

Abstract
In this study, a simple formulation of softwood-derived glycol lignin (GL)-based epoxy resin with a high GL content of greater than 50 wt % was demonstrated by direct mixing with poly(ethylene glycol) diglycidyl ether (PEGDGE), an aliphatic epoxide, without using any solvent. Because the GL powder produced from poly(ethylene glycol) (PEG400) solvolysis of Japanese cedar softwood meal was a PEG400-modified lignin (GL400), a strong affinity between PEG counterparts facilitates the uniform mixing of GL400 with PEGDGE, and one component uncured GL400/PEGDGE epoxy resin was prepared at a relatively lower temperature (100 °C) than the curing temperature (130 °C). The epoxy curing reaction was monitored by 1H NMR and Fourier transform infrared spectroscopies. The physical and mechanical properties of the epoxy resins with different GL400 contents were then evaluated. The developed resins exhibited good flexibility and elasticity depending on the GL400 content.
Introduction
Lignin is one of the main components of wood cells, in addition to cellulose and hemicellulose, and is referred to as the most abundant natural resources of aromatic biopolymer.1,2 On the other hand, lignin derivatives obtained from traditional pulp industries typically exhibit excess denaturation and contain many impurities because degradation of lignin by product was allowed to keep pulp quality to the maximum so that the direct use of the lignin derivatives is slightly difficult as industrial raw materials. In addition, the nonuniformity of lignin due to different plant species also hinders its widespread use as a chemical raw material.3 Recently, a novel method to extract lignin under mild chemical conditions using unutilized Japanese cedar forest thinning samples has been reported.4 A strategy for the large-scale production of lignin derivatives by the acid-catalyzed poly(ethylene glycol) (PEG) solvolysis of softwood to commercialize the cedar–lignin-based fabrication of advanced functional materials has been reported.5 The use of the nonvolatile PEG solvent promotes the solvolysis process under atmospheric pressure as well as the chemical introduction of PEG into the isolated lignin macromolecules, resulting in PEG-modified lignin derivatives called glycol lignin (GL).4,5 In addition, cell walls of softwoods are exclusively composed of guaiacyl (G) units in terms of the aromatic unit compositions as a lignin component. Thus, GLs produced from the direct isolation of softwood Japanese cedar have less structural heterogeneity and very low impurities,5 which has been considered as potential feedstock for a wide range of industrial applications. For example, we have recently reported the GL/clay nanocomposite film having high thermal durability, high oxygen gas, and moisture vapor barrier properties, applicable as printed electronic substrates to fabricate touch sensor devices.6−8
Here, we focused on the development of GL-based epoxy resin using GL as a natural phenol compound. The utilization of renewable resources for sustainable technology is attracting attention because of their availability and meeting environmental requirements such as low ecotoxicity and biodegradability. Therefore, significant effort on research and development of bio-based epoxy resins to substitute petrochemical-based resins has been explored to meet the increasing demands of green industries.1,9 Epoxy resins are widely used in a variety of industries, such as adhesives, coatings, insulations, and high-performance composites because of their versatility in manipulating the ultimate properties and performances in terms of strength, durability, and thermal and chemical resistances as provided by high cross-linked structures. To transform epoxy resins into cross-linked networks with desirable properties, a raw epoxide material is cured with a curing agent. The use of lignin as the curing agent has been studied because of its interesting characteristics, including heat resistance and chemical structural robustness.10−13 Like other commercially available lignin derivatives, the GL used in this work is a powder form having a thermal flow temperature (Tf) of >140 °C,5 which is typically higher than the curing polymerization temperature of epoxy resins. The higher Tf prevents uniform mixing with resins in their liquid form before curing. In one of our reports, the sealing material was fabricated by dissolving GL in some polar organic solvents, such as N-methylpyrrolidone, N,N-dimethylformamide, and PEG, to mix with epoxy chemicals.14 However, the use of solvents poses issues including the decrease in the GL content to less than 50% in the resulting resin and the deterioration in its physical properties due to the residual solvent within the resin. Because the boiling points of such solvents are higher than the curing temperature, complete removal of the residual solvent by drying at a certain temperature before curing needs to be done. In this study, a solvent-free epoxy resin with high lignin content was prepared by thermal curing using GL as the curing agent. As GL is a PEG-modified lignin derivative, a counter epoxy chemical containing PEG polymer, like poly(ethylene glycol) diglycidyl ether (PEGDGE), was selected to solve the solubility and uniform mixing between the curing agent (GL) and the epoxy chemical (PEGDGE). PEGDGE is a commercially available epoxide material in its liquid state and is composed of terminal epoxy groups in its PEG molecular structure.
The working strategy is the chemistry of “like-dissolves-like” rule. Mixing PEG containing the curing agent in the powder form and the epoxy chemical in the liquid state at a certain temperature lower than the curing temperature without using additional organic solvent was done as a preliminary test, and suitable thermal mixing temperature of 100 °C was set for this study. The available GL content that can be dissolved in PEGDGE was carried out to optimize the best epoxy resin compositions. The chemical equivalent weights of the phenolic hydroxyl groups of GL and the epoxy curing reaction were determined by 1H NMR and Fourier transform infrared (FTIR) spectroscopy, respectively. The mechanical properties of GL/PEGDGE epoxy resins with various GL contents were then investigated.
Results and Discussion
Fabrication of the GL/PEGDGE Epoxy Resin
The assumed molecular structures of GL (GL400) and PEGDGE used in the present work are illustrated in Figure 1. GL400 is one of the cedar-derived GL materials, in which lignin from cedar is modified by poly(ethylene glycol) with an average molecular weight of ca. 400. The structure of GL400 in Figure 1 is a model that has been redesigned so that the molecular weight and functional groups of the material used in the present work can be understood, according to the structural characterization of glycol lignin investigated in the previous co-authors’ literature.5 The structure of PEGDGE is simply designed as is according to the information on this commercially available substance.
Figure 1.

Molecular structures of GL400 and PEGDGE.
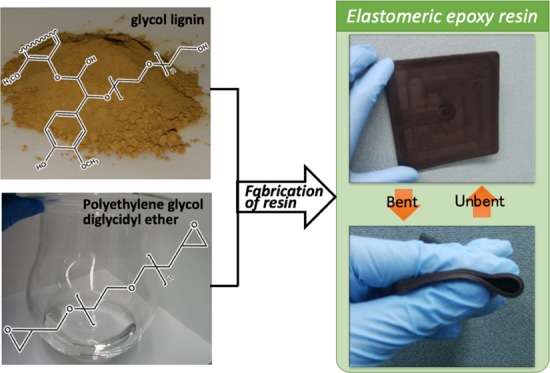
The illustration of the preparation of elastomeric GL400-epoxy resin is shown in Figure 2. The employed mixing process allows the preparation of GL/PEGDGE ratios with GL400 content as high as 71 wt %. Uniform mixing could not be performed if the GL content was higher than 71 wt %, due to the very high viscosity of the mixture (Table 1). Since it was confirmed that GL400 or PEGDGE alone could not react to give any cured resin under the same preparation conditions, as shown in Figure 2, such resin is clearly decided to be a product of polymerization between GL400 and PEGDGE. Notably, elastomeric GL-based epoxy resin with GL content of more than half was successfully obtained by this solvent-free simple formulation. The developed GL-epoxy resin was extremely flexible and exhibited elastic repulsion when bent (Figure 2), probably due to the flexible molecular nature of the aliphatic epoxide, PEGDGE. On the other hand, the GL-epoxy resin was not sufficiently cured when GL400 content of less than or equal to 50 wt % was applied even after thermal curing at 130 °C. DSC was employed to check the curing behavior of the GL-epoxy resin and the result is shown in the Supporting Information (Figure S1). There were no clear exo- and endothermic peaks, but quite broad ones that seemed to be exothermic at around 70 and 120 °C, respectively. Because the GL-epoxy mixture was still liquid at 70 °C in the actual resin curing process, no curing reaction still occurred, suggesting that the peak indicates a kind of a reaction of GL itself by heating. At above 120 °C, the GL-epoxy mixture was cured to a solid, which reveals that the second broad peak seems to be an exothermic reaction of curing. From this result, an oven curing temperature of 130 °C was suitable for the GL-epoxy resin.
Figure 2.

Schematic illustration of the preparation of GL-epoxy resin.
Table 1. Mixing Ratios of GL400 and PEGDGE and the Visual Observation of the Nature of the Uncured and Cured GL-Epoxy Resin.
| GL400: PEGDGE (w/w) | GL400 content of the GL-epoxy resin (wt %) | appearance of the uncured GL-epoxy resin (at 25 °C) | appearance of the cured GL-epoxy resin (at 25 °C) |
|---|---|---|---|
| 1.0:1.4 | 41.2 | viscous liquid | insufficient curing |
| 1.0:1.2 | 45.4 | viscous liquid | insufficient curing |
| 1.0:1.0 | 50.0 | viscous liquid | fragile flexible resin |
| 1.0:0.8 | 55.5 | viscous liquid | fragile flexible resin |
| 1.0:0.7 | 58.8 | semisolid | elastomer resin |
| 1.0:0.6 | 62.5 | semisolid | elastomer resin |
| 1.0:0.4 | 71.4 | solid | firm flexible resin |
| 1.0:0.2 | 83.3 | a |
It could not be uniformly mixed because of the high viscosity of the mixture.
Mechanism of the Curing Reaction and Determination of the Phenolic Hydroxyl Group of GL400
ATR-FTIR spectra of the GL-epoxy resin with 62.5 wt % GL400 content before and after thermal curing as well as control samples are shown in Figure 3a. The absorption peak at 911 cm–1 corresponding to the characteristic of an epoxide (oxirane ring)15,16 observed in uncured GL-epoxy resin was remarkably diminished in ATR-FTIR spectra of cured GL-epoxy resin, whereas the hydroxyl (O–H) absorption at 3350 cm–1 did not change significantly. On the other hand, the characteristic epoxide absorption peak at 911 cm–1 was observed in the ATR-FTIR spectra of the cured GL-epoxy resin with lower GL400 content of 41.2 wt % (Figure S2). The presence of the epoxide absorption peak even after thermal curing for this composition ratio indicated that the formulation of the composition ratio is integral to achieve suitable curing conditions. Owing to the reactive functional groups of GL400 (phenolic hydroxyl group) and PEGDGE (epoxide), nucleophilic addition of epoxy occurred during the thermal curing of the GL–PEGDGE mixture. Hence, it is assumed that the phenoxide generated from the phenolic hydroxyl group of GL serves as a nucleophile for nucleophilic addition to the epoxide (Figure 3b).10,17,18
Figure 3.

(a) ATR-FTIR spectra of the uncured and cured GL-epoxy resins with GL400 content of 62.5 wt %, PEGDGE, and GL400, as well as a magnified epoxide absorption region (1200–800 cm–1) to specify the epoxide absorption peak at 911 cm–1. (b) Curing mechanism of GL400 and PEGDGE.
To quantify the phenolic hydroxyl groups of GL400 involved in the curing reaction, quantitative 1H NMR measurements were performed. All GL400 samples were subjected to acetylation to improve the quantification of the phenolic hydroxyl groups before 1H NMR measurement.19,20Figure 4 shows the ATR-FTIR and 1H NMR spectra of acetylated GL400. The ATR-FTIR spectra of acetylated and nonacetylated GL400 were compared. The acetylation of the hydroxyl groups of GL400 was confirmed by the decrease in the absorption of the hydroxyl group (O–H) at around 3350 cm–1 and an increase in the absorption of the carbonyl group (C=O) at ∼1737 cm–1 (Figure 4a). In the 1H NMR spectrum of acetylated GL400, the signals corresponding to the acetyl groups were observed at 2.00 and 2.23 ppm (Figure 4b). The signal at 2.00 ppm was caused by the formation of an ester by the acetylation of an aliphatic alcoholic hydroxyl group such as PEG, whereas the signal at 2.23 ppm was caused by the formation of a phenyl ester by the acetylation of a phenolic hydroxyl group.19,20 The 1H NMR integral ratio of the phenyl ester of the acetylated GL400 to PFA revealed that 2.2–2.6 mmol of the phenolic hydroxyl group is present per gram21 of GL400, according to eqs 1 and 2,22,23 and the weight per functional equivalent of the phenolic hydroxyl group of GL400 is 385–455 g/equiv (Table 2).
| 1 |
| 2 |
where I, N, W, M, P, n, and m represent the integral area, number of protons, gravimetric weight, molar mass, purity, amount of substance, and molar amounts of the phenolic hydroxyl group in 1 g of GL400, standard (std), analyte functional group (X), analyte, and acetylated GL400 (sample), respectively. Because lignin might have a quite complex structure without unified molecular composition, five deviations were emphasized rather than showing the average value. From the calculation, a GL400 to PEGDGE weight ratio of 59–63 wt % was possibly indicative of the chemical equivalent of the epoxy curing reaction assumed in Figure 3b. This assumption was in agreement with the results summarized in Table 1. The GL400 content of >50 wt % was crucial to achieve a sufficient curing condition and GL400 content of >58.8 wt % led to the elastomeric nature of the resin.
Figure 4.

(a) ATR-FTIR spectra of GL400 and acetylated GL400. (b) 1H NMR spectrum of acetylated GL400; the inset represents the magnified region from 1.6 to 2.4 ppm to specify the chemical shift of aromatic and aliphatic ester.
Table 2. Characterization of the Functional Groups and Weight per Functional Equivalent of Samples.
| functional group |
|||
|---|---|---|---|
| samples | phenolic-OH (mmol/g) | epoxide (mmol/g) | weight per functional equivalent (g/equiv) |
| GL400 | 2.2–2.6 | 385–455 | |
| PEGDGE | 3.7a | 270a | |
This value is quoted from the test result table provided by Kyoeisha Chemical Co., Ltd., and the epoxy equivalent weight was estimated by the hydrochloric acid–dioxane method.
Mechanical Properties of the GL-Epoxy Resin
For a more detailed characterization on the flexible and elastomeric nature of the resins, the mechanical properties of the GL-epoxy resins with various compositions were investigated through hardness and tensile tests. Figure 5 summarizes the durometer hardness, tensile modulus and strength, and elongation at the break of the GL-epoxy resins with various GL400 contents. The mechanical properties totally tend to be low compared with those of traditional hard epoxy resins but the strength against elongation is only remarkable. Therefore, the developed GL-epoxy resins in this work are elastomeric type and the values are comparable with those of typical elastomeric and flexible epoxy materials.24,25 The flexible nature of GL-epoxy resins originated not only from PEGDGE but also partially from GL400 as GL itself was PEG-substituted lignin macromolecules, where the substituted PEG chains provide as a soft segment in GL macromolecules. By examining the physical and mechanical properties of GL-epoxy resins as a function of GL400 content, the hardness of the GL-epoxy resins gradually increased with the GL400 content and achieved rubber-like hardness at GL400 contents of 71–55 wt % (Figure 5a). On the other hand, the tensile modulus and strength of the GL-epoxy resins rapidly increased at a high GL400 content (Figure 5a,b). The tensile elongation at break was greater than 20% in all samples, indicative of a certain amount of reversible flexibility for the GL-epoxy resin (Figure 5b), and it increased with the GL content similar to hardness but it rapidly increased at a GL content of around 60% and did not increase beyond this point. These tendencies of physical properties are related to the equivalence point of GL400 and PEGDGE to achieve sufficient curing polymerization as described above. The ideal ratio of GL400 to PEGDGE estimated from NMR quantification for the polymerization reaction was 59–63 wt %, which is similar to the boundary value that existed in Figure 5, suggesting that mechanical properties are strongly influenced by the curing reaction between both molecules being influenced by the composition. The tensile properties were apparently linked directly to the flexibility of the polymer chains in the resins. As shown in Figure 5, the lower the content of the flexible aliphatic PEGDGE molecules, the higher the value of the tensile properties, particularly at a GL400 content greater than 60 wt %. On the other hand, the hardness of the resins was not directly related to the extension of polymer molecules but instead related to the nature of polymer components themselves. Considering the proportional increase in hardness with increase in the GL content, GL400 served as a curing agent as well as a filler to enhance the hardness of the GL-epoxy resin itself.26 Taking advantage of the features of the flexible and thermoplastic GL-epoxy resin that has been found in the present work, plastic products that require adequate strength and elasticity can contribute to the widespread use as valuable wood-derived natural materials.
Figure 5.

Mechanical properties of (a) hardness, tensile modulus, and (b) elongation and tensile strength of the GL-epoxy resins comprising various composition ratios of GL400. The gray area represents an equivalent weight ratio between GL400 and PEGDGE.
In summary, a simple formulation of bio-based elastomeric epoxy resin containing biomass resources of more than half was demonstrated by solvent-free mixing of PEG-modified glycol lignin produced from softwood meal and aliphatic epoxide material, PEGDGE, at a moderate curing temperature of 130 °C. The sufficient curing condition was monitored by the disappearance of characteristic epoxide absorption band (911 cm–1) using ATR-FTIR spectroscopy over a wide range of composition ratios. 1H NMR analysis further confirmed that the chemical equivalent, that is, weight per functional equivalent of the phenolic hydroxyl groups of the curing agent GL400 and PEGDGE resin, is crucial to determine the sufficient curing conditions of the GL-epoxy resins. The mixing ratio of GL400 of 59−63 wt % was found to be the chemical equivalent composition of the sufficient epoxy curing reaction, which also indicated as a boundary for the abrupt change in tensile properties of the GL-epoxy resins. On the other hand, the properties of hardness depend on the GL400 content, where GL400 served as a curing agent as well as a filler to enhance the hardness of the GL-epoxy resin. The GL-epoxy resins formulated in this work are expected to be used in the production of sealing materials as well as structural matrix material, which is then reinforced by fibers. As a large-scale GL production technology from softwood has already been developed,5 the development of GL-based functional materials other than GL-epoxy resins are also expected to meet the need of the future green industries.
Experimental Section
Materials
GL was prepared according to a previously reported method.4,5 The details of the synthesis method, the composition of lignin, and the characteristics of the molecular structure have already been given, so refer to these reports. In the present work, PEG400-modified glycol lignin (GL400) was used in which poly(ethylene glycol) with an average molecular weight of 380–420 is used for the modification. The GL400 used in this work is not exactly the same as in the previous papers because there are a lot of differences on cedar and molecular weight of poly(ethylene glycol) used, but such slight differences are negligible for the chemical process in the present work. PEGDGE with an average molecular weight of 540 and a purity of greater than 99% was purchased from Kyoeisha Chemical Co., Ltd. For 1H NMR measurements, analytical grade acetic anhydride, pyridine, and pentafluorobenzaldehyde (PFA, 98%) were purchased from FUJIFILM Wako Pure Chemicals Corporation. All reagents were used without further purification.
Fabrication of the GL-Epoxy Resin
GL400/PEGDGE epoxy resins containing 41–83 wt % of GL400 were prepared by mixing with an appropriate amount of GL400 and PEGDGE as shown in Table 1. In addition, it was also confirmed whether GL400 or PEGDGE alone would react under the same heating conditions. Briefly, a designated amount of GL400 powder was first mixed with PEGDGE in a 300 mL flask at room temperature. The mixture was stirred (60 rpm) at 100 °C for 4 h under the sealed condition to obtain uncured GL400-epoxy resin. The uncured GL-epoxy resin was then cast into a Teflon mold (110 × 110 mm2) and thermally cured in an oven at 130 °C for 60 h. The elastomeric GL-epoxy resin sheet, 1.4–1.6 mm thick, was finally obtained after thermal curing.
Characterizations of the GL-Epoxy Resin
ATR-FTIR spectra were recorded using Perkin Elmer Spectrum 100 FTIR spectrometer equipped with a diamond attenuated total reflection (ATR) attachment over the range of 4000–550 cm–1.
Thermogravimetric and differential thermal analysis (TG–DTA) profiles of the GL-epoxy resins were determined using Rigaku Thermo Olus EVO2 TG–DTA analyzer. The samples were loaded onto a Pt pan and heated from room temperature to 400 °C at a heating rate of 2 °C/min under the airflow (500 mL/min). Differential scanning calorimetry (DSC) profiles were recorded using Rigaku Thermo Plus DSC 8230. For DSC measurements, no reference lignin material as a standard was used. The samples were loaded onto Al pan and heated from room temperature to 250 °C at a heating rate of 5 °C/min under nitrogen flow (100 mL/min). Shore A hardness was measured on a Shore durometer GS-719G (Teclock Corporation). Samples with a thickness of up to 6 mm were used for the measurement. An average of five measurements was performed. Tensile tests were carried out on a Tokyo Koki Testing Machine Co., Ltd. using an LSC-005/30 force gauge equipped with a load cell of 50 N. The epoxy resin after curing was cut into strips (10 × 110 mm2) as the test sample. The sample strip was fixed from both sides using a gauge with a length of 40 mm and pulled with a load rate of 1 mm/min. All tests were conducted at room temperature (25 °C). An average of six measurements was carried out.
Determination of the Phenolic Hydroxyl Group of GL400 by 1H NMR Spectroscopy
First, 1.0 g of GL400 was added into 20 mL mixture of acetic anhydride and pyridine (1:1, v/v) in a 100 mL round-bottom flask. Second, the mixture was stirred (60 °C, 200 rpm) for 8 h under reflux at 20 °C. Third, the mixture was reprecipitated by dropwise addition into distilled water at 0 °C. The resulting precipitate was filtered and washed with distilled water to neutral pH. Finally, the precipitate was dried at room temperature, affording acetylated GL400. For 1H NMR measurements, 10 mg of the acetylated GL400 samples and 10 mg of PFA (internal standard) were dissolved in 0.8 mL of DMSO-d6. 1H NMR spectra were recorded on a Bruker AV400N instrument at a frequency of 400 MHz with an acquisition time of 4.0. All spectra were recorded at 26 °C. Acetylation and 1H NMR measurements were carried out five times.
Acknowledgments
This work was supported by SIP-Lignin Project, Technologies for Creating Next-Generation Agriculture, Forestry and Fisheries under the Cross-Ministerial Strategic Innovation Promotion Program (SIP) administered by the Council for Science Technology and Innovation (CSTI), Japan. The authors would like to thank Prof. Takashi Yamashita (Tokyo University of Technology) for valuable advice on NMR analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01884.
DSC profiles of GL-epoxy resin during the curing process; FTIR spectra of GL-epoxy resin with another GL400 content (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sun Z.; Fridrich B.; de Santi A.; Elangovan S.; Barta K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. 10.1021/acs.chemrev.7b00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. L.; Luo W.; Ciesielski P. N.; Fang Z. Q.; Zhu J. Y.; Henriksson G.; Himmel M. E.; Hu L. B. Wood-derived materials for green electronics, biological devices, and energy applications. Chem. Rev. 2016, 116, 9305–9374. 10.1021/acs.chemrev.6b00225. [DOI] [PubMed] [Google Scholar]
- Rabemanolontsoa H.; Saka S. Comparative study on chemical composition of various biomass species. RSC Adv. 2013, 3, 3946–3956. 10.1039/c3ra22958k. [DOI] [Google Scholar]
- Nge T. T.; Takata E.; Takahashi S.; Yamada T. Isolation and thermal characterization of softwood-derived lignin with thermal flow properties. ACS Sustainable Chem. Eng. 2016, 4, 2861–2868. 10.1021/acssuschemeng.6b00447. [DOI] [Google Scholar]
- Nge T. T.; Tobimatsu Y.; Takahashi S.; Takata E.; Yamamura M.; Miyagawa Y.; Ikeda T.; Umezawa T.; Yamada T. Isolation and characterization of polyethylene glycol (PEG)-modified glycol lignin via PEG solvolysis of softwood biomass in a large-scale batch reactor. ACS Sustainable Chem. Eng. 2018, 6, 7841–7848. 10.1021/acssuschemeng.8b00965. [DOI] [Google Scholar]
- Kaneko H.; Ishii R.; Suzuki A.; Nakamura T.; Ebina T.; Nge T. T.; Yamada T. Flexible clay glycol lignin nanocomposite film with heat durability and high moisture-barrier property. Appl. Clay Sci. 2016, 132–133, 425–429. 10.1016/j.clay.2016.07.009. [DOI] [Google Scholar]
- Takahashi K.; Ishii R.; Nakamura T.; Suzuki A.; Ebina T.; Yoshida M.; Kubota M.; Nge T. T.; Yamada T. Flexible electronic substrate film fabricated using natural clay and wood components with cross-linking polymer. Adv. Mater. 2017, 29, 1606512 10.1002/adma.201606512. [DOI] [PubMed] [Google Scholar]
- Suzuki A.; Ishii R.; Yoshida H.; Ebina T.; Nge T. T.; Yamada T. Microstructure and Properties of Clay Glycol Lignin Nanocomposite Films Using Different Mixed Solvents. Clay Sci. 2018, 22, 71–78. 10.11362/jcssjclayscience.22.3_71. [DOI] [Google Scholar]
- Kumar S.; Samal S. K.; Mohanty S.; Nayak S. K. Recent development of biobased epoxy resins: A review. Polym.-Plast. Technol. Eng. 2018, 57, 133–155. 10.1080/03602559.2016.1253742. [DOI] [Google Scholar]
- Li R. J.; Gutierrez J.; Chung Y. L.; Frank C. W.; Billington S. L.; Sattely E. S. A lignin-epoxy resin derived from biomass as an alternative to formaldehyde-based wood adhesives. Green Chem. 2018, 20, 1459–1466. 10.1039/C7GC03026F. [DOI] [Google Scholar]
- Qin J. L.; Woloctt M.; Zhang J. W. Use of polycarboxylic acid derived from partially depolymerized lignin as a curing agent for epoxy application. ACS Sustainable Chem. Eng. 2014, 2, 188–193. 10.1021/sc400227v. [DOI] [Google Scholar]
- Komiya G.; Imai T.; Happoya A.; Fukumoto T.; Sagae H.; Sone N.; Takahashi A. Effects of lignin derivatives on cross-link density and dielectric properties in the epoxy-based insulating materials for printed circuit boards. IEEE Trans. Compon., Packag., Manuf. Technol. 2013, 3, 1057–1062. 10.1109/TCPMT.2013.2253836. [DOI] [Google Scholar]
- Koike T. Progress in development of epoxy resin systems based on wood biomass in Japan. Polym. Eng. Sci. 2012, 52, 701–717. 10.1002/pen.23119. [DOI] [Google Scholar]
- Osamu T.; Ryo I.; Takashi N.; Takashi Y.; Takeo E.; Tatsuhiko Y.; Nge T. T.; Katsuro T.; Masato O.; Yuhi H.; Akihiko O.; Shinichi I.; Yuki I. Jpn. Patent P2018-104688A, July 5, 2018.
- Ma S. Q.; Liu X. Q.; Jiang Y. H.; Tang Z. B.; Zhang C. Z.; Zhu J. Bio-based epoxy resin from itaconic acid and its thermosets cured with anhydride and comonomers. Green Chem. 2013, 15, 245–254. 10.1039/C2GC36715G. [DOI] [Google Scholar]
- Chen J.; Nie X. A.; Liu Z. S.; Mi Z.; Zhou Y. H. Synthesis and application of polyepoxide cardanol glycidyl ether as biobased polyepoxide reactive diluent for epoxy resin. ACS Sustainable Chem. Eng. 2015, 3, 1164–1171. 10.1021/acssuschemeng.5b00095. [DOI] [Google Scholar]
- Laurichesse S.; Averous L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. 10.1016/j.progpolymsci.2013.11.004. [DOI] [Google Scholar]
- Ha Q. P.; Maurice J. M.. Epoxy resins. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; Vol. 13, pp 155–244. [Google Scholar]
- Li J. J.; Zhang J. Z.; Zhang S. F.; Gao Q.; Li J. Z.; Zhang W. Alkali lignin depolymerization under eco-friendly and cost-effective NaOH/urea aqueous solution for fast curing bio-based phenolic resin. Ind. Crops Prod. 2018, 120, 25–33. 10.1016/j.indcrop.2018.04.027. [DOI] [Google Scholar]
- Tejado A.; Pena C.; Labidi J.; Echeverria J. M.; Mondragon I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. 10.1016/j.biortech.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Matsushita Y. Conversion of technical lignins to functional materials with retained polymeric properties. J. Wood Sci. 2015, 61, 230–250. 10.1007/s10086-015-1470-2. [DOI] [Google Scholar]
- Al Deen T. S.; Hibbert D. B.; Hook J. M.; Wells R. J. Quantitative nuclear magnetic resonance spectrometry: II. Purity of phosphorus-based agrochemicals glyphosate (N-(phosphonomethyl)-glycine) and profenofos (O-(4-bromo-2-chlorophenyl) O-ethyl S-propyl phosphorothioate) measured by 1H and 31P QNMR spectrometry. Anal. Chim. Acta 2002, 474, 125–135. 10.1016/S0003-2670(02)01017-6. [DOI] [Google Scholar]
- Bharti S. K.; Raja R. Quantitative 1H NMR spectroscopy. TrAC, Trends Anal. Chem. 2012, 35, 5–26. 10.1016/j.trac.2012.02.007. [DOI] [Google Scholar]
- Kalkornsurapranee E.; Nakason C.; Kummerlowe C.; Vennemann N. Development and preparation of high-performance thermoplastic vulcanizates based on blends of natural rubber and thermoplastic polyurethanes. J. Appl. Polym. Sci. 2013, 128, 2358–2367. 10.1002/app.38201. [DOI] [Google Scholar]
- Polyzois G. L.; Tarantili P. A.; Frangou M. J.; Andreopoulos A. G. Physical properties of a silicone prosthetic elastomer stored in simulated skin secretions. J. Prosthet. Dent. 2000, 83, 572–577. 10.1016/S0022-3913(00)70017-5. [DOI] [PubMed] [Google Scholar]
- Shikinaka K.; Sotome H.; Kubota Y.; Tominaga Y.; Nakamura M.; Navarro R. R.; Otsuka Y. A small amount of nanoparticulated plant biomass, lignin, enhances the heat tolerance of poly(ethylene carbonate). J. Mater. Chem. A 2018, 6, 837–839. 10.1039/C7TA09216D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.