

Abstract
Mice deficient in glucose-6-phosphate dehydrogenase (G6PD) cannot replenish the cellular antioxidant glutathione, which detoxifies neurodegenerative reactive oxygen species (ROS). To determine the functional consequences of G6PD deficiency, young and aging G6PD-deficient mice were evaluated for brain G6PD activity, DNA damage (comets, γH2AX), Purkinje cell loss, brain function (electrophysiology, behaviour) and lifespan. DNA comet formation was increased and Purkinje cell counts were decreased in a G6pd gene dose-dependent fashion. γH2AX formation varied by age, sex and brain region, with increased levels in G6PD-deficient young and aging females, and in aging males. Aging male G6PD-deficient mice exhibited synaptic dysfunction in hippocampal slices. G6PD-deficient young and aging females exhibited deficits in executive function, and young deficient mice exhibited deficits in social dominance. Conversely, median lifespan in G6PD-deficient females and males was enhanced. Enhanced ROS-initiated brain damage in G6PD deficiency has functional consequences, suggesting that G6PD protects against ROS-mediated neurodegenerative disorders.
Keywords: Aging, Glucose-6-phosphate dehydrogenase (G6PD), Reactive oxygen species (ROS), DNA damage, 8-Oxo-2′-deoxyguanine (8-oxodG), Comet, Gamma-H2AX (γH2AX), Neurodegeneration, Behavioural disorders, Electrophysiology, Lifespan
Graphical abstract
Highlights
-
•
Glucose-6-phosphate dehydrogenase (G6PD) deficiencies are globally prevalent.
-
•
Brain deficiencies enhance G6pd gene dose-dependent oxidative DNA damage.
-
•
Deficient brains exhibit lower Purkinje cell numbers and synaptic dysfunction.
-
•
G6PD-deficient mice exhibit cognitive and motor abnormalities.
-
•
G6PD-dependent changes vary by age, sex and brain region.
1. Introduction
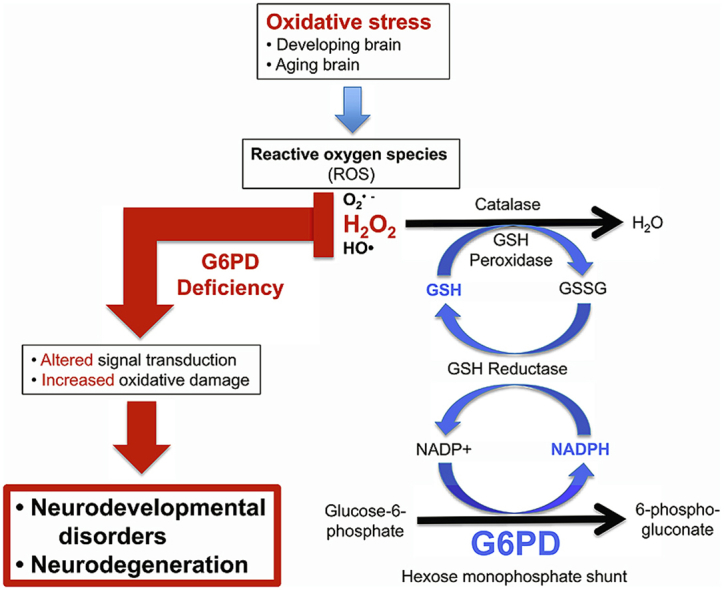
Glucose-6-phosphate dehydrogenase (G6PD) is the first and rate-limiting enzyme in the pentose phosphate (hexose monophosphate) pathway, and is important for its roles in the regeneration of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) and the production of pentoses and ribose-5-phosphate, a component of nucleotides [1,2]. NADPH plays a central role in protecting against cellular oxidative stress caused by reactive oxygen species (ROS), in particular by maintaining the cellular antioxidant glutathione in its reduced form (GSH), which is essential for the detoxification of potentially pathogenic reactive free radicals (superoxide, hydroxyl radicals) and hydroperoxides including hydrogen peroxide [3]. NADPH also is a key cofactor for the related catalytic activities of thioredoxin and peroxiredoxin reductases [4] and is important for maintaining the catalytic activity of catalase [5,6]; all these enzymes are critical for the detoxification of hydrogen peroxide and regulation of redox signaling.
It is generally believed that G6PD deficiencies constitute a problem only for mature red blood cells, which undergo lysis when acutely exposed to oxidative stress enhanced by drugs, infection or certain foods like fava beans [7]. However, recent studies suggest a broader protective role for this enzyme. G6PD plays a critical role in protecting the embryo from in utero death and birth defects caused by both physiological and drug-enhanced ROS levels [8]. The hepatic tissue of G6PD-deficient mice exhibits enhanced sensitivity to menadione-induced oxidative stress, with increases in the oxidation of DNA, lipids and proteins [9]. In another G6PD-deficient mouse model engineered to express a transgenic shuttle vector for measuring mutagenesis in vivo, the brains of deficient mice exhibited an increase in oxidative mutagenesis with accumulation of promutagenic etheno DNA adducts and increased somatic mutation rates, suggesting the potential for an increased risk of cancer [10]. Recently, we observed that the brains of untreated aged G6PD-deficient mice have increased levels of oxidatively damaged DNA, measured as 8-oxo-2′-deoxyguanine (8-oxodG), and histological examination of brain tissues revealed enhanced degenerative changes [11]. The changes associated with G6PD deficiency included qualitative and/or quantitative cellular degeneration or cell loss in the frontal cortex, cerebellum and hippocampus, and increased DNA oxidation in the same regions.
The risk of ROS-mediated damage to brain cells in G6PD-deficient mice may have important clinical implications, since this is the most common human enzymopathy, affecting over 400 million people and up to 60% of some populations [2]. The high incidence of G6PD deficiencies is believed to reflect an evolutionary adaptation to the widespread prevalence of malaria, as G6PD-deficient host red blood cells are inhospitable to the parasite Plasmodium falciparum [7], although our lifespan results herein suggest other mechanisms.
In light of our previous observations of oxidatively damaged DNA and related cellular degeneration in the brains of aging G6PD-deficient mice [11], we sought to confirm the quantitative nature of cerebellar Purkinje cell (PC) loss using a specific cellular marker (calbindin D-28K), and to determine if DNA oxidation was associated with a functional measure of DNA damage in the form of single-strand breaks and double-strand breaks, and most importantly with electrophysiological and behavioural consequences. Our results show that G6PD is neuroprotective in young as well as aging mice at the molecular, cellular and functional levels, although its functional impact may be discrete. However, the biological role of G6PD in aging is complex, as median lifespan was enhanced in G6PD-deficent mice, suggesting alternative hypotheses to malarial resistance for the high prevalence of this deficiency in humans.
2. Methods
2.1. Animals
Female and male mutant G6PD-deficient mice [12] (Medical Research Council, UK) were housed three or four per plastic cage with ground corn cob bedding. The G6PD deficiency mutation is an ethylnitrosourea-induced A-T transversion mutation in exon 1 of the G6pd gene, which causes a splicing error during post-translational processing resulting in decreased protein expression [8,13]. Food (2018 Teklad Global 18% Protein Rodent Diet, Envigo) and tap water were provided to the mice ad libitum. All animals were monitored regularly by a veterinarian, and the studies were approved by the University of Toronto Animal Care Committee in accordance with the standards of the Canadian Council on Animal Care. The G6pd gene is X-linked, and heterozygous (+/def) females were bred with wild-type (+/y) or hemizygous (def/y) males to yield progeny of all possible genotypes for females (+/+, +/def, def/def) and males (+/y, +/def), where “def” indicates a mutated gene resulting in deficient G6PD activity. The wild-type (WT) G6PD-normal genotypes for females and males respectively are G6pd+/+ and G6pd+/y. G6pd genotypes were confirmed by the TaqMan assay [14] performed by The Centre for Applied Genomics (TCAG) at the Toronto Hospital for Sick Children.
2.2. Activity of G6PD in young and aging mice
G6PD activity was measured as previously published [15] and detailed with some minor modifications in the accompanying supplementary online data.
2.3. Brain dissection
Young (2–5 months) and aging (12–18 months) mice were sacrificed using cervical dislocation and the brains were extracted and washed with distilled water. The brains were dissected to obtain the following individual regions: cerebellum, hippocampus, striatum and cerebral cortex. The dissected brain regions were snap frozen in liquid nitrogen and stored at -80 °C.
2.4. Comet assay: measurement of DNA single strand breaks in aging mice
DNA damage was measured by alkaline single-cell gel electrophoresis (SCGE), also known as the comet assay, as previously described [16] with minor modifications, detailed in the accompanying supplementary online data.
2.5. γH2AX in individual brain regions: measurement of DNA double strand breaks in young and aging mice
Western blotting was performed to detect the formation of DNA double strand breaks by the presence of H2AX phosphorylated at Ser139 (γH2AX) [84,85]. Nuclear protein was purified from brain samples using a modified protocol [86], the details of which are provided in the accompanying supplementary online data.
2.6. Purkinje cell analysis and calbindin immunoblotting in aging mice
H&E staining [17] were carried out at the Centre for Modeling Human Disease (CMHD) Pathology Core at the University of Toronto. Immunoblotting for calbindin was conducted using a mouse monoclonal anti-Calbindin-D-28K primary antibody (1:3000, Sigma-Aldrich, Oakville, ON) and a donkey anti-mouse IgG (H + L) HRP-conjugated secondary antibody (1:25000, Jackson ImmunoResearch Laboratories Inc., West Grove, PA), the details of which are in the accompanying supplementary online data.
2.7. Hippocampal function
The procedural details for electrophysiological recordings were described previously [[18], [19], [20]] with some minor modifications detailed in the accompanying supplementary online data.
2.8. Tube test for social dominance
Social dominance was assessed in both the young and aging mice using the tube test [87], the details of which are provided in the accompanying supplemental online data.
2.9. Puzzle box test
Executive function was assessed using the puzzle box test [21,22], the details of which are provided in the accompanying supplemental online data.
2.10. Marble burying test
Stereotypic behaviour was assessed in mice using the marble burying test [23], the details of which are provided in the accompanying supplemental online data.
2.11. Rotarod performance
Motor coordination was measured in aging mice using an accelerating rotarod apparatus [24,25], the details of which are provided in the accompanying supplemental online data.
2.12. Ledge balance test
The ledge balance test was used to assess balance in aging mice [26], the details of which are provided in the accompanying supplemental online data.
2.13. Hindlimb clasp test
The hindlimb clasp test was performed as described previously [27], with minor modifications, detailed in the accompanying supplementary online data.
2.14. Passive avoidance test
Passive avoidance represents a form of single-pass learning in which mice experience a brief unpleasant stimulus (mild foot shock) upon exposure to a one set of environmental cues (dark versus light chamber) [28]. The details of the experiment are provided in the accompanying supplementary online data.
2.15. Taste aversion test
Taste aversion is a cognitive behavioural test that measures the ability of the animal to learn and later recall an association of a specific taste (sweetness) with feelings of malaise [29]. The details of the experiment are provided in the accompanying supplementary online data.
2.16. Statistical analyses
Statistical significance was calculated using GraphPad Prism®, Version 5 (GraphPad Software, Inc., La Jolla, CA). The minimum level of significance used was p < 0.05. Significance between paired data was determined by the two-tailed Student's t-test, including the comet assay (Fig. 3), γH2AX comparison (Fig. 4), Purkinje cell (PC) counts (Fig. 5), hippocampal function (Fig. 6) and marble burying (Fig. S9). Fisher's exact test was used for the tube test (Figs. 7a and S4). Multiple comparisons among groups for immunoblots were analyzed by one-way ANOVA (Fig. 5). For data with two independent variables, two-way ANOVA was performed followed by a Bonferroni post-test, including G6PD activity in brain regions of young and adult mice (Fig. 1), ledge balance test (Fig. 7b), hind limb clasp test (Fig. S6), passive avoidance test (Fig. S7) and taste aversion test (Fig. S8). Linear regression was used to analyze data for the relation of cerebellar vs. blood G6PD activities (Fig. 2), and the correlation of rotarod performance with G6PD activity (Fig. S5). The Mantel-Cox log-rank test was used to analyze puzzle box data (Fig. 7c-f, Tables S1 and S2, Fig. S3) and median lifespan (Fig. 8).
Fig. 3.

Increase in DNA damage in the cerebellum and hippocampus of aging G6PD-deficient mice measured by the comet assay. Cells were obtained from the hippocampus and cerebellum and prepared and visualized as described in the Methods. Upper pictures depict representative “comet tails” resulting from DNA single strand breaks (SSBs) in single cells from each brain area and genotype. Lower graphs represent for each G6pd genotype the mean olive tail moment, or amount of SSBs, for 3 samples in each group. Statistical analyses were performed using Student's t-test. Asterisks indicate a difference from wild-type G6PD-normal mice for the same brain region (*p < 0.05, **p < 0.01).
Fig. 4.

Effect of G6PD deficiency on double strand breaks. The level of γH2AX, a marker of DNA double-strand breaks in various brain regions, was measured using western blotting. Panels A: Young G6PD wild-type (+/+ or +/y) normal mice compared with mutant G6PD-deficient (def/def or def/y) sex-matched counterparts. Panels B: Aging G6PD wild-type (+/+ or +/y) normal mice compared with mutant G6PD-deficient (def/def or def/y) sex-matched counterparts. Significant differences were found in the striatum of young males as well as the hippocampus of young and aged males and young females. The number of mice for each genotype is shown in parentheses. Statistical analyses were performed using Student's t-test. Asterisks indicate a difference from G6PD-normal mice of the same sex (def/y or +/+) (p < 0.05).
Fig. 5.

Decrease in Purkinje cell (PC) numbers and PC-related calbindin-D-28K expression in aging G6PD-deficient mice. Upper panel: PCs from aging mice (12–18 months) were counted in 10 distinct fields at 40X to give a total of cells for each sample. Statistical analysis was performed using the two-tailed Student's t-test. Asterisks indicate a difference from G6PD-normal controls of the same sex (p < 0.05). Lower panel: Densitometric analysis of calbindin-D-28K protein, a marker for PCs, was performed for cerebellar tissues from G6PD-normal mice (males: +/y; females: +/+) and G6PD-deficient mice (males: def/y; females: +/def, def/def). Statistical analysis was performed using one-way ANOVA. The number of mice for each genotype is shown in parentheses. Asterisks indicate a difference from G6PD-normal wild-type controls (p < 0.05).
Fig. 6.

Hippocampal electrophysiological function is altered in aging G6PD-deficient mice. Upper panel: Increased presynaptic function in aging hemizygous (def/y) G6PD-deficient male mice. Paired-pulse facilitation ratios (second fEPSP/first fEPSP) were plotted as a function of the interpulse intervals between the two stimulations, showing a significant enhancement in PPF in the aged G6PD-deficient mice compared to G6PD-normal wild-type controls (p < 0.05). Middle panel: Enhanced basal synaptic transmission in aging hemizygous G6PD-deficient male mice. The slope of evoked field excitatory postsynaptic potentials (fEPSPs) was plotted as a function of stimulus intensity, showing significant increases in both the slope and maximal responses in G6PD deficient mice compared to the age-matched G6PD-normal wild-type control (p < 0.05). The distance between the stimulating and recording electrodes was kept constant between slices and mice. Lower panel: Normal long-term potentiation (LTP) in aging hemizygous G6PD-deficient male mice. LTP was induced by theta burst stimulation (TBS) and showed no differences between genotypes. Traces in the inset above the graph were taken from the recordings immediately before and 40 min after delivery of the TBS given at a 20 min time point. The data were analyzed by Student's t-test.
Fig. 1.

G6PD activity in different brain regions of young (2–5 months) and aging (12–18 months) G6PD-normal and G6PD-deficient male and female mice. Panel A: Young G6PD wild-type (+/y) normal males compared to G6PD-deficient (def/y) counterparts. Panel B: Young G6PD wild-type (+/+) normal females compared to G6PD-deficient (def/def) counterparts. Panel C: Aging G6PD wild-type (+/y) normal males compared to mutant G6PD-deficient (def/y) counterparts. Panel D: Aging G6PD wild-type (+/+) normal females compared to mutant G6PD-deficient (def/def) counterparts. Panels E-H: Only mutant G6PD-deficient mice. Panel E: Young males compared to young females. Panel F: Aging males compared to aging females. Panel G: Young males compared to aging males. Panel H: Young females compared to aging females. Asterisks indicate significant differences between the G6PD-normal (+/y or +/+) group and the G6PD-deficient (def/y or def/def) group, or between young and aging females, for the same brain region (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). α indicates a difference from cortex (p < 0.0001 in Panels B and D, p < 0.01 in Panel C, p < 0.05 in Panels F – H); β indicates a difference from striatum (p < 0.0001 in cerebellum in Panel B, and in hippocampus in Panel D, p < 0.001 in hippocampus in Panel B, p < 0.05 in cerebellum in Panel D); χ indicates differences from hippocampus (p < 0.0001 in Panel D, p < 0.01 in Panel H). The differences in G6PD activity between age- and sex-matched mice depended upon the brain region analyzed. Statistical analyses were performed using two-way ANOVA, followed by a Bonferroni post-hoc test conducted independently for genotype-dependent differences vs. brain region-dependent differences (Panels A–D), and independently for sex-dependent differences (Panels E–F), or age-dependent differences (Panels G–H) vs. brain region-dependent differences (Panels E− H). For all groups, n = 3. The minimum significance level used throughout was p < 0.05.
Fig. 7.

Effect of G6PD deficiency on social dominance in young mice, motor coordination with age, and executive function in both young and aging mice. Social dominance (A): The tube test was performed to assess dysfunction in social aggression. Columns indicate % wins of unique matchups for each group and the X-axis represents the G6pd genotype along with the number of matchups performed for each group. G6PD wild-type (+/y and +/+) normal mice won more matchups against sex- and age-matched (within 20 days) G6PD-deficient (def/y and def/def, respectively) counterparts. The number (n) above each bar denotes the number of mice of each genotype used for testing. Fisher's exact test was used to determine whether the scores were significantly different from the expected 50:50 win/loss outcome by chance. Asterisks indicate a difference from the expected 50:50 outcome (**p < 0.01, ****p < 0.0001). Motor coordination (B): Ledge balance test. The ability of mice to walk across a narrow ledge was scored on a scale of 1–4, with 4 indicating the worst performance. The final results were grouped according to ages divided into 200-day intervals. Statistical analyses were performed using two-way ANOVA, followed by a Bonferroni post-hoc test. No sex differences were apparent, so the data for males and females were combined for analysis. The number of animals per group were as follows: G6pd +/+, +/y combined (no alleles mutated) = 33, G6pd +/def (one allele mutated) = 52, G6pd def/def, def/y combined (all alleles mutated) = 37. Performance in all animals progressively declined with age (p < 0.0001), but the decline was accelerated in homozygous G6PD-deficient mice (γ = p < 0.05), with a significant difference occurring at an earlier age (400 days). Executive function (C–F): The puzzle box test was performed to assess executive function and problem solving in G6PD-deficient mice. Performance was analyzed using a curve analysis relating the latencies to enter with the rate of incomplete trials at a given point in time. Each box compares the performance of the two groups in trial 1 of the puzzle box test. The number of mice for each genotype is shown in parentheses beside the respective survival curve. Panel C: Young G6PD wild-type (+/+) normal females performed significantly better than G6PD-deficient (def/def) counterparts. Panel D: Aging G6PD wild-type (+/+) normal females performed significantly better than mutant G6PD-deficient (def/def) counterparts. Panel E: Young G6PD wild-type (+/+) normal females also performed significantly better than aged counterparts. Panel F: Young G6PD wild-type (+/+) normal females performed significantly better than young G6PD males (wild-type, +/y, and deficient, def/y). Statistical analysis was performed using a Kaplan-Meier curve analysis followed by the Mantel-Cox log-rank test. Probability (p) values are shown in each panel.
Fig. 2.

Positive correlation of G6PD activity in erythrocytes and cerebellar tissues, and G6PD activities throughout the lifespan. Upper panel: G6PD activity in red blood cells and cerebellar tissue from the same mice. Lower panel: G6PD activity in red blood cells from all G6pd genotypes was measured repetitively in the same mice throughout their lifespan (about 3–24 months). The relationship was assessed by Pearson's correlation analysis.
Fig. 8.

Enhanced median lifespan in G6PD-deficient mice. Untreated mice were monitored throughout their lifetime and times and apparent causes for death were recorded. Differences in survival were assessed by the log rank Mantel-Cox test. The number of mice for each genotype is shown in parentheses beside the respective survival curve. The median lifespans of homozygous female and hemizygous male G6PD-deficient mice were indistinguishable, and greater than those for either heterozygous G6PD-deficient females (+/def) (p < 0.01) or G6PD-normal wild-type males (def/y) or females (+/+) (p < 0.01).
The puzzle box test (Fig. 7c-f, Tables S1 and S2, and Fig. S3), marble burying test (Fig. S9) and tube test (Figs. 7a and S4) were conducted in the same group of animals for both young and aging mice in the order listed. Mice were then sacrificed and used for biochemical analyses (G6PD activity levels in brain regions (Fig. 1) and yH2AX levels (Fig. 4).
Another group of aging animals was tested using the rotarod test (Fig. S5) followed by the taste aversion test (Fig. S8). A separate group of animals was assessed for behavioural deficits using the ledge balance test (Fig. 7b) followed by the hind limb clasp test (Fig. S6) and passive avoidance test (Fig. S7). Blood collected from the tail veins of all aging mice as well as rest of the colony was used to measure G6PD activity in red blood cells (Fig. 2). Survival curves were based on both the behaviourally tested mice and the entire colony (Fig. 8). Following behavioural testing, mice were sacrificed, and brains were assessed for cerebellar G6PD activity (Fig. 2), comet assay (Fig. 3), PC counts (Fig. 5) and hippocampal function (Fig. 6).
3. Results
3.1. G6PD activity varies by brain regions in young and aging normal and G6PD-deficient mice
The activity of G6PD was measured in different brain regions of both male and female young mice (2–5 months) and aging mice (12–18 months) to compare the effect of sex and age on the activity of G6PD. Regional differences in brain G6PD activity were found in G6PD-normal mice for young females and aging males and females.
In young G6PD-normal females, activity ranged 5.0-fold among the cerebellum, hippocampus, striatum and cortex in order of increasing activity (Fig. 1B). The aging G6PD-normal males (Fig. 1C) and females (Fig. 1D) had smaller respective 3.0-fold and 2.2-fold range in activity, generally increasing in activity from cerebellum, striatum and hippocampus to cortex (see figure legend for more details and specific differences).
G6PD activity in the striatum and cortex of the young G6PD-normal females was respectively 2.3- and 4.3-fold higher than that in the young G6PD-normal males, while activities in the cerebellum and hippocampus were similar (Fig. 1A and B). G6PD activity was similar between aging G6PD-normal males and females (Fig. 1C and D respectively) in all brain regions except the hippocampus, where the females had 1.5-fold higher activity.
G6PD activities in all brain regions were about 2- to 6-fold higher in aging G6PD-normal mice compared to young G6PD-normal mice of the same sex (Fig. 1A–D), but the increases were much greater in the males.
G6PD activity in deficient animals was 40–90% lower than in G6PD-normal animals, depending upon the number of G6pd alleles mutated. Among young males (Fig. 1A), G6PD-deficient mice had lower G6PD activity in the cerebellum (p < 0.05) and striatum (p < 0.01). Among young females (Fig. 1B), G6PD-deficient mice had lower G6PD activity in the hippocampus (p < 0.01), striatum (p < 0.0001) and cortex (p < 0.0001). Among aging males (Fig. 1C), G6PD-deficient mice had lower G6PD activity in the hippocampus (p < 0.05), striatum (p < 0.01) and cortex (p < 0.001). Among aging females (Fig. 1D), G6PD-deficient mice had lower G6PD activity in all four brain regions (p < 0.0001).
Among G6PD-deficient mice, sex-dependent differences in G6PD activity were not observed in either young or aging mice (Fig. 1E and F). Remarkably, age-dependent differences in regional brain G6PD activities were observed, ranging from 2.0- to 7-fold across brain regions in aging mice vs. young mice. Among G6PD-deficient males (Fig. 1G), aging mice had 3-fold higher activity in the cortex (p < 0.01) than young mice. A similar pattern of increased G6PD activity with aging was observed in G6PD-deficient females (Fig. 1H), with the aging mice having 7-fold higher activity in the hippocampus (p < 0.0001), 3.5-fold higher in the striatum (p < 0.01) and 2-fold higher in the cortex (p < 0.01).
The activity of G6PD in red blood cells correlated highly with that in cerebellar tissue from the same mice (r = 0.91, p < 0.0001) (Fig. 2, upper panel). This correlation allowed repetitive blood sampling of the same mice to determine changes in G6PD activity throughout their lifespan, and to provide estimates of activity in the brains of mice being tested for behavioural performance. In male (+/y) and female (+/+) G6PD-normal mice, and in female heterozygous (+/def) G6PD-deficient mice, red blood cell G6PD activity varied about 3-fold throughout their lifespan, approximating a “U”-shaped curve with highest activities below 200 days of age, lowest activities between 200 and 400 days of age, returning to higher activities after 400 days of age (Fig. 2, lower panel). In contrast, erythrocyte G6PD activity for both sexes remained uniformly low throughout the lifespan of both female and male mice with all G6pd alleles mutated (def/def, def/y).
3.2. DNA damage: increase in single strand breaks and altered double strand breaks in G6PD-deficient mice
Oxidative DNA damage reflected by single strand breaks (SSBs) was measured using the comet assay. Compared to age-matched wild-type controls, aging G6PD-deficient mice with either one or both G6pd alleles mutated exhibited increased SSB levels in both the cerebellum and hippocampus, with up to over 2-fold higher levels of damage in the latter region (Fig. 3). The variably enhanced comet levels in the cerebellar and hippocampal regions of the aging G6pddef/def females could not be fully explained by the magnitude of the decrease in the regional G6PD activity, as hippocampal comet formation was higher than that in the cerebellum despite having a lesser decrease in G6PD activity (Fig. 1D). Maximal levels of SSBs in the hippocampus occurred with the mutation of only a single G6pd allele (+/def), and SSB levels in the cerebellum were similarly increased in heterozygous (+/def) and homozygous (def/def) G6PD-deficient mice.
DNA double strand breaks (DSBs) in various brain regions of both young and aging mice were assessed by the formation of γH2AX [84,85] using western blotting. The effect of G6PD deficiency on the levels of γH2AX was highly dependent on age, sex and brain region. In young mice, G6PD-deficient (def/def) females had higher levels of γH2AX in the hippocampus (p < 0.05) than G6PD +/+ females, with no difference in the remaining regions (Fig. 4A). In contrast, young G6PD-deficient (def/y) males had lower levels of γH2AX in the striatum and hippocampus than the wild-type male G6PD-normal (+/y) mice (p < 0.05), while no difference was observed in the cortical and cerebellar regions (Fig. 4A). In aging mice, G6PD-deficient (def/y) males had higher levels of γH2AX in the hippocampus (p < 0.05) than their G6PD wild-type (+/y) counterparts, with no differences in the remaining regions (Fig. 4B). In aging females, there was a non-significant trend for increased γH2AX formation in the hippocampus, similar to the aging G6pddef/y males (Fig. 4B). As with comet formation, the sex-, age- and brain region-dependent increases in γH2AX formation could not be fully explained by regional G6PD activity. A substantial decrease in G6PD activity was necessary, but not sufficient, for enhanced γH2AX formation. Although brain regions of G6PD-deficient mice in the same age group exhibited similar G6PD activities (Fig. 1), only the hippocampus and striatum were susceptible to enhanced γH2AX formation, which was sex-, age- and brain region-dependent (Fig. 4).
The sex, age and brain region of G6PD-deficient mice exhibiting enhanced comet (SSBs) or γH2AX (DSBs) formation were not completely congruent, suggesting differences in the biochemical processes leading to these two forms of DNA damage, and perhaps in DNA repair. While the hippocampus from G6PD-deficient aging male exhibited increases in both SSBs and DSBs, aging G6PD-deficient female cerebella exhibited enhanced SSBs but not DSBs.
3.3. Reduced Purkinje cell (PC) counts and calbindin staining in brains of G6PD-deficient mice
The number of cerebellar PCs determined by cell counting was substantially reduced in aging G6PD-deficient males (def/y, 25%) and females (+/def, 28%; def/def, 37%) compared to wild-type controls (p < 0.05), (Fig. 5, upper panels). Homozygous G6PD-deficient females (def/def) exhibited about a 1.5-fold greater loss than hemizygous (def/y) deficient males (37% vs. 25% compared to wild-type), but this difference was not significant (Fig. 5, upper panels). A reduction in PC number with G6PD deficiency was confirmed by western analysis for the PC marker calbindin-D-28K, with a 47% decrease in aging G6PD-deficient mice with either a homozygous (females, def/def) or hemizygous (males, def/y) G6pd mutation, and a lesser 28% reduction in heterozygous G6PD-deficient females (p < 0.05), revealing a G6pd gene dose-dependent relationship (Fig. 5, lower panel).
3.4. Altered hippocampal function in G6PD-deficient mice
To determine the effect of G6PD deficiency on the functional properties of synaptic transmission and plasticity, we conducted electrophysiological recordings in the CA1 region of the hippocampus using an established hippocampal slice model [18]. First, paired-pulse facilitation (PPF) reflecting short-term synaptic plasticity was examined as an indicator of presynaptic function (Fig. 6, upper panel). The magnitude of PPF over the range of 25–100 m s was substantially and significantly increased in the G6PD-deficient mice. Second, basal synapse strength was determined by stimulating the Schaffer collaterals in the CA3 region and recording field excitatory postsynaptic potentials (fEPSPs) at the CA1 synapse (Fig. 6, middle panel). Analysis of evoked fEPSPs revealed a substantial and significant enhancement in both the stimulus intensity-response curve and the maximal response in def/y G6PD-deficient mice compared to +/y wild-type G6PD-normal controls. Finally, we analyzed long-term potentiation (LTP), an extensively studied form of long-lasting synaptic plasticity widely regarded as a synaptic model for learning and memory [30] (Fig. 6, lower panel). We induced LTP by theta burst stimulation (TBS) because it is considered to be a more physiologically relevant protocol to elicit plasticity. There was no difference in the magnitude of LTP between the G6PD-deficent mice and wild-type controls.
3.5. G6PD deficiency affects social dominance in young but not aging mice and executive function in young and aging female mice
The tube test assesses social dominance and aggression in mice without allowing them to physically injure each other [87]. Young G6PD wild-type (+/y) males won more matchups (78%) than their G6PD-deficient (def/y) counterparts (p < 0.0001). Young G6PD wild-type (+/+) female mice also won more matchups (68%) than their G6PD-deficient (def/def) counterparts (p < 0.01) (Fig. 7A). In aging G6PD-deficient mice, no differences were observed in either the males or females (Supplementary Fig. S4).
The puzzle box tests executive functions in mice by placing increasingly difficult obstacles between the brightly lit entry chamber and a dark goal chamber [22]. The test consists of 9 trials, split evenly over three consecutive days, as described in the Methods and the supplementary online data (Supplementary Tables S1 and S2). The puzzle box test was conducted in young and aging mice, and performance was compared for effects of age, sex and G6pd genotype.
Young G6PD +/+ females performed better in the puzzle box test than their G6PD def/def counterparts in trials 1 (p = 0.0011) (Figs. 7C) and 2 (p = 0.0397) (Supplementary Table S2). Among young males, there were no G6PD-dependent differences in performance in any trials (Supplementary Table S2, Fig. S3A). The similarity in performance between young G6PD +/y and def/y males allowed for them to be combined into one group and then compared to young G6PD +/+ females to test for a sex effect (Fig. 7F). In this comparison, the G6PD-normal female mice outperformed the male mice in each of the 9 trials, including six where the difference was significant (p < 0.05) and two more where the difference was marginally significant (0.05 < p < 0.1) (Supplementary Table S2).
Among aging mice, aging G6PD +/+ females performed better than their G6PD def/def counterparts in trial 1 (p = 0.0011) (Fig. 7D), with a similar but non-significant trend observed in the remaining trials (Supplementary Table S2). In contrast, G6PD-deficient males appeared to perform better than their G6PD-normal counterparts in most trials, although the difference in performance was significant only in trial 4 (p = 0.0123), with a non-significant trend in trial 2 (p = 0.0942) (Supplementary Table S2).
To look for a possible effect of age on performance in the puzzle box test, young G6PD +/+ and def/def females were compared to their respective aging counterparts. Young G6PD +/+ females performed better than their aging wild-type counterparts in only trial 1 (p = 0.001) (Fig. 7E), with a similar but only marginally significant difference in trial 2 (p = 0.0694) (Supplementary Table S2). Young G6PD def/def females performed better than their aging counterparts in only trial 1 (p = 0.037) (Supplementary Table S2). In none of these comparisons did one group perform significantly better than the other groups in all of the nine trials (Supplementary Table S2).
3.6. Motor function: rotarod, ledge balance and hindlimb clasping tests
In the rotarod test, the performance of female wild-type, G6PD-normal mice declined slightly but significantly with age (Fig. S5, upper panel). This decline with age was not observed in wild-type males, nor in G6PD-deficient females or males of any genotype. When groups were divided by age (<7 months, 7–18 months, > 18 months) or analyzed together, there was no correlation between G6PD activity and rotarod performance at any age (Supplementary Fig. S5, lower panel).
In the ledge balance test, performance declined with age for all G6pd genotypes, but did so more quickly with age for G6PD-deficient mice with all alleles mutated (def/def or def/y) compared to heterozygous (+/def) G6PD-deficient mice and wild-type G6PD-normal animals (+/+, +/y) (Fig. 7B). The G6PD-normal mice performed worse at 600 days (p < 0.01) and at 800 days (p < 0.01) compared to at 200 days, with no observable change at 400 days. The heterozygous (+/def) G6PD-deficient mice performed significantly worse at 600 days (p < 0.0001) and at 800 days (p < 0.0001) compared to at 200 days, with no significant change at 400 days. The G6PD-deficient mice (def/def, def/y) performed significantly worse at 400 days (p < 0.0001), at 600 days (p < 0.0001) and at 800 days (p < 0.01) compared to at 200 days. The results were analyzed via two-way ANOVA and a Bonferroni post-hoc test.
There were no G6PD-dependent differences with age in the hindlimb clasping test (Supplementary Fig. S6), although all genotypes showed a progressive decline. The G6PD-normal mice performed worse at 600 days (p < 0.01) and 800 days (p < 0.05) compared to at 200 days, with no significant change at 400 days. The heterozygous (+/def) G6PD-deficient mice performed worse at 400 days (p < 0.01), at 600 days (p < 0.0001) and at 800 days (p < 0.01) compared to at 200 days. The G6PD-deficient mice with all alleles mutated (def/y, def/def) performed worse at 400 days (p < 0.01), at 600 days (p < 0.0001) and at 800 days (p < 0.0001) compared to at 200 days. The results were analyzed via two-way ANOVA and a Bonferroni post-hoc test.
3.7. No effect of G6PD-deficiency in passive avoidance and taste aversion tasks
No G6PD-dependent differences in cognition with aging were observed as measured by either the passive avoidance or taste aversion tests (Supplementary Figs. S7 and S8).
3.8. G6PD deficiency does not increase marble burying behaviour
There were no differences in marble burying behaviour observed between G6PD wild-type and G6PD-deficient mice among any of the groups tested: young G6PD males and females, and aging G6PD males and females (Supplementary Fig. S9).
3.9. Improved survival in G6PD-deficient mice
The lifespan of the colony was monitored, and G6PD-deficient females (def/def) and males (def/y) with all alleles mutated showed a small but significantly improved median lifespan compared to heterozygous G6PD-deficient mice (+/def) and wild-type G6PD-normal animals (+/+, +/y) (Fig. 8).
4. Discussion
This is the first examination of G6PD activity in brain regions of both young and aging male and female G6PD-deficient mice, along with a lifespan study of G6PD activity and survival in a subset of mice. Enhanced oxidative DNA damage and cellular loss in selective brain regions of G6PD-deficient mice was differentially associated in a sex- and age-dependent fashion with disorders in cellular synaptic function and cognitive behavioural disorders. Conversely, median lifespan was increased in homozygous female and hemizygous male G6PD-deficient mice.
4.1. G6PD activity varies by brain regions in young and aging normal and G6PD-deficient mice
While regional differences in brain G6PD activity in aging mice were consistent with a previous report in aging mice [11], the current study showed that activities in all brain regions were up to about 6-fold higher in aging mice (12–18 months) than in young mice (2–5 months), the latter of which had not been previously evaluated (Fig. 1). Since G6PD gene expression is believed to be coordinately regulated along with that of other antioxidative enzymes, and particularly those components of the glutathione system [31,32], the results herein suggest that the increase in G6PD activity with age may in part constitute an attempt to compensate for antioxidative enzymes in the brain such as glutathione reductase and SOD, which decline with age [33,34]. This pattern was corroborated in a subgroup of mice followed over their lifespan, which found that G6PD activity in wild-type red blood cells, which correlated highly with cerebellar activity, was substantially higher in their aging period (greater than 400 days) than during their “middle age” period (200–400 days) (Fig. 2). Conversely, however, during at their youngest age (less than 200 days of age), erythrocyte G6PD activity was substantially higher than during their middle age, and similar to that in their aging period, suggesting an attempt to meet higher metabolic demands, including those necessary for combatting oxidative stress, during postnatal development, as has been shown for prenatal development [8,35].
4.2. DNA damage: increase in single strand breaks and altered double strand breaks in G6PD-deficient mice
In G6PD-deficient aging mouse brains, we previously reported an increase in oxidatively damaged DNA reflected by the 8-oxo-2′-deoxyguanosine lesion (8-oxodG) caused by hydroxyl radicals [11]. The alkaline comet assay used herein measures DNA single-strand breaks (SSBs) and alkali-labile sites [36], providing a complementary functional outcome in single cells, and possibly a more sensitive measure of DNA damage. The increased level of DNA damage in multiple brain regions of aging G6PD-deficient mice revealed in the comet assay corroborated our previous measures of the oxidative DNA lesion 8-oxodG, indicating a consistent neuroprotective role for G6PD in aging at the molecular level (Fig. 3). The approximately 2-fold greater level of DNA damage in hippocampal tissues compared to the cerebellum suggests that the former tissue may be at even greater risk of oxidative damage during aging, possibly due to a tissue-specific increased level of ROS formation and/or relatively lower levels of pathways for ROS detoxification and/or DNA repair. Maximally enhanced DNA damage was observed in aging mice with the loss of only one G6pd allele, indicating that even modest reductions in this enzyme may have important neurodegenerative consequences, as G6PD activity in the heterozygous mice is decreased to about 40% of normal [11].
To corroborate the comet assay at the molecular level, we measured the levels of γH2AX, a phosphorylated marker of sublethal DNA damage that is elevated in the presence of DNA double strand breaks (DSBs) and is associated with the initiation of cell death processes [37,38]. γH2AX levels were measured in young (3–5 months) as well as aging (12–18 months) mice to investigate whether there was evidence of oxidative damage in the brains of G6PD-deficient mice at a younger age that may have long-term consequences (Fig. 4). G6PD-dependent differences in γH2AX levels varied greatly by age, sex and brain region. Consistent with the comet assay results, there was increased hippocampal γH2AX in young female G6PD def/def mice, and similarly increased hippocampal γH2AX in aging male def/y mice. In contrast, young male def/y mice exhibited decreased striatal and hippocampal γH2AX levels. The pattern of changes observed with γH2AX measurement were not completely congruent with our measures of SSBs reflecting oxidative damage, suggesting that other factors, possibly including sex-, age- and/or brain region-dependent modulation of pathways for ROS formation and detoxification, and/or DNA repair, may modulate the consequences of G6PD deficiency. For example, enzymes involved in ROS formation like prostaglandin H synthases (PHS) have been associated with enhanced brain region-dependent oxidative DNA damage in cell culture and in aging Phs1 knockout mice [39,40]. Similarly, during development, oxidative DNA damage in fetal brains and postnatal neurodevelopmental deficits initiated by physiological ROS levels have been found to be modulated via genetic and pharmacological changes in numerous embryonic and fetal pathways for ROS formation and detoxification, and for the repair of oxidative DNA damage [35,41]. Our focus herein upon oxidatively damaged DNA as a potential molecular mechanism of neurodegeneration initiated by physiological brain levels of ROS is based upon related studies of neurodevelopmental deficits, and does not necessarily exclude other ROS-relevant mechanisms such as oxidative damage to other cellular macromolecules (lipids, proteins) in the brain, and/or ROS-mediated signal transduction. In relevant developmental studies, in utero exposure of the fetal brain to physiological ROS levels results in enhanced oxidative DNA damage in fetal brains and postnatal neurodevelopmental deficits in the progeny of untreated knockout mice with deficient enzymes/proteins critical to the repair of oxidative DNA damage, including oxoguanine glycosylase 1 (OGG1) [41] and breast cancer protein 1 (BRCA1) [42,43], implicating DNA damage as an important pathogenic molecular mechanism.
Similar developmental studies in our mutant G6pd mouse model found that untreated G6PD-deficient progeny exhibited enhanced oxidative DNA damage and G6pd gene dose-dependent increases in in utero death and birth defects [8]. The disorders of social dominance (both sexes) and executive function (females only) exhibited in young mice herein suggests that the initiating damage may have occurred in utero and was exacerbated postnatally in G6PD-deficient progeny by an ongoing deficit in protection against physiological levels of ROS formation.
4.3. Reduced Purkinje cell (PC) counts and calbindin staining in brains of G6PD-deficient mice
The previously reported loss of cerebellar Purkinje cells (PCs) in female heterozygous G6PD-deficient mice was determined by cell counting [11]. Herein, in addition to cell counting, we definitively identified PC numbers by immunoblotting for calbindin-D-28K, which is a specific marker for PCs [44,45]. We found a calbindin-related decrease in PCs in the cerebella of aging G6PD-deficient mice similar to that previously determined by cell counting (Fig. 5). The loss of PCs was exhibited in both male and female aging G6PD-deficient mice, and a maximal loss was observed with the mutation of only a single G6pd allele, as was similarly observed with the comet assay. The remarkable maximal cellular loss in aging G6PD-deficient mice indicates that even a modest reduction in the expression of this enzyme has neurodegenerative consequences for cellular loss in aging. Interestingly, G6PD expression is reported to be highest in PCs [46], and is associated with increased activity of NADPH-dependent reactions and enzymes in the brain of rats [47,48]. The congruency of maximal DNA damage and PC loss in heterozygous as well as homozygous and hemizygous aging G6PD-deficient mice observed herein is consistent with a causal association of low levels of G6PD leading to enhanced oxidative DNA damage and cellular neurodegeneration.
4.4. Altered hippocampal function in G6PD-deficient mice
The effect of G6PD deficiency on cellular brain function was investigated using electrophysiological studies, which showed that the excitatory synaptic function of glutaminergic neurons is substantially altered in G6PD deficiency. Hippocampal slices from aging G6PD-deficient mice exhibited remarkable increases in both presynaptic function and basal synaptic transmission, although with no apparent change in long-term potentiation (LTP) (Fig. 6), which is associated with learning and memory [49]. Activation of protein kinase C (PKC) is thought to involve a reaction with superoxide in both the presynaptic and postsynaptic neurons [50]. Activated PKC in the presynaptic nerve terminal prolongs the release of neurotransmitters [50], therefore increased ROS in G6PD-deficient mice may be enhancing presynaptic function. Similarly, activation of PKC in the postsynaptic neuron may be amplifying synaptic strength. The lack of an effect on LTP could arise from a selective loss of inhibitory neurons in the hippocampus, which would cause an increase in synaptic transmission in the remaining excitatory neurons, thereby nullifying an effect on LTP. These increased outcomes in hippocampal synaptic function in aging G6PD-deficient brains are completely different from the enhanced cerebellar cell death evidenced by reduced PC number, suggesting regional and/or cellular differences in the response to oxidative stress, including the up- and down-regulation of ROS-related compensatory pathways. As a proof of principle, our studies of molecular and biochemical outcomes focused upon the hippocampus and cerebellum because of their known roles in cognitive and motor functions [49,51], but it would not be surprising if other brain regions and neural pathways were altered in G6PD-deficient mice.
4.5. G6PD deficiency affects social dominance in young but not aging mice and affects executive function in young and aging female mice
The tube test was performed to measure social dominance, as this property is believed to be dependent on cortical function [52], which may be disturbed in G6PD-deficient mice as a result of enhanced oxidative DNA damage, assessed as the 8-oxoguanine lesion in G6PD-deficient mice [11]. The remarkable decline in tube test performance observed herein in both female (def/def) and male (def/y) young mice suggests that cortical function is indeed impaired by physiological levels of ROS when G6PD activity is impaired (Fig. 7A). Interestingly, this was the only behavioural test of cognition or motor function in our battery, along with the accelerated decline in the ledge balance test (Fig. 7B), that found a G6PD-dependent difference in both sexes. The lack of cortical DNA damage in G6PD-deficient mice measured herein as γH2AX formation, in contrast to the reported cortical increase in the 8-oxoguanine lesion [11], suggests that there is regional heterogeneity in the amounts of different types of oxidative DNA damage, and that reliance on the measurement of a single type of DNA damage can miss enhanced damage in some regions. In contrast to young mice, the tube test found no difference in social dominance between aging G6PD wild-type and deficient mice for either males or females (Supplementary Fig. S4). The loss of a G6PD-dependent impact on social dominance in aging mice may be due in part to a general decline in this function with age in even wild-type mice, as was observed with the ledge balance test (Fig. 7B) and the hindlimb clasp test (Supplementary Fig. S6). In this case, it would be more difficult to detect a subtle G6PD-dependent decline in an already reduced performance. This possibility contributed to our decision to include young mice in several other behavioural tests.
Executive function was measured using the puzzle box test, in which the mice must employ an increasing range of executive functions including working and contextual memory, spatial navigation and problem solving [88,89] in order to negotiate increasingly difficult obstacles in their path from the start chamber to the goal chamber. Mice are expected to want to leave the lighted start chamber and enter the dark goal chamber because they prefer a dark environment [21,22]. The effect of G6PD deficiency appeared to be sexually dimorphic with aging female G6PD-deficient (def/def) mice performing worse that their age-matched G6PD-normal (+/+) counterparts, while a reverse trend was observed in aging males (Fig. 7D and Table S2).
The remarkable deficit in executive function observed using the puzzle box test in both young and aging G6PD-deficient (def/def) females compared to their age-matched wild-type counterparts corroborated the role of G6PD in protecting the brain from cognitive neurodegenerative deficits caused by physiological levels of ROS. The greater deficits observed in both the female G6PD-normal (+/+) and G6PD-deficient (def/def) aging mice compared to their respective young counterparts suggests that the pathogenic effects of physiological ROS levels accumulate with age and are exacerbated by G6PD-deficiencies. Although a significant deficit in executive function was not observed in young G6PD-deficient male mice, they exhibited the same trends as did the females, suggesting only a relative sex-dependent reduction in risk. Since the two male genotypes were not different in their puzzle box performance, the data for the +/y and def/y genotypes were combined and compared to the young G6PD +/+ females, revealing a highly significant sex-dependent difference in executive function (Fig. 7F), whereby the young G6PD +/+ females were able to navigate the puzzle box more capably than the young G6PD +/y and def/y males.
4.6. Motor function: rotarod, ledge balance and hindlimb clasping tests
A reduction in PCs has been associated with motor function deficits in mouse models such as the Lurcher mouse [53] and the Purkinje Cell Degradation (PCD) mouse [54]. We have previously found that the rotarod test revealed motor coordination deficits caused by enhanced in utero oxidative stress in the fetal brain following maternal treatment with the ROS-initiating teratogen methamphetamine in normal [55] and DNA repair-deficient pregnant mice [56]. In the aging G6PD-deficient mice herein (Supplementary Fig. S5), we did not observe a decline in rotarod performance, although there was a small decline in performance in the aging G6PD-normal female mice. The basis for the decline only in female G6PD-normal mice is not known but did not correlate with declining G6PD activity (Supplementary Fig. S5). In G6PD-deficient mice with all alleles mutated (def/def, def/y), G6PD activity in all brain regions was increased up to 6-fold in aging compared to young mice (Fig. 1F), albeit still much lower than in wild-type mice, which may have contributed to the absence of any decline in rotarod performance in aging mice. Also, the maximal loss of PC cells in females (37%) and males (25%) with all G6pd alleles mutated may not have been sufficient to cause motor function deficits. In other related models, the Lurcher mouse and the PCD mouse show an almost total degeneration of PCs as well as other cells in the cerebellum, and these mice display major motor deficits in old age [53,54,57]. Interestingly, the Lurcher mice, even with almost complete loss of PCs, were still able to learn to match their wild-type littermate controls in a tilted platform test, which is similar to the rotarod test [58]. Other studies found that the degree of PC loss did not correlate with motor coordination deficits in several mouse models including Staggerer mice, Lurcher mice and Hot-foot mutants, as well as with some specific null mutations affecting PCs [59]. These studies may explain at least in part the absence of a decrease in rotarod performance in aging G6PD-deficient mice herein, despite their significant loss of PCs. Conversely, a transgenic mouse model overexpressing G6PD protein and activity exhibited significantly improved rotarod performance in females, and a non-significant improvement in males [60], so the absence of G6PD-dependent deficits in our G6PD-deficient mice may reflect the modest effects of physiological ROS levels on this particular outcome, and/or strain differences in the mouse models. In addition, G6PD-deficient mice also may differentially alter their regulation of cerebellar pathways for ROS formation and detoxification, and/or DNA repair.
In a recently published study of transgenic mice, G6PD overexpression protected various murine tissues, including the brain, from oxidative damage [60]. Furthermore, aging transgenic G6PD mice demonstrated greater neuromuscular fitness, as measured by the rotarod test, than their wild-type counterparts. This study cannot be directly compared to ours because its authors utilized a transgenic G6PD model, which overexpresses G6PD and may also have other effects on cellular metabolism, whereas we have used a G6PD-deficient mouse model and compared it to matched controls with normal G6PD wild-type expression. However, the study shows that enhanced levels of G6PD can boost the antioxidative system in the brain and help to protect it from the various endogenous sources of ROS.
In light of the decrease in PCs observed in G6PD-deficient mice, the ledge balance test and hindlimb clasping test were employed to discern deficits in motor function not detected by the rotarod test. The ledge balance test showed a general decline in performance with aging in all genotypes, and revealed that homozygous (def/def) and hemizygous (def/y) G6PD-deficient mice exhibited a maximal decline earlier in life, beginning at 400 days, whereas the heterozygous G6PD-deficient mice and wild-type normal mice did not exhibit the same maximal decline until 600 days (Fig. 7B). Similar results have been reported for mouse models of Huntington's disease [61,62], which show damage specifically to PCs in the cerebellum [63,64]. The hindlimb clasping test showed a decrease in motor function with aging in all genotypes, possibly indicative of specific damage in the substantia nigra, but no G6PD-dependent differences (Supplementary Fig. S6). Animal models for Parkinson's disease with specific damage in the substantia nigra exhibit deficits in the hindlimb clasping test but not in balance tests [27]. The absence of a G6PD-dependent impact on this outcome may be explained in part by a previous study in which we found no increase in DNA oxidation or morphological changes in the substantia nigra or striatum in aging G6PD-deficient mice [11]. The G6PD-dependent decline in motor function measured in the ledge test is consistent with the G6PD-dependent decline in PCs, suggesting a discrete impact on motor function not detected by the hindlimb clasping and rotarod tests herein.
4.7. No effect of G6PD-deficiency in passive avoidance, taste aversion and marble burying tasks
G6PD-deficient mice were tested for passive avoidance (Supplementary Fig. S7) and taste aversion (Supplementary Fig. S8) to assess cognition, and for marble burying (Supplementary Fig. S9) to assess repetitive behaviour. These three tests had been found to be sensitive in revealing functional changes in mouse models where a modest genetic or morphological change had occurred in the brain [[65], [66], [67]]. However, no G6PD-dependent changes in performance were observed in either sex at any age, possibly due in part to the enhanced presynaptic function and synaptic transmission that we observed in hippocampal slices from G6PD-deficient mice. Disorders in these cognitive behaviours may require more severe damage in the relevant brain regions, similar to the substantial loss of dopaminergic neurons required for overt behavioural changes in Parkinson's disease, where the onset of symptoms in humans is associated with a 50% decrease in cell number in the substantia nigra [68]. These tests may still reveal increased cognitive deficits in aging G6PD-deficient mice concurrently exposed to environmental conditions that enhance brain ROS formation, and/or reduce pathways of antioxidative defense or DNA repair.
4.8. Improved survival in G6PD-deficient mice
G6PD is primarily studied for its importance in preventing the hemolysis of red blood cells, and the high human prevalence of G6PD deficiencies is generally believed to result from the evolutionary pressure from enhanced survival of G6PD-deficient people with malaria [2]. However, we unexpectedly found that both male and female homozygous and hemizygous G6PD-deficient mice had an increased rather than decreased median lifespan compared to their heterozygous G6PD-deficient or wild-type G6PD-normal counterparts (Fig. 8), suggesting that the basis for the high prevalence of human G6PD deficiencies may be due to factors other than malarial resistance. The mechanism underlying this novel observation is unknown, and could include alterations in NADPH-dependent pathways aside from G6PD, and/or other biochemical changes due to a reduction in the pentose phosphate pathway, perhaps analogous to the increased survival observed in animals on a severely restricted diet [[69], [70], [71], [72]]. One proximate mechanism might involve decreased activity of ROS-producing NADPH oxidases (NOXs) in critical cells/tissues of G6PD-deficient mice, leading to reduced oxidative stress in cells/tissues conducive to lifespan enhancement. NOX activity depends upon NADPH provided by G6PD, and male hemizygous (def/y) G6PD-deficient mice have about a 50% decrease in NADPH levels [73]. Such a protective mechanism would likely be cell/tissue-specific, which is consistent with the variability herein in oxidative DNA damage both among brain regions in wild-type mice, and in the extent of enhanced oxidative DNA damage in different brain regions of G6PD-deficient mice.
In contrast to the lifespan-extending consequences of G6PD-deficiency observed in our mutant mouse model, transgenic mice expressing conversely increased levels of G6PD protein and activity also exhibited an extended median lifespan, although this occurred only in females [60], suggesting that strain differences and/or unappreciated differences resulting from the generation of the respective transgenic and mutant mouse models can substantially alter both health outcomes and their sex-dependence in mice. Relevant to our mutant mouse model, G6PD deficiency has recently been reported to have apparently contradictory health benefits, perhaps due at least in part to contrasting roles of this enzyme. For example, G6PD deficiency has been reported to reduce: (1) human retinopathy and mortality from cardiovascular disease in a discreet Mediterranean population [74]; (2) cholesterol synthesis, ROS formation and reductive stress in mice [75]; and, (3) angiotensin II-dependent hypertension in mice [73]. In contrast, other studies have reported that G6PD deficiency in mice results in a spectrum of cardiomyopathies including age-associated cardiac hypertrophy [75,76], which would not be consistent with the enhanced median lifespan observed in our aging G6PD-deficient mice. It has been postulated that G6PD deficiency may decrease the development of cardiovascular disease, but may aggravate already established disease [77]. One study reported no change in survival for G6PD-deficient mice [75], but this study ended at week 35 (day 245) in contrast to our study that followed the mice until their death (around 800 days). A population of humans with increased longevity in Sardinia, Italy, has been found to highly conserve a mutant variant of G6PD resulting in an enzymatic deficiency [[78], [79], [80]]. This population exhibited a decrease in deaths from ischemic heart disease, cerebrovascular disease and liver cirrhosis, which is consistent with the extended lifespan we found in aging G6PD-deficient mice. On the other hand, this same population exhibited an increase in mortality from non-Hodgkin's lymphoma in individuals with G6PD-deficency, which we did not observe in our aging G6PD-deficient mice. The importance of environmental factors affecting G6PD, and thereby potentially altering health, is virtually unknown. Interestingly, analogs of dehydroepiandrosterone (DHEA), a potent inhibitor of G6PD [81,82], as well as DHEA itself, are being evaluated in animal models and in human clinical trials as potential therapeutic agents for numerous diseases including cancer and cardiovascular disease [74,83], although these agents likely have confounding effects in addition of G6PD inhibition. However, our studies [8,15] and the results herein, along with other studies discussed above, suggest that G6PD inhibition may have adverse consequences in addition to any anticipated therapeutic benefits.
5. Conclusion
G6PD deficiencies constitute the most common human enzymopathy, affecting over 400 million people and up to 60% of some racial populations. Although medical interest in deficiencies in this enzyme has been largely confined to red blood cells, more recent evidence suggests broader implications of G6PD deficiencies for health, including an expanding list of potential outcomes that include birth defects, heart disease, cancer and, relevant the study herein, neurodegeneration and possibly neurodevelopmental deficits.
Footnotes
Preliminary reports of this research were presented at the 2011 annual meeting of the Society of Toxicology of Canada (STC) (Loniewska MM and Wells PG, Proceedings of the 43rd annual STC symposium, Abstract No. 24, December 2011), and at the 9th meeting of the Canadian Oxidative Stress Consortium (Gupta A, Bhatia S, MacKay-Clackett I and Wells PG, Proceedings, page 55, Abstract No. PPA08, June 2016). These studies were supported by grants from the Canadian Institutes of Health Research (CIHR) and the Faculty of Pharmacy, University of Toronto.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101332.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Buehler E. Study of glucose-6-phosphate dehydrogenase: history and molecular biology. Am. J. Hematol. 1993;42:53–58. doi: 10.1002/ajh.2830420111. [DOI] [PubMed] [Google Scholar]
- 2.Luzzatto L., Mehta A., Vulliamy T. Glucose-6-phosphate dehydrogenase deficiency. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 4517–4553. [Google Scholar]
- 3.Halliwell B., Gutteridge L.M.C. fifth ed. Oxford University Press; New York: 2015. Free Radicals in Biology and Medicine. [Google Scholar]
- 4.Hanschmann E.-M., Godoy J.R., Berndt C., Hudemann C., Lillig C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxidants Redox Signal. 2013;19(13):1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkman H.N., Galiano S., Gaetani G.F. The function of catalase-bound NADPH. J. Biol. Chem. 1987;262(2):660–666. [PubMed] [Google Scholar]
- 6.Kirkman H.N., Rolfo M., Ferraris A.M., Gaetani G.F. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J. Biol. Chem. 1999;274(20):13908–13914. doi: 10.1074/jbc.274.20.13908. [DOI] [PubMed] [Google Scholar]
- 7.Cappellini M.D., Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 8.Nicol C.J., Zielenski J., Tsui L.C., Wells P.G. An embryoprotective role for glucose-6-phosphate dehydrogenase in developmental oxidative stress and chemical teratogenesis. FASEB (Fed. Am. Soc. Exp. Biol.) J. 2000;14(1):111–127. doi: 10.1096/fasebj.14.1.111. [DOI] [PubMed] [Google Scholar]
- 9.Nichols K.D., Kirby G.M. Expression of cytochrome P450 2A5 in a glucose-6-phosphate dehydrogenase-deficient mouse model of oxidative stress. Biochem. Pharmacol. 2008;75(5):1230–1239. doi: 10.1016/j.bcp.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 10.Felix K., Rockwood L.D., Pretsch W., Nair J., Bartsch H., Bornkamm G.W., Janz S. Moderate G6PD deficiency increases mutation rates in the brain of mice. Free Radic. Biol. Med. 2002;32(7):663–673. doi: 10.1016/s0891-5849(02)00756-6. [DOI] [PubMed] [Google Scholar]
- 11.Jeng W., Loniewska M.M., Wells P.G. Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 2013;4(7):1123–1132. doi: 10.1021/cn400079y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pretsch W., Charles D.J., Merkle S. X-linked glucose-6-phosphate dehydrogenase deficiency in Mus musculus. Biochem. Genet. 1988;26(1–2):89–103. doi: 10.1007/BF00555491. [DOI] [PubMed] [Google Scholar]
- 13.Sanders S., Smith D.P., Thomas G.A., Williams E.D. A glucose-6-phosphate dehydrogenase (G6PD) splice site consensus sequence mutation associated with G6PD enzyme deficiency. Mutat. Res. 1997;374:79–87. doi: 10.1016/s0027-5107(96)00222-9. [DOI] [PubMed] [Google Scholar]
- 14.Holland P.M., Abramson R.D., Watson R., Gelfand D.H. Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 1991;88(16):7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeng W., Loniewska M.M., Wells P.G. Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 2013;4(7):1123–1132. doi: 10.1021/cn400079y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh N.P., McCoy M.T., Tice R.R., Schneider E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988;175(1):184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 17.Fishcher A.H., Jacobson K.A., Rose J., Zeller R. CSH Protocols; 2008. Hematoxylin and Eosin Staining of Tissue and Cell Sections. pdb.prot4986. [DOI] [PubMed] [Google Scholar]
- 18.Meng Y., Zhang Y., Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39(1):163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- 19.Meng Y., Zhang Y., Tregoubov V., Janus C., Cruz L., Jackson M., Lu W.Y., MacDonald J.F., Wang J.Y., Falls D.L., Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35(1):121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Z., Hu J., Passafaro M., Xie W., Jia Z. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J. Neurosci. 2011;31(3):819–833. doi: 10.1523/JNEUROSCI.3869-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milenkovic M., Mielnik C.A., Ramsey A.J. NMDA receptor-deficient mice display sexual dimorphism in the onset and severity of behavioural abnormalities. Genes Brain Behav. 2014;13(8):850–862. doi: 10.1111/gbb.12183. [DOI] [PubMed] [Google Scholar]
- 22.Ben Abdallah N.M., Fuss J., Trusel M., Galsworthy M.J., Bobsin K., Colacicco G., Deacon R.M., Riva M.A., Kellendonk C., Sprengel R., Lipp H.P., Gass P. The puzzle box as a simple and efficient behavioral test for exploring impairments of general cognition and executive functions in mouse models of schizophrenia. Exp. Neurol. 2011;227(1):42–52. doi: 10.1016/j.expneurol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Angoa-Pérez M., Kane M.J., Briggs D.I., Francescutti D.M., Kuhn D.M. Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J. Vis. Exp. : J. Vis. Exp. 2013;82 doi: 10.3791/50978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deacon R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013;75:2609. doi: 10.3791/2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiotsuki H., Yoshimi K., Shimo Y., Funayama M., Takamatsu Y., Ikeda K., Takahashi R., Kitazawa S., Hattori N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods. 2010;189(2):180–185. doi: 10.1016/j.jneumeth.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q., Bardgett M.E., Wong M., Wozniak D.F., Lou J., McNeil B.D., Chen C., Nardi A., Reid D.C., Yamada K., Ornitz D.M. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35(1):25–38. doi: 10.1016/s0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- 27.Lieu C.A., Chinta S.J., Rane A., Andersen J.K. Age-related behavioral phenotype of an astrocytic monoamine oxidase-B transgenic mouse model of Parkinson's disease. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crawley J.N. Wiley-Liss; New York, N.Y.: 2000. What's Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice. [Google Scholar]
- 29.Welzl H., D'Adamo P., Lipp H.P. Conditioned taste aversion as a learning and memory paradigm. Behav. Brain Res. 2001;125(1–2):205–213. doi: 10.1016/s0166-4328(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 30.Bliss T.V., Richter-Levin G. Spatial learning and the saturation of long-term potentiation. Hippocampus. 1993;3(2):123–125. doi: 10.1002/hipo.450030203. [DOI] [PubMed] [Google Scholar]
- 31.Ninfali P., Aluigi G., Pompella A. Postnatal expression of glucose-6-phosphate dehydrogenase in different brain areas. Neurochem. Res. 1998;23(9):1197–1204. doi: 10.1023/a:1020734203128. [DOI] [PubMed] [Google Scholar]
- 32.Ninfali P., Cuppini C., Marinoni S. Glucose-6-phosphate dehydrogenase and glutathione reductase support antioxidant enzymes in nerves and muscles of rats during nerve regeneration. Restor. Neurol. Neurosci. 1996;10(2):69–75. doi: 10.3233/RNN-1996-10202. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Y., Carvey P.M., Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006;1090(1):35–44. doi: 10.1016/j.brainres.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semsei I., Rao G., Richardson A. Expression of supreoxide dismutase and catalase in rat brain as a function of age. Mech. Ageing Dev. 1991;58(1):13–19. doi: 10.1016/0047-6374(91)90116-h. [DOI] [PubMed] [Google Scholar]
- 35.Wells P.G., Miller-Pinsler L., Shapiro A.M. Impact of oxidative stress on development. In: Dennery P.A., Buonocore G., Saugstad O., editors. Perinatal and Prenatal Disorders. Humana Press, Springer Science; Berlin: 2014. pp. 1–37. [Google Scholar]
- 36.Tice R.R., Agurell E., Anderson D., Burlinson B., Hartmann A., Kobayashi H., Miyamae Y., Rojas E., Ryu J.C., Sasaki Y.F. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000;35(3):206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y.Y., Yu Y.B., Gunawardena H.P., Xie L., Chen X. BCLAF1 is a radiation-induced H2AX-interacting partner involved in gammaH2AX-mediated regulation of apoptosis and DNA repair. Cell Death Dis. 2012;3:e359. doi: 10.1038/cddis.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solier S., Pommier Y. The nuclear gamma-H2AX apoptotic ring: implications for cancers and autoimmune diseases. Cell. Mol. Life Sci. : CMLS. 2014;71(12):2289–2297. doi: 10.1007/s00018-013-1555-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goncalves L.L., Ramkissoon A., Wells P.G. Prostaglandin H synthase-1-catalyzed bioactivation of neurotransmitters, their precursors, and metabolites: oxidative DNA damage and electron spin resonance spectroscopy studies. Chem. Res. Toxicol. 2009;22:842–852. doi: 10.1021/tx800423s. [DOI] [PubMed] [Google Scholar]
- 40.Ramkissoon A., Wells P.G. Human prostaglandin H synthase (hPHS)-1 and hPHS-2 in amphetamine analog bioactivation, DNA oxidation, and cytotoxicity. Toxicol. Sci. 2011;120(1):154–162. doi: 10.1093/toxsci/kfq377. [DOI] [PubMed] [Google Scholar]
- 41.Miller-Pinsler L., Pinto D.J., Wells P.G. Oxidative DNA damage in the in utero initiation of postnatal neurodevelopmental deficits by normal fetal and ethanol-enhanced oxidative stress in oxoguanine glycosylase 1 knockout mice. Free Radical Biol. Med. 2015;78:23–29. doi: 10.1016/j.freeradbiomed.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 42.Drake D.M., Wells P.G. Proceedings of the 9th Meeting of the Canadian Oxidative Stress Consortium. 2016. Reduced subcellular levels of breast cancer 1 protein (BRCA1) and increased DNA damage in embryonic tissue and fetal brain of untreated and ethanol-exposed Brca1 knockout progeny; pp. 21–22. (Abstract No. GS04) [Google Scholar]
- 43.Drake D.M., Wells P.G. Intrinsic and ethanol-enhanced fetal DNA damage and neurodevelopmental deficits in Brca1 knockout progeny associated with decreased brain BRCA1 levels (Abstract) Birth Defects Res. 2018;110(9):755. No. 3. [Google Scholar]
- 44.Baurle J., Grusser-Cornehls U. Calbindin D-28k in the lateral vestibular nucleus of mutant mice as a tool to reveal Purkinje cell plasticity. Neurosci. Lett. 1994;167(1–2):85–88. doi: 10.1016/0304-3940(94)91033-2. [DOI] [PubMed] [Google Scholar]
- 45.Jande S.S., Maler L., Lawson D.E. Immunohistochemical mapping of vitamin D-dependent calcium-binding protein in brain. Nature. 1981;294(5843):765–767. doi: 10.1038/294765a0. [DOI] [PubMed] [Google Scholar]
- 46.Biagiotti E., Guidi L., Capellacci S., Ambrogini P., Papa S., Del Grande P., Ninfali P. Glucose-6-phosphate dehydrogenase supports the functioning of the synapses in rat cerebellar cortex. Brain Res. 2001;911(2):152–157. doi: 10.1016/s0006-8993(01)02615-4. [DOI] [PubMed] [Google Scholar]
- 47.Biagiotti E., Guidi L., Del Grande P., Ninfali P. Glucose-6-phosphate dehydrogenase expression associated with NADPH-dependent reactions in cerebellar neurons. Cerebellum. 2003;2(3):178–183. doi: 10.1080/14734220310016123. [DOI] [PubMed] [Google Scholar]
- 48.Ferri P., Biagiotti E., Ambrogini P., Santi S., del Grande P., Ninfali P. NADPH-consuming enzymes correlate with glucose-6-phosphate dehydrogenase in Purkinje cells: an immunohistochemical and enzyme histochernical study of the rat cerebellar cortex. Neurosci. Res. 2005;51(2):185–197. doi: 10.1016/j.neures.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Bliss T.V., Collingridge G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 50.Massaad C.A., Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxidants Redox Signal. 2011;14(10):2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desmond J.E., Fiez J.A. Neuroimaging studies of the cerebellum: language, learning and memory. Trends Cogn. Sci. 1998;2(9):355–362. doi: 10.1016/s1364-6613(98)01211-x. [DOI] [PubMed] [Google Scholar]
- 52.van den Berg W.E., Lamballais S., Kushner S.A. Sex-specific mechanism of social hierarchy in mice. Neuropsychopharmacology. 2015;40(6):1364–1372. doi: 10.1038/npp.2014.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vogel M.W., Caston J., Yuzaki M., Mariani J. The Lurcher mouse: fresh insights from an old mutant. Brain Res. 2007;1140:4–18. doi: 10.1016/j.brainres.2005.11.086. [DOI] [PubMed] [Google Scholar]
- 54.Wang T., Morgan J.I. The Purkinje cell degeneration (pcd) mouse: an unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007;1140:26–40. doi: 10.1016/j.brainres.2006.07.065. [DOI] [PubMed] [Google Scholar]
- 55.Jeng W., Wong A.W., Ting A.K.R., Wells P.G. Methamphetamine-enhanced embryonic oxidative DNA damage and neurodevelopmental deficits. Free Radic. Biol. Med. 2005;39(3):317–326. doi: 10.1016/j.freeradbiomed.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 56.Wong A.W., McCallum G.P., Jeng W., Wells P.G. Oxoguanine glycosylase 1 protects against methamphetamine-enhanced fetal brain oxidative DNA damage and neurodevelopmental deficits. J. Neurosci. 2008;28(36):9047–9054. doi: 10.1523/JNEUROSCI.2557-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caddy K.W., Biscoe T.J. Preliminary observations on the cerebllum in the mutant mouse Lurcher. Brain Res. 1975;91(2):276–280. doi: 10.1016/0006-8993(75)90548-x. [DOI] [PubMed] [Google Scholar]
- 58.Lalonde R. Motor learning in lurcher mutant mice. Brain Res. 1994;639(2):351–353. doi: 10.1016/0006-8993(94)91753-1. [DOI] [PubMed] [Google Scholar]
- 59.Caston J., Lalonde R., Delhaye-Bouchaud N., Mariani J. The cerebellum and postural sensorimotor learning in mice and rats. Behav. Brain Res. 1998;95(1):17–22. doi: 10.1016/s0166-4328(97)00205-2. [DOI] [PubMed] [Google Scholar]
- 60.Nóbrega-Pereira S., Fernandez-Marcos P.J., Brioche T., Gomez-Cabrera M.C., Salvador-Pascual A., Flores J.M., Viña J., Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016;7 doi: 10.1038/ncomms10894. 10894-10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heng M.Y., Duong D.K., Albin R.L., Tallaksen-Greene S.J., Hunter J.M., Lesort M.J., Osmand A., Paulson H.L., Detloff P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010;19(19):3702–3720. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heng M.Y., Tallaksen-Greene S.J., Detloff P.J., Albin R.L. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J. Neurosci. 2007;27(34):8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dougherty S.E., Reeves J.L., Lesort M., Detloff P.J., Cowell R.M. Purkinje cell dysfunction and loss in a knock-in mouse model of Huntington disease. Exp. Neurol. 2013;240:96–102. doi: 10.1016/j.expneurol.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dougherty S.E., Reeves J.L., Lucas E.K., Gamble K.L., Lesort M., Cowell R.M. Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp. Neurol. 2012;236(1):171–178. doi: 10.1016/j.expneurol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bai X., Fermandez E., Gould G., Strong R. Homozygous deletion of glutathione peroxidase 1 and aldehyde dehydrogenase 1a1 genes is not associated with schizophrenia-like behavior in mice. J. Biochem. Pharmacol. Res. 2013;1(4):228–235. [PMC free article] [PubMed] [Google Scholar]
- 66.Goldman J.G., Williams-Gray C., Barker R.A., Duda J.E., Galvin J.E. The spectrum of cognitive impairment in Lewy body diseases. Mov. Disord. 2014;29(5):608–621. doi: 10.1002/mds.25866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang F., Zhu J., Zhu H., Zhang Q., Lin Z., Hu H. Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science. 2011;334(6056):693. doi: 10.1126/science.1209951. [DOI] [PubMed] [Google Scholar]
- 68.Kordower J.H., Olanow C.W., Dodiya H.B., Chu Y., Beach T.G., Adler C.H., Halliday G.M., Bartus R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. 1460-2156 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pifferi F., Terrien J., Marchal J., Dal-Pan A., Djelti F., Hardy I., Chahory S., Cordonnier N., Desquilbet L., Hurion M., Zahariev A., Chery I., Zizzari P.A.-O., Perret M., Epelbaum J., Blanc S., Picq J.L., Dhenain M.A.-O., Aujard F. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun. Biol. 2018;1:30. doi: 10.1038/s42003-018-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abbott A. Reduced-calorie diet shows signs of slowing ageing in people. Nature. 2018;5555(7698):570–571. doi: 10.1038/d41586-018-03431-x. [DOI] [PubMed] [Google Scholar]
- 71.Bartke A., Wright J.C., Mattison J.A., Ingram D.K., Miller R.A., Roth G.S. Extending the lifespan of long-lived mice. Nature. 2001;414(6862):412. doi: 10.1038/35106646. [DOI] [PubMed] [Google Scholar]
- 72.Sohal R.S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsui R., Xu S., Maitland K.A., Hayes A., Leopold J.A., Handy D.E., Loscalzo J., Cohen R.A. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation. 2005;112(2):257–263. doi: 10.1161/CIRCULATIONAHA.104.499095. [DOI] [PubMed] [Google Scholar]
- 74.Gupte S.A. Targeting the pentose phosphate pathway in syndrome X-related cardiovascular complications. Drug Dev. Res. 2010;71(3):161–167. doi: 10.1002/ddr.20359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hecker P.A., Mapanga R.F., Kimar C.P., Ribeiro R.F., Jr., Brown B.H., O'Connell K.A., Cox J.W., Shekar K.C., Asemu G., Essop M.F., Stanley W.C. Effects of glucose-6-phosphate dehydrogenase deficiency on the metabolic and cardiac responses to obesogenic or high-fructose diets. Am. J. Physiol. Endocrinol. Metabol. 2012;303(8):E959–E972. doi: 10.1152/ajpendo.00202.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hecker P.A., Lionetti V., Ribeiro R.F., Jr., Rastogi S., Brown B.H., O'Connell K.A., Cox J.W., Shekar K.C., Gamble D.M., Sabbah H.N., Leopold J.A., Gupte S.A., Recchia F.A., Stanley W.C. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circulation: Heart Fail. 2013;6(1):118–126. doi: 10.1161/CIRCHEARTFAILURE.112.969576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hecker P.A., Leopold J.A., Gupte S.A., Recchia F.A., Stanley W.C. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013;304(4):H491–500. doi: 10.1152/ajpheart.00721.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cocco P., Todde P., Fornera S., Manca M.B., Manca P., Sias A.R. Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood. 1998;91(2):706–709. [PubMed] [Google Scholar]
- 79.Long W.K., Wilson S.W., Frenkel E.P. Associations between red cell glucose-6-phosphate dehydrogenase variants and vascular diseases. Am. J. Hum. Genet. 1967;19(1):35–53. [PMC free article] [PubMed] [Google Scholar]
- 80.Meloni L., Manca M.R., Loddo I., Cioglia G., Cocco P., Schwartz A., Muntoni S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J. Inherit. Metab. Dis. 2008;31(3):412–417. doi: 10.1007/s10545-008-0704-5. [DOI] [PubMed] [Google Scholar]
- 81.Raineri R., Levy H.R. On the specificity of steroid interaction with mammary glucose 6-phosphate dehydrogenase. Biochemistry. 1970;9(11):2233–2243. doi: 10.1021/bi00813a003. [DOI] [PubMed] [Google Scholar]
- 82.Gordon G., M M.C., Levy H.R. On the mechanism of interaction of steroids with human glucose 6-phosphate dehydrogenase. Arch. Biochem. Biophys. 1995;318(1):25–29. doi: 10.1006/abbi.1995.1199. [DOI] [PubMed] [Google Scholar]
- 83.Schwartz A.G., Pashko L.L. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res. Rev. 2004;3(2):171–187. doi: 10.1016/j.arr.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 84.Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. ATM phopshorylates Histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276(45):42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 85.Kinner A., Wu W., Staudt C., IIiakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36(17):5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dimauro I., Pearson T., Caporossi D., Jackson M.J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes. 2012;5 doi: 10.1186/1756-0500-5-513. Article No. 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miczek K.A., Barry H., 3rd What does the tube test measure? Behav. Biol. 1975;13(4):537–539. doi: 10.1016/s0091-6773(75)91249-3. [DOI] [PubMed] [Google Scholar]
- 88.Buckner R.L. Memory and executive function in aging and AD: multiple factors that cause decline and reserve factors that compensate. Neuron. 2004;44(1):195–208. doi: 10.1016/j.neuron.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 89.Diamond A. Executive functions. Ann. Rev. Psychol. 2013;64:135–168. doi: 10.1146/annurev-psych-113011-143750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.