The extract was centrifuged at maximum speed for 15 min at 4°C. Supernatant containing approximately 2 mg total protein was incubated with 6.6 g rabbit polyclonal anti-DPY-27 (rb699) (Chuang et al., 1994), rabbit polyclonal anti-SDC-3 antibody (Rb3779) directed to amino acids 1,068–1,342, or random IgG antibodies overnight at 4°C in a volume of at least 500 μl. 50 μl of Protein A Dynabeads (ThermoFisher Scientific, 10002D) were washed in FA buffer three times, added to the immunoprecipitation, and mixed at 4°C for at least 2 hr. Beads were then washed, DNA eluted, and libraries prepared as in (Kruesi et al., 2013). Libraries were sequenced with 50 bp single-end reads on an Illumina HiSeq4000 platform.
ChIP-seq Analysis
Adapters were trimmed using cutadapt version 1.2.1 (Martin, 2011), and reads were then aligned to the ce11 genome using bowtie2 version 2.3.0 (Langmead and Salzberg, 2012) with default settings. For comparisons involving strains with rex site insertions, the reference genome was modified to incorporate the rex insertions. Reads were sorted using SAMtools version 1.3.1 (Li et al., 2009) and read coverage was calculated by normalizing the read number in each 50 bp bin to the total read number using the bamCoverage function in deepTools version 2.5.0.1 (Ramírez et al., 2016) with the “normalizeUsingRPKM” option. Analysis of data from wild-type and 8rexΔ strains was performed with two combined biological replicates, and one replicate was used for data from TY5872 8rexΔ plus rex-32 & rex-8 and TY5867 3rexΔ plus rex-14, rex-8 & rex-47 strains.
To calculate the number of reads mapping to each rex site, SDC-3 peaks were first identified using macs2 version 2.1.1 (Zhang et al., 2008) with IgG as the control and the mfold parameter set to 3 50. The total SDC-3 reads in a 400 bp window surrounding each peak were summed and the IgG reads in the same window were subtracted. The peaks were then ranked based on this read number and the strongest 30 peaks (which were all on chromosome X) were used for analyses (rex-48, rex-8, rex-23, rex-14, rex-32, rex-33, rex-43, rex-35, rex-16, rex-40, Prex-30, rex-45, rex-47, rex-34, Prex-7, rex-41, Prex-22, rex-6, rex-2, Prex-31, rex-24, rex-44, rex-36, Prex-6, rex-1, rex-42, Prex-1, rex-46, rex-39).
To quantify DCC binding at inserted rex sites, reads were mapped to reference genomes modified to include the insertions (and deletions of the endogenous sites). Reads were summed in a 400 bp window around the peak of the inserted rex site as well as around four strong rex sites on X (rex-40, rex-23, rex-16, and Prex-30). Binding was normalized by dividing binding at the inserted rex site by the average binding at the other four rex sites.
The bigwigCompare function in deepTools version 2.5.0.1 (Ramírez et al., 2016) was used to calculate the log2 ratio of DCC binding between genotypes, and the median ratio in a 1 kb sliding window was plotted. Ratios were not calculated for the basepairs in the deletions or insertions.
ChIP-qPCR
ChIP-qPCR was used to quantify DCC binding at rex-32 inserted in the wild-type background. The rex-32 insert strain (TY5854) was grown on MYOB plates with HB101 bacteria. ChIP using anti-SDC-3 antibody was performed on three biological replicates of mixed-stage TY5854 embryos as described above. After elution, DNA was diluted 4-fold and qPCR was performed using SYBR green (BioRad iQ SYBR Green Supermix 170–8886) on a BioRad CFX384 Real-Time System. The oligos used to measure binding 50 bp from the rex-32 insertion, at the endogenous rex-32 site, and at other strong rex sites for normalization (rex-8, rex-16, rex-48, and rex-35) are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-DPY-27 | Chuang et al., 1994 | Rb699 |
| Rabbit polyclonal anti-SDC-3 | This paper | Rb3779 |
| AffiniPure Rabbit Anti-Rat IgG | Jackson ImmunoResearch Laboratories | Cat#312–005-003; AB_2339800 |
| Alexa Fluor-488 AffiniPure IgG | Jackson ImmunoResearch Laboratories | Cat#111–545-003; AB_2338046 |
| Bacterial and Virus Strains | ||
| E. coli OP50 | Caenorhabditis Genetics Center | OP50–1 |
| E. coli HT115 (DE3) | Caenorhabditis Genetics Center | HT115(DE3) |
| E. coli HB101 | Caenorhabditis Genetics Center | HB101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cas9-NLS purified protein | UC Berkeley QB3 MacroLab | N/A |
| Protein A Dynabeads | ThermoFisher Scientific | Cat#10002D |
| iQ SYBR Green Supermix | BioRad | Cat#170–8886 |
| Cell-Tak | Corning | Cat#354240 |
| Tunicamycin | Sigma-Aldrich | Cat#T7765; CAS: 11089–65-9 |
| IPTG-Dioxane free | Denville Scientific | Cat#CI8280–4; CAS: 367–93-1 |
| Paraquat Dichloride Hydrate | Millipore Sigma | Cat#36541;CAS: 75365–73-0 |
| Deposited Data | ||
| Hi-C, ChIP-seq, RNA-seq datasets | This paper | GEO:GSE128568 |
| Experimental Models: Organisms/Strains | ||
| C. elegans N2: Wild-type strain from Bristol | Caenorhabditis Genetics Center | Wild Type (WT) |
| C. elegans TY0810 grown on sdc-2 RNAi bacteria: sdc-2(y93, RNAi) X | Nusbaum and Meyer, 1989 | DCC mutant |
| C. elegans TY3936: dpy-21(e428) V | Yonker and Meyer, 2003 | dpy-21 (null) |
| C. elegans TY5648: rex-32(y572) X | Lo et al., 2013 | rex-32Δ |
| C. elegans TY5815: rex-47(y671) X | Crane et al., 2015 | rex-47Δ |
| C. elegans TY5826: rex-32(y572) rex-33 (y743) X | This paper | 2rexΔ |
| C. elegans TY5818: rex-14(y738) rex-47(y671) rex-8(y737) X | This paper | 3rexΔ |
| C. elegans TY5824: rex-14(y738) rex-47(y671) rex-8(y737) rex-43(y741) rex-48(y742) rex35(y740) X | This paper | 6rexΔ |
| C. elegans TY5827: rex-32(y572) rex-33(y743) rex-14(y738) rex-47(y671) rex-8(y737) rex-43(y741) rex-48(y742) rex-35(y740) X | This paper | 8rexΔ |
| C. elegans TY5910: rex-32(y572) X | This paper | rex-32Δ |
| C. elegans TY5911: rex-33(y743) X | This paper | rex-33Δ |
| C. elegans TY5755: xol-1(y684) X | This paper | xol-1Δ |
| C. elegans TY5828: rex-32(y572) rex-33(y743) rex-14(y738) xol-1(y744) rex-47(y671) rex-8(y737) rex-43(y741) rex-48(y742) rex-35(y740) X | This paper | 8rexΔ xol-1Δ |
| C. elegans TY5868: rex-32(y572) rex-33(y743) yIs197(rex-32) rex-14(y738) rex-47(y671) rex-8(y737) rex-43(y741) rex-48(y742) rex-35(y740) X | This paper | 8rexΔ plus rex-32 |
| C. elegans TY5872: rex-32(y572) rex-33(y743) yIs197(rex-32) rex-14(y738) yIs201(rex-8) rex-47(y671) rex-8(y737) rex-43(y741) rex-48(y742) rex-35(y740) X | This paper | 8rexΔ plus rex-32 & rex-8 |
| C. elegans TY5854: dpy-27(y679 FLAG) III; yIs187(rex-32) X | This paper | rex-32 insert |
| C. elegans TY5859: yIs190(rex-47) I; rex-14(y738) rex-47(y671) rex-8(y737) X | This paper | 3rexΔ plus rex-47 on Chr I |
| C. elegans TY5860: yIs190(rex-47) yIs192(rex-8) I; rex-14(y738) rex-47(y671) rex-8(y737) X | This paper | 3rexΔ plus rex-47 & rex-8 on Chr I |
| C. elegans TY5867: yIs196(rex-14) yIs190(rex-47) yIs192(rex-8) I; rex-14(y738) rex-47(y671) rex-8(y737) X | This paper | 3rexΔ plus rex-14, rex-47 & rex-8 on Chr I |
| Oligonucleotides | ||
| CAGCGTAGTTGCTGACACTTAATGGTTC | This paper | chrX 15574551F |
| CTTTTAAGCAGTCGTCATGTACGTGTTCG | This paper | chrX15574627R |
| CACTCCCCAGCTAATTTGGA | This paper | rex-32F |
| TTCCCTTGTTGCGGAGATAG | This paper | rex-32R |
| TTTATCCACCAACATGCATAAG | This paper | rex-8 F |
| CAGTGGATAACTACACAAGGG | This paper | rex-8 R |
| GTACAAACGCAGGGAAGAGA | This paper | rex-16F |
| GACGCTACCACACCTTCAATA | This paper | rex-16R |
| CTGCGCGATAGGCAATAGT | This paper | rex-48F |
| GCACAATTCCAAGTCATCCATAC | This paper | rex-48R |
| CCATATGTTGCCCAATGTTCC | This paper | rex-35F |
| CGCAGGGAACATCAAATTAGTC | This paper | rex-35R |
| Recombinant DNA | ||
| RNAi plasmid for C. elegans sdc-2 | Source BioScience | N/A |
| RNAi plasmid for C. elegans daf-2 | Source BioScience | N/A |
| L4440 control plasmid for RNAi experiments | Source BioScience | RRID:Addgene_1654 |
| Software and Algorithms | ||
| hiclib | Imakaev et al., 2012 | https://bitbucket.org/mirnylab/hiclib/ |
| cworld | Job Dekker Lab | https://github.com/dekkerlab/cworld-dekker |
| cutadapt version 1.2.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| SAMtools version 1.3.1 | Li et al., 2009 | http://www.htslib.org/ |
| deepTools version 2.5.0.1 | Ramfrez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| ImageJ version 1.52J | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| TopHat version 2.1.1 | Trapnell et al., 2012 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks version 2.2.1 | Trapnell et al., 2012 | https://github.com/cole-trapnell-lab/cufflinks |
| cummeRbund version 2.20.0 | Trapnell et al., 2012 | http://compbio.mit.edu/cummeRbund/ |
| Prism7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Choreography | Swierczek et al., 2011 | https://github.com/lchoran/choreography |
Standard curves were generated using input DNA, and binding at each rex site was calculated as a percentage of input. Binding at the inserted rex-32 in Figure S4B was normalized to the average binding at five strong rex sites (rex-32, rex-8, rex-16, rex-48, and rex-35).
Measuring X Chromosome Volume
Intestines of adults (2 days post-L4) grown on OP50 bacteria were stained with anti-DPY-27 antibody (rb699) (Chuang et al., 1994) and DAPI, confocal images were scanned and deconvolved, and X chromosome volume was calculated as in (Brejc et al., 2017).
Measurements of X Chromosome Nuclear Position
To measure the position of X chromosomes within the nucleus, X chromosomes were visualized in wild-type and 8rexΔ embryos using indirect immunofluorescence for the DCC component DPY-27. The proportion of the chromosome located in each of three concentric zones of equal area was calculated as in (Snyder et al., 2016). Embryos at the ~120–150-cell stage were stained as in (Brejc et al., 2017) using rabbit polyclonal anti-DPY-27 (rb699) (Chuang et al., 1994) and Alexa Fluor-488 goat anti-rabbit antibodies (Jackson Immunoresearch Laboratories, Inc). Images were acquired and deconvolved as in (Brejc et al., 2017) with 50 nm XY pixel size and 80 nm Z step size. For each embryo, 0.4 μm Z projections were made by summing five Z slices. For each nucleus, the projection with the highest total DPY-27 signal was selected, and a circular or oval region of interest was drawn by hand around the nucleus in ImageJ. The average DPY-27 signal in a cytoplasmic region was calculated, and this background value was subtracted from the nuclear DPY-27 signal. The nucleus was divided into three concentric ovals of equal areas. The total DPY-27 signal (after background subtraction) in each zone was calculated using ImageJ. The average percentage of X signal located in each of the three zones and the standard error of the mean were plotted for 41 wild-type nuclei from four embryos and 45 8rexΔ nuclei from four embryos.
RNA-seq Library Preparation and Analysis
For each RNA-seq replicate, worms were grown, embryos collected, and RNA-seq performed for all genotypes in parallel. Worms were grown on RNAi plates with HT115 bacteria with either sdc-2 RNAi (for DCC mutant) or pL4440 empty vector (for wild type and 8rexΔ). For some replicates, mixed-stage embryos (majority pre-comma stage) were isolated by bleaching gravid hermaphrodites as above. Other replicates were skewed towards older embryos by bleaching young adults (68–70 hr after plating starved L1s) to isolate young embryos, letting the embryos develop at 20°C for 7–8 hr until the majority were at the three-fold stage, and then freezing. RNA was isolated and RNA-seq libraries were prepared as in (Brejc et al., 2017), and libraries were sequenced with 50 bp reads on an Illumina HiSeq4000 platform. Reads were processed by trimming adapters using cutadapt version 1.2.1, aligning to the ce11 reference genome using TopHat version 2.1.1, and assembling using Cufflinks version 2.2.1 (Trapnell et al., 2012).
The wild-type and 8rexΔ replicates that were most closely stage-matched were computationally identified. A published dataset that measured gene expression in single embryos through embryogenesis (whole embryo time course from (Hashimshony et al., 2015)) was used to select autosomal genes that had at least 1 transcript per million at some point in the time course and had at least a 20-fold change from their lowest to highest expression level. The expression levels for each of these 12,750 genes were scaled by subtracting the gene’s mean expression and dividing by its standard deviation. The genes were k-means clustered into five clusters with similar temporal expression patterns using the kmeans function in R (Figure S5D). For each paired replicate of wild-type and 8rexΔ RNA-seq data, expression of genes in each of the five clusters was compared between genotypes, and the five replicates in which the two genotypes had similar expression levels for genes in each cluster were selected. Two replicates of mixed-stage embryos and three replicates of late embryos were included, and other replicates in which the wild-type embryos clearly had higher expression of late genes and lower expression of early genes compared to 8rexΔ were excluded. For the sdc-2(y93, RNAi) to wild-type comparison, two replicates of mixed-stage embryos and one replicate of older embryos were used.
Replicates were combined using Cuffmerge. Expression levels (fpkm) and fold changes were analyzed using cummeRbund version 2.20.0. The p values comparing X to autosomal expression were calculated for genes with greater than 1 FPKM using a two-sided Wilcoxon rank-sum test.
xol-1 Rescue
A 3,035 bp deletion of the xol-1 open reading frame (X:8041318–8043793) was made in the rex deletion background using Cas9 with two sgRNAs (GCTTCAACCTGCATTTCTGG and TGATTGATATGGGAAATGG). Ten 8rexΔ xol-1Δ hermaphrodites were crossed to wild-type males, and all cross progeny were either healthy hermaphrodites or inviable embryos, the phenotype of xol-1 mutant XO animals. No viable male F1s were observed in the ten broods (approximately 3000 progeny), indicating that 8rexΔ does not rescue xol-1Δ males.
Imaging the Timing of Embryogenesis
To measure the timing of embryogenesis in wild-type and 8rexΔ strains, embryos of both genotypes were imaged simultaneously. 3.5 μL of a 1:10 dilution of Cell-Tak (Corning, catalog # 354240) and 1.8 μL of 1 N NaOH were combined, and 3 μL were pipetted onto a coverslip. After the coverslips dried, 2 μL of poly-L-lysine solution were added and allowed to dry, and coverslips were rinsed with ethanol and then water. Two-cell embryos of the first genotype in M9 buffer were transferred onto the Cell-Tak spot on the coverslip and aligned using an eyebrow hair. After the embryos settled, the coverslip was rinsed with M9 and imaged to document the positions of embryos. Two-cell embryos of the second genotype were then placed on a 2% agarose pad. The coverslip having embryos of the first genotype was then placed on the agarose pad having embryos of the second genotype and sealed with rubber cement. Each embryo was imaged every 3 min for 13 hr using the Mark and Find function on a Leica SP8 WLL confocal microscope with a 20x objective in a room that was 22–23°C. The image of the coverslip with the embryos of the first genotype was used to identify the genotypes of all embryos. The times when each embryo reached comma stage, completed two-fold stage, and hatched were determined. A total of 19 embryos of each genotype were imaged over three separate days.
Thermotolerance
For each genotype, starved L1 larvae were grown to day 1 of adulthood on RNAi plates with HT115 bacteria. Ten day 1 adults were then placed on each of five RNAi plates with HT115 bacteria. The plates were incubated at 37°C, and the number of living and dead worms were counted after 5, 7, and 9 hr. Any worms that crawled onto the sides of the plates were censored. Generally, at 5 hr, most worms were still alive, and by 9 hr, nearly all were dead. The percentage of worms alive at 7 hr was reported for eleven replicates of wild-type and 8rexΔ animals performed on separate days and six replicates of 2rexΔ and 6rexΔ animals. In four additional replicates, the percentage alive was reported at 5, 6, or 9.5 hours to accommodate slight variability in timing of demise due to heat stress (Table S4). P values were calculated using a paired two-tailed t test.
Sensitivity to Mitochondrial Stress
Starved L1 wild-type, 8rexΔ, 2rexΔ, and 6rexΔ worms were grown to day 1 of adulthood at 20°C (three days) on RNAi plates with HT115 bacteria carrying an empty vector (pL4440). Wild-type worms were also grown on HT115 bacteria carrying daf-2(RNAi) plasmid as a positive control for increased resistance to oxidative stress. For each condition, five adult day 1 hermaphrodites were placed in each of 12 wells containing 50 μL of freshly prepared 0.2 M paraquat dichloride hydrate (Millipore Sigma 36541) in S-basal media (0.1 M NaCl, 0.05 M potassium phosphate pH 6, 5 μg/mL cholesterol), for a total of 60 worms. Worms were incubated at 20°C in a dark box. Every 2 hr, the number of surviving worms was scored by observing each worm for movement (thrashing, pharyngeal pumping, etc.) for 15–20 sec. Results were averaged from three replicates for 8rexΔ, 2rexΔ, and 6rexΔ, four replicates for daf-2(RNAi), and five replicates for wild type. Error bars show standard error of the mean.
Lifespan Analysis
For lifespan experiments, starved L1s were grown at 20°C on RNAi plates with HT115 bacteria carrying empty vector (pL4440). Starting at day 1 of adulthood until the animals stopped laying embryos (day 7–9), the worms were transferred daily to fresh plates away from their progeny. The number of living and dead worms were then counted every two days. Worms that crawled off the plate or exhibited an exploded gonad or internal hatching of progeny were censored from the experiment. For all lifespan studies, worms were grown at 20°C in a dark box, an d the experimenter was blind to the animals’ genotypes. Statistical analysis was performed using Prism 6 software.
For male lifespan analysis, each day 1 adult male was placed on an individual plate and scored every two days. The majority of males crawled off the plate before dying and were censored.
Hermaphrodite sensitivity to ER stress was assayed by measuring survival in the presence of tunicamycin. Wild-type, 8rexΔ, 6rexΔ, and 2rexΔ starved L1 larvae were grown to day 1 of adulthood on RNAi plates with HT115 bacteria carrying empty vector plasmid pL4440. Day 1 adults were moved to RNAi plates with either 1% DMSO (control) or 20 ng/μL tunicamycin, and their lifespans were assayed.
Worm Tracking
Worm movement was measured using a Multi-Worm Tracker as in (Podshivalova et al., 2017). Briefly, animals were grown on OP50 bacteria, and 50 μM 5-fluoro-2’-deoxyuridine (FUDR) was added to the plates when animals reached the late L4 stage to prevent progeny from developing. Tracking was performed daily or every other day for 1260 sec with a tap stimulus delivered after 900 sec. Explorative locomotion was measured over a 50 sec time window after allowing animals to recover from handling for 650 sec. Stimulated movement was measured over a 1 sec time window following the delivery of a single tap stimulus using a solenoid plate tapper. Animals that moved by less than half a body length within a 1 min time interval were not detected. After detection, tracking continued even if the animal was still. For each genotype, eight plates each with 50 worms were tracked on eight parallel trackers. Animals that had exploded or would otherwise be censored during analysis of lifespan were removed from plates prior to tracking. Data analysis was performed using Choreography software (Swierczek et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantification and statistical analyses are explained in the relevant sections of the STAR Methods. Statistics are presented in the text and figure legends.
DATA AND SOFTWARE AVAILABILITY
Hi-C, ChIP-seq, and RNA-seq raw and processed data are available at GEO, accession number GSE128568.
Supplementary Material
Highlights.
A DCC recruitment site (rex) is necessary and sufficient to form a TAD boundary on X
Destroying X TADs by deleting only 8 rex sites does not disrupt dosage compensation
DCC establishes other X structures and promotes X compaction independently of TADs
Destroying X TADs reduces thermotolerance, accelerates aging, and shortens lifespan
ACKNOWLEDGEMENTS
We thank D. Stalford for assistance with figures, S. Uzawa for measurements of X chromosome position, A. Uebersohn for viability measurements, A. Goloborodko for advice on Hi-C analysis, and T. Cline, J. Dekker, G. Fudenberg, G. Karpen, E. Nora, J. Rine, and the Meyer lab for discussions. This work used the Vincent J. Coates Genomics Sequencing Laboratory at U.C. Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. R.H.S was supported by NIH grant 5F32AG053023 and a Glenn Foundation for Aging Postdoctoral Fellowship. K.P. and C.K. were supported by Calico Life Sciences. B.J.M. was supported in part by NIH grants R01 GM030702 and R35 GM131845. B.J.M. and A.D. are investigators of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albritton SE, Kranz A-L, Winterkorn LH, Street LA, and Ercan S (2017). Cooperation between a hierarchical set of recruitment sites targets the X chromosome for dosage compensation. ELife 6, e23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Q, Anderson EC, Brejc K, and Meyer BJ (2018). Dynamic Control of Chromosome Topology and Gene Expression by a Chromatin Modification. Cold Spring Harb Symp Quant Biol 82, 034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, Bian Q, Uzawa S, Wheeler BS, Anderson EC, King DS, Kranzusch PJ, Preston CG, and Meyer BJ (2017). Dynamic Control of X Chromosome Conformation and Repression by a Histone H4K20 Demethylase. Cell 171, 85–102.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunquell J, Morris S, Lu Y, Cheng F, and Westerheide SD (2016). The genome-wide role of HSF-1 in the regulation of gene expression in Caenorhabditis elegans. BMC Genomics 17, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, Flynn RA, Swigut T, Chang HY, Attardi LD, et al. (2018). Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RA-J, Down TA, Stempor P, Chen QB, Egelhofer TA, Hillier LW, Jeffers TE, and Ahringer J (2013). The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res. 23, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang P-T, Albertson DG, and Meyer BJ (1994). DPY-27: A chromosome condensation protein homolog that regulates C. elegans dosage compensation through association with the X chromosome. Cell 79, 459–474. [DOI] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, McDonel P, and Meyer BJ (2004). Recruitment and Spreading of the C. elegans Dosage Compensation Complex Along X Chromosomes. Science 303, 1182–1185. [DOI] [PubMed] [Google Scholar]
- Daugherty AC, Yeo RW, Buenrostro JD, Greenleaf WJ, Kundaje A, and Brunet A (2017). Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 27, 2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes HE, Berlin DS, Lapidus DM, Nusbaum C, Davis TL, and Meyer BJ (1999). Dosage Compensation Proteins Targeted to X Chromosomes by a Determinant of Hermaphrodite Fate. Science 284, 1800–1804. [DOI] [PubMed] [Google Scholar]
- de Wit E, Vos ESM, Holwerda SJB, Valdes-Quezada C, Verstegen MJAM, Teunissen H, Splinter E, Wijchers PJ, Krijger PHL, and de Laat W (2015). CTCF Binding Polarity Determines Chromatin Looping. Molecular Cell 60, 676–684. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, and Lieb JD (2007). X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nature Genetics 39, 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B, and Meyer BJ (2015). Dramatic Enhancement of Genome Editing by CRISPR/Cas9 Through Improved Guide RNA Design. Genetics 199, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B, Severson AF, and Meyer BJ (2019). Strategies for Efficient Genome Editing Using CRISPR-Cas9. Genetics 211, 431–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong Y, Bender L, Wang W, and Strome S (2002). Regulation of the Different Chromatin States of Autosomes and X Chromosomes in the Germ Line of C. elegans. Science 296, 2235–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan W-L, et al. (2016). Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269. [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, and Mirny LA (2018). Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb Symp Quant Biol 82, 034710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, and Dekker C (2018). Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, and Dekker J (2013). The Hierarchy of the 3D Genome. Molecular Cell 49, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, et al. (2018). A pathway for mitotic chromosome formation. Science 359, eaao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, and Lee SS (2005). A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Hsu A-L, Dillin A, and Kenyon C (2005). New Genes Tied to Endocrine, Metabolic, and Dietary Regulation of Lifespan from a Caenorhabditis elegans Genomic RNAi Screen. PLOS Genetics 1, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Feder M, Levin M, Hall BK, and Yanai I (2015). Spatiotemporal transcriptomics reveals the evolutionary history of the endoderm germ layer. Nature 519, 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee S-J, Cary M, and Kenyon C (2010). Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. PNAS 107, 9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, Frankino PA, Paul JW, Tronnes SU, and Dillin A (2018). A Futile Battle? Protein Quality Control and the Stress of Aging. Developmental Cell 44, 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T (2016). Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 164, 847–857. [DOI] [PubMed] [Google Scholar]
- Hocquet C, Robellet X, Modolo L, Sun X-M, Burny C, Cuylen-Haering S, Toselli E, Clauder-Münster S, Steinmetz L, Haering CH, et al. (2018). Condensin controls cellular RNA levels through the accurate segregation of chromosomes instead of directly regulating transcription. ELife 7, e38517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SE, Shevkoplyas SS, McGuigan AP, Apfeld J, Fontana W, and Whitesides GM (2010). Lifespan-on-a-chip: microfluidic chambers for performing lifelong observation of C. elegans. Lab Chip 10, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Meth 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänes J, Dong Y, Schoof M, Serizay J, Appert A, Cerrato C, Woodbury C, Chen R, Gemma C, Huang N, et al. (2018). Chromatin accessibility dynamics across C. elegans development and ageing. ELife 7, e37344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt RR, Eisen MB, and Meyer BJ (2009). A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev. 23, 602–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WG, Schaner CE, Dernburg AF, Lee M-H, Kim SK, Villeneuve AM, and Reinke V (2002). X-chromosome silencing in the germline of C. elegans. Development 129, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, and Meyer BJ (2013). Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. ELife 2, e00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AC, Nabeshima K, and Csankovszki G (2014). The C. elegans dosage compensation complex mediates interphase X chromosome compaction. Epigenetics & Chromatin 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TB, and Laub MT (2016). Transcription rate and transcript length drive formation of chromosomal interaction domain boundaries. The EMBO Journal 35, 1582–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, and Kenyon C (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nature Genetics 28, 139–145. [DOI] [PubMed] [Google Scholar]
- Lo T-W, Pickle CS, Lin S, Ralston EJ, Gurling M, Schartner CM, Bian Q, Doudna JA, and Meyer BJ (2013). Precise and Heritable Genome Editing in Evolutionarily Diverse Nematodes Using TALENs and CRISPR/Cas9 to Engineer Insertions and Deletions. Genetics 195, 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal 17, 10–12. [Google Scholar]
- McDonel P, Jans J, Peterson BK, and Meyer BJ (2006). Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature 444, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ (2018). Sex and death: from cell fate specification to dynamic control of X-chromosome structure and gene expression. MBoC 29, 2616–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LM, Plenefisch JD, Casson LP, and Meyer BJ (1988). xol-1: A gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell 55, 167–183. [DOI] [PubMed] [Google Scholar]
- Morales C, and Losada A (2018). Establishing and dissolving cohesion during the vertebrate cell cycle. Current Opinion in Cell Biology 52, 51–57. [DOI] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum C, and Meyer BJ (1989). The Caenorhabditis elegans gene sdc-2 controls sex determination and dosage compensation in XX animals. Genetics 122, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, and Amon A (2014). Aneuploidy: implications for protein homeostasis and disease. Disease Models & Mechanisms 7, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MR, Markowitz TE, Hochwagen A, and Ercan S (2018). Condensin Depletion Causes Genome Decompaction Without Altering the Level of Global Gene Expression in Saccharomyces cerevisiae. Genetics 210, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pferdehirt RR, Kruesi WS, and Meyer BJ (2011). An MLL/COMPASS subunit functions in the C. elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes Dev. 25, 499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus Z, Smith-Vikos T, and Slack FJ (2011). MicroRNA Predictors of Longevity in Caenorhabditis elegans. PLOS Genetics 7, e1002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podshivalova K, Kerr RA, and Kenyon C (2017). How a Mutation that Slows Aging Can Also Disproportionately Extend End-of-Life Decrepitude. Cell Reports 19, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Hilaire BGS, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind NR, Miller LM, Kopczynski JB, and Meyer BJ (1995). xo1–1 acts as an early switch in the C. elegans male/hermaphrodite decision. Cell 80, 71–82. [DOI] [PubMed] [Google Scholar]
- Samuelson AV, Carr CE, and Ruvkun G (2007). Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 21, 2976–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. PNAS 112, E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizay J, and Ahringer J (2018). Genome organization at different scales: nature, formation and function. Current Opinion in Cell Biology 52, 145–153. [DOI] [PubMed] [Google Scholar]
- Snyder MJ, Lau AC, Brouhard EA, Davis MB, Jiang J, Sifuentes MH, and Csankovszki G (2016). Anchoring of Heterochromatin to the Nuclear Lamina Reinforces Dosage Compensation-Mediated Gene Repression. PLOS Genet 12, e1006341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczek NA, Giles AC, Rankin CH, and Kerr RA (2011). High-throughput behavioral analysis in C. elegans. Nature Methods 8, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swygert SG, Kim S, Wu X, Fu T, Hsieh T-H, Rando OJ, Eisenman RN, Shendure J, McKnight JN, and Tsukiyama T (2019). Condensin-Dependent Chromatin Compaction Represses Transcription Globally during Quiescence. Molecular Cell 73, 533–546.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, and Dillin A (2013). XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzur YB, Winter E, Gao J, Hashimshony T, Yanai I, and Colaiácovo MP (2018). Spatiotemporal Gene Expression Analysis of the Caenorhabditis elegans Germline Uncovers a Syncytial Expression Switch. Genetics 210, 587–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton A-L, and Dekker J (2016). TAD disruption as oncogenic driver. Current Opinion in Genetics & Development 36, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Brandão HB, Le TBK, Laub MT, and Rudner DZ (2017). Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 355, 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler BS, Anderson E, Frøkjær-Jensen C, Bian Q, Jorgensen E, and Meyer BJ (2016). Chromosome-wide mechanisms to decouple gene expression from gene dose during sex-chromosome evolution. ELife 5, e17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonker SA, and Meyer BJ (2003). Recruitment of C. elegans dosage compensation proteins for gene-specific versus chromosome-wide repression. Development 130, 6519–6532. [DOI] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The Three-Dimensional Organization of Mammalian Genomes. Annual Review of Cell and Developmental Biology 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Hi-C, ChIP-seq, and RNA-seq raw and processed data are available at GEO, accession number GSE128568.