

Trichomoniasis is a sexually transmitted disease with hundreds of millions of annual cases worldwide. Approved treatment options are limited to two related nitro-heterocyclic compounds, yet resistance to these drugs is an increasing concern. New antimicrobials against the causative agent, Trichomonas vaginalis, are urgently needed.
KEYWORDS: Trichomonas vaginalis, proteasome, protozoa
ABSTRACT
Trichomoniasis is a sexually transmitted disease with hundreds of millions of annual cases worldwide. Approved treatment options are limited to two related nitro-heterocyclic compounds, yet resistance to these drugs is an increasing concern. New antimicrobials against the causative agent, Trichomonas vaginalis, are urgently needed. We show here that clinically approved anticancer drugs that inhibit the proteasome, a large protease complex with a critical role in degrading intracellular proteins in eukaryotes, have submicromolar activity against the parasite in vitro and on-target activity against the enriched T. vaginalis proteasome in cell-free assays. Proteomic analysis confirmed that the parasite has all seven α and seven β subunits of the eukaryotic proteasome although they have only modest sequence identities, ranging from 28 to 52%, relative to the respective human proteasome subunits. A screen of proteasome inhibitors derived from a marine natural product, carmaphycin, revealed one derivative, carmaphycin-17, with greater activity against T. vaginalis than the reference drug metronidazole, the ability to overcome metronidazole resistance, and reduced human cytotoxicity compared to that of the anticancer proteasome inhibitors. The increased selectivity of carmaphycin-17 for T. vaginalis was related to its >5-fold greater potency against the β1 and β5 catalytic subunits of the T. vaginalis proteasome than against the human proteasome subunits. In a murine model of vaginal trichomonad infection, proteasome inhibitors eliminated or significantly reduced parasite burden upon topical treatment without any apparent adverse effects. Together, these findings validate the proteasome of T. vaginalis as a therapeutic target for development of a novel class of trichomonacidal agents.
INTRODUCTION
Trichomonas vaginalis is the causative agent of the most common, nonviral sexually transmitted global infection, with an estimated incidence of 276 million new cases each year (1, 2). In the United States alone, over 3 million cases are estimated to occur annually, with overall prevalence rates of 1.8 to 3.1% in females and 0.5% in males and even higher rates among African American women (8%) and men (4%) (3, 4). In addition to urogenital infections, trichomoniasis increases the risk of adverse pregnancy outcomes (5, 6) and HIV transmission (7), and 15% of HIV-infected women carry concurrent T. vaginalis infections (3). Trichomoniasis also increases the incidence and severity of cervical and prostate cancers (8).
Only two drugs are currently FDA approved for the treatment of trichomoniasis, the nitro drugs metronidazole and tinidazole. Both are prodrugs that must be activated to reactive intermediates by reductive processes present in susceptible microbes (9). The intermediates form covalent adducts with DNA and protein targets, but the nature and relative importance of these targets in mediating cell killing are not well understood. Oral administration of either drug leads to clinical and microbiological cure in the majority of cases although treatment failures occur in 1 to 17% of patients (10, 11). A recent survey in the United States found that 4.3% of 538 T. vaginalis isolates showed metronidazole resistance (12). Higher oral metronidazole doses can sometimes lead to cure of refractory infections but tend to be poorly tolerated (13). Given the prevalence of T. vaginalis infections and the increase in resistance (12, 14), an urgent need exists to develop new antimicrobials against trichomoniasis, particularly agents with new targets and mechanisms of action (15, 16).
Proteasomes are large protease complexes that degrade intracellular proteins in all eukaryotes and several prokaryotes. These multisubunit complexes are formed by two rings of seven β subunits (β1 to β7) sandwiched between two rings of seven α subunits (17). Proteins targeted for degradation are first unfolded and then threaded into a barrel-shaped core of the proteasome, where the catalytic subunits, β1, β2, and β5, degrade the proteins into peptides. Each of the catalytic β subunits has a distinct substrate specificity that varies between species (18). Because normal proteasome function is required for cell survival, proteasome inhibitors have been developed as agents against cancer, namely, multiple myeloma, in which degradation of large amounts of misfolded proteins is particularly important for cell survival (19). More recently, proteasome inhibition has been explored as a new strategy for antimicrobial drugs against eukaryotic pathogens, particularly Plasmodium falciparum, Trypanosoma cruzi, Leishmania donovani, and Babesia divergens (18, 20–24).
The genome of T. vaginalis encodes a number of proteolytic enzymes that play a role in host-microbe interactions (25–27). The parasite, like all eukaryotic cells, also produces proteasome subunits, but their importance for parasite growth and survival is poorly defined. Short-term incubation with a proteasome inhibitor leads to autophagy in T. vaginalis under glucose-rich growth conditions (28), suggesting a vital function of the proteasome under specific nutrient conditions. In another trichomonad, Tritrichomonas foetus, the proteasome inhibitor lactacystin A inhibits parasite growth (29). Based on these initial observations and data from other parasites, we hypothesized that the proteasome of T. vaginalis is a valuable target for the development of a novel class of trichomonacidal agents.
RESULTS
Trichomonacidal activity of proteasome inhibitors.
T. vaginalis encodes ∼440 putative proteases in five major classes that include serine, threonine, cysteine, aspartic acid, and metalloproteases (27, 30). To determine whether any of these protease classes may be suitable as novel targets for trichomonacidal agents, we tested the activity of several class-specific inhibitors, including AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride; specific for serine proteases], MG132 (threonine), E-64 (cysteine), pepstatin (aspartic acid), and phenanthroline (metalloproteases), against T. vaginalis trophozoites in a 24-h growth and survival assay. Only MG132 showed significant activity against the parasite, with a 50% effective concentration (EC50) value of 4.2 μM, whereas the other four inhibitors had no detectable activity at the highest tested concentration of 20 μM (Fig. 1A). These data suggest that threonine-specific proteases may be new targets for agents against T. vaginalis.
FIG 1.

Trichomonacidal activity of proteasome inhibitors. (A and B) Activity of the indicated protease and proteasome inhibitors and of metronidazole as the reference drug against T. vaginalis F1623 trophozoites was evaluated in vitro in a 24-h growth and survival assay with ATP content as readout. Results are shown as the EC50, the effective compound concentration that resulted in 50% inhibition of growth and survival. Data are means + standard errors of three separate experiments. *P < 0.05, for results with the drugs versus those with the DMSO control (by analysis of variance). (C) Immunoblot analysis of ubiquitin-labeled proteins in T. vaginalis lysates after 1 h treatment with a 1 μM concentration of the indicated proteasome inhibitors or vehicle alone (DMSO). Total protein was analyzed by staining a parallel gel with Coomassie brilliant blue.
Threonine proteases, such as those in the proteasome, have important functions in eukaryotic cells to maintain proteostasis (31). In fact, MG132, a reversible peptide-aldehyde inhibitor of proteasomes, has been used extensively as a research tool to investigate the ubiquitin-proteasome system in mammalian cells and was the lead compound for the development of an anticancer agent (32). Several of these agents have undergone extensive preclinical and clinical development in regard to target specificity and tolerability for cancer indications (33, 34), which afforded us the opportunity to utilize them for further exploration of the proteasome as a new trichomonacidal target. We evaluated three covalent reversible inhibitors containing boronic acid-reactive groups and three irreversible inhibitors with epoxyketone-reactive groups (Fig. 1B). All but one of the six proteasome inhibitors displayed significant activity against T. vaginalis trophozoites, with EC50 values that were 2- to 5-fold better than the value for the reference drug metronidazole. These initial studies showed that proteasome inhibitors with different chemically reactive warheads were effective against T. vaginalis with potencies that match or exceed the current standard drug.
On-target activity of proteasome inhibitors in T. vaginalis.
In eukaryotes, proteins destined for degradation by the proteasome are first conjugated with one or more ubiquitin chains and then transported to the proteasome, where the ubiquitin chains are removed and the protein substrate is degraded. Consequently, inhibition of the proteasome leads to accumulation of ubiquitinated proteins in the cell, providing a functional readout for proteasome inhibitors (35). Therefore, we treated T. vaginalis with three of the proteasome inhibitors and examined the abundance of ubiquitin-labeled proteins by immunoblotting with an antibody against ubiquitin. Treatment with ixazomib and bortezomib at 1 μM (equivalent to about 2× to 5× the EC50 for these agents) for 1 h led to a substantial increase in ubiquitinated proteins (Fig. 1C). In contrast, addition of carfilzomib at 1 μM, which represented only 0.1× the EC50, did not alter the abundance of ubiquitin-labeled proteins compared to level with the solvent (dimethyl sulfoxide [DMSO]) control (Fig. 1C). These results indicate that ubiquitin labeling of proteins correlated with the potency of the proteasome inhibitors, suggesting that the drugs had the expected on-target activity in T. vaginalis even though they were not developed for this application.
Enrichment of T. vaginalis 20S proteasome.
To further explore the biochemical specificity of the proteasome inhibitors in T. vaginalis, we developed a two-step fractionation protocol to enrich for the proteasome of the parasite. Cellular lysates were first fractionated by DEAE anion exchange column chromatography, and enzymatic activity in each of the eluted fractions was monitored with a standard fluorescent substrate, Suc-LLVY-AMC (succinyl-Leu-Leu-Val-Tyr-amino-4-methylcoumarin) (Fig. 2A), which detects chymotrypsin-type activity in human, Saccharomyces cerevisiae, parasite, and bacterial proteasomes (21, 36, 37). Fractions with the highest activity were pooled, concentrated, and further separated by gel filtration chromatography using a Sepharose-6 size exclusion column. Activity assays of the eluted fractions with the same Suc-LLVY-AMC substrate yielded two well-defined activity peaks (Fig. 2B). Proteolytic activity in fractions 12 to 14 was inhibited by the proteasome inhibitor bortezomib, whereas activity in fractions 16 to 18 was insensitive to bortezomib but sensitive to the cysteine protease inhibitor E-64 (Fig. 2C). These data show that our fractionation strategy could separate the large proteasome complex of T. vaginalis with chymotrypsin-type activity from lower-molecular-weight cysteine proteases.
FIG 2.

Enrichment of T. vaginalis proteasome. Total lysates of T. vaginalis F1623 were prepared and enriched for the 20S proteasome by two-step column chromatography. (A) Lysates were first fractionated with a DEAE anion exchange column, and fractions were eluted with a NaCl gradient and assayed for total protein (blue line) and enzymatic activity with the substrate Suc-LLVY-AMC (black line). AU, arbitrary units. (B) Fractions 33 to 37 were then pooled and further separated by gel filtration using a Superose 6 column. Fractions were again assayed for total protein (blue line) and enzymatic activity (black line). (C) Fractions 12 to 14 and 16 to 18 of the second fractionation were pooled, and the pools were tested for sensitivity to the proteasome inhibitor, bortezomib (BTZ), and the cysteine protease inhibitor, E-64. Data are expressed relative to the activity in vehicle-treated controls (means + standard errors, n = 3; *P < 0.05, for results with the inhibitors versus those with the respective vehicle controls). (D) The pool of fractions 12 to 14 and purified preparations of the human constitutive proteasome (c20S) and human immunoproteasome (i20S) were run on a denaturing SDS-PAGE gel and stained with silver. Proteins shown in the boxed area were excised and subjected to proteomic analysis.
The proteins in pooled fractions 12 to 14 representing the proteasome were visualized by silver staining following denaturing gel electrophoresis. As controls, the human constitutive proteasome (c20S) and human immunoproteasome (i20S) were run in parallel. The α and β subunits of c20S and i20S migrated in the expected molecular-weight range of 22 to 38 kDa (Fig. 2D) (38). Several T. vaginalis proteins were visible in the same mass range although a number of higher-molecular-weight proteins were also detected (Fig. 2D). We excised the region from the T. vaginalis lane that corresponded in mass to the human α and β subunits and analyzed the proteins with proteomic methods. Seven putative α and β subunits were identified in the gel section, confirming that we had successfully enriched the 20S proteasome from the T. vaginalis (Tv20S) protein extract.
Proteasomes are structurally highly conserved, consisting of two rings of seven α and two rings of seven β subunits that assemble into a core complex of 28 proteins. The β1, β2, and β5 subunits generally have catalytic activity, with the active sites facing the inner channel of the proteasome (Fig. 3A). In the mammalian proteasome, the three catalytic β subunits have different substrate preferences, as illustrated by the differently shaped active sites in Fig. 3A. The proteasome subunits of T. vaginalis are poorly annotated, so we performed protein sequence alignments with the human α and β subunits. The resulting dendrogram shows that each T. vaginalis subunit aligned to a single c20S subunit with between 28% and 52% sequence identity (Fig. 3B). In addition, we confirmed that the T. vaginalis proteins A2E7Z2, A2F2T6, and A2DD57 (UniProt accession numbers) closely align with the human β1, β2, and β5 subunits, respectively, and that each of these subunits contains the critical active-site triad of Thr-1, Asp-17 and Lys-33 (mature protein numbering) (Fig. 3C). Therefore, we conclude that T. vaginalis possesses the same three catalytically active proteasome β subunits as other eukaryotic cells.
FIG 3.

Bioinformatic analysis of T. vaginalis proteasome subunits. (A) Scheme of the mammalian 20S proteasome in three dimensions (left) and as a planar cross-section of a β ring containing the three enzymatically active subunits (right). The β1, β2, and β5 active sites face the inner core of the proteasome barrel. Each subunit has a unique substrate and inhibitor-binding preference, as indicated by differently shaped active sites. (B) Alignment dendrogram of putative T. vaginalis proteasome subunits (UniProt accession numbers in blue) and α and β subunits of the human proteasome (UniProt accession numbers in black). The percentage of sequence identity for each pair is noted. (C) Sequence alignment of the N-terminal amino acids of the mature human β1, β2, and β5 subunits (black) with the homologous T. vaginalis proteins (blue). The active-site residues in positions 1 (Thr), 17 (Asp), and 33 (Lys) are boxed.
Differential activity of ixazomib against human and T. vaginalis proteasome β subunits.
To identify a candidate proteasome inhibitor for further biochemical studies and in vivo efficacy testing, we evaluated each of the proteasome inhibitors in a HeLa human cytotoxicity assay. Not surprisingly, all of the tested inhibitors, which were originally developed as anticancer drugs, were more potent against HeLa cells than T. vaginalis, with selectivity ratios (i.e., ratios of human cytotoxicity over trichomonacidal activity) ranging from 0.01 to 0.23 (Fig. 4A). Ixazomib had the most favorable selectivity among these drugs, so we focused on this drug for further functional evaluation. Because this compound preferentially targets two of the proteolytically active subunits, β1 and β5, in the human c20S proteasome, we tested the potency of ixazomib against the enriched Tv20S proteasome (corresponding to pooled fractions 12 to 14 in Fig. 2C) using substrates specific for the β1 and β5 subunits. For the β5 subunit, we assayed c20S and Tv20S proteasomes for sensitivity to ixazomib with a β5-selective substrate, Suc-LLVY-AMC. The drug was a potent inhibitor of both the human and parasite β5 subunits, with similar 50% inhibitory concentration (IC50) values (Fig. 4B). In contrast, using the β1 substrate Z-LLE-AMC (carboxybenzyl-Leu-Leu-Gly-7-amino-4-methylcoumarin), ixazomib was found to be a weak inhibitor of the β1 subunit of Tv20S, whereas it inhibited the human β1 subunit as effectively as the β5 subunit (Fig. 4C). These studies suggest that human cytotoxicity of ixazomib is associated with dual inhibition of the c20S β1 and β5 subunits, whereas inhibition of the β5 subunit alone may be sufficient for trichomonacidal activity.
FIG 4.

Selectivity and β subunit specificity of the proteasome inhibitor ixazomib. (A) Proteasome inhibitors were tested for T. vaginalis activity (EC50) and HeLa cytotoxicity (CC50), and selectivity indices (CC50/EC50) were calculated from the results. (B and C) Ixazomib, which displayed the best selectivity among the tested compounds, was assayed for inhibition of the β5 subunit and β1 subunit of the enriched Tv20S (blue lines; corresponding to fractions 12 to 14 in Fig. 2C) and purified human c20S (black lines), using the indicated subunit-specific substrates. Data are expressed relative to activity without inhibitor, and are shown as means ± standard errors (n = 3). (D) Female BALB/c mice (3 to 4 weeks old) were infected intravaginally with T. foetus trophozoites. After 1 day, mice were treated intravaginally with a 0.5% (14 mM) suspension of ixazomib in PBS or with PBS alone, for a total of five or three times over 3 days (top). Live parasites were enumerated in vaginal washes 1 day after the last treatment (bottom). Each point represents an individual mouse, and horizontal bars show geometric means (*, P < 0.05, for results with inhibitor versus those in controls, by a Kruskal-Wallis test and Dunn’s post hoc test). The dashed line shows the detection limit of the assay.
In vivo efficacy of ixazomib in a murine trichomonad infection model.
To test for in vivo efficacy of ixazomib, we employed T. foetus, a trichomonad species related to T. vaginalis but with more robust vaginal infectivity in mice (39). We first confirmed that ixazomib was active against T. foetus trophozoites in vitro (EC50 of 4.3 ± 0.3 μM). Female BALB/c mice (3 to 4 weeks old) were then infected intravaginally with T. foetus trophozoites and subsequently treated with five intravaginal doses of ixazomib or vehicle control over a 3-day period (Fig. 4D, top). Treatment with ixazomib eradicated the parasites, whereas >85% of vehicle-treated mice had between 5 × 103 and 2 × 105 trophozoites per mouse (Fig. 4D). Furthermore, treatment with only three doses of ixazomib also led to a marked reduction in parasite numbers, indicating that fewer than five doses can be therapeutic (Fig. 4D). No apparent adverse effects were observed in any of the ixazomib-treated mice, demonstrating that proteasome inhibition is a viable therapeutic strategy against vaginal trichomonad infection and has the potential for reduced systemic adverse effects through effective topical application.
Carmaphycins as proteasome inhibitors with increased T. vaginalis selectivity.
Although ixazomib had marked trichomonacidal activity in vitro and in vivo, its significant human cytotoxicity and lack of selectivity (Fig. 4A) would presumably preclude its use as an anti-infective agent. Alternative compounds with greater selectivity are likely to be needed for therapy of trichomoniasis. In previous work by our group, we isolated a compound from a marine cyanobacterium, carmaphycin B (CPB) (Fig. 5A), which displays potent inhibitory activity against human and Plasmodium proteasomes (40). In an effort to make a selective Plasmodium proteasome inhibitor with reduced mammalian cytotoxicity, we synthesized 20 derivatives of CPB with the general structure of an N-terminal hexanoic acid group, a tripeptide sequence (P3-P2-P1), and a C-terminal epoxyketone group (21) (Fig. 5A). We evaluated this targeted compound library for activity against T. vaginalis and HeLa cells. The parent compound, CPB, was active against T. vaginalis, with an EC50 of 490 nM, but was also highly cytotoxic in HeLa cells (50% cytotoxic concentration [CC50], 5 nM) (Fig. 5B). In contrast, the derivatives showed a spectrum of activities against T. vaginalis and HeLa cells (Fig. 5B). For example, carmaphycin-17 (CP-17), consisting of bulky aromatic groups (indole and phenyl) in the P1, P2, and P3 positions, had 493-fold reduced HeLa cytotoxicity compared to that of CPB but a 2-fold increase in potency for T. vaginalis (EC50 of 217 nM), resulting in a dramatic 1,113-fold improvement in selectivity (Fig. 5B). On the other hand, carmaphycin-18 (CP-18), which mostly differs from CPB in the P2 and P3 positions (Fig. 5A), had no effect on T. vaginalis or HeLa cells at 20 μM (Fig. 5B). Consistent with the cell killing data, incubation of T. vaginalis with CP-17 resulted in an increase in ubiquitinated proteins similar to that found with ixazomib, whereas CP-18 had no effect (Fig. 5C).
FIG 5.

Carmaphycin derivatives as proteasome inhibitors with improved selectivity against T. vaginalis. (A) Structures of the marine natural product carmaphycin B (CPB) and two synthetic derivatives, carmaphycin-17 (CP-17) and carmaphycin-18 (CP-18). (B) A library of 20 carmaphycin derivatives was tested for activity (EC50) against T. vaginalis and cytotoxicity (CC50) against human HeLa cells. Each data point represents the mean values of EC50 and CC50 of three independent experiments; data for CPB, CP-17, and CP-18 are highlighted in red. The horizontal and vertical dashed lines represent the assay sensitivities. Compounds that had no detectable activity are shown below or to the left of these lines. For comparison, the diagonal dashed line depicts a selectivity ratio (CC50/EC50) of 1, so compounds located below that line are more active against human cells than against the parasite, while compounds above the line are more active against the parasite than against human cells. (C) Immunoblot analysis of ubiquitin-labeled proteins in T. vaginalis lysates after 1-h treatment with a 1 μM concentration of the indicated proteasome inhibitors or vehicle alone (DMSO). Total protein was visualized by Coomassie brilliant blue staining. (D and E) CP-17 was assayed for inhibition of the β5 subunit and β1 subunit of the enriched Tv20S (blue lines; corresponding to fractions 12 to 14 in Fig. 2C) and purified human c20S (black lines), using subunit-specific substrates. Data are expressed relative to activity without inhibitor and are shown as means ± standard errors (n = 3). Potency was calculated by extrapolation and is shown as IC50, the inhibitory concentration that inhibited 50% of the enzyme activity in the assay. For c20S in panel E, none of the data points showed more than 50% inhibition, so the IC50 could not be calculated; instead, it is shown as exceeding the highest tested drug concentration.
To explore the biochemical basis of the improved selectivity of CP-17, we assayed its activity against the β1 and β5 subunits of Tv20S and c20S. The compound was >5-fold more active against the β5 subunit of the parasite proteasome than that of the human proteasome (Fig. 5D). Furthermore, CP-17 was also more potent at targeting the Tv20S β1 subunit than the human c20S equivalent (Fig. 5E) even though the potency against this subunit was much lower than that against the β5 subunit. These results suggest that the T. vaginalis proteasome is functionally distinct from the human constitutive proteasome and that this difference can be exploited for the development of more selective proteasome inhibitors against the parasite.
Carmaphycin-17 can overcome metronidazole resistance in T. vaginalis and has significant in vivo efficacy against trichomonads.
An important rationale for developing new classes of drugs against trichomoniasis is the increasing occurrence of resistance to the most commonly used trichomonacidal drug, metronidazole (12). Therefore, we tested the activity of CP-17, as well as ixazomib, against a metronidazole-resistant strain of T. vaginalis (41). Both compounds were equally or even slightly more effective against the resistant line than against the susceptible strain of T. vaginalis, in contrast to metronidazole, which showed the expected ∼10-fold decrease in potency against the resistant strain (Fig. 6A) (41). Furthermore, ixazomib and CP-17 showed similar potencies against a third, unrelated T. vaginalis strain (Fig. 6A). These data show that metronidazole resistance does not impact the activity of proteasome inhibitors against T. vaginalis and that such inhibitors are active against several different strains, thus underlining that these compounds represent a mechanistically novel class of trichomonacidal drugs.
FIG 6.
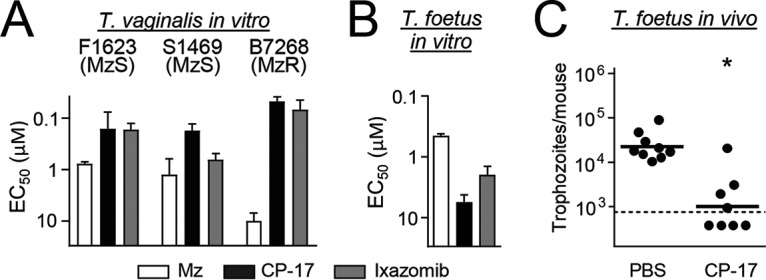
Activity of carmaphycin derivative against metronidazole-resistant T. vaginalis and vaginal trichomonad infection. (A) The proteasome inhibitors CP-17 and ixazomib (as a control) were tested for activity (EC50) against metronidazole (Mz)-sensitive (MzS) and Mz-resistant (MzR) strains of T. vaginalis in a 24-h growth and survival assay, with ATP content as readout. Data are means + standard errors of three separate experiments. (B) CP-17 and ixazomib were also tested for activity (EC50) against T. foetus (mean + standard error; n = 3). (C) T. foetus-infected mice were given five intravaginal treatments with a 1% (15 mM) suspension in CP-17 or PBS as outlined in the legend of Fig. 4D, and live parasites were enumerated in vaginal washes 1 day after the last treatment. Each point represents an individual mouse, and horizontal bars show geometric means (*, P < 0.05 for results with the drugs versus those with the controls by a Mann-Whitney U test). The dashed line shows the detection limit of the assay.
To further evaluate utility of CP-17 as a new lead compound, we investigated its in vivo efficacy against vaginal trichomonad infection. We first confirmed that CP-17, like ixazomib, had significant in vitro activity against the model parasite, T. foetus (Fig. 6B). Subsequently, T. foetus-infected mice were given five topical administrations of the compound over 3 days, and parasite numbers were evaluated in vaginal washes. CP-17 led to a significant 20-fold reduction in trophozoite numbers compared to levels for the vehicle-treated controls (Fig. 6C). None of the drug-treated mice showed any apparent adverse effects. These data demonstrate that CP-17 has significant in vivo efficacy against trichomonads, further supporting its potential as a promising new drug lead.
DISCUSSION
Our studies advance the T. vaginalis proteasome as a new target for development of potent antimicrobials against trichomoniasis. Proteasome inhibitors were lethal to the parasite in vitro and in vivo, and bioactivity was associated with engagement of the predicted drug target. Other recent reports have also validated the proteasome as a drug target for disease-causing microbes (23, 42). For example, saturation mutagenesis revealed that many essential genes of P. falciparum are in the proteasome degradation pathway (43), and proteasome inhibitors have shown promise as new antimalarial agents (21, 44–46). Similarly, inhibition of the proteasome in the kinetoplastid parasites, Trypanosoma and Leishmania, is toxic to the parasites in vitro and clears infection in murine models (23). Thus, our results and those in other parasites add to the emerging concept that the proteasome is a drug target against a wide range of eukaryotic pathogens.
Key to the development and ultimate utility of proteasome inhibitors as anti-infectives will be maximizing selectivity relative to human cells. We found that the improved selectivity of the most promising compound, CP-17, against T. vaginalis over human cells was paralleled by a >5-fold greater potency against the β1 and β5 proteasome subunits of the parasite than that against the human subunits. In contrast, another inhibitor, CP-18, which blocked the β5 proteasome subunit in Plasmodium falciparum and had the best antimalarial potency and selectivity of a group of carmaphycin derivatives (21), was inactive against T. vaginalis. Together, these observations indicate that the proteasomes of different species have unique substrate specificities, a contention generally supported by analysis of cleavage specificities and structural modeling (36, 47–49). As a practical consequence, proteasome inhibitors developed for a particular infectious indication may not be easily repurposed for other indications, so separate medicinal chemistry efforts will probably be needed for further development of the current lead, CP-17, into an optimal trichomonacidal agent. Such development would also have to consider optimal pharmacokinetic characteristics since CP-17 has so far proved to be less efficacious in vivo than ixazomib despite its slightly greater in vitro potency. However, our dosing experiments with ixazomib suggest that it might be possible to develop high-potency compounds that could be therapeutic with small numbers and perhaps only a single drug administration.
The mechanisms by which proteasome inhibition causes cell death in parasites are not well defined. In human cancer cells, proteasome inhibitors prevent clearance of unfolded or misfolded proteins that are cytotoxic to the cells (19). They also inhibit IκB kinase degradation, thereby preventing the induction of gene products with prosurvival functions and leading to caspase-mediated cell death (50). In P. falciparum, treatment with proteasome inhibitors results in accumulation of polyubiquitinated proteins that activate an endoplasmic reticulum stress response, which ultimately leads to cell death (51). Interestingly, another antimalarial drug, dihydroartemisinin, also causes accumulation of polyubiquitinated proteins and synergizes with proteasome inhibitors to kill cells, underlining the utility of the proteasome as a drug target (52). As for P. falciparum, treatment of T. vaginalis for only 1 h with proteasome inhibitors resulted in a significant accumulation of ubiquitinated proteins, suggesting that cell death in this parasite may occur by a mechanism similar to that in P. falciparum. Furthermore, depending on growth conditions, inhibition of the proteasome may also induce autophagy in T. vaginalis, which, if excessive, could induce cell death (28).
The trichomonacidal activity of proteasome inhibitors was not impacted by resistance to the most commonly used drug for treating trichomoniasis, metronidazole. This suggests that proteasome inhibition holds promise as a rescue therapy for infections refractory to nitro drugs (11, 53). Nitro drugs act in a two-step sequence, involving initial activation by reduction to reactive intermediates, followed by covalent binding of intermediates to and inactivation of multiple target molecules. Resistance is caused by loss of the necessary reductases (9), a process that is evidently unrelated to the ubiquitin-proteasome system. This molecular target independence raises the future possibility that nitro drugs and proteasome inhibitors may exhibit synergistic activity in T. vaginalis killing.
Both proteasome inhibitors that were tested in vivo, ixazomib and CP-17, were active against vaginal trichomonad infection after topical administration, albeit with differences in their efficacies. Treatment of multiple myeloma with proteasome inhibitors is by intravenous or oral drug administration (19, 54), so a suitable drug against T. vaginalis may also be effective through those routes. However, topical administration has potential advantages over systemic routes, including the possibility of reduced systemic exposure and thus a lower risk of adverse effects. Although currently approved trichomonacidal drugs are only given orally, partly because of insufficient efficacy with achievable topical doses (55), other vaginal infections are treated or protected against with vaginal creams, gels, dissolvable tablets, or suppositories (56). For those drugs, tissue levels in the mid-micromolar range are achievable (57, 58), which are well above the range of potencies we found for proteasome inhibitors. It remains to be determined whether topical treatment is feasible in males infected with T. vaginalis, but it is noteworthy that the urgency for alternative treatment strategies is greater in females, where infection can persist for months or even years, than in males, where infection generally lasts less than 10 days (59). Overall, our findings demonstrate that proteasome inhibition is a viable new therapeutic strategy for trichomoniasis and thus, provides an impetus to the systematic development of suitable inhibitors with improved potency, selectivity, and in vivo efficacy.
MATERIALS AND METHODS
Drugs and chemicals.
Protease inhibitors were purchased from Selleckchem (Houston, TX, USA). Carmaphycin B and analogs were synthesized as described previously (21). Human 20S constitutive proteasome, human 20S immunoproteasome, and fluorescent substrates were purchased from Boston Biochem (Cambridge, MA).
Antimicrobial and cytotoxicity assays.
The following trichomonad strains were used: metronidazole-sensitive T. vaginalis F1623 and S1469 (60), metronidazole-resistant T. vaginalis B7268 (41, 61), and metronidazole-sensitive T. foetus D1 (62). Cells were grown at 37°C in TYM (trypticase, yeast extract, maltose) Diamond’s medium supplemented with 180 μM ferrous ammonium sulfate. Drug susceptibility assays were done as described previously (41). Briefly, stocks of the test compounds were diluted in phosphate-buffered saline (PBS) to 75 μM, and 1:3 serial dilutions were made. Trophozoites (3 × 103/well) were added to 96-well plates, and cultures were incubated for 24 h at 37°C under anaerobic conditions (AnaeroPack-Anaero system). Growth and viability were determined with an ATP assay by adding BacTiter-Glo microbial cell viability assay reagent (Promega) and measuring ATP-dependent luminescence in a microplate reader. The 50% effective concentration (EC50) was derived from the concentration-response curves using BioAssay software (CambridgeSoft Software).
Proteasome enrichment.
T. vaginalis lysates were prepared in a buffer of 100 mM NaCl, 20 mM Tris (pH 7.5), 0.01% Tween 20, 0.5% glycerol, and 100 μM E-64 by sonication for 30 s and three subsequent freeze-thaw cycles. Protein amounts were determined by bicinchoninic acid assay. Tv20S was enriched from the lysates by two chromatographic steps. In a first enrichment step, lysate (∼8.5 mg of protein in 2 ml) was loaded onto a 5-ml anion exchange HiTrap DEAE FF column (GE Healthcare) connected to an AKTA Pure instrument (GE Healthcare). Unbound proteins were removed by washing with a buffer of 25 mM Tris (pH 7.5), 1 mM dithiothreitol (DTT), and 10% glycerol. Bound proteins were eluted with 25 ml of a linear gradient from 0 to 0.5 M NaCl, with collection of 1.5-ml fractions. Aliquots of the fractions were analyzed for proteasome activity with a 25 μM concentration of the substrate succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC) in a buffer of 20 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 1 mM ATP, and 0.02% SDS, with and without 10 μM bortezomib or 10 μM E-64. Fractions containing bortezomib-sensitive activity were pooled and concentrated to 0.5 ml using a 100-kDa centrifugal filter (Amicon). In a second step, pooled fractions were loaded onto a Superose 6 10/300 gel filtration column (GE Healthcare). Proteins were eluted with 1.5 column volumes of 25 mM Tris (pH 7.5), 1 mM DTT, 10% glycerol, and 125 mM NaCl into 1-ml fractions, which were assayed again for proteolytic activity with Suc-LLVY-AMC, with and without 10 μM bortezomib or 10 μM E-64. Bortezomib-sensitive fractions without detectable E-64 sensitive activity (fractions 12 to 14) (Fig. 2C) were pooled and used for all biochemical and structural characterization of Tv20S.
Proteomics.
Tv20S was analyzed by electrophoresis together with 100 ng of c20S and i20S. Samples were treated with NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Thermo Fisher Scientific), denatured at 85°C for 10 min, run on a 4 to 12% Bis-Tris Plus gel (Thermo Fisher Scientific), and stained with silver. The appropriate bands were excised from the gel with a clean scalpel, diced into 1-mm cubes, and placed into 0.6-ml tubes (Axygen). In-gel trypsin digestion was performed as described previously (63). Following digestion and extraction, the peptides were desalted with C18 LTS tips (Rainin) and dried in a vacuum centrifuge. Peptides were resuspended in 12 μl of 0.1% trifluoroacetic acid (TFA), and 5 μl was analyzed on a Q Exactive mass spectrometer (MS) (Thermo Scientific). Peptides were first separated by reverse-phase chromatography on an UltiMate 3000 high-performance liquid chromatography (HPLC) system (Thermo Scientific) equipped with an in-house packed C18 column (1.7-μm bead size, 75 μm by 25 cm, heated to 65°C) at a flow rate of 300 nl/min over a 76-min linear gradient from 5% solvent A to 25% solvent B, where solvent A and B correspond to 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Survey scans were recorded at a resolution of 35,000 at 200 m/z over a range of 350 to 1,500 m/z with the automatic gain control (AGC) at 1 × 106 and a maximum injection time of 300 ms. Tandem MS (MS/MS) was performed in data-dependent acquisition mode with higher-energy collision dissociation fragmentation (28 normalized collision energy) on the 20 most intense precursor ions at a resolution of 17,500 at 200 m/z, with the AGC at 5 × 106 and a maximum injection time of 50 ms.
Data analysis was performed using the PEAKS, version 8.5, software (Bioinformatics Solutions, Inc.). RAW files were searched against the reference proteome of T. vaginalis strain ATCC PRA-98 from UniProt (https://www.uniprot.org/proteomes/UP000001542) with 50,190 entries using the following settings: 15 ppm precursor mass tolerance, 0.01 Da MS/MS mass tolerance, and an enzyme digest of trypsin with a maximum of three missed cleavages. Carbamidomethylation of cysteines was included as a fixed modification, and oxidation of methionine and protein N-terminal acetylation were included as variable modifications. The false discovery rate for protein identification was set to 1%. Annotation of the putative T. vaginalis proteasome subunits with α and β subunit nomenclature was performed by comparing sequence alignments against the human 20S proteasome subunits with BLAST, version 2.6.0+, command line tools.
Proteasome assays.
Proteasome activity was assayed using 0.29 nM c20S or Tv20S in 20 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 1 mM ATP, 0.02% SDS, and 25 μM Suc-LLVY-AMC or 25 μM carboxybenzyl-Leu-Leu-Gly-7-amino-4-methylcoumarin (Z-LLE-AMC) in 50-μl reaction mixtures in black-walled 96-well plates. AMC fluorophore release was monitored at excitation 360 nm and emission 460 nm at 24°C using a Synergy HTX multimode reader (Biotek). For inhibition assays, substrate and inhibitor were added simultaneously to the enzyme, and the reaction rate, expressed in relative fluorescence units (RFU) per second, was determined for 4 h. The 50% inhibitory concentration (IC50) was calculated in GraphPad Prism, version 6, by normalizing activity to that of the DMSO controls and interpolating the data using the log(inhibitor)-variable slope curve fitting algorithm.
Immunoblot analysis.
T. vaginalis F1623 was incubated for 1 h with 1 μM proteasome inhibitor or solvent control, and cells were lysed using radioimmunoprecipitation assay (RIPA) buffer with added Halt protease inhibitors (Thermo Scientific). Protein concentrations were measured with detergent-compatible (DC) protein assay (Bio-Rad) and adjusted to 10 mg/ml. Proteins were fractionated by SDS-PAGE, transferred onto a nitrocellulose membrane (Bio-Rad), which was blocked with 5% nonfat dry milk in TBST buffer (20 mM Tris base, 150 mM NaCl, 0.05% Tween 20, pH adjusted to 7.6 with HCl) for 1 h, and probed with primary anti-ubiquitin K48 antibody (Boston Biochem) at a 1:5,000 dilution, followed by incubation with anti-rabbit horseradish peroxidase (HRP)-conjugated IgG (Jackson laboratory) at a 1:10,000 dilution. Immunoreactions were visualized by chemiluminescence (Immobilon, Millipore).
Murine trichomonad infection model.
Weanling (3- to 4-week-old) female BALB/cJ mice were inoculated intravaginally with 5 μl of a suspension containing 106 T. foetus D1 trophozoites in TYM medium. For drug treatment studies, mice were given 5 μl/dose of 0.5% (14 mM) ixazomib or 1% (15 mM) of CP-17 in 0.1% hypromellose (Sigma), or vehicle controls, intravaginally five or three times for 3 days beginning 1 day after infection. On day 4, live trophozoites were enumerated in vaginal washes. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Data availability.
The complete mass spectrometry data and annotations can be accessed on the Mass Spectrometry Interactive Virtual Environment (MassIVE) public data repository under accession number MSV000083474. The FTP download link is ftp://massive.ucsd.edu/MSV000083474.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI146387 (A.J.O. and L.E.), AI133393 (A.J.O. and C.R.C.), CA100851 (W.H.G.), AI114671 (L.E.), and DK120515 (L.E.) and a UCSD Academic Senate Research Grant (A.J.O. and C.R.C.). Z.J. is supported by the UCSD Chancellor’s Research Excellence Scholarship. J.A. is grateful to the University of Jordan and the Scientific Research Support Fund for financial support. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Elaine Hanson and Lucia Hall for expert technical support.
REFERENCES
- 1.Newman L, Rowley J, Vander Hoorn S, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. 2015. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One 10:e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menezes CB, Frasson AP, Tasca T. 2016. Trichomoniasis—are we giving the deserved attention to the most common non-viral sexually transmitted disease worldwide? Microb Cell 3:404–419. doi: 10.15698/mic2016.09.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poole DN, McClelland RS. 2013. Global epidemiology of Trichomonas vaginalis. Sex Transm Infect 89:418–422. doi: 10.1136/sextrans-2013-051075. [DOI] [PubMed] [Google Scholar]
- 4.Patel EU, Gaydos CA, Packman ZR, Quinn TC, Tobian A. 2018. Prevalence and correlates of Trichomonas vaginalis infection among men and women in the United States. Clin Infect Dis 67:211–217. doi: 10.1093/cid/ciy079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swygard H, Sena AC, Hobbs MM, Cohen MS. 2004. Trichomoniasis: clinical manifestations, diagnosis and management. Sex Transm Infect 80:91–95. doi: 10.1136/sti.2003.005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fichorova RN. 2009. Impact of T. vaginalis infection on innate immune responses and reproductive outcome. J Reprod Immunol 83:185–189. doi: 10.1016/j.jri.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClelland RS, Sangare L, Hassan WM, Lavreys L, Mandaliya K, Kiarie J, Ndinya-Achola J, Jaoko W, Baeten JM. 2007. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 195:698–702. doi: 10.1086/511278. [DOI] [PubMed] [Google Scholar]
- 8.Marous M, Huang WY, Rabkin CS, Hayes RB, Alderete JF, Rosner B, Grubb RL III, Winter AC, Sutcliffe S. 2017. Trichomonas vaginalis infection and risk of prostate cancer: associations by disease aggressiveness and race/ethnicity in the PLCO Trial. Cancer Causes Control 28:889–898. doi: 10.1007/s10552-017-0919-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upcroft P, Upcroft JA. 2001. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin Microbiol Rev 14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Upcroft JA, Dunn LA, Wal T, Tabrizi S, Delgadillo-Correa MG, Johnson PJ, Garland S, Siba P, Upcroft P. 2009. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex Health 6:334–338. doi: 10.1071/SH09011. [DOI] [PubMed] [Google Scholar]
- 11.Cudmore SL, Delgaty KL, Hayward-McClelland SF, Petrin DP, Garber GE. 2004. Treatment of infections caused by metronidazole-resistant Trichomonas vaginalis. Clin Microbiol Rev 17:783–793. table of contents. doi: 10.1128/CMR.17.4.783-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkcaldy RD, Augostini P, Asbel LE, Bernstein KT, Kerani RP, Mettenbrink CJ, Pathela P, Schwebke JR, Secor WE, Workowski KA, Davis D, Braxton J, Weinstock HS. 2012. Trichomonas vaginalis antimicrobial drug resistance in 6 US cities, STD Surveillance Network, 2009–2010. Emerg Infect Dis 18:939–943. doi: 10.3201/eid1806.111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobel JD, Nyirjesy P, Brown W. 2001. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 33:1341–1346. doi: 10.1086/323034. [DOI] [PubMed] [Google Scholar]
- 14.Snipes LJ, Gamard PM, Narcisi EM, Beard CB, Lehmann T, Secor WE. 2000. Molecular epidemiology of metronidazole resistance in a population of Trichomonas vaginalis clinical isolates. J Clin Microbiol 38:3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Ambrogi M. 2017. Turning the spotlight on sexually transmitted infections. Lancet Infect Dis 17:792–793. doi: 10.1016/S1473-3099(17)30363-8. [DOI] [PubMed] [Google Scholar]
- 16.Klausner JD, Broutet N. 2017. Health systems and the new strategy against sexually transmitted infections. Lancet Infect Dis 17:797–798. doi: 10.1016/S1473-3099(17)30361-4. [DOI] [PubMed] [Google Scholar]
- 17.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. 2012. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148:727–738. doi: 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 18.Bibo-Verdugo B, Jiang Z, Caffrey CR, O'Donoghue AJ. 2017. Targeting proteasomes in infectious organisms to combat disease. FEBS J 284:1503–1517. doi: 10.1111/febs.14029. [DOI] [PubMed] [Google Scholar]
- 19.Manasanch EE, Orlowski RZ. 2017. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14:417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, O’Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PC, Bogyo M. 2016. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530:233–236. doi: 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaMonte GM, Almaliti J, Bibo-Verdugo B, Keller L, Zou BY, Yang J, Antonova-Koch Y, Orjuela-Sanchez P, Boyle CA, Vigil E, Wang L, Goldgof GM, Gerwick L, O’Donoghue AJ, Winzeler EA, Gerwick WH, Ottilie S. 2017. Development of a potent inhibitor of the Plasmodium proteasome with reduced mammalian toxicity. J Med Chem 60:6721–6732. doi: 10.1021/acs.jmedchem.7b00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirkman LA, Zhan W, Visone J, Dziedziech A, Singh PK, Fan H, Tong X, Bruzual I, Hara R, Kawasaki M, Imaeda T, Okamoto R, Sato K, Michino M, Alvaro EF, Guiang LF, Sanz L, Mota DJ, Govindasamy K, Wang R, Ling Y, Tumwebaze PK, Sukenick G, Shi L, Vendome J, Bhanot P, Rosenthal PJ, Aso K, Foley MA, Cooper RA, Kafsack B, Doggett JS, Nathan CF, Lin G. 2018. Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc Natl Acad Sci U S A 115:E6863–E6870. doi: 10.1073/pnas.1806109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khare S, Nagle AS, Biggart A, Lai YH, Liang F, Davis LC, Barnes SW, Mathison CJ, Myburgh E, Gao MY, Gillespie JR, Liu X, Tan JL, Stinson M, Rivera IC, Ballard J, Yeh V, Groessl T, Federe G, Koh HX, Venable JD, Bursulaya B, Shapiro M, Mishra PK, Spraggon G, Brock A, Mottram JC, Buckner FS, Rao SP, Wen BG, Walker JR, Tuntland T, Molteni V, Glynne RJ, Supek F. 2016. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jalovecka M, Hartmann D, Miyamoto Y, Eckmann L, Hajdusek O, O'Donoghue AJ, Sojka D. 2018. Validation of Babesia proteasome as a drug target. Int J Parasitol Drugs Drug Resist 8:394–402. doi: 10.1016/j.ijpddr.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bozner P, Demes P. 1991. Proteinases in Trichomonas vaginalis and Tritrichomonas mobilensis are not exclusively of cysteine type. Parasitology 102:113–115. doi: 10.1017/S0031182000060418. [DOI] [PubMed] [Google Scholar]
- 26.Arroyo R, Cárdenas-Guerra RE, Figueroa-Angulo EE, Puente-Rivera J, Zamudio-Prieto O, Ortega-López J. 2015. Trichomonas vaginalis cysteine proteinases: iron response in gene expression and proteolytic activity. Biomed Res Int 2015:946787. doi: 10.1155/2015/946787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mancilla-Olea MI, Ortega-López J, Figueroa-Angulo EE, Avila-González L, Cárdenas-Guerra RE, Miranda-Ozuna JFT, González-Robles A, Hernández-García MS, Sánchez-Ayala L, Arroyo R. 2018. Trichomonas vaginalis cathepsin D-like aspartic proteinase (Tv-CatD) is positively regulated by glucose and degrades human hemoglobin. Int J Biochem Cell Biol 97:1–15. doi: 10.1016/j.biocel.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Huang KY, Chen RM, Lin HC, Cheng WH, Lin HA, Lin WN, Huang PJ, Chiu CH, Tang P. 2018. Potential role of autophagy in proteolysis in Trichomonas vaginalis. J Microbiol Immunol Infect doi: 10.1016/j.jmii.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Pereira-Neves A, Gonzaga L, Menna-Barreto RF, Benchimol M. 2015. Characterisation of 20S proteasome in Tritrichomonas foetus and its role during the cell cycle and transformation into endoflagellar form. PLoS One 10:e0129165. doi: 10.1371/journal.pone.0129165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, Wortman JR, Bidwell SL, Alsmark UC, Besteiro S, Sicheritz-Ponten T, Noel CJ, Dacks JB, Foster PG, Simillion C, Van de Peer Y, Miranda-Saavedra D, Barton GJ, Westrop GD, Muller S, Dessi D, Fiori PL, Ren Q, Paulsen I, Zhang H, Bastida-Corcuera FD, Simoes-Barbosa A, Brown MT, Hayes RD, Mukherjee M, Okumura CY, Schneider R, Smith AJ, Vanacova S, Villalvazo M, Haas BJ, Pertea M, Feldblyum TV, Utterback TR, Shu CL, Osoegawa K, de Jong PJ, Hrdy I, Horvathova L, Zubacova Z, Dolezal P, Malik SB, Logsdon JM Jr, Henze K, Gupta A, et al. . 2007. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315:207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra R, Upadhyay A, Prajapati VK, Mishra A. 2018. Proteasome-mediated proteostasis: novel medicinal and pharmacological strategies for diseases. Med Res Rev 38:1916–1973. doi: 10.1002/med.21502. [DOI] [PubMed] [Google Scholar]
- 32.Goldberg AL. 2012. Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol 199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogl DT, Martin TG, Vij R, Hari P, Mikhael JR, Siegel D, Wu KL, Delforge M, Gasparetto C. 2017. Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leuk Lymphoma 58:1872–1879. doi: 10.1080/10428194.2016.1263842. [DOI] [PubMed] [Google Scholar]
- 34.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. 2007. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK. 2007. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67:6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 36.Luan B, Huang X, Wu J, Mei Z, Wang Y, Xue X, Yan C, Wang J, Finley DJ, Shi Y, Wang F. 2016. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc Natl Acad Sci U S A 113:2642–2647. doi: 10.1073/pnas.1601561113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. 2009. Inhibitors selective for mycobacterial versus human proteasomes. Nature 461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrader J, Henneberg F, Mata RA, Tittmann K, Schneider TR, Stark H, Bourenkov G, Chari A. 2016. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 353:594–598. doi: 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- 39.Cobo ER, Eckmann L, Corbeil LB. 2011. Murine models of vaginal trichomonad infections. Am J Trop Med Hyg 85:667–673. doi: 10.4269/ajtmh.2011.11-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pereira AR, Kale AJ, Fenley AT, Byrum T, Debonsi HM, Gilson MK, Valeriote FA, Moore BS, Gerwick WH. 2012. The carmaphycins: new proteasome inhibitors exhibiting an α,β-epoxyketone warhead from a marine cyanobacterium. Chembiochem 13:810–817. doi: 10.1002/cbic.201200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto Y, Kalisiak J, Korthals K, Lauwaet T, Cheung DY, Lozano R, Cobo ER, Upcroft P, Upcroft JA, Berg DE, Gillin FD, Fokin VV, Sharpless KB, Eckmann L. 2013. Expanded therapeutic potential in activity space of next-generation 5-nitroimidazole antimicrobials with broad structural diversity. Proc Natl Acad Sci U S A 110:17564–17569. doi: 10.1073/pnas.1302664110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Totaro KA, Barthelme D, Simpson PT, Jiang X, Lin G, Nathan CF, Sauer RT, Sello JK. 2017. Rational design of selective and bioactive inhibitors of the Mycobacterium tuberculosis proteasome. ACS Infect Dis 3:176–181. doi: 10.1021/acsinfecdis.6b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, Brown J, Li S, Swanson J, Rayner JC, Jiang RHY, Adams JH. 2018. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360:eaap7847. doi: 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie SC, Gillett DL, Spillman NJ, Tsu C, Luth MR, Ottilie S, Duffy S, Gould AE, Hales P, Seager BA, Charron CL, Bruzzese F, Yang X, Zhao X, Huang SC, Hutton CA, Burrows JN, Winzeler EA, Avery VM, Dick LR, Tilley L. 2018. Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J Med Chem 61:10053–10066. doi: 10.1021/acs.jmedchem.8b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoo E, Stokes BH, de Jong H, Vanaerschot M, Kumar T, Lawrence N, Njoroge M, Garcia A, Van der Westhuyzen R, Momper JD, Ng CL, Fidock DA, Bogyo M. 2018. Defining the determinants of specificity of Plasmodium proteasome inhibitors. J Am Chem Soc 140:11424–11437. doi: 10.1021/jacs.8b06656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Ponder EL, Verdoes M, Asbjornsdottir KH, Deu E, Edgington LE, Lee JT, Kirk CJ, Demo SD, Williamson KC, Bogyo M. 2012. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem Biol 19:1535–1545. doi: 10.1016/j.chembiol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter MB, La Greca F, Arastu-Kapur S, Caiazza F, Cimermancic P, Buchholz TJ, Anderl JL, Ravalin M, Bohn MF, Sali A, O'Donoghue AJ, Craik CS. 2017. Immunoproteasome functions explained by divergence in cleavage specificity and regulation. Elife 6. doi: 10.7554/eLife.27364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber EM, de Bruin G, Heinemeyer W, Paniagua Soriano G, Overkleeft HS, Groll M. 2015. Systematic analyses of substrate preferences of 20S proteasomes using peptidic epoxyketone inhibitors. J Am Chem Soc 137:7835–7842. doi: 10.1021/jacs.5b03688. [DOI] [PubMed] [Google Scholar]
- 49.Lei B, Abdul Hameed MD, Hamza A, Wehenkel M, Muzyka JL, Yao XJ, Kim KB, Zhan CG. 2010. Molecular basis of the selectivity of the immunoproteasome catalytic subunit LMP2-specific inhibitor revealed by molecular modeling and dynamics simulations. J Phys Chem B 114:12333–12339. doi: 10.1021/jp1058098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. 1994. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 51.Bridgford JL, Xie SC, Cobbold SA, Pasaje CFA, Herrmann S, Yang T, Gillett DL, Dick LR, Ralph SA, Dogovski C, Spillman NJ, Tilley L. 2018. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun 9:3801. doi: 10.1038/s41467-018-06221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, Chotivanich K, Kenny S, Gnadig N, Straimer J, Bozdech Z, Fidock DA, Simpson JA, Dondorp AM, Foote S, Klonis N, Tilley L. 2015. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 13:e1002132. doi: 10.1371/journal.pbio.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kung E, Furnkranz U, Walochnik J. 2019. Chemotherapeutic options for the treatment of human trichomoniasis. Int J Antimicrob Agents 53:116–127. doi: 10.1016/j.ijantimicag.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 54.Gupta N, Hanley MJ, Xia C, Labotka R, Harvey RD, Venkatakrishnan K. 2018. Clinical Pharmacology of ixazomib: the first oral proteasome inhibitor. Clin Pharmacokinet doi: 10.1007/s40262-018-0702-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.duBouchet L, McGregor JA, Ismail M, McCormack WM. 1998. A pilot study of metronidazole vaginal gel versus oral metronidazole for the treatment of Trichomonas vaginalis vaginitis. Sex Transm Dis 25:176–179. doi: 10.1097/00007435-199803000-00012. [DOI] [PubMed] [Google Scholar]
- 56.Baloglu E, Senyigit ZA, Karavana SY, Bernkop-Schnürch A. 2009. Strategies to prolong the intravaginal residence time of drug delivery systems. J Pharm Sci 12:312–336. [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan P, Zhang J, Martin A, Kelley K, McNicholl JM, Buckheit RW Jr, Smith JM, Ham AS. 2016. Safety and pharmacokinetics of quick-dissolving polymeric vaginal films delivering the antiretroviral IQP-0528 for preexposure prophylaxis. Antimicrob Agents Chemother 60:4140–4150. doi: 10.1128/AAC.00082-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mashingaidze F, Choonara YE, Kumar P, Du Toit LC, Maharaj V, Buchmann E, Pillay V. 2017. Submicron matrices embedded in a polymeric caplet for extended intravaginal delivery of zidovudine. AAPS J 19:1745–1759. doi: 10.1208/s12248-017-0130-4. [DOI] [PubMed] [Google Scholar]
- 59.Kissinger P. 2015. Epidemiology and treatment of trichomoniasis. Curr Infect Dis Rep 17:484. doi: 10.1007/s11908-015-0484-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown DM, Upcroft JA, Dodd HN, Chen N, Upcroft P. 1999. Alternative 2-keto acid oxidoreductase activities in Trichomonas vaginalis. Mol Biochem Parasitol 98:203–214. doi: 10.1016/s0166-6851(98)00169-8. [DOI] [PubMed] [Google Scholar]
- 61.Upcroft JA, Upcroft P. 2001. Drug susceptibility testing of anaerobic protozoa. Antimicrob Agents Chemother 45:1810–1814. doi: 10.1128/AAC.45.6.1810-1814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skirrow SZ, BonDurant RH. 1990. Induced Tritrichomonas foetus infection in beef heifers. J Am Vet Med Assoc 196:885–889. [PubMed] [Google Scholar]
- 63.Shevchenko A, Tomas H, Havli J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The complete mass spectrometry data and annotations can be accessed on the Mass Spectrometry Interactive Virtual Environment (MassIVE) public data repository under accession number MSV000083474. The FTP download link is ftp://massive.ucsd.edu/MSV000083474.