

Abstract
Background and Purpose
Osteoarthritic pain is a chronic disabling condition lacking effective treatment. Continuous use of opioid drugs during osteoarthritic pain induces tolerance and may result in dose escalation and abuse. Sigma‐1 (σ1) receptors, a chaperone expressed in key areas for pain control, modulates μ‐opioid receptor activity and represents a promising target to tackle these problems. The present study investigates the efficacy of the σ1 receptor antagonist E‐52862 to inhibit pain sensitization, morphine tolerance, and associated electrophysiological and molecular changes in a murine model of osteoarthritic pain.
Experimental Approach
Mice received an intra‐knee injection of monoiodoacetate followed by 14‐day treatment with E‐52862, morphine, or vehicle, and mechanical sensitivity was assessed before and after the daily doses.
Key Results
Monoiodoacetate‐injected mice developed persistent mechanical hypersensitivity, which was dose‐dependently inhibited by E‐52862. Mechanical thresholds assessed before the daily E‐52862 dose showed gradual recovery, reaching complete restoration by the end of the treatment. When repeated treatment started 15 days after knee injury, E‐52862 produced enhanced short‐term analgesia, but recovery to baseline threshold was slower. Both a σ1 receptor agonist and a μ receptor antagonist blocked the analgesic effects of E‐52862. An acute, sub‐effective dose of E‐52862 restored morphine analgesia in opioid‐tolerant mice. Moreover, E‐52862 abolished spinal sensitization in osteoarthritic mice and inhibited pain‐related molecular changes.
Conclusion and Implications
These findings show dual effects of σ1 receptor antagonism alleviating both short‐ and long‐lasting antinociception during chronic osteoarthritis pain. They identify E‐52862 as a promising pharmacological agent to treat chronic pain and avoid opioid tolerance.
Abbreviation
- BDNF
brain‐derived neurotrophic factor
- DRG
dorsal root ganglia
- MIA
monoiodoacetate
- NPY
neuropeptide Y
- WDR
wide dynamic range
- σ1 receptor
sigma‐1 receptor
What is already known
σ1 receptor antagonists modulate μ‐opioid receptor activity and acutely alleviate inflammatory and neuropathic pain.
What this study adds
Repeated E‐52862 promotes gradual normalization of mechanical sensitivity inhibiting neuroinflammation, critical for osteoarthritic pain.
E‐52862 antinociception involves μ‐opioid receptors and reverses morphine tolerance during chronic osteoarthritic pain.
What is the clinical significance
E‐52862 could dampen deleterious side effects of opioid and be an alternative for long‐term treatments.
E‐52862 provides acute and long‐lasting pain‐relieving effects during osteoarthritic pain.
1. INTRODUCTION
Osteoarthritis is the most frequent chronic musculoskeletal pain condition (Breivik, Collett, Ventafridda, Cohen, & Gallacher, 2006), characterized by progressive destruction of articular cartilage (Sutton et al., 2009; Zhang, Ren, & Dubner, 2013). Pain is the major symptom of osteoarthritis and the reason for presentation of patients to clinical services. However, available therapeutic approaches do not control the progression of the disease and do not provide satisfactory analgesia (Wieland, Michaelis, Kirschbaum, & Rudolphi, 2005). Opioids are potent analgesics widely used for severe pain management. However, prolonged administration has been associated with tolerance, abuse liability, and hyperalgesia (Vowles et al., 2015). Long‐term opioid prescriptions increased importantly over the last decade, mainly in the United States, becoming a public health problem with devastating consequences including overdose‐related deaths (Lyden & Binswanger, 2019). Therefore, there is an urgent need to develop safer therapeutic alternatives for chronic pain.
The shttp://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2552 is a ligand‐regulated chaperone located mainly in the endoplasmic reticulum (Alonso et al., 2000). It has been proposed as an amplifier of signal transduction cascades that modulate a variety of receptors and ion channels (Su, Hayashi, Maurice, Buch, & Ruoho, 2010; Zamanillo, Romero, Merlos, & Vela, 2013). The σ1 receptors are expressed in areas of the nervous system crucial for pain transmission such as the dorsal root ganglia (DRG), dorsal horn, and periaqueductal grey (Alonso et al., 2000; Bangaru et al., 2013). Behavioural studies showed their involvement in models of neuropathic and inflammatory pain (Romero, Merlos, & Vela, 2016).
Osteoarthritis involves inflammatory and neuropathic pain mechanisms (Ivanavicius et al., 2007; Wylde, Hewlett, Learmonth, & Dieppe, 2011) at distinct points of the disease (Arendt‐Nielsen et al., 2010), but the role of σ1 receptors in these different stages has not been yet investigated. Moreover, functional interactions have been reported between σ1 receptors and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319s (Kim et al., 2010). Indeed, σ1 receptor antagonists enhance opioid‐induced analgesia in rodent models of acute nociception and inflammation (Chien & Pasternak, 1995; Montilla‐García et al., 2019; Vidal‐Torres et al., 2013) and did not potentiate opioid‐induced side effects such as tolerance or physical dependence (Vidal‐Torres et al., 2013), which could represent an important advantage for long‐term opioid treatment. However, it is not known if σ1 receptors modulate opioid analgesia and tolerance during osteoarthritis chronic pain. Interestingly, σ1 receptors are up‐regulated and redistributed from the endoplasmic reticulum to other subcellular locations under cellular stress (Hayashi & Su, 2007; Zamanillo et al., 2013), which is associated to chronic pain and morphine tolerance (Inceoglu et al., 2015; Liu et al., 2018). Hence, characterizing the role of σ1 receptors in osteoarthritic pain and opioid tolerance could provide insights into improving the available therapeutic strategies.
This study investigated the role of σ1 receptors in the monoiodoacetate (MIA) mouse model of osteoarthritic pain and its participation on opioid tolerance. We assessed the antinociceptive effects of acute and chronic treatment with the σ1 receptor antagonist E‐52862 (also named S1RA [Romero et al., 2012] and MR309 [Castany, Gris, Vela, Verdú, & Boadas‐Vaello, 2018]) and evaluated its effects in vivo on spinal electrophysiological recordings performed in animals with osteoarthritic pain. Involvement of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 in the analgesic effects of E‐52862 and its participation on morphine tolerance were also investigated. Pain and morphine‐associated biochemical alterations were also investigated in spinal cord and DRG of mice with osteoarthritic pain.
2. METHODS
2.1. Animals
All experimental procedures and animal husbandry were conducted following the ARRIVE guidelines and according to the ethical principles of the International Association for the Study of Pain (IASP) for the evaluation of pain in conscious animals (Zimmermann, 1986) and the European Parliament and the Council Directive (2010/63/EU) and were approved by the Animal Care and Use Committees of the PRBB and Departament de Territori I Habitatge of Generalitat de Catalunya. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
Swiss albino male mice (Charles River, Lyon, France) 8 to 12 weeks old were used in all the experiments. Mice weighed 22 to 24 g at the beginning of the experiments and were housed in groups of 3–4 with free access to water and food. Housing conditions were maintained at 21 ± 1°C and 55 ± 10% relative humidity in a controlled light/dark cycle (light on between 8:00 a.m. and 8:00 p.m.). All experiments were performed under blinded conditions.
2.2. Intra‐articular injection of MIA
Osteoarthritic pain was induced in mice briefly anaesthetized with isoflurane (2% v/v) vaporized in oxygen. The knee joint was shaved and flexed at a 90o angle and 10 μl of MIA (10 mg·ml−1, Sigma‐Aldrich, UK) dissolved in sterile saline (NaCl 0.9%) were injected into the joint space with a 30‐gauge needle, as previously described (Negrete, García Gutiérrez, Manzanares, & Maldonado, 2017). Sham mice received the same volume of sterile saline.
2.3. Nociceptive behaviour
Hypersensitivity to punctate stimuli (which will be referred as mechanical allodynia) was used as outcome measure of osteoarthritic pain by measuring the hind paw withdrawal response to von Frey filaments stimulation (Chaplan, Bach, Pogrel, Chung, & Yaksh, 1994). Briefly, animals were placed in Plexiglas cylinders (20 cm high, 9 cm diameter) positioned on a grid surface through which calibrated von Frey filaments (North Coast Medical, USA) were applied following the up‐down paradigm as previously reported (Chaplan et al., 1994). The 0.4‐g filament was used first, and the strength of the next filament was decreased or increased according to the response following this sequence 0.07, 0.16, 0.4, 0.6, 1, 2 g. The 2‐g filament was used as a cut‐off. The mechanical threshold (in grams) was then calculated by the up‐down Excel program (Dixon, 1965). Animals were habituated for 1 hr before testing to allow an appropriate behavioural immobility. Clear paw withdrawal, shaking, or licking was considered as a nociceptive response. Both ipsilateral and contralateral hind paws were tested. Only ipsilateral responses are shown since contralateral sides showed no significant alteration of the mechanical thresholds. The percentage of inhibition of mechanical nociception for the dose–response curves was calculated based on the hypersensitivity before the drug administration (0%) and the maximal possible mechanical thresholds (100%).
2.4. Drug preparation
The selective σ1 receptor antagonist E‐52862 (4‐(2‐[5‐methyl‐1‐(naphthalen‐2‐yl)‐1H‐pyrazol‐3‐yloxy]ethyl) morpholine) and the μ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 were dissolved in an aqueous solution (0.5% hydroxypropylmethyl cellulose, HPMC) and were administered by the i.p. route at a volume of 10 ml·kg−1 30 min before behavioural testing. For spinal electrophysiological recordings, the doses of E‐52862 were selected based on previous studies showing antinociceptive effects after intrathecal administration (Vidal‐Torres et al., 2014). The σ1 receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6678 ([2‐(4‐morpholinethyl)1‐phenylcyclohexanecarboxylate]) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638 were diluted in physiological saline (0.9% sodium chloride) and were administered s.c. at a volume of 5 ml·kg−1, 5 min before E‐52862.
2.5. In vivo spinal cord electrophysiology
In vivo electrophysiology was performed 13–17 days following MIA/sham injection. Animals were anaesthetized with 3.5% v/v isoflurane delivered in 3:2 ratio of nitrous oxide and oxygen and were maintained on 1.5% v/v for the whole duration of the experiment (approximately 5–7 hr), core body temperature was regulated with a homeothermic blanket, and respiratory rate was visually monitored. Mice were secured in a stereotaxic frame and a laminectomy was performed exposing L3‐L5 of the spinal cord. Extracellular recordings were performed using 127‐μm‐diameter parylene‐coated tungsten electrodes. Single wide dynamic range (WDR) neurons receiving inputs from the hind paw were isolated in the ipsilateral dorsal horn, and the receptive field was then stimulated using a wide range of stimuli: brush, von Frey filaments (0.16, 0.4, 0.6, 1, 2, 4, 6, 8, 15, 26, 60 g), static pressure (pinch), and heat (48°C) applied over a period of 10 s per stimulus. Stimuli were applied starting with the stimulus of lowest intensity in the following order: brush, von Frey, pressure, and heat; 1–2 WDR cells were isolated from each animal, and the average response was calculated. A sample size of six to eight animals per group was obtained except for the sham group where only four animals could be recorded due to casualties during the process of neuron isolation, where animals are maintained under anaesthesia for long periods of time.
Baseline recordings were made with 15‐μl vehicle (0.9% saline) applied to the dorsal part of the spinal cord. After obtaining three to four baseline responses (with 5 min between each set of trials, data were averaged to obtain control values), the vehicle was removed and 90 and 180 μg E‐52862 diluted in saline were applied to the spinal cord in a volume of 15 μl. Firing frequency was recorded 10 and 30 min after application of the drug. All mice were terminally anaesthetized with isoflurane after the experiment. Data were captured and analysed using a CED 1401 interface coupled to a computer running Spike 2 software.
2.6. Tissue isolation
Mice were killed by cervical dislocation 1 or 15 days after sham or induction of osteoarthritis. The dorsal lumbar region of the spinal cord and L3‐L5 DRG ipsilateral to the side of the knee injection were rapidly isolated. Tissues were fresh frozen and stored at −80°C until use.
2.7. Immunoblot analysis
The antibody‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). Frozen tissues were processed to obtain the total solubilized fraction, as previously described (Ozaita, Puighermanal, & Maldonado, 2007). Briefly, tissues were homogenized (Dounce homogenizer) in 30 volumes of lysis buffer (50 mmol·L−1 Tris–HCl pH 7.4, 150 mmol·L−1 NaCl, 10% glycerol, 1 mmol·L−1 EDTA, 1 μg·ml−1 aprotinin, 1 μg·ml−1 leupeptin, 1 μg·ml−1 pepstatin, 1 mmol·L−1 sodium fluoride, 5 mmol·L−1 sodium pyrophosphate, and 40 mmol·L−1 β‐glycerolphosphate) with 1% Triton X‐100. After 10 min of incubation at 4°C, samples were centrifuged at 16,000 g for 30 min to remove insoluble debris. Protein contents in the supernatants were determined by DC‐micro plate assay following manufacturer's instructions. Blots containing equal amounts of protein samples were probed for different primary antibodies: anti‐Iba1 (1:500), anti‐GluA2 (1:2,000), anti‐phosphoGluN1 (1:500), anti‐ GluN1 (1:2,000), anti‐phospho GluN2B (1:500), anti‐ GluN2B (1:1,000), anti‐mGlu5 (1:2,000), and anti‐GAPDH (1:10,000), and anti‐actin (1:10,000). Bound primary antibodies were detected using HRP‐conjugated antibodies to rabbit (1:2,000) or mouse antibodies (1:2000) and visualized by enhanced chemiluminescence detection (Clarity Western ECL Substrate). When necessary, Immobilon‐P membranes were stripped in buffer containing 62.5‐mM Tris pH 6.5, 2% SDS (v/v), and 0.1‐M β‐mercaptoethanol for 30 min at 37°C, followed by extensive washing in 100‐mM NaCl, 10‐mM Tris, and 0.1% Tween 20 (pH 7.4) before re‐blocking and re‐probing. The optical density of the relevant immunoreactive bands was quantified after acquisition on a ChemiDoc MP Imaging System controlled by Image Lab Touch Software. For quantitative purposes, the optical density values of phospho‐specific antibodies were normalized to the detection of non‐phospho antibodies in the same sample, and GAPDH or actin was used as the housekeeping control. Data were expressed as a fold change of the control sham‐vehicle group.
2.8. Gene expression analysis by real‐time PCR
Total RNA was isolated from frozen (−80°C) spinal cords and DRG with TRIzol reagent plus RNA purification kit and subsequently retrotranscribed to cDNA with the High‐capacity cDNA reverse transcription kit. Gene expression of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1504, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4872, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074, and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 in both tissues obtained 1 and 15 days after MIA injection was assessed by real‐time PCR. Quantitative analysis of gene expression was measured with TaqMan Gene Expression assays “Mm01410146_m1” for NPY, “Mm01334043_m1” for BDNF, “Mm00443258_m1” for TNF‐α, and “Mm00434228_m1” for IL‐1β as a double‐stranded DNA‐specific fluorescent dye and performed on the ABI Prism 7900 HT. HPRT was used as housekeeping gene, detected with TaqMan gene expression assay “Mm00446968_m1.” Data for each target gene were normalized to HPRT, and the fold change in target gene mRNA abundance was determined by the 2(−∆∆Ct) method (Livak & Schmittgen, 2001).
2.9. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Sample size was based on previous behavioural studies with similar number of factors under the same experimental conditions that achieved statistically significant results (Gris, Merlos, Vela, Zamanillo, & Portillo‐Salido, 2014; Negrete et al., 2017). Mice were randomly assigned to each surgery and/or treatment group, generating groups with similar initial number of animals for each experiment. The minor variations in the final number among the groups in each experiment are due to the loss of one to two animals during the whole experimental sequence mainly as a result of surgical procedures and subsequent consequences. Animals showing behavioural abnormalities after surgical procedures were killed following the ethical committee criteria. Animal casualties during the electrophysiological recordings were higher, and a group of four mice was obtained, which was not subjected to statistical analysis. Outliers were not excluded, and the declared group size is the number of independent values. A three‐way repeated measure ANOVA with “surgery” as between factor and “day” and “pre versus post” as within‐subject factors was used to analyse von Frey data during chronic treatment. To analyse the differences between early and late treatments and pre and post values, the slopes of the regression lines were calculated for each animal and the average slope for each group. Afterwards, a two‐way ANOVA was used with “early and late” as between factor and “pre and post” as within‐subject factor. For von Frey assessment on Day 14 before and after acute treatments, a two‐way repeated measures ANOVA was used, with “treatment” as between‐subjects factor and “pre versus post” as within‐subject factor. For dose–response curves, an F test was used to compare the non‐linear regression fittings. Electrophysiological data were analysed with an F test to compare non‐linear regressions and a two‐way repeated measure ANOVA (“surgery” as between‐subject factor and “stimulus” as within‐subject factor). A two‐way ANOVA (“surgery” and “treatment”) was used to analyse data from molecular studies. In all comparisons, Fisher least significant difference (LSD) post hoc analysis was applied when appropriate (significant interaction between factors). STATISTICA 6.0 software was used. The differences were considered statistically significant when the P value was below .05.
2.10. Materials
The selective σ1 receptor antagonist E‐52862 (4‐(2‐[5‐methyl‐1‐(naphthalen‐2‐yl)‐1H‐pyrazol‐3‐yloxy]ethyl) morpholine) and the σ1 receptor agonist PRE‐084 ([2‐(4‐morpholinethyl)1‐phenylcyclohexanecarboxylate]) were developed and supplied by Laboratories Esteve (Barcelona, Spain). The μ receptor agonist morphine hydrochloride was obtained from the General Directorate of Pharmacy and Drugs, Spanish Ministry of Health (Madrid, Spain), and the μ receptor antagonist naloxone hydrochloride was purchased from Sigma‐Aldrich (Saint Louis, United States). HPMC was obtained from Sigma‐Aldrich (Saint Louis, United States). For the electrophysiological study, parylene‐coated tungsten electrodes from A‐M Systems (Sequim, United States) and Spike 2 software from Cambridge Electronic Design (Cambridge, United Kingdom, RRID:SCR_000903) were used. DC‐micro plate assay was purchase from Bio‐Rad (Hercules, United States), and Immobilon‐P membranes from Merck Millipore (Burlington, United States; Cat# IPVH09120). In the molecular studies, the following antibodies were used: anti‐Iba1 (Wako Pure Chemical Industries, Osaka, Japan; Cat# 016‐20001, RRID:AB_839506), anti‐GluR2 (Merck Millipore; Cat# AB1768, RRID:AB_2313802), anti‐phosphoNR1 (Cell Signalling, Danvers, United States; Cat# 3381, RRID:AB_2294781), anti‐NR1 (Novus Biologicals, Centennial, United States; Cat# NB300‐118, RRID:AB_10002447), anti‐phosphoNR2B (Sigma‐Aldrich, Saint Louis, United States; Cat# M2442, RRID:AB_262150), anti‐NR2B (Merck Millipore; Cat# AB1557P, RRID:AB_90772), anti‐mGluR5 (Merck Millipore; Cat# AB5675, RRID:AB_2295173), anti‐GAPDH (Santa Cruz Technologies, Dallas, United States; Cat# sc‐32233, RRID:AB_627679), anti‐Actin (Sigma‐Aldrich, Saint Louis, United States; Cat# A5441, RRID:AB_476744), anti‐rabbit (Cell signalling; Cat# 7074, RRID:AB_2099233) and anti‐mouse (Santa Cruz Technologies; Cat# sc‐2005, RRID:AB_631736). The optical density of the relevant immunoreactive bands was visualized using Clarity Western ECL Substrate (Cat# 1705061) and the ChemiDoc MP Imaging System from Bio‐Rad. For the gene expression analysis by real‐time PCR, the following reagents were used: TRIzol reagent plus RNA purification kit from Ambion (Waltham, United States; Cat# AM1924), and high‐capacity cDNA reverse transcription kit (Cat# 4368814), TaqMan Gene Expression assays, and ABI Prism 7900 HT from Applied Biosystems (Waltham, United States). The statistical analysis was performed with the STATISTICA 6.0 software was used (StatSoft, Inc., Tulsa, United States, RRID:SCR_014213).
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos, et al., 2017; Alexander, Kelly et al., 2017; Alexander, Peters et al., 2017).
3. RESULTS
3.1. The σ1 receptor antagonist E‐52862 produces acute antinociception and a gradual normalization of mechanical sensitivity during osteoarthritic pain
To assess the therapeutic potential of E‐52862 in pain due to chronic osteoarthritis, we evaluated its effects on mechanical allodynia in mice subjected to joint pain induced by MIA. Mice received vehicle or different i.p. E‐52862 doses (5–10–20 mg·kg−1) twice a day during 14 days, starting the first day after MIA or sham injection. Mechanical thresholds were measured before (PRE values) and 30 min after (POST) the first daily administration (Figure 1a). Baseline mechanical thresholds were similar in all groups, and sham animals did not show nociceptive changes during the experiment, regardless of the treatment (Figure 1b,g). Intra‐knee injection of MIA led to a marked decrease of withdrawal thresholds to mechanical stimuli when compared with sham. Mechanical allodynia was shown from the first day after MIA until the end of the experiment in vehicle‐treated mice (Figure 1b,f). Acute E‐52862 doses of 5 and 10 mg·kg−1 were sub‐effective, whereas 20 mg·kg−1 clearly alleviated MIA‐induced hypersensitivity, demonstrating dose‐dependent analgesic effects of E‐52862 after single administration (POST values compared with PRE values at Day 1; Figure 1c,e). Interestingly, E‐52862 also induced a gradual recovery of the mechanical thresholds measured before its daily administration. This recovery was significant after the seventh day of treatment for all doses tested (Figure 1c,e). Hence, the sustained recovery of mechanical thresholds was independent of the E‐52862 doses tested.
Figure 1.
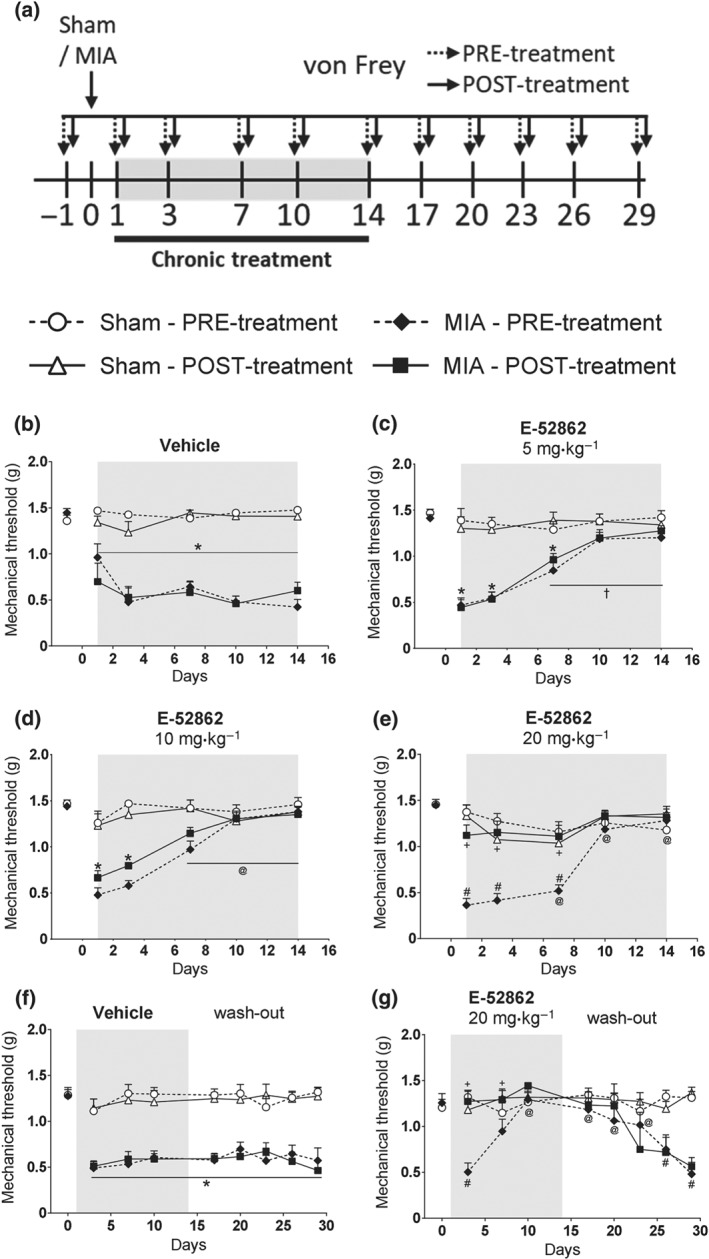
The σ1 receptor antagonist E‐52862 produces both acute anti‐allodynia and a gradual normalization of mechanical thresholds in a model of osteoarthritic pain. (a) Mice received an intra‐knee injection of MIA or saline and were treated with vehicle (0.5% HMPC) or E‐52862 (5, 10, or 20 mg·kg−1) twice a day from Day 1 to Day 14. Mechanical allodynia was assessed with the von Frey test before (PRE) and 30 min after (POST) the first daily dose, under basal conditions and 1, 3, 7, 10, 14, and 17, 20, 23, 26, and 29 days after the intra‐articular injection. (b) MIA‐injected mice treated with vehicle showed a persistent decrease on mechanical thresholds. (c–e) E‐52862 produced acute dose‐dependent antiallodynic effects (POST values) and a gradual recovery of mechanical thresholds observed before the daily doses (PRE values). (f) MIA‐induced reduction of mechanical thresholds persisted up to 29 days in vehicle‐treated mice. (g) The sustained recovery induced by chronic E‐52862 was maintained for 9 days after interrupting the repeated treatment. The von Frey pressure (g) required to elicit the paw withdrawal is expressed as mean ± SEM. The number of animals is as follows (first value represents sham groups and second value represents MIA groups): (b) 8, 8; (c) 8, 8; (d) 8, 8; (e) 8, 10; (f) 8, 16; and (g) 8, 8. *P<.05, significant difference between MIA and sham; † P<.05, significant difference between MIA‐PRE and MIA‐PRE at Day 1; #P<.05, significant difference between MIA‐PRE and Sham‐PRE; + P<.05, significant difference between MIA‐PRE and MIA‐POST; three‐way repeated measures ANOVA plus Fisher least significant difference test. MIA, monoiodoacetate
To evaluate the persistence of E‐52862‐induced antiallodynic effects, we continued the evaluation of mechanical thresholds after ending the repeated treatment (wash‐out period; Figure 1f,g). Mice previously treated with vehicle or E‐52862 (20 mg·kg−1, 14 days) were evaluated for 15 additional days after ending the treatment. The reduction of mechanical allodynia induced by the 14‐day treatment was maintained for 9 days after interrupting E‐52862 administration (Figure 1g). Therefore, the blockade of σ1 receptors had sustained antiallodynic effects that lasted for several days after treatment discontinuation.
3.2. The analgesic efficacy of acute or repeated treatments with E‐52862 depends on the stage of the osteoarthritic pain sensitization
Treatment with the σ1 receptor antagonist showed acute and long‐lasting efficacy inhibiting osteoarthritic pain at stages in which inflammatory and neuroplastic changes may have not been fully developed. Thus, we wanted to assess its analgesic efficacy once these changes were already present. We compared the efficacy of acute and chronic E‐52862 treatments starting 1 (Early) or 15 (Late) days after MIA injection (Figure 2a). MIA‐injected mice showed similar decrease of mechanical thresholds 1 and 15 days after MIA (Figures 1b and 2b). We observed a significant shift to the left of the dose–response curve of E‐52862 measured 15 days after MIA injection, when compared with the curve assessed on Day 1 (Figure 2c). This was reflected in lower median effective dose (ED50) of the σ1 receptor antagonist 15 days after MIA (ED50 = 7.024 mg·kg−1) than on Day 1 (ED50 = 14.10 mg·kg−1). Thus, the dose needed to induce pain relief on Day 15 was half of the dose required on Day 1.
Figure 2.
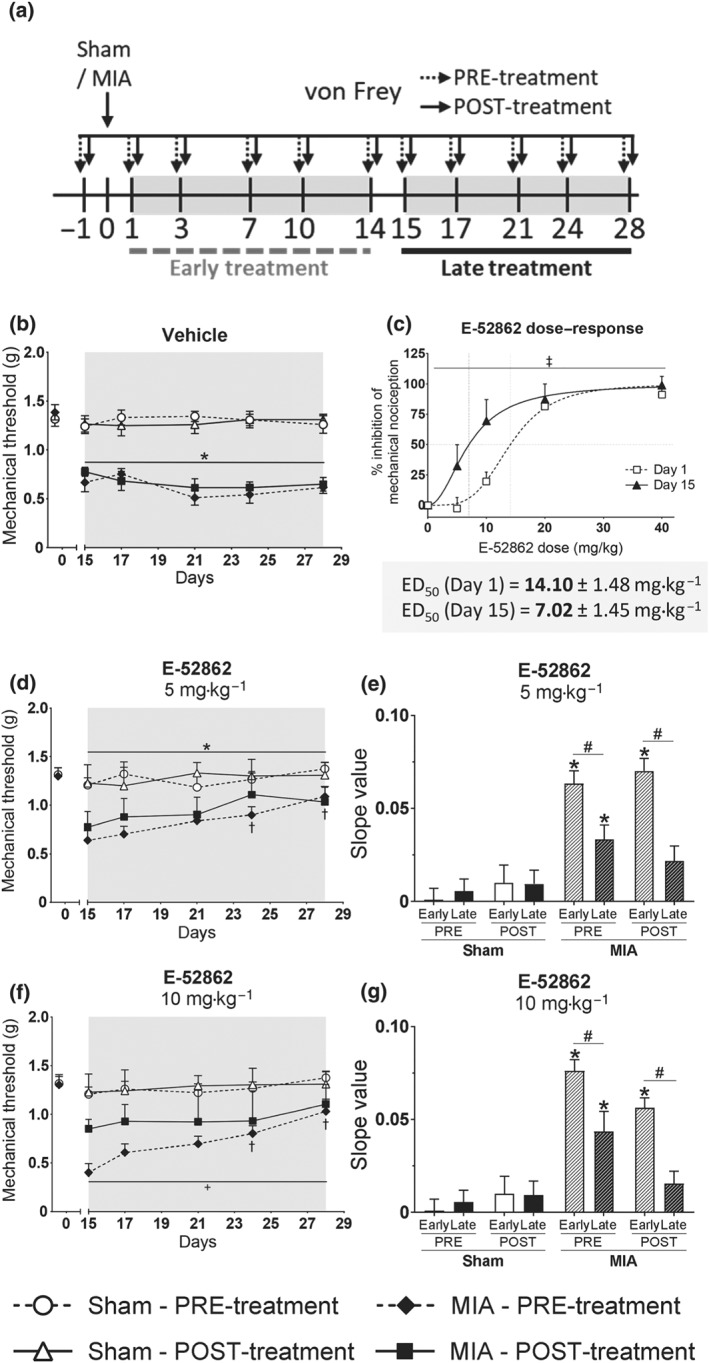
Acute and repeated E‐52862 treatments starting 1 or 15 days after MIA injection differ in their analgesic efficacy. (a) The analgesic effects of acute and chronic E‐52862 (5 or 10 mg·kg−1, twice a day during 14 days) were compared between treatments starting 1 (early) or 15 (late) days after the injection of MIA. (b) MIA‐induced sensitization was stable in vehicle‐treated mice from Day 15 to Day 28 after intra‐knee injection. (c) Lower doses were needed to induce acute relief of mechanical hypersensitivity 15 days than 1 day after the intra‐knee injection of MIA. Mice receiving chronic late treatments with E‐52862 5 mg·kg−1 (d) and 10 mg·kg−1 (f) also showed a recovery of the mechanical thresholds. (e,g) The restoration of mechanical sensitivity was slower in the late than in the early treatment protocol, as reflected on reduced slopes of the time‐course curves of mechanical allodynia. Data are expressed as mean ± SEM. The number of animals is the following (first value represents Day 1 or sham groups and second value represents Day 15 or MIA groups): (b) 9, 8; (c) 10, 8; and (d) 8, 8; (f) 8, 8. For panels (b) and (d–g), *P<.05, significant difference between MIA and Sham; †P<.05, significant difference between MIA and MIA at Day 1; + for MIA‐PRE versus MIA‐POST (three‐way repeated measures ANOVA plus Fisher least significant difference test). For panel (c), ‡P<.05, significant difference between Day 1 and Day 15 (F test of non‐linear regression). ED50, median effective dose; MIA, monoiodoacetate
Comparison between early and late repeated treatments with E‐52862 (5 and 10 mg·kg−1) revealed that the time period in which the σ1 receptor antagonist was applied had a significant effect on the gradual recovery of mechanical thresholds. MIA‐induced sensitization was stable in vehicle‐treated mice up to 28 days after intra‐knee injection (Figure 2b). Mice receiving the late chronic E‐52862 treatment also showed the sustained restoration of mechanical sensitivity observed in the previous treatment schedule (Figure 2d,f), but this improvement was slower compared to the treatment starting at Day 1, as reflected on reduced slopes of the time‐course curves of mechanical allodynia (Figure 2e,g). Therefore, late repeated treatments with E‐52862 required a longer duration of treatment to restore mechanical thresholds than that for early treatments, even if single administrations at late time points showed higher acute antinociceptive effects.
3.3. Spinal administration of the σ1 receptor antagonist reduces evoked firing frequency of spinal neurons from mice with osteoarthritic pain
To distinguish whether E‐52862 could modulate central sensitization in the spinal cord of mice with osteoarthritic pain (Harvey & Dickenson, 2009), in vivo electrophysiological recordings were performed in WDR neurons of lamina V of the dorsal horn. These neurons respond to mechanical and thermal stimuli, including punctate stimulation, dynamic brush, static pressure, and heat (Figure 3a). In this exploratory experiment, evoked responses to punctate mechanical stimuli were facilitated in osteoarthritic mice (Figure 3b). However, firing frequency evoked by the other stimuli was similar in MIA and sham animals (Figure 3c). Application of 90 μg of E‐52862 to the exposed dorsal horn of mice with osteoarthritis significantly reduced firing frequency in response to mechanical and thermal stimuli 30 min after administration (Figure 3d,e). Application of 180 μg induced earlier inhibitory effects that were evident 10 and 30 min after E‐52862 (Figure 3f,g), indicating dose‐dependent drug effects. Hence, MIA induced central sensitization characterized by increased firing frequency of spinal WDR neurons that was normalized by the pharmacological blockade of σ1 receptors. This effect is associated with the short‐term antiallodynic effects of E‐52862 observed in mice with osteoarthritic pain.
Figure 3.
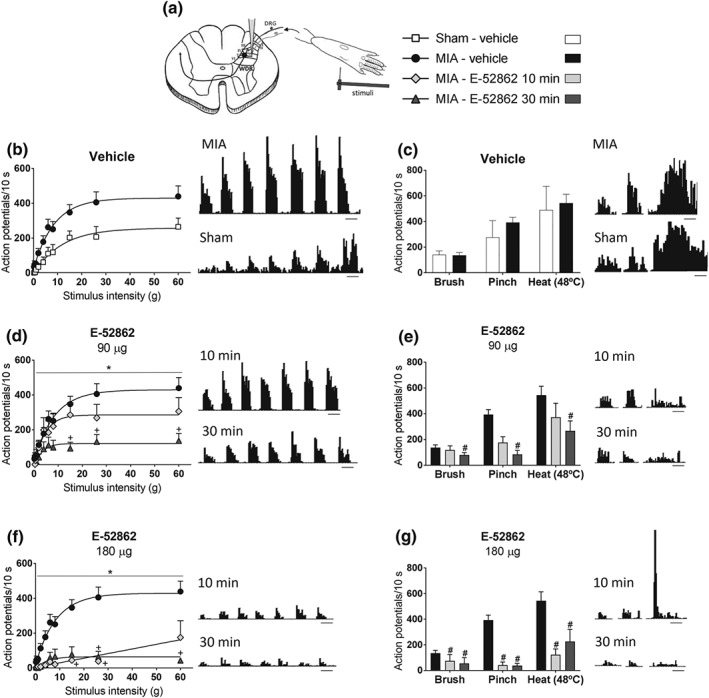
E‐52862 reduces osteoarthritic pain‐facilitated firing frequency of WDR spinal neurons. (a) Extracellular recordings were assessed from ipsilateral WDR neurons in the lamina 5 of the dorsal horn. The receptive field in the hind paw was stimulated using von Frey filaments, dynamic brush, static pressure (pinch), and heat (48°C). (b) MIA injection facilitated the evoked responses to punctate mechanical stimuli. (c) Firing frequency evoked by brush, pinch, or heat was similar in MIA and sham‐injected animals. (d,e) 90 μg of E‐52862 significantly reduced the evoked responses to mechanical and thermal stimuli 30 min after intrathecal application in mice with osteoarthritis pain. (f,g) Application of 180 μg of E‐52862 inhibited the firing frequency in response to stimulation of the hind paw 10 and 30 min after administration. Stimuli were applied for 10 s, and responses are presented as mean ± SEM. Histogram traces for single unit responses of WDR neurons representative for each group are presented. The number of animals is the following: sham‐vehicle = 4; MIA‐vehicle = 8; and MIA‐E‐52862 (10 and 30 min) = 6. Panels (d) and (f): *P<.05, significant difference between MIA‐E‐52862 (10 and 30 min) and MIA‐vehicle (F test of non‐linear regression) and +P<.05, significant difference between MIA‐E‐52862 and MIA‐vehicle (two‐way repeated‐measures ANOVA plus Fisher least significant difference test). Panels (e) and (g): # P<.05, significant difference between MIA‐E‐52862 and MIA‐vehicle (two‐way repeated‐measures ANOVA plus Fisher least significant difference test). MIA, monoiodoacetate
3.4. σ1 receptors and μ receptors show reciprocal modulation in chronic osteoarthritic pain
The σ1 receptors have been shown to modulate opioid tolerance during acute nociception (Chien & Pasternak, 1995; Mei & Pasternak, 2002), but this effect has not been investigated under chronic pain conditions. To address this question, we induced opioid tolerance in mice with osteoarthritic pain using a chronic treatment with 2.5 mg·kg−1 morphine administered twice a day for 14 days. Afterwards, mice received acute morphine (2.5 mg·kg−1), E‐52862 (5–20 mg·kg−1), or combinations of both, and mechanical sensitivity was assessed (Figure 4a). Osteoarthritic mice developed tolerance to the antinociceptive effects of morphine, revealed by the absence of an opioid response at the end of the repeated treatment (Figure 4b,d). This tolerance persisted for at least 9 days, as morphine antinociception was still absent 17, 20, and 23 days after MIA (Figure 4b). In this context, a single sub‐effective dose of E‐52862 (5 mg·kg−1), combined with morphine, restored opioid analgesia (Days 17 and 23, Figure 4c). This restorative effect was not observed when morphine was administered alone 3 days after the first morphine‐E‐52862 combination, suggesting that may be a transient effect (Day 20; Figure 4c). To assess the effect of higher doses of the σ1 receptor antagonist, we co‐administered E‐52862 20 mg·kg−1 with morphine. As expected, this combination produced complete restoration of mechanosensitivity to baseline levels (Days 17 and 23, Figure 4d). Interestingly, this dose induced a long‐lasting recovery, revealed by increased mechanical thresholds when morphine was given alone 3 days after the first morphine‐E‐52862 combination (Day 20, Figure 4d). This was not a residual effect of the earlier E‐52862 dose, because the mechanical sensitivity assessed before the administration of morphine showed regular nociceptive sensitization (Figure 4d). Therefore, these results suggest that σ1 receptors participate in morphine tolerance during chronic osteoarthritic pain and its antagonism can restore opioid analgesia.
Figure 4.
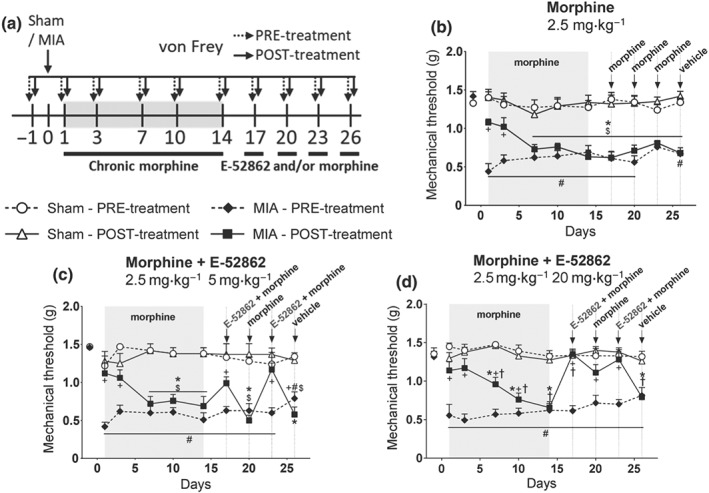
A single administration of the σ1 receptor antagonist restores morphine analgesia in opioid‐tolerant mice. (a) After a 14‐day treatment with morphine (2.5 mg·kg−1, twice a day during 14 days), mice received acute administrations of morphine (2.5 mg·kg−1), E‐52862 (5–20 mg·kg−1), or combinations of both drugs. (b–d) Mice repeatedly treated with the opioid developed analgesic tolerance. (b) The antinociceptive effect of morphine was not recovered for at least 9 days after the end of the repeated treatment. Single sub‐effective (5 mg·kg−1) (c) and effective doses (20 mg·kg−1) (d) of E‐52862 combined with morphine produced a restoration of opioid analgesia in morphine‐tolerant mice. Mechanical thresholds expressed as mean ± SEM. The number of animals is the following (first value represents sham groups and second value represents MIA groups): (b) 8, 8; (c) 8, 8; and (d) 8, 10. *P<.05, significant difference between MIA‐POST and Sham‐POST; #P<.05, significant difference between MIA‐PRE and Sham‐PRE; +P<.05, significant difference between MIA‐PRE and MIA‐POST; †P<.05, significant difference between MIA‐POST and MIA‐POST at Day 1 (three‐way repeated‐measures ANOVA plus Fisher least significant difference test). MIA, monoiodoacetate
The σ1 receptor antagonist showed efficacy increasing the antinociceptive effect of morphine. However, it is not known whether μ receptors contribute to the analgesic effects of σ1 receptor ligands. Thus, we evaluated the involvement of σ1 receptors and μ receptors on the analgesic efficacy of E‐52862. Mice with osteoarthritic pain were treated with vehicle or E‐52862 for 13 days and on Day 14 received acute doses of E‐52862 (20 mg·kg−1), naloxone (1 mg·kg−1), the σ1 receptor agonist PRE‐084 (32 mg·kg−1), or combinations of these drugs (Figure 5a). MIA‐injected mice receiving vehicle showed reduced mechanical thresholds before drug administration (PRE values; Figure 5b). Acute injection of E‐52862 decreased mechanical allodynia, whereas acute naloxone or PRE‐084 did not induce significant responses. As expected, PRE‐084 reduced the acute antiallodynic effect of the σ1 receptor antagonist (Figure 5b). Interestingly, co‐administration of E‐52862 with naloxone revealed a trend towards reduced analgesic effects of the σ1 receptor antagonist (Figure 5b), suggesting μ receptors also contribute to these acute responses. We also investigated whether μ receptors participate in the sustained recovery of mechanical thresholds induced by repeated E‐52862 (PRE values; Figure 5c). In these conditions, both naloxone and PRE‐084 injections decreased the mechanical thresholds (Figure 5c). Hence, the results indicate that the effects of E‐52862 were selective for the σ1 receptors and a possible participation of μ receptors on the acute and sustained effects of E‐52862 was noted.
Figure 5.
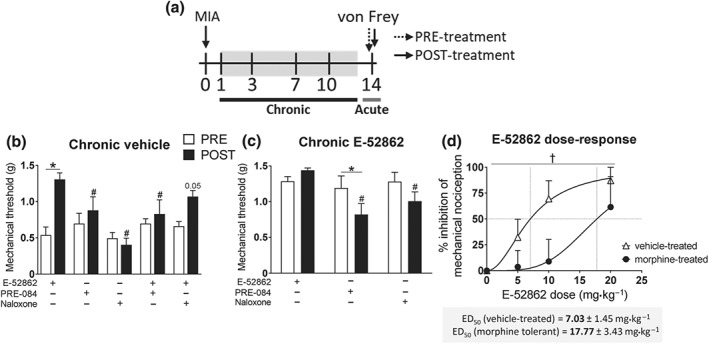
Participation of μ receptors in the antiallodynic effect of E‐52862. (a) Osteoarthritic mice were treated with vehicle (0.5% HPMC), E‐52862 (20 mg·kg−1), or morphine (2.5 mg·kg−1, twice a day during 14 days). In the last day of treatment, mice received acute doses of E‐52862 (5–20 mg·kg−1), PRE‐084 (32 mg·kg−1), naloxone (1 mg·kg−1), or combinations of these drugs. (b) Acute E‐52862 (20 mg·kg−1) showed reduced anti‐allodynic effect when co‐administered with PRE‐084 and a trend towards a decreased antinociception when given together with naloxone. (c) Once mechanical allodynia was normalized after chronic E‐52862, acute administration of PRE‐084 and naloxone induced reduction of the mechanical thresholds. (d) E‐52862 administered to morphine‐tolerant mice showed reduced antinociceptive effects. Data are expressed as mean ± SEM. The number of animals is the following: (b) 6 per group; (c) 6 per group; (d) 8 for vehicle‐treated, 6 for morphine‐treated. For panels (b) and (c): *P<.05, significant difference between PRE and POST; # P<.05, significant difference from E‐52862 POST (two‐way repeated measures ANOVA plus Fisher least significant difference test). For panel (d): † P<.05, significant difference between vehicle‐treated and morphine‐treated (F test of non‐linear regression). MIA, monoiodoacetate. †
An additional experiment was conducted to assess whether chronic stimulation of μ receptors could in turn modulate E‐52862‐induced antinociception. Different E‐52862 doses were administered to mice with osteoarthritic pain after chronic morphine or vehicle (Figure 5a,d). Interestingly, we observed a significant shift to the right of the E‐52862 dose–response curve when the drug was administered to morphine‐tolerant mice. The σ1 receptor antagonist produced higher antinociceptive effects when mice were not exposed to morphine (ED50 = 7.028 mg·kg−1) than when mice were chronically treated with the opioid (ED50 = 17.77 mg·kg−1). Thus, the antinociceptive effect of E‐52862 was 2.5 times lower when μ receptors were desensitized after repeated morphine. Hence, the results suggest bidirectional modulation of σ1 receptors and μ receptors during chronic osteoarthritic pain.
3.5. Molecular changes associated with chronic osteoarthritic pain are reversed by repeated treatment with the σ1 receptor antagonist E‐52862
The σ1 receptor antagonist induced acute and long‐lasting analgesic effects in mice with osteoarthritis pain, in contrast with morphine, which was devoid of antinociceptive effects after 14‐day repeated administration. To compare the effects of these drugs at the molecular level, expression of markers associated with chronic pain was assessed in DRG and spinal cord samples of sham‐ or MIA‐injected mice treated with vehicle, morphine, or E‐52862 (Figure 6). One day after MIA, BDNF and NPY were over‐expressed in the DRG (Figure 6b,c). This over‐expression was reduced by acute morphine administration, but not by acute E‐52862 (Figure 6b,c). This suggests peripheral effects of acute morphine treatments at early time points of the intra‐knee injury. At spinal level, MIA‐injected mice also showed early over‐expression of TNF‐α (Figure 6d), whereas IL‐1β and NPY were unchanged (Figure 6e,f). No effect of the treatments was observed on these spinal markers at this early time point (Figure 6d,f).
Figure 6.
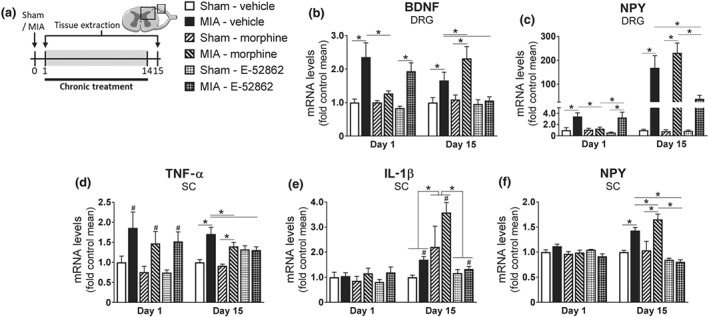
Repeated E‐52862 reduces the expression of neuroinflammatory mediators associated with osteoarthritis pain and chronic morphine administration. (a) Spinal cord and DRG were extracted 1 or 15 days after sham or MIA injection from mice treated with vehicle (0.5% HPMC), morphine (2.5 mg·kg−1), or E‐52862 (20 mg·kg−1). The first day after the intra‐knee injection, MIA induced over‐expression of BDNF (b) and NPY (c) in the DRG, which were reduced by the acute administration of morphine. In the spinal cord, MIA‐injected mice showed increased levels of TNF‐α (d), whereas IL‐1β (e) and NPY (f) were not altered; 15 days after MIA or sham injection, BDNF (b) and NPY (c) in the DRG, and spinal TNF‐α (d), IL‐1β (e), and NPY (f) were increased in osteoarthritic mice and normalized by E‐52862 chronic treatment. Morphine treatment further increased expression of BDNF (b), IL‐1β (e), and spinal NPY (f). Data are expressed as mean ± SEM. The number of animals is the following: Day 1 = 5 per group; Day 15 = 6 per group. #P<.05, main effect, significant difference between MIA and sham, *P<.05, significantly different as indicated; two‐way ANOVA followed by Fisher least significant difference test for each time point. BDNF, brain‐derived neurotrophic factor; DRG, dorsal root ganglia; MIA, monoiodoacetate; NPY, neuropeptide Y; SC, spinal cord
After the 14‐day treatment with vehicle, BDNF and TNF‐α were still increased in osteoarthritic mice (Figure 6b,d), whereas NPY levels in the DRG showed a marked over‐expression (Figure 6c). At this time point, spinal IL‐1β and NPY levels were also enhanced (Figure 6e,f). Thus, additional molecular changes were established at later stages of osteoarthritic pain. Continuous blockade of σ1 receptors normalized the over‐expression of all these markers (Figure 6b,f), whereas chronic morphine further increased BDNF, IL‐1β, and spinal NPY expression (Figure 6b,e,f). Thus, the σ1 receptor antagonist disrupted the expression of pain signalling‐related molecules after chronic treatment, whereas repeated morphine promoted the expression of neuroinflammatory mediators.
To further investigate the neuroinflammatory mechanisms underlying the long‐term effects of E‐52862 and morphine, we also analysed protein levels of the macrophage and microglial marker Iba‐1 in DRG and spinal cord (Figure 7a). We did not observe significant differences in DRG (Figure 7b). In contrast, increased spinal Iba‐1 levels were detected in osteoarthritic mice and after repeated morphine (Figure 7c). Interestingly, E‐52862 reversed the enhancement of Iba‐1 expression associated with osteoarthritic pain, suggesting a role for σ1 receptors in modulating glial reactivity. As microgliosis is associated with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1369 excitotoxicity on neurons (Takeuchi, 2013), we decided to explore the spinal levels of glutamate receptors. We observed that chronic morphine significantly increased the levels of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=445 subunit (Figure 7d), whereas MIA enhanced phosphorylation of the NMDA receptor subunits https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=455 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=457 and the expression of metabotropic glutamate mGlu5 receptor (Figure 7e,g). While the increases in phospho‐GluN1 and phospho‐GluN2B were independent of the treatment, the results showed that over‐expression of mGlu5 receptors was completely reversed by E‐52862 (Figure 7g), revealing a novel effect of σ1 receptor antagonism in modulating glutamatergic signalling.
Figure 7.
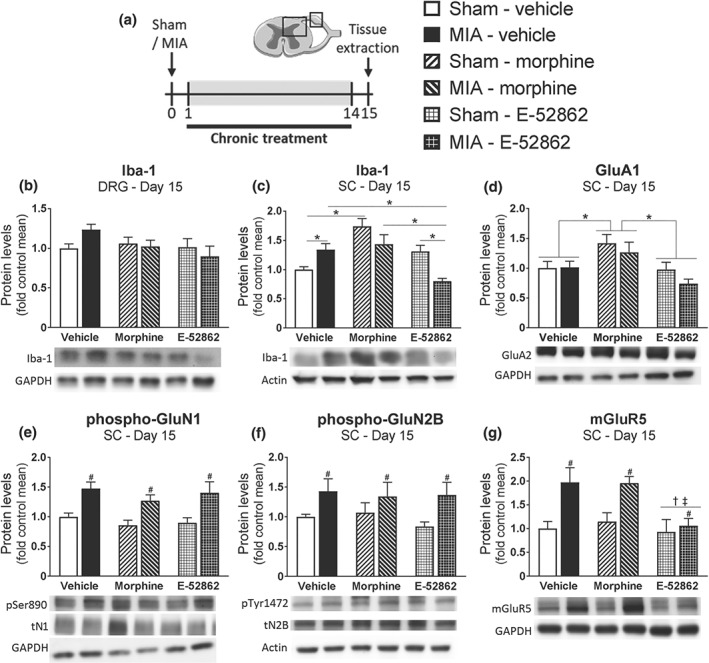
Repeated treatment with E‐52862 decreases pain‐induced microgliosis and mGluR5 up‐regulation. (a) Fifteen days after sham or MIA injection, spinal cord, and DRG were extracted from vehicle‐ (0.5% HPMC), morphine‐ (2.5 mg·kg−1), or E‐52862‐ (20 mg·kg−1) treated mice. (b) Protein levels of Iba‐1 showed no significant differences in the DRG. (c) At spinal level, Iba‐1 was increased after MIA or chronic morphine and reduced by E‐52862. (d) Morphine induced an up‐regulation of GluA2 AMPA receptor subunit. Phosphorylation levels of GluN1 (e) and GluN2B subunits (f) of the NMDA receptor were increased after MIA injection regardless of the treatment received. (g) MIA injection significantly increased the protein levels of mGlu5, which were reduced after E‐52862. GAPDH or actin was used as a housekeeping control. Data are expressed as mean ± SEM. The number of animals is 6 per group. #P <.05, significant difference between MIA and sham; †P <.05, significant difference between E‐52862 and vehicle; ‡P <.05, significant difference between E‐52862 and morphine; *P <.05, significantly different as indicated; two‐way ANOVA followed by Fisher least significant difference test: MIA, monoiodoacetate; SC, spinal cord; DRG, dorsal root ganglia; pSer890, phosphorylation of Ser890 of the N1 subunit of the NMDA receptor; tN1, total N1 subunit; pTyr1472, phosphorylation of Tyr1472 of the N2B subunit of the NMDA receptor; tN2B, total N2B subunit
Overall, acute administration of morphine decreased expression of peripheral nociceptive markers, but chronic administration aggravated the pain‐related molecular changes. On the contrary, E‐52862 did not affect the expression of these markers after acute administration, but repeated treatment abolished central and peripheral long‐term alterations associated with chronic osteoarthritic pain.
4. DISCUSSION
This work reveals that the selective σ1 receptor antagonist E‐52862 has short‐ and long‐term analgesic effects and reverses morphine tolerance in mice subjected to a model of chronic osteoarthritic pain. Our findings indicate dose‐dependent, short‐term antinociception observed 30 min after E‐52862 and sustained recovery of the mechanical thresholds when the drug was repeatedly given. Importantly, electrophysiological recordings revealed that E‐52862 inhibited central sensitization of spinal WDR neurons in osteoarthritic mice. These effects partly involved μ receptors and σ1 receptor antagonism reversed morphine tolerance during osteoarthritic pain, demnstrating a crosstalk between σ1 receptors and the opioid system. Biochemical assays identified common alterations of neuroinflammatory markers and glutamatergic signalling associated with chronic pain and repeated opioid exposure, both specifically inhibited by E‐52862.
Acute E‐52862 induced short‐term inhibition of MIA‐induced hypersensitivity, confirming the analgesic efficacy of the σ1 receptor antagonist observed in different pain models (Romero et al., 2016). E‐52862 or vehicle treatments did not induce significant changes in mechanical sensitivity of sham mice, although minor variations associated with the intra‐knee saline injections were occasionally observed. Thus, normal mechanosensitivity remains intact following σ1 receptor antagonism. In agreement, it was proposed that σ1 receptors do not have a primary role in physiological conditions (Su et al., 2010; Zamanillo et al., 2013). The repeated treatment with E‐52862 also produced a gradual recovery of sensitivity, which was observed with all the doses tested and maintained after interrupting the treatment. Previous studies in mice investigated acute antinociceptive effects of E‐52862 (González‐Cano, Merlos, Baeyens, & Cendán, 2013; Gris et al., 2014; Romero et al., 2012) and its effects during chronic administration without testing mechanical allodynia before the daily dose of the compound (Bura, Guegan, Zamanillo, Vela, & Maldonado, 2013; Romero et al., 2012). Our work shows long‐lasting restorative effects of the σ1 receptor antagonist, suggesting an additional benefit for long‐term chronic pain treatments.
E‐52862 showed different efficacy depending on the stage of the osteoarthritic pain sensitization. It was proposed that the initial mechanical hyperalgesia in osteoarthritis is caused by inflammatory processes, while later stages involve neuropathic mechanisms (Thakur, Rahman, Hobbs, Dickenson, & Bennett, 2012). In agreement, rat models showed an increase of the nerve injury marker ATF‐3 in the DRG between 8 and 14 days after MIA (Ivanavicius et al., 2007; Orita et al., 2011), and our mice increased expression of neuroinflammatory markers 15 days after intra‐knee injury. The doses of E‐52862 needed to exert acute antiallodynic effect 15 days after MIA were lower than on 1 day post‐injection. Thus, acute effects of E‐52862 were more prominent once the neuropathic component of osteoarthritic pain was established. Such intra‐model differences agree with previous results showing higher efficacy of E‐52862 after neuropathic injuries than during inflammatory pain (Gris et al., 2014; Romero et al., 2012). The long‐lasting alleviation of pain required longer exposure to the σ1 receptor antagonist when persistent neuroinflammatory changes were present, although E‐52862 was still able to restore normal sensitivity. Thus, the σ1 receptor antagonist showed higher short‐term analgesic efficacy when pain had become chronic, while preserving its long‐term effects on chronic pain sensitization.
In vivo electrophysiological recordings showed facilitation of the firing frequency of spinal WDR neurons after MIA, revealing central sensitization in mice with osteoarthritic pain. Our result in CD1 mice agrees with previous work using C57Bl/6 mice, which revealed similar MIA‐induced responses to mechanical stimuli (Harvey & Dickenson, 2009). Furthermore, the lack of increased firing in response to thermal stimuli is in agreement with previous behavioural studies showing that MIA‐injected mice do not exhibit consistent heat hyperalgesia (La Porta, Bura, Aracil‐Fernández, Manzanares, & Maldonado, 2013). Interestingly, we found that intrathecal application of E‐52862 reduced these MIA‐facilitated responses. This is in line with previous ex vivo electrophysiological studies showing that E‐52862 inhibited wind‐up responses elicited after repeated nociceptor stimulation (Romero et al., 2012). In agreement, spinal cords of σ1 receptor knockout mice exhibited reduced wind‐up responses compared to wild‐type mice (de la Puente et al., 2009). Therefore, our experiments demonstrate that spinal central sensitization associated with osteoarthritis pain in live animals was reversed by the σ1 receptor antagonist.
It has been suggested that σ1 receptor agonists induce phosphorylation of μ receptors, a process involved in opioid tolerance (Rodríguez‐Muñoz et al., 2015; Rodríguez‐Muñoz, Cortés‐Montero, Pozo‐Rodrigálvarez, Sánchez‐Blázquez, & Garzón‐Niño, 2015). We found modulation, by σ1 receptors, of opioid analgesia during chronic osteoarthritic pain, showing that a single sub‐effective dose of E‐52862 co‐administered with morphine restored opioid antinociception. Earlier preclinical research had shown this modulatory role in the absence of chronic pain, using σ1 receptor knockout mice and σ1 receptor antagonists (Chien & Pasternak, 1994; Sánchez‐Fernández et al., 2013; Sánchez‐Fernández et al., 2014). Moreover, E‐52862 demonstrated efficacy restoring morphine analgesia in tolerant animals with acute nociceptive and inflammatory pain (Montilla‐García et al., 2019; Rodríguez‐Muñoz, Sánchez‐Blázquez, et al., 2015; Vidal‐Torres et al., 2013). Surprisingly, there were no previous studies assessing the role of σ1 receptors in modulation of opioid analgesia under conditions of chronic pain. Our data suggest that σ1 receptor antagonists could be efficient not only alleviating pain by themselves but also restoring opioid analgesia in tolerant individuals. Taking into account that opioid tolerance drives dose escalation and abuse, and E‐52862 did not produce tolerance development during this long‐term treatment and it is void of reinforcing effects in animals without pain (Bura et al., 2013), the σ1 receptor antagonist could represent a valuable alternative for chronic pain treatment. Considering the lifespan of mice (2.5 years) and humans (80 years), the length of the treatment used in this study could be compared to the duration of chronic pain treatments that produce adverse events in patients.
We observed inhibition of the acute and sustained effects of E‐52862 after administration of the σ1 receptor agonist PRE‐084. Previous studies using pharmacological and genetic approaches demonstrated that acute analgesic effects of E‐52862 are selectively mediated by σ1 receptors (Gris et al., 2014; Sánchez‐Fernández et al., 2014). Our work also showed that long‐term restoration of mechanical thresholds is also σ1 receptor‐dependent. Interestingly, naloxone also diminished the acute and sustained antinociceptive effects of E‐52862 revealing opioid participation. In addition, morphine‐tolerant mice showed decreased E‐52862 efficacy. antagonism of σ1 receptors facilitates σ1 receptor‐ μ receptor binding and protects μ receptors from phosphorylation, thus preserving their activity. Such phosphorylation is enhanced after persistent stimulation of μ receptors (Rodríguez‐Muñoz, Cortés‐Montero, et al., 2015, Rodríguez‐Muñoz, Sánchez‐Blázquez, et al., 2015). As both naloxone and μ receptor desensitization after chronic morphine decreased the antinociceptive effects of E‐52862, it can be concluded that part of the analgesic effects of the σ1 receptor antagonist rely on the enhancement of endogenous μ receptor activity.
We investigated pain‐related molecular alterations involved in morphine and E‐52862 analgesia during osteoarthritis. One day after MIA, BDNF and NPY over‐expression was observed in the DRG and TNF‐α expression increased in the spinal cord. Previous studies reported similar changes after nerve injuries, including immediate increases in BDNF and TNF‐α expression that persisted for long‐term periods (Ohtori, Takahashi, Moriya, & Myers, 2004; Uchida, Matsushita, & Ueda, 2013). Several researchers also showed prominent NPY up‐regulation in nerve‐injured primary afferents (Benoliel, Eliav, & Iadarola, 2001; Son et al., 2007). This synthesis de novo could represent an adaptive response to nociceptive sensitization (Munglani et al., 1995). While acute morphine inhibited pain‐related over‐expression of BDNF and NPY, a single E‐52862 administration did not provoke such effects, suggesting that its acute effects do not rely on these molecules.
Later stages of osteoarthritic pain involved pronounced increases of IL‐1β and NPY levels in spinal cord or DRG, accompanied by enhancement of microglial marker Iba1. In addition, the enhanced levels of BDNF and TNF‐α were maintained in DRG and spinal cord respectively. Thus, the intra‐knee injury induced persistent changes in neuroinflammatory mediators possibly contributing to the osteoarthritic phenotype. These pain‐related changes were reported to increase glutamate release and stimulate the glutamatergic system (Takeuchi, 2013; Vaz, Lérias, Parreira, Diógenes, & Sebastião, 2015). In agreement, osteoarthritic mice showed over‐expression of mGlu5 receptors, which is associated with increased glutamate levels in the nervous system (Wang, Wang, Zhong, Li, & Cong, 2012). As previously described, chronic morphine further increased neuroinflammation and glutamatergic signalling (Cabañero et al., 2013; Johnston et al., 2004), characterized by exacerbated BDNF, IL‐1β, spinal microgliosis, and AMPA receptor expression. Interestingly, increased BDNF/TrkB signalling contributes to chronic pain by eliciting microglial reactivity and glutamate release (Zhou et al., 2011). At the same time, IL‐1β and TNF‐α are mainly released by activated microglia, which also liberate BDNF after chronic morphine (Takayama & Ueda, 2005). Thus, repeated morphine contributed to an overall increase of spinal neuroinflammation. In sharp contrast, chronic E‐52862 normalized the expression of BDNF and proinflammatory cytokines. These findings agree with previous studies revealing potentiation of BDNF effects after over‐expression of σ1 receptors (Yagasaki et al., 2006) or chronic σ1 receptor activation (Kikuchi‐Utsumi & Nakaki, 2008). Likewise, recent experiments showed normalization of TNF‐α and IL‐1β expression in spinal cord‐injured σ1 receptor knockout mice (Castany et al., 2018). E‐52862 also reduced MIA‐induced microgliosis, consistent with the effects of the σ1 receptor antagonist BD1047 attenuating spinal microgliosis in a model of bone cancer pain (Zhu et al., 2015) and with the high levels of σ1 receptors reported in microglia (Gekker et al., 2006). Interestingly, E‐52862 was effective in preventing the increased microglial density induced by MIA in supraspinal structures (Carcolé et al., 2019). In addition, over‐expression of mGlu5 receptors was completely abolished by E‐52862. Hence, contrary to chronic morphine, repeated treatment with the σ1 receptor antagonist normalized the expression of neuroinflammatory mediators and glutamate receptors involved in chronic osteoarthritic pain.
In summary, the present study shows a dual effect of the σ1 receptor antagonist E‐52862 alleviating pain in our model of osteoarthritis. On the one hand, the σ1 receptor antagonist reduced acute mechanical allodynia, involving inhibition of spinal sensitization without modification of pain‐related molecular alterations. On the other hand, repeated E‐52862 exposure induced gradual recovery of the mechanical thresholds in osteoarthritic mice without inducing tolerance. This effect was associated with inhibition of biochemical changes related to osteoarthritic pain and opioid tolerance. Such alterations involve neuroinflammatory mediators, microglial reactivity and glutamatergic signalling, which could constitute a common pathway by which the σ1 receptor antagonist provided relief of chronic pain and restoration of opioid analgesia in tolerant individuals. Hence, the σ1 receptor antagonist could dampen deleterious side effects of opioid prescription drugs, which has reached now dramatic consequences in the United States (Lyden & Binswanger, 2019), and represents a promising alternative to opioids in chronic pain conditions requiring long‐term treatment with analgesics.
AUTHOR CONTRIBUTIONS
All authors listed above have contributed sufficiently to be included as authors. M.C. conducted the behavioural, electrophysiological, and molecular experiments and wrote the manuscript. S.K. conducted the molecular studies. L.G. supervised all the electrophysiological experiments. D.Z. and M.M. participated in the experimental design. A.H.D. provided the electrophysiological equipment. B.F‐P. supervised the project and participated in the experimental design. D.C. conceptualized and supervised the project, participated in the experimental design, and wrote the manuscript. R.M. conceptualized, supervised, and funded the project, participated in the experimental design, and wrote the manuscript. All the authors have revised the work critically for important intellectual content and approved the final version to be published.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
We acknowledge the financial support of the European Commission (Seventh Framework Programme, NeuroPain #2013‐602891), the Catalan Government (AGAUR, #SGR2017‐669), the Spanish Instituto de Salud Carlos III (RTA, #RD16/0017/0020), and AGAUR (ICREA Academia Award 2015). M.C. is the recipient of an Industrial Doctorate contract from the Catalan Government and Laboratorios Esteve (AGAUR, #2014‐DI‐040). Partial support from FEDER funds is also acknowledged.
Carcolé M, Kummer S, Gonçalves L, et al. Sigma‐1 receptor modulates neuroinflammation associated with mechanical hypersensitivity and opioid tolerance in a mouse model of osteoarthritis pain. Br J Pharmacol. 2019;176:3939–3955. 10.1111/bph.14794
REFERENCES
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The concise guide to PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso, G. , Phan, V. , Guillemain, I. , Saunier, M. , Legrand, A. , Anoal, M. , & Maurice, T. (2000). Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience, 97, 155–170. 10.1016/S0306-4522(00)00014-2 [DOI] [PubMed] [Google Scholar]
- Arendt‐Nielsen, L. , Nie, H. , Laursen, M. B. , Laursen, B. S. , Madeleine, P. , Simonsen, O. H. , & Graven‐Nielsen, T. (2010). Sensitization in patients with painful knee osteoarthritis. Pain, 149, 573–581. 10.1016/j.pain.2010.04.003 [DOI] [PubMed] [Google Scholar]
- Bangaru, M. L. , Weihrauch, D. , Tang, Q.‐B. , Zoga, V. , Hogan, Q. , & Wu, H. (2013). Sigma‐1 receptor expression in sensory neurons and the effect of painful peripheral nerve injury. Molecular Pain, 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoliel, R. , Eliav, E. , & Iadarola, M. J. (2001). Neuropeptide Y in trigeminal ganglion following chronic constriction injury of the rat infraorbital nerve: is there correlation to somatosensory parameters? Pain, 91, 111–121. 10.1016/S0304-3959(00)00417-6 [DOI] [PubMed] [Google Scholar]
- Breivik, H. , Collett, B. , Ventafridda, V. , Cohen, R. , & Gallacher, D. (2006). Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. European Journal of Pain, 10, 287–287. 10.1016/j.ejpain.2005.06.009 [DOI] [PubMed] [Google Scholar]
- Bura, A. S. , Guegan, T. , Zamanillo, D. , Vela, J. M. , & Maldonado, R. (2013). Operant self‐administration of a sigma ligand improves nociceptive and emotional manifestations of neuropathic pain. European Journal of Pain, 17, 832–843. 10.1002/j.1532-2149.2012.00251.x [DOI] [PubMed] [Google Scholar]
- Cabañero, D. , Baker, A. , Zhou, S. , Hargett, G. L. , Irie, T. , Xia, Y. , … Morón, J. A. (2013). Pain after discontinuation of morphine treatment is associated with synaptic increase of GluA4‐containing AMPAR in the dorsal horn of the spinal cord. Neuropsychopharmacology, 38, 1472–1484. 10.1038/npp.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcolé, M. , Zamanillo, D. , Merlos, M. , Fernández‐Pastor, B. , Cabañero, D. , & Maldonado, R. (2019). Blockade of the sigma‐1 receptor relieves cognitive and emotional impairments associated to chronic osteoarthritis pain. Frontiers in Pharmacology, 10 10.3389/fphar.2019.00468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castany, S. , Gris, G. , Vela, J. M. , Verdú, E. , & Boadas‐Vaello, P. (2018). Critical role of sigma‐1 receptors in central neuropathic pain‐related behaviours after mild spinal cord injury in mice. Scientific Reports, 8, 3873 10.1038/s41598-018-22217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan, S. R. , Bach, F. W. , Pogrel, J. W. , Chung, J. M. , & Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods, 53, 55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chien, C. C. , & Pasternak, G. W. (1994). Selective antagonism of opioid analgesia by sigma system. The Journal of Pharmacology and Experimental Therapeutics, 271, 1583–1590. [PubMed] [Google Scholar]
- Chien, C. C. , & Pasternak, G. W. (1995). Sigma antagonists potentiate opioid analgesia in rats. Neuroscience Letters, 190, 137–139. 10.1016/0304-3940(95)11504-P [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Puente, B. , Nadal, X. , Portillo‐Salido, E. , Sánchez‐Arroyos, R. , Ovalle, S. , Palacios, G. , … Vela, J. M. (2009). Sigma‐1 receptors regulate activity‐induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain, 145, 294–303. [DOI] [PubMed] [Google Scholar]
- Dixon, W. J. (1965). The up‐and‐down method for small samples. Journal of the American Statistical Association, 60, 967–978. 10.1080/01621459.1965.10480843 [DOI] [Google Scholar]
- Gekker, G. , Hu, S. , Sheng, W. S. , Rock, R. B. , Lokensgard, J. R. , & Peterson, P. K. (2006). Cocaine‐induced HIV‐1 expression in microglia involves sigma‐1 receptors and transforming growth factor‐β1. International Immunopharmacology, 6, 1029–1033. 10.1016/j.intimp.2005.12.005 [DOI] [PubMed] [Google Scholar]
- González‐Cano, R. , Merlos, M. , Baeyens, J. M. , & Cendán, C. M. (2013). σ1 Receptors are involved in the visceral pain induced by intracolonic administration of capsaicin in mice. Anesthesiology, 118, 691–700. 10.1097/ALN.0b013e318280a60a [DOI] [PubMed] [Google Scholar]
- Gris, G. , Merlos, M. , Vela, J. M. , Zamanillo, D. , & Portillo‐Salido, E. (2014). S1RA, a selective sigma‐1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund's adjuvant models in mice. Behavioural Pharmacology, 25, 226–235. 10.1097/FBP.0000000000000038 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey, V. L. , & Dickenson, A. H. (2009). Behavioural and electrophysiological characterisation of experimentally induced osteoarthritis and neuropathy in C57Bl/6 mice. Molecular Pain, 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, T. , & Su, T.‐P. (2007). Sigma‐1 receptor chaperones at the ER‐mitochondrion interface regulate Ca2+ signaling and cell survival. Cell, 131, 596–610. 10.1016/j.cell.2007.08.036 [DOI] [PubMed] [Google Scholar]
- Inceoglu, B. , Bettaieb, A. , Trindade da Silva, C. A. , Lee, K. S. S. , Haj, F. G. , & Hammock, B. D. (2015). Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proceedings of the National Academy of Sciences, 112, 9082–9087. 10.1073/pnas.1510137112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanavicius, S. P. , Ball, A. D. , Heapy, C. G. , Westwood, F. R. , Murray, F. , & Read, S. J. (2007). Structural pathology in a rodent model of osteoarthritis is associated with neuropathic pain: Increased expression of ATF‐3 and pharmacological characterisation. Pain, 128, 272–282. 10.1016/j.pain.2006.12.022 [DOI] [PubMed] [Google Scholar]
- Johnston, I. N. , Milligan, E. D. , Wieseler‐Frank, J. , Frank, M. G. , Zapata, V. , Campisi, J. , … Watkins, L. R. (2004). A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. The Journal of Neuroscience, 24, 7353–7365. 10.1523/JNEUROSCI.1850-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi‐Utsumi, K. , & Nakaki, T. (2008). Chronic treatment with a selective ligand for the sigma‐1 receptor chaperone, SA4503, up‐regulates BDNF protein levels in the rat hippocampus. Neuroscience Letters, 440, 19–22. 10.1016/j.neulet.2008.05.055 [DOI] [PubMed] [Google Scholar]
- Kim, F. J. , Kovalyshyn, I. , Burgman, M. , Neilan, C. , Chien, C.‐C. , & Pasternak, G. W. (2010). σ1 Receptor modulation of G‐protein‐coupled receptor signaling: Potentiation of opioid transduction independent from receptor binding. Molecular Pharmacology, 77, 695–703. 10.1124/mol.109.057083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Porta, C. , Bura, S. A. , Aracil‐Fernández, A. , Manzanares, J. , & Maldonado, R. (2013). Role of CB1 and CB2 cannabinoid receptors in the development of joint pain induced by monosodium iodoacetate. Pain, 154, 160–174. 10.1016/j.pain.2012.10.009 [DOI] [PubMed] [Google Scholar]
- Liu, D. , Zhou, Y. , Peng, Y. , Su, P. , Li, Z. , Xu, Q. , … Gao, F. (2018). Endoplasmic reticulum stress in spinal cord contributes to the development of morphine tolerance. Frontiers in Molecular Neuroscience, 11, 72 10.3389/fnmol.2018.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT Method. Methods, 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lyden, J. , & Binswanger, I. A. (2019). The United States opioid epidemic. Seminars in Perinatology, 43, 123–131. 10.1053/j.semperi.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, J. , & Pasternak, G. W. (2002). Sigma1 receptor modulation of opioid analgesia in the mouse. The Journal of Pharmacology and Experimental Therapeutics, 300, 1070–1074. 10.1124/jpet.300.3.1070 [DOI] [PubMed] [Google Scholar]
- Montilla‐García, Á. , Tejada, M. Á. , Ruiz‐Cantero, M. C. , Bravo‐Caparrós, I. , Yeste, S. , Zamanillo, D. , & Cobos, E. J. (2019). Modulation by sigma‐1 receptor of morphine analgesia and tolerance: Nociceptive pain, tactile allodynia and grip strength deficits during joint inflammation. Frontiers in Pharmacology, 10, 136 10.3389/fphar.2019.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munglani, R. , Bond, A. , Smith, G. D. , Harrison, S. M. , Elliot, P. J. , Birch, P. J. , & Hunt, S. P. (1995). Changes in neuronal markers in a mononeuropathic rat model relationship between neuropeptide Y, pre‐emptive drug treatment and long‐term mechanical hyperalgesia. Pain, 63, 21–31. 10.1016/0304-3959(95)00013-I [DOI] [PubMed] [Google Scholar]
- Negrete, R. , García Gutiérrez, M. S. , Manzanares, J. , & Maldonado, R. (2017). Involvement of the dynorphin/KOR system on the nociceptive, emotional and cognitive manifestations of joint pain in mice. Neuropharmacology, 116, 315–327. 10.1016/j.neuropharm.2016.08.026 [DOI] [PubMed] [Google Scholar]
- Ohtori, S. , Takahashi, K. , Moriya, H. , & Myers, R. R. (2004). TNF‐α and TNF‐α receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila pa 1976), 29, 1082–1088. [DOI] [PubMed] [Google Scholar]
- Orita, S. , Ishikawa, T. , Miyagi, M. , Ochiai, N. , Inoue, G. , Eguchi, Y. , … Ohtori, S. (2011). Pain‐related sensory innervation in monoiodoacetate‐induced osteoarthritis in rat knees that gradually develops neuronal injury in addition to inflammatory pain. BMC Musculoskeletal Disorders, 12, 134 10.1186/1471-2474-12-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaita, A. , Puighermanal, E. , & Maldonado, R. (2007). Regulation of PI3K/Akt/GSK‐3 pathway by cannabinoids in the brain. Journal of Neurochemistry, 102, 1105–1114. 10.1111/j.1471-4159.2007.04642.x [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Muñoz, M. , Cortés‐Montero, E. , Pozo‐Rodrigálvarez, A. , Sánchez‐Blázquez, P. , & Garzón‐Niño, J. (2015). The ON:OFF switch, σ1R‐HINT1 protein, controls GPCR‐NMDA receptor cross‐regulation: implications in neurological disorders. Oncotarget, 6, 35458–35477. 10.18632/oncotarget.6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Muñoz, M. , Sánchez‐Blázquez, P. , Herrero‐Labrador, R. , Martínez‐Murillo, R. , Merlos, M. , Vela, J. M. , & Garzón, J. (2015). The σ1 receptor engages the redox‐regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxidants & Redox Signaling, 22, 799–818. 10.1089/ars.2014.5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, L. , Merlos, M. , & Vela, J. M. (2016). Antinociception by sigma‐1 receptor antagonists: Central and peripheral effects. Advances in Pharmacology (San Diego, Calif.), 75, 179–215. [DOI] [PubMed] [Google Scholar]
- Romero, L. , Zamanillo, D. , Nadal, X. , Sánchez‐Arroyos, R. , Rivera‐Arconada, I. , Dordal, A. , … Vela, J. M. (2012). Pharmacological properties of S1RA, a new sigma‐1 receptor antagonist that inhibits neuropathic pain and activity‐induced spinal sensitization. British Journal of Pharmacology, 166, 2289–2306. 10.1111/j.1476-5381.2012.01942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Fernández, C. , Montilla‐Garcia, A. , Gonzalez‐Cano, R. , Nieto, F. R. , Romero, L. , Artacho‐Cordon, A. , … Cobos, E. J. (2014). Modulation of peripheral μ‐opioid analgesia by σ1 receptors. The Journal of Pharmacology and Experimental Therapeutics, 348, 32–45. [DOI] [PubMed] [Google Scholar]
- Sánchez‐Fernández, C. , Nieto, F. R. , González‐Cano, R. , Artacho‐Cordón, A. , Romero, L. , Montilla‐García, Á. , … Cobos, E. J. (2013). Potentiation of morphine‐induced mechanical antinociception by σ1 receptor inhibition: Role of peripheral σ1 receptors. Neuropharmacology, 70, 348–358. 10.1016/j.neuropharm.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Son, S.‐J. , Lee, K.‐M. , Jeon, S.‐M. , Park, E.‐S. , Park, K.‐M. , & Cho, H.‐J. (2007). Activation of transcription factor c‐jun in dorsal root ganglia induces VIP and NPY upregulation and contributes to the pathogenesis of neuropathic pain. Experimental Neurology, 204, 467–472. 10.1016/j.expneurol.2006.09.020 [DOI] [PubMed] [Google Scholar]
- Su, T.‐P. , Hayashi, T. , Maurice, T. , Buch, S. , & Ruoho, A. E. (2010). The sigma‐1 receptor chaperone as an inter‐organelle signaling modulator. Trends in Pharmacological Sciences, 31, 557–566. 10.1016/j.tips.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, S. , Clutterbuck, A. , Harris, P. , Gent, T. , Freeman, S. , Foster, N. , … Mobasheri, A. (2009). The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Veterinary Journal, 179, 10–24. 10.1016/j.tvjl.2007.08.013 [DOI] [PubMed] [Google Scholar]
- Takayama, N. , & Ueda, H. (2005). Morphine‐induced chemotaxis and brain‐derived neurotrophic factor expression in microglia. The Journal of Neuroscience, 25, 430–435. 10.1523/JNEUROSCI.3170-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, H. (2013). Microglia and glutamate. Advances in Neuroimmune Biology, 4(2), 77–83. [Google Scholar]
- Thakur, M. , Rahman, W. , Hobbs, C. , Dickenson, A. H. , & Bennett, D. L. H. (2012). Characterisation of a peripheral neuropathic component of the rat monoiodoacetate model of osteoarthritis. PLoS ONE, 7, e33730 10.1371/journal.pone.0033730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, H. , Matsushita, Y. , & Ueda, H. (2013). Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: Implications in the development of neuropathic pain. Neuroscience, 240, 147–154. 10.1016/j.neuroscience.2013.02.053 [DOI] [PubMed] [Google Scholar]
- Vaz, S. H. , Lérias, S. R. , Parreira, S. , Diógenes, M. J. , & Sebastião, A. M. (2015). Adenosine A2A receptor activation is determinant for BDNF actions upon GABA and glutamate release from rat hippocampal synaptosomes. Purinergic Signal, 11, 607–612. 10.1007/s11302-015-9476-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal‐Torres, A. , de la Puente, B. , Rocasalbas, M. , Touriño, C. , Bura, S. A. , Fernández‐Pastor, B. , … Vela, J. M. (2013). Sigma‐1 receptor antagonism as opioid adjuvant strategy: Enhancement of opioid antinociception without increasing adverse effects. European Journal of Pharmacology, 711, 63–72. 10.1016/j.ejphar.2013.04.018 [DOI] [PubMed] [Google Scholar]
- Vidal‐Torres, A. , Fernández‐Pastor, B. , Carceller, A. , Vela, J. M. , Merlos, M. , & Zamanillo, D. (2014). Effects of the selective sigma‐1 receptor antagonist S1RA on formalin‐induced pain behavior and neurotransmitter release in the spinal cord in rats. Journal of Neurochemistry, 129, 484–494. 10.1111/jnc.12648 [DOI] [PubMed] [Google Scholar]
- Vowles, K. E. , McEntee, M. L. , Julnes, P. S. , Frohe, T. , Ney, J. P. , & van der Goes, D. N. (2015). Rates of opioid misuse, abuse, and addiction in chronic pain: A systematic review and data synthesis. Pain, 156, 569–576. 10.1097/01.j.pain.0000460357.01998.f1 [DOI] [PubMed] [Google Scholar]
- Wang, J.‐W. , Wang, H.‐D. , Zhong, W.‐Z. , Li, N. , & Cong, Z.‐X. (2012). Expression and cell distribution of metabotropic glutamate receptor 5 in the rat cortex following traumatic brain injury. Brain Research, 1464, 73–81. 10.1016/j.brainres.2012.05.014 [DOI] [PubMed] [Google Scholar]
- Wieland, H. A. , Michaelis, M. , Kirschbaum, B. J. , & Rudolphi, K. A. (2005). Osteoarthritis—An untreatable disease? Nature Reviews. Drug Discovery, 4, 331–344. 10.1038/nrd1693 [DOI] [PubMed] [Google Scholar]
- Wylde, V. , Hewlett, S. , Learmonth, I. D. , & Dieppe, P. (2011). Persistent pain after joint replacement: Prevalence, sensory qualities, and postoperative determinants. Pain, 152, 566–572. 10.1016/j.pain.2010.11.023 [DOI] [PubMed] [Google Scholar]
- Yagasaki, Y. , Numakawa, T. , Kumamaru, E. , Hayashi, T. , Su, T.‐P. , & Kunugi, H. (2006). Chronic antidepressants potentiate via sigma‐1 receptors the brain‐derived neurotrophic factor‐induced signaling for glutamate release. The Journal of Biological Chemistry, 281, 12941–12949. 10.1074/jbc.M508157200 [DOI] [PubMed] [Google Scholar]
- Zamanillo, D. , Romero, L. , Merlos, M. , & Vela, J. M. (2013). Sigma 1 receptor: A new therapeutic target for pain. European Journal of Pharmacology, 716, 78–93. 10.1016/j.ejphar.2013.01.068 [DOI] [PubMed] [Google Scholar]
- Zhang, R.‐X. , Ren, K. , & Dubner, R. (2013). Osteoarthritis pain mechanisms: Basic studies in animal models. Osteoarthritis and Cartilage, 21, 1308–1315. 10.1016/j.joca.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L.‐J. , Yang, T. , Wei, X. , Liu, Y. , Xin, W.‐J. , Chen, Y. , … Liu, X. G. (2011). Brain‐derived neurotrophic factor contributes to spinal long‐term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain, Behavior, and Immunity, 25, 322–334. 10.1016/j.bbi.2010.09.025 [DOI] [PubMed] [Google Scholar]
- Zhu, S. , Wang, C. , Han, Y. , Song, C. , Hu, X. , & Liu, Y. (2015). Sigma‐1 receptor antagonist BD1047 reduces mechanical allodynia in a rat model of bone cancer pain through the inhibition of spinal NR1 phosphorylation and microglia activation. Mediators of Inflammation, 2015, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann, M. (1986). Ethical considerations in relation to pain in animal experimentation. Acta Physiologica Scandinavica. Supplementum, 554, 221–233. [PubMed] [Google Scholar]