

Abstract
Opioid ligands are a large group of G-protein-coupled receptor ligands possessing high structural diversity, along with complicated structure–activity relationships (SARs). To better understand their structural correlations as well as the related SARs, we developed the innovative template-based alignment modeling in our recent studies on a variety of opioid ligands. As previously reported, this approach showed promise but also with limitations, which was mainly attributed to the small size of morphine as a template. With this study, we set out to construct an artificial μ-agonist template to overcome this limitation. The newly constructed template contained a largely extended scaffold, along with a few special μ-features relevant to the μ-selectivity of opioid ligands. As demonstrated in this paper, the new template showed significantly improved efficacy in facilitating the alignment modeling of a wide variety of opioid ligands. This report comprises of two main parts. Part 1 discusses the general construction process and the structural features as well as a few typical examples of the template applications and Part 2 focuses on the template refinement and validation.
Introduction
Opioid ligands are an exceedingly large group of G-protein-coupled receptor (GPCR) ligands that interact with opioid receptors and possess high structural diversity, ranging from alkaloids, peptides, terpenoids, peptidomimetics, and synthetic small molecules of random scaffolds.1,2 Amazingly, all of the highly diverse ligands can effectively bind in the same pockets of opioid receptors. As is well known, opioid ligands often interact specifically with the receptors; sometimes, even one minor structural alternation can lead to a totally changed bioactivity for the ligand. So how can all of these diverse ligands bind at the same pockets? And how are these ligands structurally correlated? These kinds of questions are difficult to answer but are important for examining the medicinal chemistry of opioid ligands.
Particularly, the structural correlation between morphine (an alkaloid) and Leu-enkephalin (a peptide), two well-known prototypes of opioid ligands, the former from the plants, the latter the natural endogenous ligand, both interacting with the μ-opioid receptor, has been a focus. Understanding their structural correlation will not only help for the elucidation of the structure–activity relationships (SARs) of opioid ligands but can also greatly aid the discovery of new opioid drugs. Numerous models have been proposed in the literature for their correlation, but the majority of them seem to have only focused on matches between the scaffold of morphine and the side chains of opioid peptides.3−7
Based on a unique X-ray crystal structure of Leu-enkephalin, we recently proposed a new model for the structural correlation of morphine and Leu-enkephalin, featuring matches between morphine’s scaffold and the backbones of the peptides, instead of the side-chain groups (Figure 1).8 This new approach, referred to as the backbone alignment modeling (or now called the template-based alignment modeling (TAM)) seemed to be the first to tackle the backbone elements for structural correlations of opioid ligands.
Figure 1.
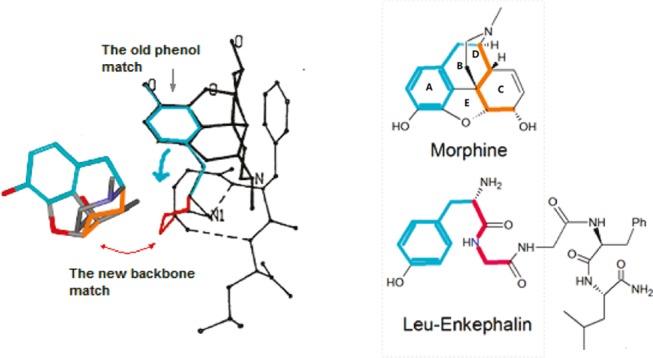
Match of morphine’s scaffold to the backbone of Leu-enkephalin.
Applying this innovative approach, we examined a large number of opioid ligands with respect to their possible structural correlations, with which, we proposed three new backbone-conformational models, corresponding to the three types of opioid ligands, μ, δ, and κ,8 (refer to Figure 10A below). These new models have significantly improved our understanding of the complicated SARs of opioid ligands.
Figure 10.

Correlations between the three backbone-conformational models of opioid peptides and the three special loops of the template (stereoview). (A) The three backbone-conformational models (μ: green, δ: red, and κ: blue); (B) the corresponding loops (the up-middle: green, the inner: red, and the in-middle: blue).
However, the original modeling displayed only limited capacity in interpreting the SARs of opioid ligands, mostly because the small-sized scaffold of morphine was used as the key template. Many opioid ligands with bulky or novel scaffolds were not able to align well with the morphine scaffold. Thus, how to upgrade to a large-sized template has been a critical issue in our ongoing studies to further explore the innovative backbone alignment modeling.
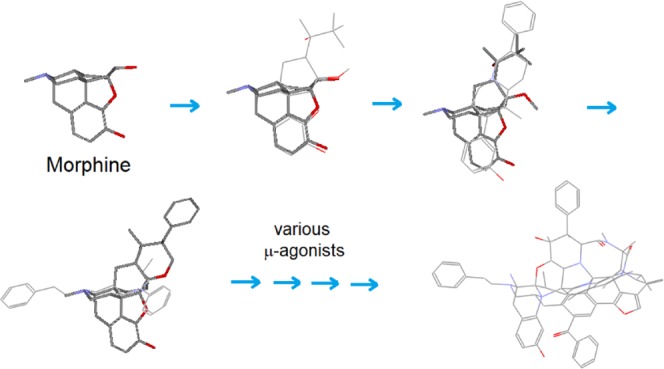
Ideally, we anticipated an inclusive template that would have a large and versatile scaffold to fully represent the structural features of diverse opioid ligands (or speaking differently, to fully cover the ligand-binding space of opioid receptors). To that end, it seemed best for us to build an artificial template using a wide variety of μ-ligands as the building blocks (to use μ-agonists for the template construction was mainly because of the abundance in structural diversity and SAR data as well as the importance and significance of this subtype for opioid ligand studies.). In fact, with a similar strategy, we had already built an artificial template for the Hsp90 ligands,9 which was of great help for us with this study.
Herein, we wish to report the results of our recent efforts in establishing an artificial μ-agonist template for opioid ligand modeling.
Results and Discussion
This section, relatively long in size, is organized into two main parts. Part 1 discusses the general construction process as well as the structural features of the template and Part 2 focuses on the template refinement and validation.
Part 1. Template Construction
Strategies for Template Construction
With the construction of an artificial template of N-terminal Hsp90 ligands,9 we gained substantial experience. Similar to opioid ligands, the structures of N-terminal Hsp90 ligands were highly diversified. So how to recognize their correlations was a challenge for us. Thus, we started by first examining a cluster of the crystal structures of a group of Hsp90 ligands, which, on quick examination, were much like a bundle of randomly tangled structures (Figure 2). With the backbone alignment concept as a guideline to closely examine the cluster, however, we were able to see high structural correlations among the ligands. It was observed that regardless of the size and the rigidity, all of the ligands in the cluster were confined within a unique three-dimensional (3D)-space and with many structures overlapping through similar-to-similar patterns (e.g., aromatic ring to aromatic ring and hetero-atoms to hetero-atoms, etc.). In particular, most of the overlapping structures were also in bond-to-bond correlation, a scenario much resembling what we observed in our alignment modeling. And the entire situation strongly suggested that all of the ligands were not randomly bound at the pocket, but their bindings were strictly following certain patterns. Consequently, through proper structural processes, it was possible for us to create a core structure (namely, an artificial template) out of the cluster to characterize all of the ligands at the binding site.
Figure 2.

Construction of an artificial template for the N-terminal Hsp90 ligands. The crystal structure cluster of Hsp90 ligands was converted into a rigid artificial template through bond merging and bond connecting.
Accordingly, we set out to process the structures of the ligands by merging and connecting the individual bonds, followed by refining the scaffold as a whole, so as to give rise to a rigid and bulky artificial template, which, by size and 3D shape as well as many other structural features, much resembled the original crystal structure cluster of the Hsp90 ligands (Figure 2).
Although Hsp90 and opioid receptor are totally different proteins with respect to their sequences and structures, presumably the fundamental ways they interact with the ligands are the same. For example, the special bond-to-bond correlation pattern seen with the Hsp90 ligands should be present with opioid ligands as well. In fact, the same patterns were well observed with many other types of protein–ligand complexes, such as protein kinase inhibitors and tubulin-binding agents (unpublished results). Hence, by following a similar process for the construction of the Hsp90-ligand template, we should be able to build an artificial template for opioid ligands as well.
For the template construction, it was essential for us to know the binding conformations of the ligands, which, for the Hsp90 ligands, were mainly derived from the crystal structures of their ligand–protein complexes (see a list of the collection at ChEMBL database).10 For opioid ligands, however, there were only a handful of crystal structures of ligand–receptor complexes available, too few to support our project. Hence, as an alternative way, we exploited template-based alignment modeling (TAM) again to deduce the ligand-binding conformations.
The principle of TAM states that different ligands that bind at the same binding site are structurally highly correlated, that we are able to align the backbones/scaffolds of the ligands with a template, with which the binding conformations of the individual ligands relevant to the template can be realized, a practice that was already illustrated in our previous publication.8
Thus, here we would first need to assemble a ligand cluster by using a group of selected μ-agonists as the building blocks (particularly those with rigid and bulky scaffolds). And then, we would convert the cluster into a preliminary template through structural processing, and then we would align a variety of μ-agonists with the template to further refine it (see Figure 3).
Figure 3.

Basic strategy for the μ-agonist template construction. Through TAM, a group of selected μ-agonists are assembled and converted into a universal template.
From the literature, we were able to obtain the structural information of a large number of μ-agonists, many of them with bulky and rigid (or semirigid) scaffolds,10,11 including a variety of bulky morphinan derivatives as well as a series of semirigid Tyr1-attached cyclic peptides, which would be exploited as efficient building blocks for the template construction.
Construction Process
In the practical process, the ligand cluster was stepwise assembled and processed (see Figure 4). First, a small group of special μ-agonists were selected as the basic building blocks based on their structural features (i.e., rigid and diversified, able to be aligned with morphine’s scaffold) (note: here EM1 was not a rigid ligand and it was selected mainly because of its large AA3 and AA4 moieties helpful for the construction of a special region of the template). And then, each of the selected ligands was subject to individual alignment with morphine (as an initial template), followed by structural processing (i.e., bond merging and bond connecting), with which a preliminary template was formed (see the stepwise additions and processes of ligands (b)–(e) in Figure 4).
Figure 4.

Stepwise construction of the template. (a) morphine; (b) norbuprenorphine; (c) Le Bourdonnec 2006;12 (d) fentanyl; (e) EM1; (f) cyclic peptides; (g) many other μ-ligands.
In the subsequent process, we further extended and refined the scaffold of the preliminary template by iterative alignments of a wide variety of μ-agonists onto it. Thereby, we were able to closely examine each of the ligands for its special matching pattern with the template so as to modify and improve the main scaffold as well as all of the substructures of the template accordingly (the detailed refinement will be discussed below in Part 2 of this report).
Along with the refining process, the template validation was concomitantly undertaken through assessing the alignments of many structurally diversified μ-ligands. As shown in Figure 5, the majority of ligands were aligned well with the template with respect to their various match patterns, e.g., the 3D-shape, the stereochemistry, the structural and conformational features, etc., suggesting that the template was able to well represent the diverse ligands (see Figure 5).
Figure 5.

Template validation. A wide variety of μ-agonists were aligned to examine their match patterns with the template.
In particular, it was observed that many of the μ-ligands coincidently aligned their binding-selectivity-related moieties to the special μ-features of the template, such as N-PhEt and Ph/Ind (see Figure 6) (for discussion of the special μ-features, see General Features of the Template below). And due to the scaffold diversity of the ligands, the co-alignments of their key moieties would, therefore, be an excellent validation to the special μ-features of the template. Moreover, it was seen that a few nonclassical μ-ligands tended to align their unique scaffolds beyond or exclusively out of the morphinan core, the classical “message” of opioid ligands, (such as Salvinorin A and CJ-15 208, see Case Discussion below). And this scenario provided us with a new vision about the diverse binding modes of opioid ligands for receptor interactions.
Figure 6.

Co-alignments to the special μ-features of the template. Many μ-ligands co-aligned their selectivity-related moieties (blue or red circled) to the special μ-features of the template, which would help also for the structural validation of the template.
Crystal Structures for Validation
The crystal structures of μ-ligands as extracted from their receptor complexes are highly valuable information for the template validation, because they can directly provide the binding conformations of ligands. However, the biggest issue was still the lack of enough crystal structures for use in this study. And there were only two μ-receptor–ligand complexes available at the time of this study: one with μ-antagonist β-FNA bound (PDB ID: 4dkl) and the other with μ-agonist Bu72 bound (PDB ID: 5clm). Additionally, there existed a δ-receptor complex with DIPP-NH2 bound (PDB ID: 4rwa), where DIPP-NH2 happened to be a mixed μ-agonist/δ-antagonist.13 Since close similarities in sequence and 3D-structure were seen between μ-and δ-receptors,14 presumably DIPP-NH2 would take the binding pose at μ-receptor similar to that at δ-receptor, so that it would be possible for us to exploit it for the template validation. Thus, we set out to cluster the three ligand–receptor complexes as a bundle to examine the binding behaviors of their bound ligands (Figure 7).
Figure 7.

Comparison between the binding poses of the experimentally determined and the modeling predicted of the three ligands (stereoview). (A) The three crystal structures of DIPP-NH2, Bu72, and β-FNA overlap at the morphinan core; (B) the similar binding poses are seen, as the three ligands are co-aligned with the template.
Figure 7 shows the superimposed crystal structures of the three receptor complexes (left panel) as well as of the three ligands as extracted out (Figure 7A). As we can see, the major scaffolds of the three ligands appear to be aligned around the morphinan core, which is consistent with the predicted binding poses in our modeling (Figure 7B). The good agreement in binding conformations between the crystal structures and the modeling results would, therefore, strongly support the template validation.
For validation purpose, we also managed to dock the template into the binding pocket of a μ-receptor complex (PDB ID: 5clm), where the template was superimposed with Bu72 (the bound ligand) at the morphinan moieties (Figure 8). For this match, the whole scaffold of the template appeared to fit by size and 3D-shape into the binding pocket pretty well, which helped further for the template validation.
Figure 8.
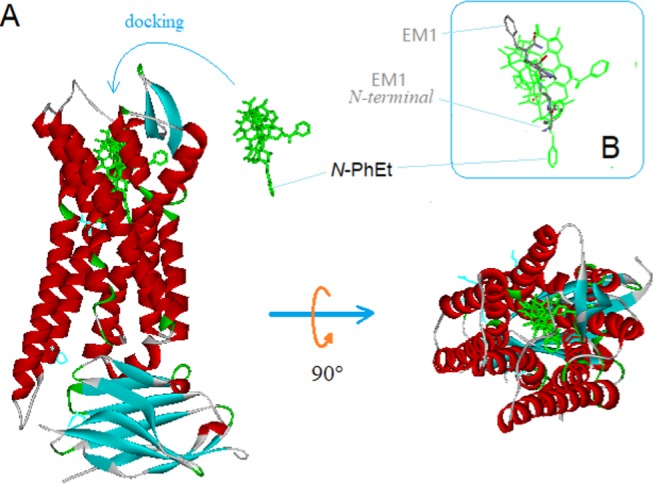
Docking the template at the binding site of μ-receptor. (A) On docking, the scaffold of the template (in green) appeared to fit well by size and 3D-shape to the binding pocket. (B) The alignment of EM1 shows the downward orientation of the N-terminal.
The template docking helped to reveal the binding conformations of linear μ-opioid peptides, where the N-terminal of the peptides was oriented toward the bottom of the binding site, while the C-terminal toward the opening (see the alignment of EM1, a μ-agonistic peptide, Figure 8B) (note: prior to the docking modeling, our modeling was only able to show the relative conformation of the peptide in reference to the template). Indeed, the binding conformation was shown to be correct with the cryo-electron microscopy structure of DAMGO recently15 (PDB ID: 6dde).
In addition, the docking pose of the template was also able to help for the understanding of some classical SARs of opioid ligands. For example, the N-PhEt moiety of the template at the docking is inserted deeply into the bottom of the binding pocket (see Figure 8A), which helps to account for the significantly enhanced binding affinities often associated with the N-PhEt-bearing ligands.16 Thus, the template’s docking modeling was supporting the template validation.
General Features of the Template
Morphinan Core and the Extended Scaffold
The template consists of a morphinan core along with a largely extended scaffold. Overall, the template appears to be a rigid structure embodying a number of fused rings, along with many other structural features, such as aromatic zones, hetero-atoms, and μ-selectivity-related moieties (see Figure 9).
Figure 9.

Structural features of the template. The template possesses a unique scaffold consisting of a morphinan core along with a largely extended scaffold, where identified are several μ-selectivity-related features as noted with letters a–d as well as a few special loops in different colors: the inner (red), the in-middle (blue), and the up-middle (green).
Designated Loops and Special μ-Features
We have designated several special loops of the template (Figure 9): the inner (red), the in-middle (blue), and the up-middle (green). Meanwhile, we have also identified a few structural features of the template relevant to the μ-selectivity of the ligands: (a) Ph/Ind; (b) N-PhEt; (c) up-middle loop; (d) front benzoyl (see Figure 9). In addition, there are also some other features of the template, such as the embedded backbones of the peptide ligands, which will be discussed in Part 2 of this report. All of these features will be of help to facilitate the discussions below.
In our previous study, by aligning a variety of opioid ligands with morphine, we proposed three backbone-conformational models, corresponding to the three types of opioid peptides8 (Figure 10A). In this study, the three conformational models are correlated, respectively, to three special loops of the template: the up-middle (green), the inner (red), and the in-middle loops (blue) (Figure 10B). As shown in the figure, the previous backbones models and the three special loops are pretty consistent with each other in terms of their relative positions and orientations. This comparison helps to reveal the intrinsic correlations of the template with the backbones/scaffolds of opioid ligands relative to the binding selectivity. And the correlations would, thus, allow us to exploit this μ-agonist-specialized template for δ- and κ-ligands’ modeling, as well (see related discussions in Examples of Applications below).
Examples of Application
As anticipated initially, the new template worked indeed much better in facilitating our alignment modeling of a wide variety of opioid ligands. And the extended scaffold of the template was able to help for us to improve the alignments of several special μ-ligands as proposed in our previous study (e.g., Em-Mm,17 etonitazene, salvinorin A, and CJ-15 208), and the updated alignments were shown to account well for the related SARs (see examples in Case Studies below).
With the new template, many potential applications in medicinal chemistry studies can be expected, such as ligand–structure correlations, binding-pose predictions, SAR interpretations, recognition of the structural features of antagonists vs agonists, and so on.
General Guidelines for the Alignment Modeling
For an aligning process, we will need first to examine the whole scaffold of a ligand so as to recognize and match the key moieties to the corresponding parts of the template, followed by matching the rest of the molecule. Meanwhile, we will need to observe a number of match patterns in the aligning process (see a list of the match patterns in “Supporting Information” at the end of this article). However, the rule of thumb for the ligand aligning is straightforward, just “SIMILAR to SIMILAR”. In addition, as discussed in our previous study,8 the alignment modeling requires only the approximate conformational fit of a ligand with the template, while taking into account most of the special structural match patterns between them.
Due to the structural complexity of the template, it is not always straightforward to determine a meaningful alignment of ligands. In fact, we need to handle the confusing cases pretty often, such as with ligands having unique scaffolds or possessing more than one potential alignment. In many cases, however, the special μ-features as well as some other characters of the template can be utilized to assist the matching process.
There are three major approaches we take to verify the putative alignments of ligands: to match with the μ-features, to fit with SAR data, and to compare with the structures of well-verified ligands. Although largely empirical in nature, these approaches, when applied in combination, have proven to be effective in verifying the ligand alignments.
Case Studies
Interpretation of Salvinorin A as a κ-Agonist and Herkinorin as a μ-Agonist
Salvinorin A is a naturally occurring κ-agonist, while Herkinorin and Kurkinorin, the two close analogs, are pure μ-agonists18 (Figure 11).
Figure 11.

Structures of Salvinorin A, Herkinorin, and Kurkinorin.
Salvinorin A has a unique neoclerodane scaffold, which does not contain a prototypic amino group that many opioid ligands have. And also, Salvinorin A’s scaffold is rather symmetric, allowing for multiple alignments. So how to properly align Salvinorin A as well as its analogs was a complicated case in our previous modeling.
The originally proposed alignment matched Salvinorin A’s A/B rings to the morphine’s D/C rings, respectively.8 With the same alignment, Herkinorin’s 2O-benzoyl group was matched to the μ-featured N-PhEt to account for the special μ-selectivity of this ligand (see the blue-circled area in Figure 12A). However, that alignment did not seem to be a smooth one because Herkinorin’s A-ring without an amino group was not really a good match to the nitrogen-containing D-ring of morphine. And also, the 2O-benzoyl ester of Herkinorin and the N-PhEt of the template were structurally not a good match. In addition, the alignment did not seem to make sense in interpreting the enhanced μ-affinity of Kurkinorin with a double bond at the A-ring19 (Figure 11).
Figure 12.
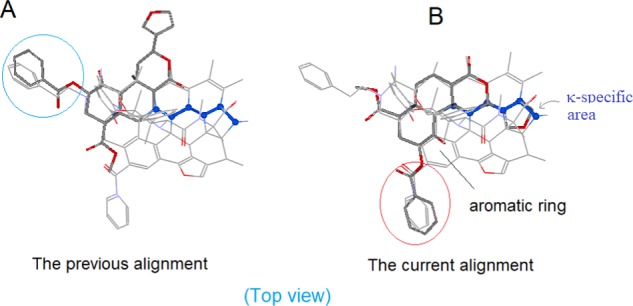
Alignment of Herkinorin. (A) The 2O-benzyl moiety was matched to the μ-featured N-PhEt in the previous alignment (blue circled). (B) With the current alignment, the 2O-benzyl is aligned to the front area (red circled) as a new μ-feature to account for the high μ-binding selectivity of this ligand.
Aided with the extended scaffold of the new template, we realigned Salvinorin A as well as the two analogs. And the updated alignments appeared to be much better for understanding the special SARs of these ligands (see Figure 12B).
-
(i)
With the new alignment, Salvinorin A’s κ-binding is well explained. As shown in Figure 12B, the central area of Salvinorin A along with the furan ring is aligned smoothly to the in-middle loop (indicated with blue ball–sticker rendering), which is the κ-specific area of the template corresponding to the κ-backbone-conformational model (Figure 10). So, this alignment can well account for the κ-activity of this ligand (this is a typical example to show how the μ-agonist template can be applicable for the alignment of δ-/κ-ligands. Another case can be seen below with the interpretation of the δ-antagonism of DIPP-NH2).
-
(ii)
At the new alignment, Herkinorin’s 2O-benzoyl moiety responsible for the μ-binding is aligned to the front area of the template, a new μ-feature hence being proposed and being verified later on with the alignments of several other special μ-agonists (see discussions below and in Part 2 of this report). Presumably, the 2O-benzoyl can also work to block Herkinorin from binding to the κ-receptor so as to account for the exclusive μ-binding of this ligand (EC50: μ/κ = 3.0/>10 000 nM). For Kurkinorin, on the other hand, its A-ring has a double bond in conjugation with the neighboring carbonyl group, which can make a better match with the local aromatic ring of the template (see Figure 12B), so as to account for the enhanced μ-affinity of this ligand (Ki = 1.2 nM) vs Herkinorin (Ki = 40 nM).19
Further Refinement of the Front Benzoyl
Herkinorin’s new alignment helped to reveal the front benzoyl of the template as a new μ-feature. Upon the recognition, we looked further at several other special μ-ligands able to align their key moieties with the front benzoyl (such as JOM-5 Mm20 and Em-Mm17), which greatly helped for validation of the front benzoyl as a new μ-feature (see the detailed discussions in Part 2 of this report).
Alignment of the Crystal Structure of DIPP-NH2
DIPP-NH2 (H-Dmt-Tic-Phe-Phe-NH2, Dmt = 2,6-dimethyltyrosine), as mentioned above, is a mixed δ-antagonist/μ-agonist,13 and its binding conformation at the μ-receptor is considered to be the same as at the δ-receptor. Thus, by aligning the crystal structure of DIPP-NH2 (PDB ID: 4rwa)21 with the template, we are set up to assess the SARs of this ligand.
As μ-Agonist
Figure 13 shows the alignment of the crystal structure of DIPP-NH2 with the template, where Dmt1 is aligned around the morphinan core, similarly to the Tyr1 of many other μ-peptides, while the rigid bicyclic moiety of Tic2 is aligned to the inner loop, a pattern commonly seen with δ-ligands (refer to Figure 15). Upon the alignments, the backbones of Phe3 and Phe4 are consequently matched to the up-middle loop of the template (the green lines). Since the up-middle loop is the important μ-feature of the template as mentioned previously, this close match can quickly account for the μ-agonist activity of DIPP-NH2.
Figure 13.

Alignment of the crystal structure of DIPP-NH2 (stereoview). The backbone segments between Phe3 and Phe4 of DIPP-NH2 are matched to the up-middle loop (green lines) of the template so as to account for the μ-agonist activity of this ligand.
Figure 15.

δ-ligands aligned at the δ-specific area. (A) Top view of the template to show the δ-specific area (green lines); (B) δ-Agonist TAN-67 aligned bond-to-bond to the δ-specific area; (C) δ-antagonist (N-CH3)-analog of NTI aligned in one-bond-less mismatch to the δ-specific area; (D) alignment of the crystal structure of δ-antagonist DIPP-NH2 with the Tic moiety in one-bond-less mismatch.
As δ-Antagonist
Backbone/Scaffold Mismatch as a Structural Basis for Opioid Antagonism
As frequently observed in our modeling, although the majority of μ-agonists display bond-to-bond match of their key backbones/scaffolds to the template, μ-antagonists often show mismatches (typically mismatched by one or more bonds). Presumably, this type of backbone/scaffold mismatch is one of the important structural bases for opioid antagonism.
Built up with μ-agonists, the template is considered to be μ-agonistic in nature, namely, the template, if placed at the binding site, would behave just like a large μ-agonist so as to keep the receptor in activated conformation. However, if some key structures of the template are altered by inserting/deleting a bond or shifting a key moiety to a nearby location of the scaffold, the original agonistic nature of the template would be disrupted, so as to result in failure in activating the receptor, a prospective mechanism for μ-antagonists associated with the backbone/scaffold mismatch.
As an example, Figure 14 shows the co-alignments with the template of both μ-antagonist Alvimopan (in red) and μ-agonist EM1 (in green). As we can see, although the backbone of EM1 is in a bond-to-bond match with the template, there is a one-bond-less mismatch with Alvimopan’s as compared to EM1’s (see the right panel, Figure 14).
Figure 14.

Co-alignments of Alvimopan and EM1. Both Alvimopan and EM1 are aligned with the template to reveal an apparent one-bond-less mismatch of Alvimopan’s key moiety vs EM1’s, (right panel: the dashed arrows indicate the correlated atoms between the two ligands).
Accounting for the δ-Antagonism of DIPP-NH2
With the bond-mismatch notion discussed above, the structural basis for DIPP-NH2’s δ-antagonism is understood. As mentioned previously, the inner loop of the template is δ-specific (see Figures 10 and 15). So δ-ligands that are bond-to-bond aligned with the loop would be agonists (such as TAN-67,22Figure 15B) or would be the antagonists if mismatched (such as the N-CH3 analog of NTI,23Figure 15C).
When the crystal structure of DIPP-NH2 is aligned to the template, a one-bond-less mismatch of its key moiety between Dmt1 and Tic2 with the inner loop (Figure 15D) is seen, with which, the structural basis for the δ-antagonism of DIPP-NH2 is accounted.
The structural basis for opioid antagonism is an often discussed topic in opioid studies.24−26 Here, our backbone/scaffold mismatch-related modeling on DIPP-NH2 and the other opioid ligands offers a new way to look at the potential structural relationships between the agonists and the antagonists (for more examples of opioid antagonism due to the backbone/scaffold mismatch, see CJ-15 208 discussion below as well as some others in the Supporting Information).
Interpretation of the SARs of CJ-15 208
CJ-15 208 [c(Phe1-d-Pro2-Phe3-Trp4)] is a cyclic opioid peptide with both κ- and μ-antagonistic activities27,28 (see Figure 16A) (note: although trans-configuration is set for the amide bond between Phe1 and d-Pro2 in Figure 16A–C, a minor population of the cis-configuration can co-exist, as indicated in conformational studies with EMs29−31).
Figure 16.

Alignment of CJ-15 208 as μ-antagonist with the template. (A) CJ-15 208, a naturally occurring cyclic opioid peptide; (B) CJ-15 208 is aligned as a μ-antagonist with the template; (C) at the alignment, the backbone of CJ-15 208 shows two-bond-less mismatch with the template (green lines).
To understand the structural basis for CJ-15 208’s μ-antagonism, we need first to align the structure with the template properly. Different from many other cyclic opioid peptides, CJ-15 208 did not have an exo-ring Tyr1 to guide the aligning process, adding difficulties in the initial match process. After rigorous searching, we eventually identified an alignment as the best one (shown in Figure 16B). With this alignment, the backbone segment between d-Pro2 to Trp4 was aligned bond-to-bond to the up-middle loop as the μ-specific area, where the Phe3 side chain was matched to the Ph/Ind moiety of the template.
Here, we decided to align Phe3 but not Trp4 with Ph/Ind mainly because of the relevant SAR data. The Ph/Ind moiety of the template, as observed in our modeling, was stereospecific in most cases, only aligning to l-AA-related structures. However, the SAR studies showed that replacement of the l-Trp4 of CJ-15 208 with d-Trp4 was tolerated, and the d-Trp4 analog was even more potent than l-Trp4 at μ-binding.27 On the other hand, Phe3 of CJ-15 208 was l-specific. So, it was suitable to match Phe3 with Ph/Ind, while Trp4 was aligned accordingly to the end of the up-middle loop, where the steric demanding as well as substituent effects were relatively low (note: most of the edge areas of the template, as observed in our modeling, were associated with relatively low steric/substituent effects). And at this setting, the less significant residue Phe1 was aligned to the front edge of the template to agree with the SAR (to be discussed below). Thus, the overall alignment seemed to well account for the μ-binding of CJ-15 208, (but not yet the μ-antagonism).
With the ligand’s alignment determined, we can now examine further the SARs of CJ-15 208.
Difference between Phe1 and Phe3 on Binding Affinities
An alanine scan in SAR studies indicated that Phe3 was necessary for high μ-affinity, while Phe1 was not,27 data for which can be interpreted readily with the current alignment. As shown in Figure 16B, Phe3 is matched to the Ph/Ind moiety with the μ-featured up-middle loop so as to account for its significant effect on μ-binding, whereas Phe1 is aligned to the front of template as an edge area, which is considered to be relatively low in sensitivity to steric or substituent effects (as mentioned above). Thus, the front area alignment can account for the less significant effect of Phe1 on the μ-affinities.
μ-Antagonism of CJ-15 208
As shown in Figure 16C, although the backbone between Phe3 and Trp4 of CJ-15 208 is bond-to-bond matched to the up-middle loop, an apparent two-bond-less mismatch exists around where Phe1 is aligned (see the red circle in Figure 16C). Although the front area is a less significant zone for μ-binding as compared to the up-middle loop, presumably the two-bond-less mismatch occurring at the front area will significantly shorten the distance between Phe1 and Trp4, so as to disturb the bond-to-bond alignment at the up-middle loop. Thus, the μ-antagonism of CJ-15 208 is interpreted.
Conversion of CJ-15 208 into a μ-Agonist
Based on the above understanding, it appears to be possible to transform CJ-15 208 from a μ-antagonist into a μ-agonist through extending its backbone segments around Phe1 to match with the template. Interestingly enough, this idea was examined already by a group in Italy recently.32 The authors used β-Ala1 to replace Phe1 so as to prolong the backbone by one bond (Figure 17A), a modification for which indeed gave rise to a full μ-agonist analog, along with high μ-selectivity and affinity.
Figure 17.

Conversion of CJ-15 208 from μ-antagonist to μ-agonist. (A) One-bond extension of CJ-15 208’s backbone resulted in a μ-agonistic β-Ala analog; (B) the one-bond-less alignment of the β-Ala analog (thick lines) appears to fit smoothly to the template (green lines), as compared to that of CJ-15 208 (gray lines).
It should be noted, however, that the one-bond extension here does not really make the β-Ala analog in bond-to-bond match with the template. Instead, a one-bond-less mismatch still exists, which otherwise would suggest it to be an antagonist (Figure 17B). For this issue, we seek a possible explanation by looking into the alignment itself.
As mentioned above, the two-bone-less mismatch of CJ-15 208 significantly shortens the distance between Phe1 and Trp4 so as to cause the μ-antagonism of CJ-15 208. On the other hand, the alignment of β-Ala analog at the front area appears to be pretty smooth, and the distance between β-Ala and Trp4 looks rather close to that of the bond-to-bond match (Figure 17B). So presumably, this one-bond-less mismatch does not cause much disturbance to the backbone aligned at the μ-featured up-middle loop, so that this ligand can still act as a μ-agonist.
In addition, the above-discussed alignments/SAR interpretations for CJ-15 208 and the analog may be applicable as well in accounting for the potent μ-binding and agonistic activities of a class of special cyclic EM1 analogs, since they are also the atypical cyclic peptides with the similar sequences.33,34
Part 2. Template Refinement
This part is focused on the template refinement and validation, an important process in the template construction. After construction of the preliminary template (see Figure 4 in Part 1), we have made great efforts into refining the scaffold, process for which is still ongoing. Herein, we will illustrate how the template is stepwise refined and validated, how it will help us understand the SARs of individual ligands, and also how we can contemplate further improvements.
As mentioned in Part 1, template refinement is an iterative process. Upon alignment of the various μ-ligands with the template, we carefully analyzed the aligning patterns of the backbone/scaffolds of ligands so as to identify their common structural features for improvement of the template scaffold. This refinement and validation process has been carried out repetitively for all parts of the template scaffold. Therefore, almost every portion of the template is derived from multiple ligands, even though only a handful of them will be discussed herein.
To facilitate the discussions below, we divide the template scaffold into six regions: A–E and the back region (differently colored in Figure 18), each having special structural features for the ligand alignment.
Figure 18.
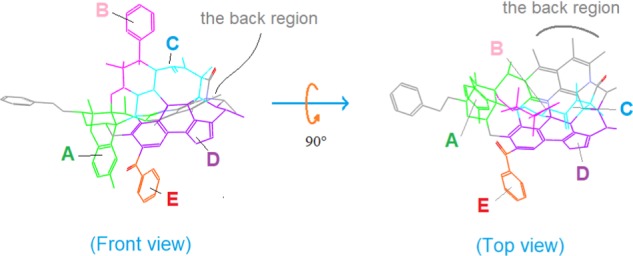
Six regions of the template. The scaffold of the template is divided into six differently colored regions: A–E and the back region, each with special structural features for the ligand alignment.
Region A (the Green Zone)
Region A is basically constituted with the core of morphine as well as many other classical morphinan derivatives. Conventionally, the morphinan core is considered as the message to achieve the opioid activity, while a special “address” is needed for opioid ligands to confer their receptor selectivity. For the peptide ligands, the address is usually related to AA2 and the residues beyond8,35 (see also discussions of Region C (the Blue Zone) below). However, there can also be other address moieties for opioid ligands, e.g., the μ-features of the template for μ-ligands (see Part 1 of this report). Typical examples of μ-ligands, which can be aligned at Region A, include both the morphinan and the nonmorphinan derivatives, such as 1–416,36 and 5–637 (Figure 19). Thus, the μ-featured N-phenethyl moiety is the address component of the ligands (see Figure 19A).
Figure 19.

Ligand alignment at Region A (the green zone). Several μ-selective morphinan derivatives are aligned in this region (e.g., 1–6), where their morphinan core is considered as the message and the N-PhEt as the address. (A) The alignment of 1 at Region A indicates the N-PhEt group as an address for the μ-binding selectivity; (B) the steric match of the 9S-OH of 5 with the template’s oxygen atom at Region A ensures the high μ-binding of this ligand; (C) the low μ-affinity of 6 is attributed to the sterically mismatched 9R-OH group.
Nature has uniquely structured the morphinan core. It seems that appropriate alignment with the core is essential for an opioid ligand to achieve good affinity and activity, whereas a misalignment as caused by a substituent, a steric, or other factors would potentially result in significant changes in binding behaviors of the ligands. 5, for example, is a potent μ-agonist [Ki(μ/δ/κ) = 0.19/13/184 nM].37 As shown in Figure 19B, 5 has a 9S-OH in its scaffold, which can make a heteroatom match with the oxygen atom at Region A of the template (see Figure 19B), so that it displays high binding. However, 6, the epimer, is a weak binder [Ki(μ/δ/κ) = 59/1570/245 nM],37 because the 9R-OH of 6 sterically does not fit (Figure 19C).
For many opioid peptides, Region A is where their first two residues at the N-terminal are aligned (often Tyr1 and Pro2 for the μ-selective peptides38). The Pro2 of μ-peptides, as proposed in the previous study, plays a critical role in keeping the AA2 backbone oriented perpendicularly to the C-ring of morphine,8 so as to promote the successive backbones to align with Region C to achieve the μ-selectivity (see Region C (the Blue Zone) discussion in the following section).
In addition, we proposed two possible backbone conformations in our previous study for the two N-terminal residues aligned at this region, the μ- and the δ-alignments.8 However, we now drop the “μ-alignment” and only keep the “δ alignment”, since the δ alignment appears to work well with all of the cases in our modeling.
Region A does not retain the 4,5-epoxy ring (the E-ring) of morphine, because this moiety does not seem to be a necessary component, while its presence would rather interfere with the template refinement. However, awareness of a potential five-membered ring existing at this position would be of help for the alignment of related ligands (e.g., the five-membered ring of EMs’ Pro2 or of CJ-15 208’s d-Pro2; see the related alignment in Part 1).
Region B (the Pink Zone)
This region contains a phenyl moiety at top of the scaffold, which is considered as one of the special μ-features (see discussions in Part 1). However, this phenyl moiety for high μ-selectivity appears to be mostly associated with special ligand types, i.e., those with rigid scaffolds and aligned at Regions A and B (such as 8 and 10 in Figure 20).
Figure 20.

Ligand alignment at Region B (the pink zone). 7 and 8 are two rigid μ-agonists and their alignments contribute to the construction of Region B; 10, on the other hand, is a naturally occurring μ-agonist with a bulky and rigid scaffold, alignment for which helps for validation of the structures of Regions A and B.
7 [Norbuprenophine, Ki(μ/δ/κ) = 0.07/3.14/0.91 nM]39 and 8 [Ki(μ/δ/κ) = 0.90/2.1/65 nM]12 were the two major building blocks for the construction of Region B (see their alignments in Figure 20).
10 (Cebranopadol) is a naturally occurring opioid ligand with potent μ-binding activity ([Ki(μ/δ/κ) = 0.7/18/2.6 nM]40), rigid and bulky scaffold for which can be aligned to Regions A and B (Figure 20) to provide a good validation of these two regions. Although 10’s scaffold contains a spiro-ring, which conformationally does not fit the template, the major scaffold with the top phenyl as well as the dimethylamine substituent matches well with the template. And because of the overall balanced alignment, this ligand can still bind effectively to the μ-receptor (see Figure 20). Thus, this case can serve as a good example to showcase the importance of an overall structural and conformational match of a ligand in the alignment modeling.
Region C (the Blue Zone)
There are a large number of opioid peptides reported in the literature, including the linear and the cyclic peptides, many of which are excellent μ-agonists with high selectivity and affinity.38,41,42 Yet, most of the linear μ-peptides were not selected as building blocks for the template construction because of the high conformational flexibility associated with their structures. However, after the template was basically constructed, the linear μ-peptides were efficiently used for the refinement and validation (see examples in the discussion below).
Our modeling has revealed that the majority of the linear μ-peptides, naturally occurring or synthetic (e.g., endomorphins, morphiceptin, dermorphin, DAMGO, DALDA, PL017, etc.), can all be aligned with the template in a similar pattern. As shown in Figure 21A, the embedded ball–stick rendering (with nitrogen atoms labeled in blue) is the common backbone alignment of AA1 through AA4 of the linear μ-peptides.
Figure 21.

Embedded backbones of μ-peptides and Region C. (A) The backbones of AA1 through AA4 of the μ-binding peptides are embedded in the template (in stereoview, with the peptide-bond nitrogens labeled), while Region C still retains some of the peptide-bond features; (B) aligned to Region C as well as the whole embedded backbones are the binding conformations of DIPP-NH2 and DAMGO.
Region C, mainly built with the backbones (AA2–AA4) of μ-peptides, still retains some of the peptide-bond features, such as the α-carbon and the carbonyl group, as well as the nitrogen atoms (see Figure 21A). The structures and conformations of Region C were well refined with the alignments of many peptide/peptidomimetic μ-ligands and were also validated with the experimentally derived binding conformations of two well-known peptide ligands: DIPP-NH221 and DAMGO15 (see Figure 21B).
Region C has been identified as one of the most important μ-features of the template, as almost all of the μ-peptides/peptidomimetics are shown to align their backbone segments to this region. On the other hand, the side-chain effects at this region do not seem to be very significant. For instance, the AA4 can be Phe4, Sar4, or β-Ala4 (see ref (38)). In addition, Phe3, often as an important residue, does not seem to be special with μ-peptides, as many δ-peptides also possess Phe3 in their structures.38
It seems that keeping the backbones aligned with Region C is a key factor for the high μ-binding of many opioid peptides. And the side chains (or some other structural factors) are considered mostly to play assisting roles for the backbones to be properly aligned, which can be illustrated with the following examples.
Pro2 is known to be an important feature of many μ-selective peptides.8,38 However, a few well-known μ-peptides without Pro2 still show high μ-selectivity, which would be attributed to some special factors involved in their structures to keep the backbones aligned to Region C. For example, DAMGO (H-Tyr-d-Ala-Gly-N-MePhe-Gly-ol) without Pro2 displays high μ-selectivity [Ki(μ/δ) = 1.22/1280 nM].43 Presumably, here the N-methylation of its Phe4 (Figure 22A) is partly responsible for the high μ-selectivity44 and its presence reinforces DAMGO’s backbone to align at Region C. The similar effect of the N-methylation to enhance μ-selectivity is seen with DAS-DER (H-Tyr-d-Arg-Phe-Sar-OH; Sar = N-MeGly).45 In addition, DAMGO’s terminal Gly5-ol moiety may also contribute to the alignment to Region C, since this moiety can be bond-to-bond matched to Region D to stabilize the backbone alignment at Region C (see Figure 22A). DALDA [H-Tyr-d-Arg-Phe-Lys-NH2; Ki(μ/δ) = 1.69/192000 nM]43 is another example of μ-peptide without Pro2. And here the d-Arg2 is considered as the special factor for the backbone to be aligned at Region C, as the side chain of d-Arg2 including the terminal guanidino group can be well aligned to Region D as well as Region E (Figure 22B), so as to facilitate the backbones aligned at Region C. In addition, the Lys4 side chain of this ligand, similar to DAMGO’s Gly-ol, can also be bond-to-bond aligned to Region D, so as to further stabilize the backbone alignment at Region C. Likewise, 11 (H-Dmt- d -Aba-Gly-NH-Bn), a μ-selective peptidomimetic [Ki(μ/δ) = 0.46/11.0 nM],46 has a special d-Aba2 to keep its Gly3-NH-Bn aligned at Region C (Figure 22C).
Figure 22.

Ligand alignment at Region C (the blue zone). Region C is one of the most important μ-features of the template, as the majority of μ-peptides/peptidomimetics align their backbone moieties at this region to assume μ-selectivity, such as DAMGO (A) and DALDA (B) as the μ-peptides, and 11 (C) as the μ-peptidomimetic; (D) a key moiety of 12 (Alvimopan) is aligned at Region C as a μ-antagonist.
In addition to μ-agonists, μ-antagonists can also be aligned to contribute to the template refinement and validation. For example, 12 (Alvimopan) is a peptidomimetic μ-antagonist,47,48 and its peptide-mimicking moiety at top of the scaffold can be aligned to Region C, where an apparent one-bond-short backbone mismatch is accountable for the μ-antagonistic activity of this ligand (Figure 22D; see also Figure 14 in Part 1). Thus, the alignment of 12 as a μ-antagonist can help for the refinement and validation, as well.
Region D (the Purple Zone)
Region D is a relatively complex zone, and a wide variety of μ-ligands were involved for its construction. As shown in our modeling, many of the μ-ligands align their μ-selectivity-related moieties there, indicating that Region D would be of rich μ-features.
Region D was mainly constructed based on a number of μ-selective cyclic peptides that contained an exo-Tyr1 attached to their macrocyclic rings.38,49,50 The Tyr1 moiety can be quickly matched to Region A, so as to facilitate the alignment of the rest of the molecules.
Most of the macrocyclic rings of the cyclic peptides are formed with 3–5 residues by connecting the side chain of AA2 to another residue at the C-terminal of the sequences. Therefore, the two opposite rims of the macrocycles are structurally distinct, with one featuring the peptide backbone, and the other featuring the side chains. Initially, it was difficult to determine the orientations of the macrocyclic rings because both Regions C and D were not formed at that point (refer to Figure 4 in Part 1). And hence, there seemed to be multiple ways for the two rims of the macrocyclic rings to be arranged: to the back region, to Region C, or to Region D.
Nevertheless, it was later found that the backbone rim would be best aligned to Region C, which would be in accordance with the μ-backbone orientation as roughly set with the preliminary template (see Figure 4), so as to leave the back region for the δ- and the κ-backbone alignment (refer to Figure 10 in Part 1). Meanwhile, the side-chain rim, often variable in length and structure, appeared appropriate to be matched with Region D. Region D was found to be an area with greater structural variety according to the alignment patterns of many μ-ligands. Thus, the backbone and the side-chain rims of the cyclic peptides were aligned to Region C and Region D, respectively, to help define the two regions.
The literature data indicated that both the μ-selectivity and affinities of the cyclic peptides were affected by the ring sizes as well as the nature of substituents,49,51 which information was of help for the refinement of Region D. For example, shown in Table 1 are four cyclic enkephalin analogs (13–16) along with their related SAR data. As we can see, all four analogs have the similar sequences (different only with AA2), featuring the variable lengths of the side chains of AA2s, thus the resulting macrocycles in different sizes. And also, all of the cyclic analogs display lower binding affinities than their linear counterparts (see Table 1; data for the linear peptides are shown in the blanket), but their μ-selectivity is more or less enhanced, especially with the 14-membered analog, which shows the highest μ-selectivity among the others.51
Table 1. Ring-Size Effects on the μ-Binding Affinity and Selectivity of the Cyclic Peptidesa.
| IC50 (nM) |
||||
|---|---|---|---|---|
| cyclic peptide | ring size | μ | δ | δ/μ |
| 13 = Tyr-c[D-A2pr-Gly-Phe-Leu] | 13 | 95.8 (3.97) | 118 (5.51) | 1.23 (1.13) |
| 14 = Tyr-c[D-A2bu-Gly-Phe-Leu] | 14 | 24.9 (10.1) | 253 (8.94) | 10.16 (0.72) |
| 15 = Tyr-c[D-Orn-Gly-Phe-Leu] | 15 | 56.6 (5.89) | 221 (6.81) | 3.90 (0.95) |
| 16 = Tyr-c[D-Lys-Gly-Phe-Leu] | 16 | 22.4 (4.08) | 32.2 (9.57) | 1.44 (1.92) |
All of the cyclic analogs display lower binding affinities than their linear counterparts, but their μ-selectivity is more or less enhanced, especially with the 14-membered analog (data in parentheses are for the linear counterparts).
These SAR data can be understood with the alignments of the analogs. For example, the linear peptides display high binding affinities but little binding selectivity, which is because their backbones can be equally aligned either at the μ-specific Region C or at the δ-specific area in the back region. For the cyclic analogs, however, although their backbone rims of rings are aligned to Region C, the side-chain rims are aligned at Region D, which would help to stabilize the backbone alignment at Region C, thus to enhance the μ-selectivity.
In addition, 14 (the 14-membered cyclic analog) can be bond-to-bond aligned at Region D (see Figure 23A). Note that the existing bond-to-bond match pattern may not be seen clearly in the figure, while 13 and 15, the d-A2pr and the d-Orn analogs, are aligned by one-bond shorter and one-bond longer, respectively. Therefore, the difference in the aligning pattern of the ligands can account for the higher μ-selectivity of 14. On the other hand, the 16-membered analog can also be bond-to-bond aligned, but via a different pattern (see Figure 23B), with which, the altered activity profile of 16 can be interpreted (i.e., with the higher μ-affinity but reduced δ/μ-ratio). Coincidently, the similar ring-size effects were reported also in a recent study, in which the 14-/16-/17-membered derivatives were displaying high μ-affinities, while the 15-membered showed only low μ-binding. And also, the 14 membered was associated with the highest μ-selectivity among all.52
Figure 23.

Alignment and SAR interpretation of cyclic peptides with different ring sizes at Region D (the purple zone). Depending on their ring sizes, μ-selective cyclic peptides can be aligned at Region D in different patterns, with which, their different binding profiles can be accounted for. (A) The 14-membered cyclic analog can be bond-to-bond aligned at Region D to account for its higher μ-binding affinity and selectivity; (B) the 16-membered analog can also be bond-to-bond aligned but in a different pattern, which can account for the higher μ-affinity but reduced δ/μ-ratio of this ligand; (C) the 18-membered analog can be aligned bond-to-bond around the edges of Region D, which can probably interpret the extremely high μ-affinity and selectivity of this ligand.
However, it should be noted that the above ring-size-related pattern may not be critical to some special cyclic peptides. For instance, the β-Ala analog of cyclic peptide CJ-15 208 as discussed in Part 1 does not appear to follow this pattern, which having a 13-membered ring instead of the best-sized 14 membered was shown to be a potent μ-agonist.32 In addition, also reported in the same article was a γ-Ala analog (14-membered), which turned out to be a δ-agonist.32 The special structural factors for those seemingly contradicted SARs of the ligands will be discussed elsewhere.
Moreover, as compared to the linear peptides, the lower affinities associated with the cyclic analogs (Table 1) can be partly attributed to the distorted conformations of the side chains of AA2 as well as the backbones of Leu5 due to their involvement in the ring formation. These two moieties would be best aligned along the left and the right edges of Region D to achieve high μ-affinities, a pattern that we have observed from the alignment modeling on a number of μ-ligands. (Below we can see such an example with 17, an 18-membered macrocyclic peptide).
Another example of a cyclic peptide for the refinement is 17 (Tyr-c[d-Lys-Trp-Phe-Glu]-Gly-NH2), which showed extremely high μ-affinity and selectivity [Ki(μ/δ/κ) = 0.68/127/5119 nM].53 As we can see, this ligand has an extra-large macrocyclic ring (18 memberred) that can be bond-to-bond aligned along the left and the right as well as the front edges of Region D (Figure 23C) to account for its potent μ-affinity and selectivity. In addition, its C-terminal residue (Gly-NH2) appears to critically contribute to the high μ-selectivity, as one of its analogs without the Gly residue showed high affinity but reduced μ-selectivity.53
Besides the peptide ligands, the nonpeptide ligands also contributed to the refinement of Region D. Many of the nonpeptide μ-ligands with their μ-selectivity-related moieties were aligned at this region, which helped further for the refinement and validation. For example, 18 (fentanyl) is a well-known opioid drug with potent μ-agonistic activity. This ligand as well as many of its analogs has a relatively simple piperidine scaffold,54 which can be aligned to Region A, where the N-phenethyl group presumably plays a major role for the μ-selectivity. Meanwhile, the N-phenyl and N-propionyl on the other side of the scaffold are matched to Region D and the back region, respectively (see Figure 24A). And the aligning pattern suggests that both the moieties would also contribute to the μ-selectivity of this ligand and is consistent with the SARs as observed with many fentanyl analogs.54
Figure 24.

Alignment of nonpeptide ligands at Region D (the purple zone). Many of the nonpeptide ligands align their μ-selectivity-related moieties at Region D. (A) Fentanyl’s N-phenyl is aligned to Region D; (B) the diethylaminoethyl group as well as part of the benzimidazole moiety of Etonitazene is matched to Region D; (C) although the DMT moiety of JOM-5 Mm is matched to Region A, its THQ scaffold is aligned to Region D; (D) Em-Mm’s scaffold is aligned at Region D as well as the back region.
19 (Etonitazene) is a well-documented μ-agonist displaying high μ-binding affinity and selectivity.55,56 A previous alignment of 19 matched the main benzimidazole scaffold with Region A.8 However, that alignment was questionable, because some of the match patterns did not fit well. With the current alignment, the p-ethoxybenzyl is matched to the aromatic zone in the back region, and the diethylaminoethyl is aligned at Region D, while the benzimidazole scaffold including the nitro substituent is matched to the aromatic zones in both Region D and Region E (Figure 24B) (region E is another important μ-featured area to be discussed below). And all of the match patterns seem to be smooth and meaningful, which would well account for this ligand’s high μ-selectivity and affinity.
20 (JOM-5 Mm) is a peptidomimetic ligand with mixed μ-agonist/δ-antagonist activity.20 This ligand’s THQ scaffold along with the substituents can be aligned across three regions of the template: Regions A, D, and E (see Figure 24C). The alignment can account for the SAR data. For example, its DMT moiety, just like the Tyr1 of the μ-cyclic peptides discussed above, is aligned to Region A, which subsequently places the THQ scaffold in Region D. The 6-benzyl shown to be important for the μ-activity of this ligand57 is aligned to Region E. The N1-benzoyl is positioned to overlap with the aromatic zone in Region D to account for its μ-affinity enhancing effect.
21 (Em-Mm), designed as the β-turn mimetic of endomorphin, was found to be a potent μ-agonist.17 Its structure, featuring a rigid bicyclic scaffold with three large substituents, is similar to that of 20. But the alignments of the two scaffolds are quite different because of their distinct substitution patterns. Although 20’s scaffold is aligned to Region D, part of 21’s is aligned to the back region. And the whole structure of 21 is aligned across three regions: Regions A, D, and E (see Figure 24D). In this alignment, the Ni-p-OH-phenethyloxyacyl, resembling the Tyr1 of a μ-peptide, is bond-to-bond matched but along the inner edge of Region A, which is different from the alignment of 20. In addition, the bicyclic scaffold of 21 involving three amide bonds is a large conjugated system, so that it can be better matched with the aromatic zone in the back region.
As we can see, there appears no significant μ-feature involved in the areas where both the scaffold and the N-p-OH-phenethyloxyacyl of 21 are aligned. So, although these two moieties seem to work merely as the message, the “μ-selectivity address” for this ligand would be found with the other two substituents. Indeed, the phenethyl substituent at “i + 2” position is aligned to Region E and the benzyl at “i + 3” is aligned to Region D, both in the μ-feature-rich zones. And the SARs data also supports the alignment: the μ-affinity of the ligand was significantly reduced when both the i + 2 and i + 3 substituents were replaced with Bn and nBu, respectively.17
Region D appears to be a special zone with rich μ-features but it still is not fully defined at this time. As shown above, several of the nonpeptide ligands align their μ-selectivity-related moieties into this region, but there seems no clear pattern seen to help further defining the regional μ-features. Therefore, continued refinement of this region is warranted.
Region E (the red zone)
Region E proves to be an important μ-feature of the template. This region was not initially recognized until the later stage of the template construction, when Herkinorin’s 2O-benzoyl moiety was found to be best aligned at this region to convey the ligand’s unique μ-selectivity (see the related discussions in Part 1 of this report).
It was found that some other μ-ligands were also best aligned when their key moieties were matched to this region, such as the above-discussed 19–21 as well as 25 and 26 (see Figure 25).
Figure 25.

Ligand alignment in Region E (the red zone). Many μ-ligands are best aligned when their key moieties are matched to this region. (A) The phenylpyrazol moiety of IQMF-4 is matched to Region E as well as Region D; (B) With the diazabicyclo-nonane scaffold matched to Region C, the 9-arylpropenyl substituent of DBN-2 is well aligned at Regions E; (C) In the alignment similar to DBN-2’s, the phenyl derivative of DBN-2 matches the phenyl group to the new linker area (see Figure 26C).
25 (IQMF-4) with a fentanyl-related scaffold is a relatively new μ-agonist.58 When aligned similarly to fentanyl, its phenylpyrazol moiety is matched to Regions D and E, which is in accordance with its μ-selectivity (Figure 25A).
26 (DBN-2) is a highly μ-selective agonist, and it has a unique diazabicyclo[3.3.1]nonane scaffold with two N-substituents attached.59 In our previous modeling, many different ways had been tried for the appropriate alignment of this ligand, but none of them seemed optimal. For instance, although the diazabicyclo-nonane scaffold could be matched to Region A by its structural features, the substituent-related SARs were hard to interpret. Now with the μ-featured Region E revealed, the new alignment of this ligand turned out to be rather meaningful to account for the related SARs. By the current alignment, the rigid scaffold along with the propionyl substituent is smoothly aligned in Region C (Figure 25B), in which one significant heteroatom match exists: N3 of the scaffold is matched to the backbone nitrogen at this region. Meanwhile, the 9-arylpropenyl substituent, with the double bond and the aryl in conjugation, is well aligned to Regions D and E. All of the alignment patterns can account for the high μ-selectivity and potency of this ligand. Particularly interesting to note is that this alignment can also account for the SARs of the phenyl derivative of 26. As shown in Figure 25C, the additional phenyl group (blue circled) is matched into a conjugated area between Region A and Region E, where a linker structure has been identified (see the discussion below) to account for the enhanced μ-affinity of this derivative (Ki = 5 nM) as compared to 26’s (Ki = 13 nM).59
Linker
The template refinement has been undertaken throughout the study, and in the latest process, a linker was added between Region A and Region E (see Figure 26C). This construction was mainly based on the alignment of a series of aryl-containing N-monosubstituted analogs of 8-carboxamidocyclazocine (8-CAC).60 Among them, N-BPE-8-CAC (27) displayed partial μ-agonist activity but with very high μ-binding affinities [Ki(μ/δ/κ) = 0.3/0.74/1.8 nM],61 while the 4′-OH analog of N-BPE-8-CAC (28) showed even higher μ-affinity and selectivity [Ki(μ/δ/κ) = 0.0056/0.81/0.49 nM]. SAR studies indicated that an ethylene spacer between the aryl and the amide nitrogen of the 8-N-arylethyl (Figure 26A) was the best for high potency.60
Figure 26.

Ligand alignment for the linker construction. (A) 8-CAC and the derivatives; (B) Alignment of 28 shows good structural as well as conformational match to both Region A (the green zone) and Region E (the red zone); (C) A linker between Regions A and E, as well as an additional p-OH-Ph moiety, is generated accordingly.
The 8-CAC scaffold of the ligands can be well matched to Region A, which, in turn, orients the N-BPE (N-bis-phenylethyl) moiety toward Region E (Figure 26B). Because of the close disposition of the two regions as well as their apparent structural correlations with the ligands, we thought that a linker construction between the two regions would be an interesting idea to facilitate the ligand alignment, with which the current linker structure was generated (Figure 26C).
As we can see, the linker formation between Region A and Region E extends each of the regions for the ligand alignment. Although it somewhat distorts the regional structures, overall the linker construction is pretty smooth, well incorporating both the structural and conformational features of the 8-CAC derivatives.
The linker structure was supported by docking modeling (Figure 27), where the whole template was docked at the binding pocket of the μ-receptor (PDB ID: 5c1m), and the morphinan core of the template was superimposed to that of Bu72 (the bound ligand). On the pose, the extended Region E of the template was found existing between the TM4 and TM5 of the receptor, with no significant steric constraints against the boundary of the binding pocket (see Figure 27) [it should be noted that for that docking, there was indeed a steric constraint of the top portion of the template against a short sequence at the N-terminal of the receptor (G1SHSLX6). However, this constraint was not considered to be significant, as this short N-terminal sequence located at the opening of the receptor’s binding pocket is thought to be highly flexible and would, therefore, undergo an induced conformational adaptation readily upon the binding of differently structured ligands (e.g., the template in this case) to avoid an unfavorable steric constraint].
Figure 27.

Docking the template at the binding site of the μ-receptor. The extended p-OH-Ph moiety of the template is found between the TM4 and TM5 of the receptor without any steric constraint (note: α-helices colored in red, β-sheets in blue).
The linker construction is of significance not only because it facilitates the alignments of both 27 and 28 as well as the phenyl derivative of 26 as mentioned above but also because it helps to reveal more inclusive ligand-binding space of the receptor’s binding pocket, which is beneficial for our continued efforts in construction of a universal μ-agonist template that will cover all of the ligand-binding space at the pocket.
Summary and Concluding Remarks
The template-based alignment modeling developed in our recent studies is an innovative approach for SAR studies of opioid ligands. As we previously reported, this approach showed promise but also with the limitation, which was mainly attributed to the small size of morphine as a template. To overcome the limitation, we set out to construct an alternative μ-agonist template with this study. The newly constructed template contained a largely extended scaffold, along with a few special μ-features relevant to the μ-selectivity of opioid ligands. As demonstrated in this paper, the new template showed significantly improved efficacy in facilitating the alignment modeling of a wide variety of opioid ligands, in terms of understanding the structural correlations as well as interpreting the related SARs.
As illustrated in Part 2, refinement is a very important process for the template construction, which has greatly contributed to the validation and improvement of the template. And the refinement process is still ongoing currently.
Besides the further validation and improvement, another major goal for the template refinement is to reveal additional μ-features on the scaffold so as to better construe the SARs of various μ-ligands. As demonstrated in Part 2, although Regions C and E appear to be the major μ-featured areas of this template, μ-features can also exist anywhere around the template. And apparently, there are still unsolved SAR problems around Regions A and D as well as at the back region. So we will continue to look into these areas for the additional μ-featured structures or patterns.
How to understand the structural diversity and specificity of GPCR ligands is a major topic in the studies of ligand–receptor interactions. As we know well, the single binding pocket of a GPCR receptor can accommodate a wide variety of structurally diverse ligands, while on the other hand, each of the individual ligands appears to be bound very specifically. As we can understand better now, this scenario is simply because all of the structures of ligands are highly correlated so that they can be superimposed and merged to form a large artificial template to represent the ligand-binding space of the receptor. And according to the template-based alignment modeling, for a ligand to bind effectively at the receptor’s binding pocket, high structural specificity has to be met, namely, the major scaffold as well as the key moieties of the ligand has to be adequately aligned with the template.
With this vision in mind, we are able to interpret many of the well-known SARs associated with the GPCR ligands, such as the structural diversity and mutual correlations, the binding specificity and selectivity, and agonism vs antagonism, etc. Hopefully, by continued exploration with the innovative template-based alignment modeling, we will be able to learn further the deep nature of GPCR–ligand interactions.
Modeling Method and General Process
Information about the ligands and the related SARs was collected from the literature. The structure drawings and aligning were carried out with Accelrys DS Visualizer, a molecular modeling software available from the Accelrys Software Inc.62 However, the modeling study was essentially based on the visual examination of the 3D structures of various opioid ligands along with the analysis of their SAR data, a typical means in conventional medicinal chemistry research, rather than by any computational process.
Template Docking
Preparation of the Protein Chains of an Agonist-Bound μ-Receptor
The file of 5c1m in pdb format was downloaded from PDB (http://www.rcsb.org/). At the interface of Accelrys DS Visualizer, by unchecking “Side chain” at “Protein Groups”, all of the side chains of the receptor’s protein chains were hidden from view with only the backbone shown, so as to increase the visibility of the bound ligand and to facilitate the subsequent docking process.
Docking the Template
With the bound ligand (Bu72) as a position reference, the template was manually docked into the binding pocket of the receptor, where the template and the bound ligand were superimposed at their common morphinan core (note: the superimposition was carried out in such a way that the orientation of the template was able to be adjusted slightly to avoid any steric constraints against the receptor).
Acknowledgments
We thank all our colleagues who have helped for the review and evaluation of the manuscript. Z.W. wishes to give special thanks to his mentors friends and family for their constant supports.
Glossary
Abbreviations
- GPCR
G-protein-coupled receptor
- SAR
structure–activity relationship
- TAM
template-based alignment modeling
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02244.
The authors declare no competing financial interest.
Supplementary Material
References
- Ghodse A. H.; Bown P. J.; Aronson J. K.. Opioid Analgesics and Narcotic Antagonists. In Side Effects of Drugs Annual; Elsevier, 2000; Vol. 23, pp 96–113. [Google Scholar]
- Pasternak G. W.; Pan Y. X. Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury A. F.; Smyth D. G.; Snell C. R. Biosynthetic origin and receptor conformation of methionine enkephalin. Nature 1976, 260, 165–166. 10.1038/260165a0. [DOI] [PubMed] [Google Scholar]
- Schiller P. W.; Yam C. F.; Lis M. Evidence for topographical analogy between methionine-enkephalin and morphine derivatives. Biochemistry 1977, 16, 1831–1838. 10.1021/bi00628a011. [DOI] [PubMed] [Google Scholar]
- Balodis Y. Y.; Nikiforovich G. V.; Grinsteine I. V.; Vegner R. E.; Chipens G. I. Enkephalin: structure--function relationships. FEBS Lett. 1978, 86, 239–242. 10.1016/0014-5793(78)80570-5. [DOI] [PubMed] [Google Scholar]
- Aubry A.; Birlirakis N.; Sakarellos-Daitsiotis M.; Sakarellos C.; Marraud M. A crystal molecular conformation of leucine-enkephalin related to the morphine molecule. Biopolymers 1989, 28, 27–40. 10.1002/bip.360280106. [DOI] [PubMed] [Google Scholar]
- Zhorov B. S.; Ananthanarayanan V. S. Similarity of Ca(2+)-bound conformations of morphine and Met-enkephalin: a computational study. FEBS Lett. 1994, 354, 131–134. 10.1016/0014-5793(94)01071-4. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Hruby V. J. Backbone Alignment Modeling of the Structure-Activity Relationships of Opioid Ligands. J. Chem. Inf. Model. 2011, 51, 1151–1164. 10.1021/ci2000852. [DOI] [PubMed] [Google Scholar]
- Wu Z. In Construction Validation and Application of an Artificial Template for the Template-based Alignment Modeling on the N-Terminal Inhibitors of Hsp90, 246th ACS National Meeting, Indianapolis, IN, Sept 8–12, 2013.
- ChEMBL Database. https://www.ebi.ac.uk/chembldb/, 2012.
- IUPHAR . Opioid Receptors; IUPHAR/BPS, 2017. [Google Scholar]
- Le Bourdonnec B.; Goodman A. J.; Michaut M.; Ye H. F.; Graczyk T. M.; Belanger S.; Herbertz T.; Yap G. P.; DeHaven R. N.; Dolle R. E. Elucidation of the bioactive conformation of the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine class of mu-opioid receptor antagonists. J. Med. Chem. 2006, 49, 7278–7289. 10.1021/jm060486f. [DOI] [PubMed] [Google Scholar]
- Schiller P. W.; Fundytus M. E.; Merovitz L.; Weltrowska G.; Nguyen T. M.; Lemieux C.; Chung N. N.; Coderre T. J. The opioid mu agonist/delta antagonist DIPP-NH(2)[Psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- Filizola M.; Devi L. A. Grand opening of structure-guided design for novel opioids. Trends Pharmacol. Sci. 2013, 34, 6–12. 10.1016/j.tips.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; Schertler G. F. X.; Granier S.; Weis W. I.; Dror R. O.; Manglik A.; Skiniotis G.; Kobilka B. K. Structure of the μ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haddou T.; Beni S.; Hosztafi S.; Malfacini D.; Calo G.; Schmidhammer H.; Spetea M. Pharmacological investigations of N-substituent variation in morphine and oxymorphone: opioid receptor binding, signaling and antinociceptive activity. PLoS One 2014, 9, e99231 10.1371/journal.pone.0099231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi M.; Shen R. Y.; Shea J. P.; Lee M. S.; Kahn M. Design, synthesis, and evaluation of opioid analogues with non-peptidic beta-turn scaffold: enkephalin and endomorphin mimetics. J. Med. Chem. 2002, 45, 1395–1398. 10.1021/jm0155897. [DOI] [PubMed] [Google Scholar]
- Harding W. W.; Tidgewell K.; Byrd N.; Cobb H.; Dersch C. M.; Butelman E. R.; Rothman R. B.; Prisinzano T. E. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J. Med. Chem. 2005, 48, 4765–4771. 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Crowley R. S.; Riley A. P.; Sherwood A. M.; Groer C. E.; Shivaperumal N.; Biscaia M.; Paton K.; Schneider S.; Provasi D.; Kivell B. M.; Filizola M.; Prisinzano T. E. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting mu Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem. 2016, 59, 11027–11038. 10.1021/acs.jmedchem.6b01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland A. A.; Bender A. M.; Griggs N. W.; Gao C.; Anand J. P.; Pogozheva I. D.; Traynor J. R.; Jutkiewicz E. M.; Mosberg H. I. Effects of N-Substitutions on the Tetrahydroquinoline (THQ) Core of Mixed-Efficacy mu-Opioid Receptor (MOR)/delta-Opioid Receptor (DOR) Ligands. J. Med. Chem. 2016, 59, 4985–4998. 10.1021/acs.jmedchem.6b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G.; Zatsepin N. A.; Betti C.; Giguere P.; Han G. W.; Ishchenko A.; Liu W.; Guillemyn K.; Zhang H.; James D.; Wang D.; Weierstall U.; Spence J. C.; Boutet S.; Messerschmidt M.; Williams G. J.; Gati C.; Yefanov O. M.; White T. A.; Oberthuer D.; Metz M.; Yoon C. H.; Barty A.; Chapman H. N.; Basu S.; Coe J.; Conrad C. E.; Fromme R.; Fromme P.; Tourwe D.; Schiller P. W.; Roth B. L.; Ballet S.; Katritch V.; Stevens R. C.; Cherezov V. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. 10.1038/nsmb.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp R. J.; Landsman R.; Waite S.; Malatynska E.; Varga E.; Haq W.; Hruby V. J.; Roeske W. R.; Nagase H.; Yamamura H. I. Properties of TAN-67, a nonpeptidic delta-opioid receptor agonist, at cloned human delta- and mu-opioid receptors. Eur. J. Pharmacol., Mol. Pharmacol. 1995, 291, 129–134. 10.1016/0922-4106(95)90134-5. [DOI] [PubMed] [Google Scholar]
- Peng Y.; Keenan S. M.; Zhang Q.; Kholodovych V.; Welsh W. J. 3D-QSAR Comparative Molecular Field Analysis on Opioid Receptor Antagonists: Pooling Data from Different Studies. J. Med. Chem. 2005, 48, 1620–1629. 10.1021/jm049117e. [DOI] [PubMed] [Google Scholar]
- Collins N.; Flippen-Anderson J. L.; Haaseth R. C.; Deschamps J. R.; George C.; Kover K.; Hruby V. J. Conformational Determinants of Agonist versus Antagonist Properties of [d-Pen2,d-Pen5]Enkephalin (DPDPE) Analogs at Opioid Receptors. Comparison of X-ray Crystallographic Structure, Solution 1H NMR Data, and Molecular Dynamic Simulations of [l-Ala3]DPDPE and [d-Ala3]DPDPE. J. Am. Chem. Soc. 1996, 118, 2143–2152. 10.1021/ja9531081. [DOI] [Google Scholar]
- Schiller P. W.; Weltrowska G.; Schmidt R.; Berezowska I.; Nguyen T. M.; Lemieux C.; Chung N. N.; Carpenter K. A.; Wilkes B. C. Subtleties of structure-agonist versus antagonist relationships of opioid peptides and peptidomimetics. J. Recept. Signal Transduction 1999, 19, 573–588. 10.3109/10799899909036673. [DOI] [PubMed] [Google Scholar]
- Goodman A. J.; Le Bourdonnec B.; Dolle R. E. Mu opioid receptor antagonists: recent developments. ChemMedChem 2007, 2, 1552–1570. 10.1002/cmdc.200700143. [DOI] [PubMed] [Google Scholar]
- Dolle R. E.; Michaut M.; Martinez-Teipel B.; Seida P. R.; Ajello C. W.; Muller A. L.; DeHaven R. N.; Carroll P. J. Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15 208. Bioorg. Med. Chem. Lett. 2009, 19, 3647–3650. 10.1016/j.bmcl.2009.04.105. [DOI] [PubMed] [Google Scholar]
- Aldrich J. V.; Kulkarni S. S.; Senadheera S. N.; Ross N. C.; Reilley K. J.; Eans S. O.; Ganno M. L.; Murray T. F.; McLaughlin J. P. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15 208. ChemMedChem 2011, 6, 1739–1745. 10.1002/cmdc.201100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlogar B. L.; Paterlini M. G.; Ferguson D. M.; Leo G. C.; Demeter D. A.; Brown F. K.; Reitz A. B. Conformational analysis of the endogenous mu-opioid agonist endomorphin-1 using NMR spectroscopy and molecular modeling. FEBS Lett. 1998, 439, 13–20. 10.1016/S0014-5793(98)01202-2. [DOI] [PubMed] [Google Scholar]
- Keller M.; Boissard C.; Patiny L.; Chung N. N.; Lemieux C.; Mutter M.; Schiller P. W. Pseudoproline-Containing Analogues of Morphiceptin and Endomorphin-2: Evidence for a Cis Tyr-Pro Amide Bond in the Bioactive Conformation. J. Med. Chem. 2001, 44, 3896–3903. 10.1021/jm000332e. [DOI] [PubMed] [Google Scholar]
- Leitgeb B. Structural investigation of endomorphins by experimental and theoretical methods: hunting for the bioactive conformation. Chem. Biodiversity 2007, 4, 2703–2724. 10.1002/cbdv.200790221. [DOI] [PubMed] [Google Scholar]
- De Marco R.; Bedini A.; Spampinato S. M.; Cavina L.; Pirazzoli E.; Gentilucci L. Versatile Picklocks to Access All Opioid Receptors: Tuning the Selectivity and Functional Profile of the Cyclotetrapeptide c[Phe-D-Pro-Phe-Trp] (CJ-15 208). J. Med. Chem. 2016, 59, 9255–9261. 10.1021/acs.jmedchem.6b00420. [DOI] [PubMed] [Google Scholar]
- Cardillo G.; Gentilucci L.; Tolomelli A.; Spinosa R.; Calienni M.; Qasem A. R.; Spampinato S. Synthesis and evaluation of the affinity toward mu-opioid receptors of atypical, lipophilic ligands based on the sequence c[-Tyr-Pro-Trp-Phe-Gly-]. J. Med. Chem. 2004, 47, 5198–5203. 10.1021/jm0498811. [DOI] [PubMed] [Google Scholar]
- Gentilucci L.; Tolomelli A.; De Marco R.; Spampinato S.; Bedini A.; Artali R. The inverse type II beta-turn on D-Trp-Phe, a pharmacophoric motif for MOR agonists. ChemMedChem 2011, 6, 1640–1653. 10.1002/cmdc.201100169. [DOI] [PubMed] [Google Scholar]
- Lipkowski A. W.; Tam S. W.; Portoghese P. S. Peptides as receptor selectivity modulators of opiate pharmacophores. J. Med. Chem. 1986, 29, 1222–1225. 10.1021/jm00157a018. [DOI] [PubMed] [Google Scholar]
- Wentland M. P.; Lou R.; Lu Q.; Bu Y.; VanAlstine M. A.; Cohen D. J.; Bidlack J. M. Syntheses and opioid receptor binding properties of carboxamido-substituted opioids. Bioorg. Med. Chem. Lett. 2009, 19, 203–208. 10.1016/j.bmcl.2008.10.134. [DOI] [PubMed] [Google Scholar]
- Hiebel A. C.; Lee Y. S.; Bilsky E.; Giuvelis D.; Deschamps J. R.; Parrish D. A.; Aceto M. D.; May E. L.; Harris L. S.; Coop A.; Dersch C. M.; Partilla J. S.; Rothman R. B.; Cheng K.; Jacobson A. E.; Rice K. C. Probes for narcotic receptor mediated phenomena. 34. Synthesis and structure-activity relationships of a potent mu-agonist delta-antagonist and an exceedingly potent antinociceptive in the enantiomeric C9-substituted 5-(3-hydroxyphenyl)-N-phenylethylmorphan series. J. Med. Chem. 2007, 50, 3765–3776. 10.1021/jm061325e. [DOI] [PubMed] [Google Scholar]
- Janecka A.; Fichna J.; Janecki T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17. 10.2174/1568026043451618. [DOI] [PubMed] [Google Scholar]
- Huang P.; Kehner G. B.; Cowan A.; Liu-Chen L.-Y. Comparison of Pharmacological Activities of Buprenorphine and Norbuprenorphine: Norbuprenorphine Is a Potent Opioid Agonist. J. Pharmacol. Exp. Ther. 2001, 297, 688–695. [PubMed] [Google Scholar]
- Linz K.; Christoph T.; Tzschentke T. M.; Koch T.; Schiene K.; Gautrois M.; Schroder W.; Kogel B. Y.; Beier H.; Englberger W.; Schunk S.; De Vry J.; Jahnel U.; Frosch S. Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J. Pharmacol. Exp. Ther. 2014, 349, 535–548. 10.1124/jpet.114.213694. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Agnes R. S. Conformation-activity relationships of opioid peptides with selective activities at opioid receptors. Biopolymers 1999, 51, 391–410. . [DOI] [PubMed] [Google Scholar]
- Gentilucci L.; Tolomelli A. Recent advances in the investigation of the bioactive conformation of peptides active at the micro-opioid receptor. conformational analysis of endomorphins. Curr. Top. Med. Chem. 2004, 4, 105–121. 10.2174/1568026043451627. [DOI] [PubMed] [Google Scholar]
- Schiller P. W.; Nguyen T. M.; Chung N. N.; Lemieux C. Dermorphin analogues carrying an increased positive net charge in their “message” domain display extremely high mu opioid receptor selectivity. J. Med. Chem. 1989, 32, 698–703. 10.1021/jm00123a035. [DOI] [PubMed] [Google Scholar]
- Handa B. K.; Land A. C.; Lord J. A.; Morgan B. A.; Rance M. J.; Smith C. F. Analogues of beta-LPH61-64 possessing selective agonist activity at mu-opiate receptors. Eur. J. Pharmacol. 1981, 70, 531–540. 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]
- Sasaki Y.; Matsui M.; Taguchi M.; Suzuki K.; Sakurada S.; Sato T.; Sakurada T.; Kisara K. d-Arg2-dermorphin Tetrapeptide Analogs: A Potent and Long-lasting Analgesic Activity after Subcutaneous Administration. Biochem. Biophys. Res. Commun. 1984, 120, 214–218. 10.1016/0006-291X(84)91435-9. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Feytens D.; Wachter R. D.; Vlaeminck M. D.; Marczak E. D.; Salvadori S.; Graaf C.; Rognan D.; Negri L.; Lattanzi R.; Lazarus L. H.; Tourwe D.; Balboni G. Conformationally constrained opioid ligands: the Dmt-Aba and Dmt-Aia versus Dmt-Tic scaffold. Bioorg. Med. Chem. Lett. 2009, 19, 433–437. 10.1016/j.bmcl.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman D. M.; Gidda J. S.; Cantrell B. E.; Schoepp D. D.; Johnson B. G.; Leander J. D. Discovery of a potent, peripherally selective trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist for the treatment of gastrointestinal motility disorders. J. Med. Chem. 1994, 37, 2262–2265. 10.1021/jm00041a003. [DOI] [PubMed] [Google Scholar]
- Neary P.; Delaney C. P. Alvimopan. Expert Opin. Invest. Drugs 2005, 14, 479–488. 10.1517/13543784.14.4.479. [DOI] [PubMed] [Google Scholar]
- Janecka A.; Kruszynski R. Conformationally restricted peptides as tools in opioid receptor studies. Curr. Med. Chem. 2005, 12, 471–481. 10.2174/0929867053362983. [DOI] [PubMed] [Google Scholar]
- Remesic M.; Lee Y. S.; Hruby V. J. Cyclic Opioid Peptides. Curr. Med. Chem. 2016, 23, 1288–1303. 10.2174/0929867323666160427123005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio J.; Nguyen T. M.; Lemieux C.; Schiller P. W. Synthesis and pharmacological characterization in vitro of cyclic enkephalin analogues: effect of conformational constraints on opiate receptor selectivity. J. Med. Chem. 1982, 25, 1432–1438. 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- Piekielna J.; Kluczyk A.; Gentilucci L.; Cerlesi M. C.; Calo G.; Tomboly C.; Lapinski K.; Janecki T.; Janecka A. Ring size in cyclic endomorphin-2 analogs modulates receptor binding affinity and selectivity. Org. Biomol. Chem. 2015, 13, 6039–6046. 10.1039/C5OB00565E. [DOI] [PubMed] [Google Scholar]
- Zadina J. E.; Nilges M. R.; Morgenweck J.; Zhang X.; Hackler L.; Fasold M. B. Endomorphin analog analgesics with reduced abuse liability, respiratory depression, motor impairment, tolerance, and glial activation relative to morphine. Neuropharmacology 2016, 105, 215–227. 10.1016/j.neuropharm.2015.12.024. [DOI] [PubMed] [Google Scholar]
- Vardanyan R. S.; Hruby V. J. Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014, 6, 385–412. 10.4155/fmc.13.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolten M. S.; Fishman J. B.; Chen J. C.; Carlson K. R. Etonitazene: an opioid selective for the mu receptor types. Life Sci. 1993, 52, PL199–203. 10.1016/0024-3205(93)90118-M. [DOI] [PubMed] [Google Scholar]
- Toll L.; Berzetei-Gurske I. P.; Polgar W. E.; Brandt S. R.; Adapa I. D.; Rodriguez L.; Schwartz R. W.; Haggart D.; O’Brien A.; White A.; Kennedy J. M.; Craymer K.; Farrington L.; Auh J. S. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res. Monogr. 1998, 178, 440–466. [PubMed] [Google Scholar]
- Mosberg H. I.; Yeomans L.; Harland A. A.; Bender A. M.; Sobczyk-Kojiro K.; Anand J. P.; Clark M. J.; Jutkiewicz E. M.; Traynor J. R. Opioid Peptidomimetics: Leads for the Design of Bioavailable Mixed Efficacy mu-Opioid Receptor (MOR) Agonist/delta-Opioid Receptor (DOR) Antagonist Ligands. J. Med. Chem. 2013, 56, 2139–2149. 10.1021/jm400050y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea C.; Sánchez E.; Cano C.; Jagerovic N.; MartÃn M. I. Analgesic activity and pharmacological characterization of N-[1-phenylpyrazol-3-yl]-N-[1-(2-phenethyl)-4-piperidyl] propenamide, a new opioid agonist acting peripherally. Eur. J. Pharmacol. 2008, 595, 22–29. 10.1016/j.ejphar.2008.07.052. [DOI] [PubMed] [Google Scholar]
- Pinna G. A.; Cignarella G.; Loriga G.; Murineddu G.; Mussinu J. M.; Ruiu S.; Fadda P.; Fratta W. N-3(9)-arylpropenyl-N-9(3)-propionyl-3,9-diazabicyclo[3.3.1]nonanes as mu-opioid receptor agonists. Effects on mu-affinity of arylalkenyl chain modifications. Bioorg. Med. Chem. 2002, 10, 1929–1937. 10.1016/S0968-0896(01)00436-9. [DOI] [PubMed] [Google Scholar]
- Wentland M. P.; VanAlstine M.; Kucejko R.; Lou R.; Cohen D. J.; Parkhill A. L.; Bidlack J. M. Redefining the structure-activity relationships of 2,6-methano-3-benzazocines. 4. Opioid receptor binding properties of 8-[N-(4′-phenyl)-phenethyl)carboxamido] analogues of cyclazocine and ethylketocycalzocine. J. Med. Chem. 2006, 49, 5635–5639. 10.1021/jm060278n. [DOI] [PubMed] [Google Scholar]
- Wentland M. P.; Jo S.; Gargano J. M.; VanAlstine M. A.; Cohen D. J.; Bidlack J. M. Redefining the structure-activity relationships of 2,6-methano-3-benzazocines. Part 8. High affinity ligands for opioid receptors in the picomolar Ki range: oxygenated N-(2-[1,1′-biphenyl]-4-ylethyl) analogues of 8-CAC. Bioorg. Med. Chem. Lett. 2012, 22, 7340–7344. 10.1016/j.bmcl.2012.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accelrys. Accelrys Discovery Studio Visualizer. http://accelrys.com/products/discovery-studio/visualization.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.