

Graphical abstract

Protocol name: A practical synthesis of a novel DPAGT1 inhibitor, aminouridyl phenoxypiperidinbenzyl butanamide (APPB) for in vivo studies
Keywords: Selective dolichyl-phosphate, N-Acetylglucosaminephosphotransferase 1, DPAGT1 inhibitor, Gram-scale synthesis
Abstract
Immunotherapy that targets N-linked glycans has not yet been developed due in large part to the lack of specificity of N-linked glycans between normal and malignant cells. N-Glycan chains are synthesized by the sequential action of glycosyl transferases in the Golgi apparatus. It is an overwhelming task to discover drug-like inhibitors of glycosyl transferases that block the synthesis of specific branching processes in cancer cells, killing tumor cells selectively. It has long been known that N-glycan biosynthesis can be inhibited by disruption of the first committed enzyme, dolichyl-phosphate N-acetylglucosaminephosphotransferase 1 (DPAGT1). Selective DPAGT1 inhibitors have the promising therapeutic potential for certain solid cancers that require increased branching of N-linked glycans in their growth progressions. Recently, we discovered that an anti-Clostridium difficile molecule, aminouridyl phenoxypiperidinbenzyl butanamide (APPB) showed DPAGT1 inhibitory activity with the IC50 value of 0.25 μM. It was confirmed that APPB inhibits N-glycosylation of β-catenin at 2.5 nM concentration. A sharp difference between APPB and tunicamycin was that the hemolytic activity of APPB is significantly attenuated (IC50 > 200 μM RBC). Water solubility of APPB is >350-times greater than that of tunicamycin (78.8 mg/mL for APPB, <0.2 mg/mL for tunicamycin). A novel DPAGT1 inhibitor, APPB selectively inhibits growth of the solid tumors (e.g. KB, LoVo, SK-OV-3, MDA-MB-432S, HCT116, Panc-1, and AsPC-1) at low μM concentrations, but does not inhibit growth of a leukemia cell (L1210) and the healthy cells (Vero and HPNE) at these concentrations. In vitro metabolic stability using rat liver microsomes indicated that a half-life (t1/2) of APPB is sufficiently long (>60 min) for in vivo studies (PK/PD, safety profiles, and in vivo efficacy) using animal models. We have refined all steps in the previously reported synthesis for APPB for larger-scale. This article summarizes protocols of gram-scale synthesis of APPB and its physicochemical data, and a convenient DPAGT1 assay.
-
•
Remember that the abstract is what readers see first in electronic abstracting & indexing services.
-
•
This is the advertisement of your article. Make it interesting, and easy to be understood.
-
•
Be accurate and specific, keep it as brief as possible.
Specifications Table
| Subject Area: | Chemistry |
| More specific subject area: | Medicinal Chemistry |
| Protocol name: | A practical synthesis of a novel DPAGT1 Inhibitor, aminouridyl phenoxypiperidinbenzyl butanamide (APPB) for in vivo studies |
| Reagents/tools: | All were operated with standard tools available in general synthetic and biochemistry lab. |
| Experimental design: | All synthetic steps were demonstrated in gram-quantity. Selectivity of all asymmetric reactions is greater than 15:1 ratio. |
| Trial registration: | N/A |
| Ethics: | N/A |
Value of the Protocol
|
Description of protocol
Synthesis of a novel DPAGT1 inhibitor, aminouridyl phenoxypiperidinbenzyl butanamide (APPB, 1)
The monomethoxytetrachlorodiphenylmethoxymethyl (MTPM)-protected uridine 2 was prepared according to the previously reported procedure [1]. The primary alcohol of 2 was oxidized by a modified Swern condition to provide the corresponding aldehyde in quantitative yield, which was then subjected to Carreira’s asymmetric alkynation reaction using (−)-N-methylephedrine [2], yielding the (S)-propargyl alcohol 3 in 80% yield with selectivity of >98:2. NIS-AgBF4 promoted ribosylation of (S)-propargyl alcohol 3 with 4 furnished the β-riboside 5 exclusively in 95% yield. The azido group of 5 was reduced with Zn metal in the presence of aq. NH4Cl, and the triple bond was partially reduced with Lindlar’s catalyst. The generated free-amine was protected with (Boc)2O to furnish 6 in 64% overall yield. The alkene moiety of 6 was subjected to a two-step procedure (osmylation and oxidative cleavage with Pb(OAc)4), providing the crude aldehyde 7. Ti(OiPr)4-mediated Strecker reaction of 7 with the 4-aminobutanamide derivatives 8 provided the S-diasteromer 9S in 70% yield with greater than 15:1 S/R ratio. The desired diastereomer, 9S was subjected to hydration reaction with HgCl2-acetoaldoxime, furnishing the amide 10 in 83% overall yield. Global deprotection of 10 was performed in one-pot two step reaction using 30% TFA in CH2Cl2 followed by 80% TFA in H2O; the crude product was purified by DOWEX 50W x 4 ion exchange resin followed by preparative HPLC to furnish 1 in 88% overall yield (Scheme 1).
Scheme 1.

Synthesis of APPB (1).
General
All chemicals were purchased from commercial sources and used without further purification unless otherwise noted. THF, CH2Cl2, and DMF were purified via Innovative Technology's Pure-Solve System. All reactions were performed under nitrogen atmosphere. Reactions were monitored by TLC using 0.25 mm coated commercial silica gel plates (EMD, Silica Gel 60F254). TLC spots were visualized by UV light at 254 nm, or developed with ceric ammonium molybdate or anisaldehyde or copper sulfate or ninhydrin solutions by heating on a hot plate. Reactions were also monitored by using SHIMADZU LCMS-2020 with solvents: A: 0.1% formic acid in water, B: acetonitrile. Flash chromatography was performed with SiliCycle silica gel (Purasil 60 Å, 230–400 Mesh). 1H NMR spectral data were recorded on 400, and 500 MHz instruments. 13C NMR spectral data were recorded on 100 and 125 MHz instruments. For all NMR spectra, chemical shifts (δH, δC) were quoted in parts per million (ppm), and J values were quoted in Hz. 1H and 13C NMR spectra were calibrated with residual undeuterated solvent (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; CD3CN: δH = 1.94 ppm, δC = 1.32 ppm; CD3OD: δH = 3.31 ppm, δC = 49.00 ppm; DMSO-d6: δH = 2.50 ppm, δC = 39.52 ppm; D2O: δH = 4.79 ppm) as an internal reference. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, dd = double doublets, t = triplet, q = quartet, quin = quintet, hept = heptet, m = multiplet, br = broad. Infrared (IR) spectra were recorded on a Perkin-Elmer FT1600 spectrometer. HPLC analyses were performed with a Shimadzu LC-20AD HPLC system. HR-MS data were obtained from a Waters Synapt G2-Si (ion mobility mass spectrometer with nanoelectrospray ionization).
Synthetic procedure for 1

3,3-Dimethylpentane-1,5-diol (16): The title compound was synthesized according to the reported procedure [1,3]: TLC (hexanes/EtOAc 20:80) Rf = 0.20; IR (thin film) νmax = 3317 (br), 2955, 2934, 1676, 1469, 1366, 1030, 1006, 990 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.73 (t, J = 7.0 Hz, 4H), 2.04 (brs, 2H), 1.57 (t, J = 7.0 Hz, 4H), 0.94 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 59.60 (2C), 44.06 (2C), 31.67, 28.08 (2C); HRMS (ESI+) m/z calcd for C7H16O2 132.1150, found 132.1144.

5-Hydroxy-3,3-dimethylpentyl acetate (17): To a stirred solution of 16 (47.5 g, 359.3 mmol) in CH2Cl2 (500 mL) were added pyridine (31.8 mL, 395.2 mmol), Ac2O (33.9 mL, 359.3 mmol) and DMAP (0.44 g, 3.59 mmol) at 0 °C. The reaction mixture was stirred for 12 h at rt, and all volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes/EtOAc 90:10 to 50:50) to gave 17 (26.3 g, 150.9 mmol, 42%): TLC (hexanes/EtOAc 67:33) Rf = 0.20; 1H NMR (400 MHz, Chloroform-d) δ 4.13 (t, J = 7.5 Hz, 2H), 3.72 (t, J = 7.5 Hz, 2H), 2.04 (s, 3H), 1.57 (dt, J = 14.8, 7.5 Hz, 4H), 0.95 (s, 6H); HRMS (ESI+) m/z calcd for C9H18O3 174.1256, found 174.1249.

3,3-Dimethyl-5-((triisopropylsilyl)oxy)pentyl acetate (18): To a stirred solution of 17 (26.3 g, 150.9 mmol) and imidazole (20.6 g, 301.8 mmol) in dry CH2Cl2 (500 mL) were added TIPSCl (48.4 mL, 226.4 mmol) and DMAP (0.18 g, 1.51 mmol) at 0 °C. The reaction mixture was warmed to rt and stirred for 16 h. The reaction was quenched with saturated NaHCO3 (aq.) and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 97:3) to obtain 18 (49.4 g, 149.4 mmol, 99%): TLC (hexanes/EtOAc 90:10) Rf = 0.70; 1H NMR (400 MHz, Chloroform-d) δ 4.12 (t, J = 7.6 Hz, 2H), 3.74 (t, J = 7.2 Hz, 2H), 2.03 (s, 3H), 1.59 (t, J = 7.6 Hz, 2H), 1.53 (t, J = 7.2 Hz, 2H), 1.11–1.02 (m, 21H), 0.94 (s, 6H); HRMS (ESI+) m/z calcd for C18H39O3Si [M+H] 331.2668, found 331.2685.

3,3-Dimethyl-5-((triisopropylsilyl)oxy)pentan-1-ol (19): To a stirred solution of 18 (49.4 g, 149.4 mmol) in MeOH/THF (4:1, 300 mL) was added [tBu2Sn(OH)Cl]2 (0.86 g, 1.50 mmol). After 20 h at rt, all volatiles were evaporated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 95:5 to 90:10) to provide 19 (38.8 g, 134.5 mmol, 90%): TLC (hexanes/EtOAc 80:20) Rf = 0.40; IR (thin film) νmax = 3343 (br), 2941, 2891, 2866, 1463, 1384, 1366, 1096, 1065, 1012, 995, 881, 745, 678, 656 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.76 (t, J = 6.9 Hz, 2H), 3.72 (t, J = 7.2 Hz, 2H), 1.57 (td, J = 7.1, 2.8 Hz, 4H), 1.12–1.03 (m, 21H), 0.94 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 60.30, 59.85, 44.31, 31.67, 28.14 (2C), 18.05 (6C), 11.95 (3C); HRMS (ESI+) m/z calcd for C16H36O2Si 288.2485, found 288.2473.

3,3-Dimethyl-5-((triisopropylsilyl)oxy)pentanoic acid (15): To a stirred solution of 19 (38.8 g, 134.5 mmol) and TEMPO (1.05 g, 6.73 mmol) in MeCN (135 mL) an phosphate buffer (pH = 6.8, 135 mL) were added NaClO2 (14.6 g, 141.4 mmol) and bleach (8.25%, 65 mL) at 35 °C. After being stirred for 4 h, the reaction mixture was extracted with EtOAc and combined organic phase was dried over Na2SO4 and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 90:10) to give 15 (40.7 g, 134.5 mmol, 100%) as an orange oil: TLC (hexanes/EtOAc 50:50) Rf = 0.50; IR (thin film) νmax = 2942, 2866, 1705, 1463, 1246, 1097, 996, 881, 738, 678 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.88 (t, J = 5.8 Hz, 2H), 2.38 (s, 2H), 1.71 (t, J = 5.8 Hz, 2H), 1.20–1.11 (m, 3H), 1.09 (s, 12H), 1.08 (s, 6H), 1.07 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 173. 9, 60.7, 46.8, 42.6, 32.4, 28.5 (2C), 17.9 (6C), 11.8 (3C); HRMS (ESI+) m/z calcd for C16H34O3NaSi [M+Na] 325.2175, found 325.2171.

(2R,3S,4S,5S)-2-(Acetoxymethyl)-5-(p-tolylthio)tetrahydrofuran-3,4-diyl diacetate (10): The title compound was synthesized according to the reported procedure [1]: TLC (hexanes/EtOAc 50:50) Rf = 0.60; [α]20D −0.411 (c = 0.51, CHCl3); IR (thin film) νmax = 1742, 1371, 1214, 1091, 1045, 1017, 899, 810 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 7.7 Hz, 2H), 5.25–5.22 (m, 1H), 5.21–5.17 (m, 2H), 4.26–4.20 (m, 2H), 4.07 (dd, J = 12.9, 5.5 Hz, 1H), 2.34 (s, 3H), 2.09 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.50, 169.63, 169.42, 138.80, 134.18 (2C), 129.81 (2C), 127.45, 87.95, 79.97, 73.67, 71.41, 63.46, 21.15, 20.75, 20.53 (2C); HRMS (ESI+) m/z calcd for C18H22O7NaS [M+Na] 405.0984, found: 405.0970.

(2R,3S,4S,5S)-2-(Hydroxymethyl)-5-(p-tolylthio)tetrahydrofuran-3,4-diyl diacetate (12): To a stirred solution of 10 (24.3 g, 62.8 mmol) in MeOH/THF (4:1, 300 mL) was added [tBu2Sn(OH)Cl]2 (0.72 g, 1.26 mmol). After 20 h at rt, all volatiles were evaporated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 67:33) to provide 11 (17.9 g, 52.7 mmol, 83%): TLC (hexanes/EtOAc 60:40) Rf = 0.40; [α]21D −0.298 (c = 1.37, CHCl3); IR (thin film) νmax = 3484 (br), 3021, 2924, 2877, 1746, 1493, 1432, 1373, 1239, 1222, 1102, 1093, 1046, 1017, 810 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.8 Hz, 2H), 7.16 (d, J = 7.8 Hz, 2H), 5.27 (d, J = 5.6 Hz, 1H), 5.24 (t, J = 4.6 Hz, 1H), 5.20 (d, J = 5.8 Hz, 1H), 4.13 (q, J = 3.7 Hz, 1H), 3.74 (dd, J = 12.3, 2.8 Hz, 1H), 3.58 (dd, J = 12.2, 3.2 Hz, 1H), 2.34 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.89, 169.39, 138.92, 133.88 (2C), 129.93 (2C), 127.45, 87.76, 83.46, 73.89, 71.40, 62.08, 21.17, 20.62, 20.57; HRMS (ESI+) m/z calcd for C16H21O6S [M+H] 341.1059, found 341.1075.

(2R,3S,4S,5S)-2-(Azidomethyl)-5-(p-tolylthio)tetrahydrofuran-3,4-diyl diacetate (13): To a stirred solution of 12 (17.9 g, 52.7 mmol) and PPh3 (27.6 g, 105.1 mmol) in dry toluene (100 mL) were added HN3 (1.0 M in toluene, 262.9 mL, 262.9 mmol) and DIAD (20.7 mL, 105.1 mmol). The reaction mixture was stirred for 8 h at rt, and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 80:20 to 70:30) to afford 13 (19.0 g, 52.0 mmol, 99%): TLC (hexanes/EtOAc 75:25) Rf = 0.40; [α]21D −0.899 (c = 3.93, CHCl3); IR (thin film) νmax = 3023, 2924, 2101, 1746, 1493, 1436, 1372, 1233, 1217, 1094, 1064, 1044, 1016, 965, 899, 810 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 7.9 Hz, 2H), 5.27 (d, J = 5.2 Hz, 1H), 5.19 (t, J = 5.3 Hz, 1H), 5.11 (t, J = 5.2 Hz, 1H), 4.15 (q, J = 5.0 Hz, 1H), 3.42 (d, J = 1.2 Hz, 1H), 3.41 (d, J = 2.2 Hz, 1H), 2.34 (s, 3H), 2.10 (s, 3H), 2.05 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.63, 169.35, 138.74, 133.86 (2C), 129.81 (2C), 127.60, 88.27, 80.97, 73.74, 71.73, 52.46, 21.14, 20.50, 20.49; HRMS (ESI+) m/z calcd for C16H20N3O5S [M+H] 366.1124, found: 366.1133.

(2R,3R,4S,5S)-2-(Azidomethyl)-5-(p-tolylthio)tetrahydrofuran-3,4-diol (13): To a stirred solution of 13 (19.0 g, 52.0 mmol) in MeOH (200 mL) was added K2CO3 (10.0 g, 72.5 mmol). After being stirred for 30 min, the reaction mixture was filtered and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 70:30 to 50:50) to afford 14 (12.9 g, 45.9 mmol, 88%): TLC (hexanes/EtOAc 33:67) Rf = 0.60; [α]21D −0.152 (c = 0.34, CHCl3); IR (thin film) νmax = 3385 (br), 2923, 2103, 1493, 1437, 1399, 1286, 1117, 1065, 1042, 1017, 809 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.7 Hz, 2H), 7.14 (d, J = 7.8 Hz, 2H), 5.19 (d, J = 4.7 Hz, 1H), 4.11 (t, J = 4.4 Hz, 1H), 4.04 (d, J = 3.7 Hz, 2H), 3.49 (dd, J = 13.0, 2.9 Hz, 1H), 3.42 (dd, J = 13.0, 4.2 Hz, 1H), 2.57 (brs, 2H), 2.34 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 138.34, 133.17 (2C), 129.82 (2C), 128.68, 90.76, 82.62, 74.88, 72.24, 52.68, 21.15; HRMS (ESI+) m/z calcd for C12H16N3O3S [M+H] 282.0912, found: 282.0928.

(2R,5S)-2-(Azidomethyl)-5-(p-tolylthio)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (4): To a stirred solution of 14 (12.9 g, 45.9 mmol) and 15 (34.7 g, 114.8 mmol) in CH2Cl2 (231 mL) were added DMAP (1.12 g, 9.17 mmol) and DIC (18.0 mL, 114.8 mmol) at 0 °C. The reaction mixture was stirred for 16 h at rt and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 95:5) to afford 4 (35.1 g, 41.2 mmol, 90%): TLC (hexanes/EtOAc 90:10) Rf = 0.60; [α]21D −0.293 (c = 1.39, CHCl3); IR (thin film) νmax = 2792, 2892, 2866, 2102, 1745, 1464, 1390, 1367, 1282, 1254, 1219, 1190, 1100, 1071, 1054, 1013, 998, 882, 809, 772, 742, 681 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 7.9 Hz, 2H), 5.26 (d, J = 5.3 Hz, 1H), 5.18 (t, J = 5.3 Hz, 1H), 5.11 (t, J = 5.0 Hz, 1H), 4.13 (q, J = 4.7 Hz, 1H), 3.76 (dt, J = 10.6, 6.9 Hz, 4H), 3.42 (d, J = 4.7 Hz, 2H), 2.34 (s, 3H), 2.31 (d, J = 10.6 Hz, 2H), 2.26 (d, J = 4.9 Hz, 2H), 1.61 (dtd, J = 17.4, 6.9, 2.1 Hz, 4H), 1.08–1.00 (m, 54H); 13C NMR (101 MHz, CDCl3) δ 170.91, 170.54, 138.67, 133.89 (2C), 129.81 (2C), 127.88, 88.58, 81.48, 73.52, 71.70, 60.02, 59.97, 52.70, 46.15, 46.03, 44.64, 44.55, 32.68, 32.60, 27.51, 27.47, 27.38, 21.17, 18.06 (6C), 18.05 (6C), 11.93 (3C), 11.92 (3C); HRMS (ESI+) m/z calcd for C44H79N3NaO7SSi2 [M+Na] 872.5075, found: 872.5088.

3-(((2,6-Dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-1-((3aR,4R,6R,6aR)-6-((S)-1-hydroxy-5-phenylpent-2-yn-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (3): Title compound was synthesized according to the reported procedure [1]: TLC (hexanes/EtOAc 50:50) Rf = 0.30; [α]22D −0.116 (c = 2.17, CHCl3); IR (thin film) νmax = 3387 (br), 2981, 2937, 1664, 1454, 1276, 1065, 1039, 856, 733, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.53 (ddd, J = 20.4, 8.5, 0.7 Hz, 1H), 7.35–7.27 (m, 4H), 7.24–7.15 (m, 4H), 6.85 (d, J = 5.1 Hz, 2H), 6.51 (d, J = 5.4 Hz, 1H), 5.68 (dd, J = 8.1, 4.1 Hz, 1H), 5.60–5.50 (m, 3H), 4.89–4.78 (m, 2H), 4.57 (ddt, J = 12.0, 4.3, 2.0 Hz, 1H), 4.24 (dd, J = 4.4, 3.1 Hz, 1H), 3.78 (d, J = 3.3 Hz, 3H), 2.83 (t, J = 7.5 Hz, 2H), 2.53 (td, J = 7.4, 2.0 Hz, 2H), 1.57 (s, 3H), 1.36 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 162.11, 162.08, 159.5, 150.87, 150.85, 141.1, 140.8, 140.30, 140.27, 136.9, 135.4, 135.3, 133.99, 133.95, 133.8, 133.6, 131.2, 129.4, 129.3, 128.41, 128.39, 126.4, 126.21, 126.18, 125.5, 125.4, 115.34, 115.32, 114.3, 114.2, 101.8, 101.7, 96.7, 96.4, 89.23, 89.19, 86.8, 86.7, 84.1, 84.0, 80.9, 69.5, 63.02, 62.99, 55.7, 34.72, 34.70, 27.2, 25.3, 20.87, 20.85; HRMS (ESI+) m/z calcd for C37H34N2O8NaCl4 [M+Na] 797.0967, found: 797.0994.

(2R,3R,4R,5R)-2-(Azidomethyl)-5-(((1S)-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-5-phenylpent-2-yn-1-yl)oxy)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (5). To a stirred suspension of 3 (5 g, 6.44 mmol), 4 (6.57 g, 7.73 mmol), MS3A (7.56 g) and SrCO3 (4.75 g, 32.2 mmol) in CH2Cl2 (260 mL) were added AgBF4 (0.63 g, 3.22 mmol) and NIS (1.88 g, 8.37 mmol) at 0 °C. After 24 h, the reaction mixture was added Et3N (2 mL) and passed through a silica gel pad (hexanes/EtOAc 1:1). The combined organic phase was concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 90:10 to 80:20 to 70:30) to afford 5 (9.19 g, 6.12 mmol, 95%): TLC (hexanes/EtOAc 67:33) Rf = 0.70; [α]21D +0.100 (c = 2.09, CHCl3); IR (thin film) νmax = 2942, 2866, 2102, 1743, 1724, 1675, 1456, 1278, 1218, 1099, 1070, 882, 772 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.54 (dd, J = 23.1, 8.5 Hz, 1H), 7.32–7.27 (m, 4H), 7.24–7.16 (m, 4H), 6.84 (d, J = 7.3 Hz, 2H), 6.51 (d, J = 3.7 Hz, 1H), 5.71–5.64 (m, 2H), 5.60–5.49 (m, 2H), 5.20–5.16 (m, 3H), 4.79 (ddd, J = 7.5, 6.5, 3.1 Hz, 1H), 4.64 (td, J = 5.9, 2.6 Hz, 1H), 4.57 (ddt, J = 11.4, 6.3, 1.9 Hz, 1H), 4.28 (dt, J = 6.2, 2.8 Hz, 1H), 4.19 (tt, J = 6.1, 3.0 Hz, 1H), 3.79–3.72 (m, 7H), 3.50 (ddd, J = 13.0, 7.6, 3.3 Hz, 1H), 3.35 (dd, J = 13.0, 3.4 Hz, 1H), 2.83 (t, J = 7.4 Hz, 2H), 2.55 (td, J = 7.4, 1.8 Hz, 2H), 2.29 (t, J = 1.6 Hz, 2H), 2.24 (dd, J = 5.1, 2.1 Hz, 2H), 1.62–1.55 (m, 7H), 1.36 (d, J = 2.0 Hz, 3H), 1.08–1.00 (m, 54H); 13C NMR (101 MHz, CDCl3) δ 175.6, 171.0, 170.9, 170.71, 170.70, 170.6, 162.2, 162.1, 159.5, 150.8, 150.7, 140.4, 140.19, 140.15, 140.13, 136.92, 136.91, 135.4, 135.3, 133.9, 133.8, 133.7, 131.2, 129.4, 129.3, 128.5 (2C), 128.4 (2C), 126.5, 126.4, 126.2, 126.1, 125.6, 125.5, 115.29, 115.25, 114.23, 114.22, 104.61, 104.55, 101.83, 101.82, 88.8, 88.2, 84.44, 84.35, 83.9, 81.4, 81.3, 80.6, 79.9, 76.5, 75.9, 75.8, 74.1, 71.8, 71.7, 71.4, 70.7, 69.6, 69.5, 68.9, 68.8, 59.97, 59.96, 55.7, 46.2, 46.0, 44.7, 44.6, 34.7, 34.51, 34.49, 32.7, 32.61, 32.57, 28.0, 27.38, 27.35, 27.3, 27.1, 25.34, 25.27, 20.9, 18.1 (12C), 11.9 (6C); HRMS (ESI+) m/z calcd for C74H106Cl4N5O15Si2 [M+H] 1500.5978, found: 1500.5992.

(2R,3R,4R,5R)-2-(Aminomethyl)-5-(((1S)-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-5-phenylpent-2-yn-1-yl)oxy)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (22): A suspended solution of 5 (7.03 g, 4.68 mmol), NH4Cl (7.50 g, 140.3 mmol) and Zn (9.17 g, 140.3 mmol) in EtOH/H2O (9:1, 50 mL) was stirred at 80 °C for 12 h and cooled to rt. The precipitates were filtered and the combined organic solution was concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 50:50 to CHCl3/MeOH 96:4) to afford the primary amine 22 (5.80 g, 3.93 mmol, 84%): TLC (CHCl3/MeOH 90:10) Rf = 0.60; [α]21D −0.013 (c = 1.35, CHCl3); IR (thin film) νmax = 2941, 2866, 1742, 1721, 1675, 1600, 1556, 1461, 1382, 1278, 1215, 1099, 1070, 1050, 999, 882 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1H), 7.50 (dd, J = 31.4, 8.4 Hz, 1H), 7.33–7.30 (m, 2H), 7.28 (d, J = 7.5 Hz, 3H), 7.24–7.15 (m, 5H), 6.85 (d, J = 9.5 Hz, 2H), 6.49 (d, J = 6.1 Hz, 1H), 5.75 (dd, J = 8.5, 1.9 Hz, 1H), 5.72–5.66 (m, 1H), 5.59–5.46 (m, 2H), 5.30 (d, J = 5.3 Hz, 1H), 5.22–5.13 (m, 2H), 4.82 (dt, J = 6.3, 3.1 Hz, 1H), 4.78 (d, J = 7.0 Hz, 1H), 4.65 (dd, J = 14.5, 7.6 Hz, 1H), 4.28 (dt, J = 7.4, 3.5 Hz, 1H), 4.17 (quin, J = 3.9 Hz, 1H), 3.87 (t, J = 5.8 Hz, 1H), 3.75 (dt, J = 15.3, 6.3 Hz, 6H), 3.14 (d, J = 13.6 Hz, 1H), 2.94–2.86 (m, 1H), 2.83 (t, J = 7.4 Hz, 2H), 2.55 (td, J = 7.2, 2.0 Hz, 2H), 2.35 (s, 1H), 2.30 (s, 2H), 2.25 (s, 2H), 2.23–2.17 (m, 1H), 1.70 (t, J = 5.9 Hz, 1H), 1.63–1.51 (m, 4H), 1.35 (d, J = 5.1 Hz, 2H), 1.25 (s, 1H), 1.12–0.95 (m, 51H); 13C NMR (101 MHz, CDCl3) δ 173.46, 171.73, 171.39, 170.66, 162.24, 159.45, 150.84, 140.16, 136.82, 135.21, 135.04, 134.04, 133.95, 133.75, 131.18, 131.16, 131.14, 129.40, 129.35, 128.52, 128.43 (2C), 128.40 (2C), 128.37, 126.42, 126.29, 126.16, 125.40, 125.26, 115.30, 115.24, 114.01, 101.82, 89.55, 84.49, 74.87, 70.13, 60.70, 59.93, 55.69, 47.00, 46.16, 45.95, 44.74, 44.64, 42.72, 34.53, 34.51, 32.62, 32.58, 32.33, 29.69, 28.47, 27.38, 27.34, 27.29, 27.03, 25.21, 25.19, 20.92, 18.04 (12C), 17.92, 11.87 (6C), 11.78; HRMS (ESI+) m/z calcd for C74H108Cl4N3O15Si2 [M+H] 1474.6073, found: 1475.6091.
(2R,3R,4R,5R)-2-(((tert-Butoxycarbonyl)amino)methyl)-5-(((1S,Z)-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-5-phenylpent-2-en-1-yl)oxy)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (6): To a stirred solution of 22 (5.80 g, 3.93 mmol) and quinoline (10 mL) in THF-MeOH (1:1, 200 mL) was added Lindlar catalyst (2.90 g). H2 gas was introduced and the reaction mixture was stirred under H2 atmosphere (1000 psi). After being stirred for 20 h, the reaction mixture was added Lindlar catalyst (2.90 g). The reaction mixture was stirred for 20 h under H2 atmosphere (1000 psi) at rt. The solution was filtered through Celite, concentrated in vacuo. The crude mixture was used for the next reaction without purification. To a stirred solution of the crude mixture in CH2Cl2 (40 mL) was Boc2O (1.29 g, 5.89 mmol). After being stirred for 12 h at rt, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic solution was washed with saturated aq. NaHCO3, dried over Na2SO4 and concentrated in vacuo. The crude mixture was passed through a silica gel pad (hexanes/EtOAc 80:20 to 70:30). to afford 6 (4.71 g, 2.99 mmol, 76%): TLC (hexanes/EtOAc 75:25) Rf = 0.40; [α]21D −0.015 (c = 0.86, CHCl3); IR (thin film) νmax = 3403 (br), 2957, 2941, 2866, 1720, 1675, 1600, 1556, 1507, 1456, 1382, 1367, 1278, 1247, 1218, 1161, 1100, 1071, 1049, 1013, 999, 882 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.54 (dd, J = 8.4, 6.7 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.30 (t, J = 2.1 Hz, 2H), 7.25–7.21 (m, 2H), 7.21–7.12 (m, 5H), 6.82 (d, J = 10.7 Hz, 2H), 6.51 (d, J = 13.6 Hz, 1H), 5.84–5.74 (m, 2H), 5.72 (d, J = 8.1 Hz, 1H), 5.62–5.50 (m, 2H), 5.47 (t, J = 8.0 Hz, 1H), 5.14 (t, J = 4.2 Hz, 1H), 5.07–4.97 (m, 2H), 4.90 (s, 1H), 4.75 (ddd, J = 24.4, 6.4, 2.0 Hz, 1H), 4.58–4.45 (m, 2H), 4.19 (dt, J = 8.4, 4.2 Hz, 1H), 4.01 (dt, J = 6.6, 4.3 Hz, 1H), 3.76 (d, J = 5.1 Hz, 4H), 3.75–3.70 (m, 4H), 3.32 (d, J = 5.0 Hz, 2H), 2.79–2.58 (m, 2H), 2.57–2.41 (m, 2H), 2.36–2.28 (m, 1H), 2.26–2.19 (m, 5H), 1.66–1.52 (m, 4H), 1.41 (s, 6H), 1.33 (d, J = 4.9 Hz, 2H), 1.05 (q, J = 2.7 Hz, 51H), 0.99 (dd, J = 9.6, 4.0 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 170.87, 159.38, 155.94, 150.85, 141.06, 136.87, 136.80, 135.57, 135.31, 135.27, 133.86, 133.66, 133.56, 131.23, 131.20, 129.29, 129.27, 128.52, 128.51, 128.37 (2C), 126.16, 126.15, 126.06, 126.03, 125.99, 125.64, 125.52, 125.46, 125.43, 115.24, 115.23, 114.17, 114.11, 84.61, 81.15, 81.03, 79.30, 79.25, 74.72, 74.29, 70.50, 69.81, 59.95, 59.91, 55.66, 55.65, 46.18, 46.17, 45.92, 44.80, 44.79, 41.64, 35.37, 35.34, 32.56, 32.55, 32.52, 32.50, 29.70, 28.34, 27.27, 27.24, 27.22, 27.10, 27.08, 25.25, 18.05 (12C), 17.88, 11.88 (6C), 11.74; HRMS (ESI+) m/z calcd for C79H118Cl4N3O17Si2 [M+H] 1576.6754, found: 1576.6771.

(4-(4-(4-(Trifluoromethoxy)phenoxy)piperidin-1-yl)phenyl)methanamine (20): The title compound was synthesized according to the reported procedure [5]: 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.2 Hz, 2H), 7.14 (d, J = 8.6 Hz, 2H), 6.97–6.87 (m, 4H), 4.43 (tt, J = 7.7, 3.8 Hz, 1H), 3.79 (s, 2H), 3.49 (ddd, J = 11.7, 7.2, 3.7 Hz, 2H), 3.09 (ddd, J = 12.2, 8.2, 3.6 Hz, 2H), 2.15–2.06 (m, 2H), 1.98–1.88 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 155.8, 150.2, 142.8, 134.6, 128.0 (2C), 122.5 (2C), 116.83 (2C), 116.76 (2C), 72.9, 46.9 (2C), 45.9, 30.4 (2C); HRMS (ESI+) m/z calcd for C19H22F3N2O2 [M+H] 367.1633, found 367.1628.

tert-Butyl (4-oxo-4-((4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)benzyl)amino)butyl)carbamate (21): To a stirred solution of 4-aminobutyric acid (2.50 g, 24.0 mmol) and NaHCO3 (6.00 g, 72.0 mmol) in THF-H2O (1:1, 24 mL) was added Boc2O (5.76 g, 26.4 mmol). After being stirred for 8 h at rt, the reaction mixture was quenched with 1N HCl and extracted with CHCl3. The combined organic solution was dried over Na2SO4 and concentrated in vacuo. To a stirred solution of the crude mixture, 20 (4.45 g, 12.14 mmol), NaHCO3 (5.10 g, 60.7 mmol) and Glyceroacetonide-Oxyma (5.54 g, 24.3 mmol) in DMF-H2O (9:1, 60 mL), was added EDCI (4.65 g, 24.3 mmol). After being stirred for 13 h at rt, the reaction mixture was quenched with H2O and extracted with EtOAc. The combined organic solution was washed with 1N HCl (aq.), saturated NaHCO3 (aq.), dried over Na2SO4 and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc 33:67 to 20:80) to afford 21 (5.35 g, 9.71 mmol, 80%) [4]: TLC (hexanes/EtOAc 20:80) Rf = 0.30; IR (thin film) νmax = 3303 (br), 2931, 1692, 1637, 1613, 1542, 1504, 1465, 1366, 1264, 1238, 1219, 1193, 1159, 1120, 1111, 1036, 918, 841, 827, 772 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.19 (dd, J = 8.4, 4.7 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 6.91 (dd, J = 9.1, 1.1 Hz, 4H), 6.22 (brs, 1H), 4.77 (brs, 1H), 4.72 (brs, 1H), 4.44 (tt, J = 7.2, 3.5 Hz, 1H), 4.35 (d, J = 5.6 Hz, 1H), 4.29 (d, J = 5.5 Hz, 1H), 3.48 (ddt, J = 11.6, 7.6, 3.8 Hz, 2H), 3.20–3.05 (m, 4H), 2.22 (t, J = 7.1 Hz, 2H), 2.18–2.07 (m, 2H), 1.98–1.89 (m, 2H), 1.81 (quin, J = 6.9 Hz, 2H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.31, 157.99, 156.40, 155.72, 155.70, 142.76, 128.90 (2C), 128.56 (2C), 122.52 (3C), 116.76 (3C), 79.32, 72.55, 46.87, 44.10, 43.12, 39.76, 33.69, 30.15, 28.38 (3C), 26.35; HRMS (ESI+) m/z calcd for C28H37F3N3O5 [M+H] 552.2685, found: 552.2701.

4-Amino-N-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)benzyl)butanamide (8): To a stirred solution of 22 (3.81 g, 6.99 mmol) in CH2Cl2 (10 mL) was added TFA (5 mL). The reaction mixture was stirred for 3 h at rt, and all volatile were evaporated in vacuo. The residue was neutralized with aq. NaHCO3 extracted with CHCl3. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude mixture of 8 was used for next reaction without purification.

(2R,3R,4R,5S)-2-(((tert-Butoxycarbonyl)amino)methyl)-5-(((1S)-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-2,3-dihydroxy-5-phenylpentyl)oxy)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (23): To a stirred solution of 6 (4.71 g, 2.99 mmol) and lepidine (2.37 mL, 17.9 mmol) in t-BuOH/THF/H2O (1:1:1, 180 mL) were added K2CO3 (2.06 g, 14.9 mmol), K3Fe(CN)6 (4.91 g, 14.9 mmol) and K2OsO4·2H2O (1.10 g, 2.99 mmol) at rt. After being stirred for 12 h, the reaction mixture were added K2CO3 (2.06 g, 14.9 mmol), K3Fe(CN)6 (4.91 g, 14.9 mmol) and K2OsO4·2H2O (1.10 g, 2.99 mmol). After 20 h, the reaction mixture was diluted with EtOAc and quenched with saturated aq. Na2SO3. The heterogeneous mixture was stirred for 30 min, and extracted with EtOAc. The combined organic solution was washed with 1N HCl, saturated aq. NaHCO3, dried over Na2SO4, and concentrated in vacuo. The crude mixture was passed through a silica gel pad (hexanes/EtOAc 75:25 to 50:50) to afford 23 (3.76 g, 2.33 mmol, 78%) as diastereomeric mixture. This mixture was used for next reaction without further purification. Data for less-polar diastereomer: TLC (hexanes/EtOAc 67:33) Rf = 0.30; [α]22D 0.210 (c = 1.62, CHCl3); IR (thin film) νmax = 3444 (br), 2941, 2866, 1741, 1719, 1675, 1600, 1556, 1457, 1382, 1367, 1278, 1249, 1216, 1160, 1098, 1070, 1049, 1013, 998, 882, 867, 754, 681 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.52 (dd, J = 8.4, 3.6 Hz, 1H), 7.31 (d, J = 2.0 Hz, 2H), 7.30–7.27 (m, 2H), 7.25–7.14 (m, 6H), 6.85 (d, J = 3.4 Hz, 2H), 6.50 (d, J = 5.9 Hz, 1H), 5.75 (dd, J = 17.6, 8.0 Hz, 1H), 5.63 (d, J = 22.1 Hz, 1H), 5.58–5.52 (m, 2H), 5.48 (d, J = 9.7 Hz, 1H), 5.21 (q, J = 7.3, 6.2 Hz, 2H), 5.11 (d, J = 6.8 Hz, 1H), 5.01 (dd, J = 8.4, 4.7 Hz, 1H), 4.85–4.78 (m, 2H), 4.25 (d, J = 5.6 Hz, 1H), 4.16 (dt, J = 8.6, 4.4 Hz, 1H), 4.03 (dd, J = 14.2, 5.1 Hz, 1H), 3.90 (d, J = 1.8 Hz, 1H), 3.78 (d, J = 1.8 Hz, 4H), 3.77–3.71 (m, 4H), 3.69–3.62 (m, 2H), 3.39–3.22 (m, 2H), 2.97–2.86 (m, 2H), 2.77–2.66 (m, 2H), 2.34–2.18 (m, 5H), 2.12–2.00 (m, 1H), 1.91–1.67 (m, 2H), 1.64–1.51 (m, 4H), 1.42 (s, 6H), 1.35 (d, J = 3.9 Hz, 3H), 1.13–0.99 (m, 41H), 0.99–0.94 (m, 6H), 0.86 (dtd, J = 9.1, 6.6, 2.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 170.85, 170.74, 170.70, 170.70, 162.10, 162.09, 159.44, 159.44, 156.05, 150.59, 150.53, 141.92, 141.89, 136.87, 136.84, 135.25, 135.09, 133.96, 133.77, 131.21, 131.17, 129.37, 129.32, 128.43 (2C), 128.38 (2C), 126.22, 126.14, 125.81, 125.36, 125.26, 115.27, 114.97, 80.36, 80.34, 79.85, 79.67, 79.58, 74.64, 74.62, 74.60, 73.82, 73.77, 73.72, 70.31, 70.31, 59.94, 59.90, 55.69, 46.13, 45.92, 44.72, 34.63, 34.50, 32.58, 32.57, 32.55, 32.54, 31.76, 29.69, 29.03, 28.35, 27.28, 27.22, 26.88, 25.32, 25.25, 20.68, 18.04 (1C), 11.88 (3C), 11.86 (3C), 11.43; HRMS (ESI+) m/z calcd for C79H120Cl4N3O19Si2 [M+H] 1610.6809, found: 1610.6827. Data for polar diastereomer: TLC (hexanes/EtOAc 67:33) Rf = 0.20; [α]22D 0.071 (c = 1.08, CHCl3); IR (thin film) νmax = 3413 (br), 2941, 2866, 1719, 1675, 1457, 1367, 1278, 1248, 1219, 1160, 1099, 1070, 1049, 882, 772 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.5 Hz, 1H), 7.31–7.29 (m, 2H), 7.28 (s, 2H), 7.24–7.11 (m, 6H), 6.84 (d, J = 1.4 Hz, 2H), 6.51 (d, J = 6.6 Hz, 1H), 5.90 (dd, J = 6.3, 2.7 Hz, 1H), 5.84 (t, J = 8.2 Hz, 1H), 5.61–5.41 (m, 2H), 5.23–5.10 (m, 2H), 5.04 (t, J = 5.8 Hz, 2H), 4.86–4.77 (m, 1H), 4.68 (ddd, J = 21.0, 6.3, 2.8 Hz, 1H), 4.57 (dt, J = 10.8, 3.8 Hz, 1H), 4.25–4.14 (m, 1H), 4.06–3.98 (m, 1H), 3.92–3.84 (m, 1H), 3.80–3.71 (m, 6H), 3.47–3.23 (m, 2H), 2.92–2.83 (m, 2H), 2.77–2.66 (m, 2H), 2.31–2.20 (m, 4H), 2.19–2.06 (m, 2H), 1.92–1.66 (m, 3H), 1.63–1.53 (m, 6H), 1.42 (d, J = 3.7 Hz, 2H), 1.36 (s, 6H), 1.10–0.94 (m, 50H), 0.91–0.81 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 171.04, 171.01, 170.93, 170.92, 162.00, 159.38, 150.79, 136.92, 136.91, 135.45, 131.30, 131.28, 129.29, 129.28, 128.46 (2C), 128.42 (2C), 126.09, 125.95, 125.93, 115.24, 81.03, 81.01, 79.95, 79.67, 75.03, 75.00, 74.98, 72.17, 70.38, 70.31, 69.52, 69.49, 59.95, 59.91, 55.69, 55.67, 46.13, 45.93, 44.86, 44.66, 35.27, 35.25, 34.64, 32.63, 32.59, 32.58, 31.95, 28.32, 27.38, 27.37, 27.36, 27.28, 27.27, 27.20, 26.89, 25.26, 18.05 (12C), 11.88 (3C), 11.87 (3C); HRMS (ESI+) m/z calcd for C79H120Cl4N3O19Si2 [M+H] 1610.6809, found: 1610.6831.
(2R,3R,4R,5S)-2-(((tert-Butoxycarbonyl)amino)methyl)-5-((1S,2R)-2-cyano-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-2-((4-oxo-4-((4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)benzyl)amino)butyl)amino)ethoxy)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (9): To a stirred suspension of 23 (3.76 g, 2.33 mmol) and NaHCO3 (0.98 g, 11.6 mmol) in CH2Cl2 (46.6 mL) was added Pb(OAc)4 (2.06 g, 4.66 mmol) at 0 °C. The reaction mixture was stirred for 2 h at 0 °C and quenched with saturated aq. NaHCO3, and extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude mixture of aldehyde 7 was used for the next reaction without purification. To a stirred solution of 7 (3.44 g, 2.33 mmol) and 8 (3.15 g, 6.99 mmol) in CH2Cl2 (30 mL) was added MS3A (7.5 g) followed by Ti(OiPr)4 (6.89 mL, 23.3 mmol). After 6 h, the reaction was added TMSCN (2.91 mL, 23.3 mmol) and stirred for 12 h at rt. After completion, the reaction mixture was quenched with saturated aq. NaHCO3, and extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 80:20 to 60:40) to afford 9S (3.15 g, 1.63 mmol, 70% for 2 steps): TLC (hexanes/EtOAc 50:50) Rf = 0.40; [α]21D +0.102 (c = 0.75, CHCl3); IR (thin film) νmax = 3342 (br), 2941, 2866, 1718, 1675, 1505, 1464, 1243, 1164, 1101, 1071, 883, 772, 688 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.49 (dd, J = 8.5, 4.3 Hz, 1H), 7.32 (d, J = 2.0 Hz, 1H), 7.22–7.11 (m, 7H), 6.94–6.88 (m, 5H), 6.86 (d, J = 6.5 Hz, 2H), 6.50 (d, J = 8.6 Hz, 1H), 6.25–6.16 (m, 1H), 5.73 (dd, J = 22.2, 8.0 Hz, 1H), 5.60 (t, J = 8.8 Hz, 1H), 5.56–5.41 (m, 3H), 5.21 (d, J = 4.4 Hz, 1H), 5.05–4.98 (m, 2H), 4.94–4.77 (m, 2H), 4.53–4.37 (m, 3H), 4.25–4.16 (m, 2H), 4.05–3.98 (m, 1H), 3.80–3.69 (m, 6H), 3.68–3.63 (m, 1H), 3.56 (dd, J = 17.3, 3.4 Hz, 1H), 3.48 (ddt, J = 11.6, 7.2, 4.0 Hz, 2H), 3.44–3.29 (m, 1H), 3.08 (dq, J = 9.5, 5.3, 4.8 Hz, 2H), 2.95 (dt, J = 11.4, 5.5 Hz, 1H), 2.47 (td, J = 12.0, 11.4, 5.7 Hz, 1H), 2.36–2.14 (m, 5H), 2.13–2.05 (m, 2H), 1.97–1.85 (m, 3H), 1.84–1.75 (m, 1H), 1.58 (t, J = 6.9 Hz, 2H), 1.55–1.50 (m, 4H), 1.40 (s, 9H), 1.33 (d, J = 4.8 Hz, 3H), 1.28–1.23 (m, 3H), 1.08–1.02 (m, 42H), 1.01 (s, 6H), 0.94 (d, J = 2.1 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 172.4, 171.0, 170.9, 159.5, 155.8, 150.9, 150.7, 142.8, 136.9, 136.8, 135.3, 135.1, 134.13, 134.05, 133.86, 133.85, 133.78, 131.2, 131.1, 129.42, 129.37, 129.0, 126.4, 126.2, 125.5, 125.2, 122.5 (2C), 121.8, 119.3, 118.4, 116.8 (2C), 116.6 (2C), 115.4, 115.3, 114.71, 114.66, 106.4, 102.3, 102.2, 84.8, 80.7, 80.6, 79.9, 79.8, 79.3, 76.2, 74.32, 74.30, 72.9, 60.38, 60.35, 60.0, 59.9, 55.72, 55.71, 52.0, 46.6, 46.2, 45.9, 44.84, 44.77, 42.99, 42.96, 42.4, 41.2, 33.53, 33.49, 32.6, 32.5, 30.3, 28.4, 27.3 (2C), 27.17, 27.16, 27.1, 25.4, 18.1 (12C), 14.2, 14.1, 11.91 (3C), 11.90 (3C); HRMS (ESI+) m/z calcd for C94H135Cl4F3N7O20Si2 [M+H] 1934.8007, found: 1934.8021.

(2S,3R,4R,5R)-2-((1S,2S)-3-Amino-1-((3aR,4R,6R,6aR)-6-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-3-oxo-2-((4-oxo-4-((4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)benzyl)amino)butyl)amino)propoxy)-5-(((tert-butoxycarbonyl)amino)methyl)tetrahydrofuran-3,4-diyl bis(3,3-dimethyl-5-((triisopropylsilyl)oxy)pentanoate) (10): To a stirred solution of 9S (3.15 g, 1.63 mmol) in EtOH/H2O (9:1, 10 mL) were added HgCl2 (0.89 g, 3.26 mmol) and acetaldoxime (0.99 mL, 16.3 mmol) at rt. After being stirred for 10 h at rt, the reaction mixture was concentrated under reduced pressure. The residue was quenched with saturated aq. NaHCO3, extracted with CHCl3. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (CHCl3/MeOH 99.5:0.5–99.2:0.8–98.8:1.2) to afford 10 (2.64 g, 1.35 mmol, 83%): TLC (CHCl3/MeOH 95:5) Rf = 0.30; [α]21D +0.144 (c = 0.53, CHCl3); IR (thin film) νmax = 3335 (br), 2940, 2866, 1719, 1676, 1505, 1464, 1367, 1242, 1162, 1101, 1070, 882, 681 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.53 (dd, J = 8.6, 5.1 Hz, 1H), 7.30 (s, 1H), 7.28–7.22 (m, 2H), 7.21–7.12 (m, 6H), 6.91 (d, J = 8.5 Hz, 4H), 6.86 (d, J = 2.6 Hz, 2H), 6.51 (d, J = 8.7 Hz, 1H), 5.94 (brs, 1H), 5.79–5.67 (m, 3H), 5.56–5.47 (m, 2H), 5.17 (brs, 1H), 5.06 (s, 1H), 4.96 (brs, 1H), 4.82–4.73 (m, 2H), 4.43 (tt, J = 7.8, 3.8 Hz, 1H), 4.39–4.28 (m, 3H), 4.21 (brs, 1H), 4.13 (brs, 1H), 3.78 (s, 3H), 3.73 (q, J = 7.4 Hz, 5H), 3.67 (brs, 1H), 3.48 (ddd, J = 11.7, 7.2, 3.7 Hz, 2H), 3.41–3.28 (m, 1H), 3.17 (s, 1H), 3.09 (ddd, J = 12.2, 8.2, 3.3 Hz, 2H), 2.80–2.60 (m, 2H), 2.38–2.15 (m, 7H), 2.13–2.05 (m, 2H), 1.93 (ddd, J = 12.8, 8.0, 3.7 Hz, 2H), 1.85–1.79 (m, 2H), 1.54 (s, 3H), 1.42 (s, 9H), 1.34 (s, 3H), 1.04 (d, J = 2.8 Hz, 42H), 1.01 (s, 6H), 0.96 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 162.1, 162.0, 159.6, 159.5, 156.2, 155.8, 150.9, 150.4, 142.80, 142.78, 136.88, 136.86, 135.23, 135.21, 133.9, 133.6, 131.33, 131.30, 131.29, 129.40, 129.37, 129.2, 129.1, 129.02, 128.98, 126.24, 126.22, 126.21, 125.40, 125.36, 124.5, 124.4, 123.20, 123.19, 122.5 (2C), 121.8, 120.1, 119.3, 116.8 (2C), 115.4, 80.4, 80.02, 79.99, 79.96, 79.95, 79.92, 79.87, 79.85, 79.83, 74.51, 74.50, 72.7, 70.4, 70.3, 69.5, 60.0, 59.9, 55.73, 55.72, 46.7, 46.19, 46.15, 46.13, 46.11, 46.10, 46.07, 46.0, 44.8, 34.7, 34.5, 32.61, 32.58, 30.2, 29.7, 29.64, 29.60, 28.50, 28.45, 28.42, 28.38, 28.34, 27.25 (2C), 27.19, 27.16, 25.31, 25.29, 25.27, 18.1 (12C), 14.1, 12.2, 11.9 (6C); HRMS (ESI+) m/z calcd for C94H137Cl4F3N7O21Si2 [M+H] 1952.8112, found: 1952.8098.

4-(((2S,3S)-1-Amino-3-(((2S,3R,4S,5R)-5-(aminomethyl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)-3-((2S,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-1-oxopropan-2-yl)amino)-N-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)benzyl)butanamide (1): To a stirred solution of 10 (2.64 g, 1.35 mmol) in CH2Cl2 (15 mL) was added TFA (10 mL). The reaction mixture was stirred for 3 h at rt, and all volatile were evaporated in vacuo. To a stirred solution of the crude mixture in H2O (5 mL) was added TFA (20 mL). The reaction mixture was stirred for 2 days at rt, and all volatile were evaporated in vacuo. The crude mixture was purified by DOWEX (50W x 4) ion exchange resin. The resin was washed with MeOH/H2O (4:1) and MeOH. The crude product (TFA salt) was dissolved in MeOH (10 mL) and absorbed on DOWEX (50W x 4): the crude 1 was not detected by TLC (CHCl3/MeOH/H2O/50% aqueous ammonia 56:42:7:3). The resins were washed with MeOH and eluted with MeOH/50% aqueous ammonia (10:1). The eluate was concentrated under reduced pressure and the resultant aqueous solution was lyophilized. The resulted mixture was purified by C18 reverse-phase HPLC [column: HYPERSIL GOLD™ (175 Å, 12 μm, 150 × 20 mm), solvents: 80:20 MeOH:0.05M NH4HCO3 in H2O, flow rate: 6.0 mL/min, UV: 254 nm, retention time: 14 min] to afford 1 (1.05 g, 1.19 mmol, 88%): TLC (n-butanol/ethanol/CHCl3/28% aqueous ammonia 4:7:2:7) Rf = 0.50; [α]21D +0.375 (c = 0.30, methanol); IR (thin film) νmax = 3352 (br), 2932, 1677, 1505, 1243, 1201, 1136, 801, 722 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.78 (d, J = 8.1 Hz, 1H), 7.18 (dd, J = 9.0, 3.5 Hz, 4H), 7.00 (dd, J = 16.0, 8.6 Hz, 4H), 5.77 (d, J = 2.9 Hz, 1H), 5.73 (d, J = 8.1 Hz, 1H), 5.14 (s, 1H), 4.57–4.50 (m, 1H), 4.28 (s, 2H), 4.22–4.13 (m, 3H), 4.10 (dd, J = 8.6, 4.4 Hz, 1H), 4.07–3.98 (m, 2H), 3.52–3.46 (m, 3H), 3.44 (d, J = 8.8 Hz, 1H), 3.17 (d, J = 13.0 Hz, 1H), 3.14–3.02 (m, 3H), 2.60 (ddq, J = 18.4, 11.8, 6.9 Hz, 2H), 2.29 (td, J = 7.3, 2.8 Hz, 2H), 2.12 (dd, J = 14.5, 5.6 Hz, 2H), 1.93–1.73 (m, 4H), 1.39–1.25 (m, 2H); 13C NMR (101 MHz, CD3OD) δ 175.6, 166.2, 157.6, 152.0, 142.6, 131.2, 129.6 (2C), 123.6 (2C), 118.11 (2C), 118.07 (2C), 110.5, 102.7, 92.3, 85.3, 81.4, 80.4, 76.5, 75.1, 74.1 (2C), 73.0, 71.3, 64.4, 43.7, 43.6, 34.7, 31.5, 26.9; HRMS (ESI+) m/z calcd for C39H51F3N7O13 [M+H] 882.3497, found: 882.3512 (Fig. 1).
Fig. 1.

HPLC analysis of 1.
Area % purity: 96.8%.
Conditions: column: Phenomenex Kinetex 5 μm XB-C18 100 Å 250 × 4.60 mm column, solvents: 85:15 MeOH:0.05M NH4HCO3 in water, UV: 254 nm, flow rate: 0.5 mL/min.
Preparation of HCl salt of 1

To a stirred solution of 1 (1.05 g, 1.19 mmol) in MeOH (50 mL) was added ice cold 1N HCl (23.8 mL, 23.8 mmol) dropwise. After being stirred for 1 h at rt, the solution was concentrated under reduced pressure and the resultant aqueous solution was lyophilized to give 1•HCl salt (Fig. 2).
Fig. 2.

Water solubility of 1•HCl in saline.
Determination of solubility of 1•HCl in 0.9% NaCl (saline)
A suspension of 1•HCl (4.0 mg) in 0.9% NaCl (30 μL) was stirred for 24 h, and the precipitate was separated by centrifugation at 10,000 × g for 5 min. The upper solution (1 μL) was analyzed via C18 reverse-phase HPLC [column: Kinetex (100 Å, 5 μm, 250 × 4.60 mm), solvents: 70:30 MeOH: 0.05 M NH4HCO3 aq., flow rate: 0.5 mL/min, UV: 254 nm, retention time: 12.0 min]. The area of the peak for 1 was quantified. The concentrations were determined via the HPLC intensity-concentration curves [[7], [8], [9]].
Determination of solubility of 1•HCl in PBS (pH7.4) buffer
A suspension of 1•HCl (3.8 mg) in phosphate buffered saline (pH 7.4, 30 μL) was stirred for 24 h, and the precipitate was separated by centrifugation at 10,000 × g for 5 min. The upper solution (1 μL) was analyzed via C18 reverse-phase HPLC [column: Kinetex (100 Å, 5 μm, 250 × 4.60 mm), solvents: 70:30 MeOH:0.05 M NH4HCO3 aq., flow rate: 0.5 mL/min, UV: 254 nm, retention time: 12.0 min]. The area of the peak for 1 was quantified. The concentrations were determined via the HPLC intensity-concentration curves [[7], [8], [9]] (Fig. 3).
Fig. 3.

Water solubility of 1•HCl in PBS (pH7.4).
Microsomal stability
Pooled Sprague-Dawley rat liver microsomes were purchased from Corning Life Sciences (Oneonta, NY, USA). Microsomes (20 mg/mL) were thawed on ice and diluted with PBS, potassium phosphate buffer (100 mM, pH: 7.4) at a 1:8 ratio in 1.5 mL Eppendorf tubes. Stock solutions of 1•HCl and verapamil (positive control) were made by diluting 10 mg/mL solutions. From the drug stock solution, 10 μL was diluted with 390 μL of buffer (0.1 mg/400 μL). The diluted microsomes (390 μL) were reacted with 10 μL of the diluted drug solution and allowed to equilibrate for 5 min while shaking at 440 rpm. NADPH (10 mg/200 μL; 1000× drug concentration) was used as a co-factor for this reaction, and 100 μL was added to the solution after equilibration. Ice cold methanol (200 μL) was used to quench the reaction mixture (50 μL aliquots) at 0, 5, 10, 20, 30, 45 and 60 min. The samples containing methanol was lyophilized to remove all volatiles. The residue was dissolved in 1N HCl aq. (10 μL) and MeOH (40 μL). The resulting solution (20 μL) was injected to LC–MS. MS solvent 90:10 acetonitrile/0.05% formic acid in water. Flow rate: 0.5 mL/min (Fig. 4).
Fig. 4.
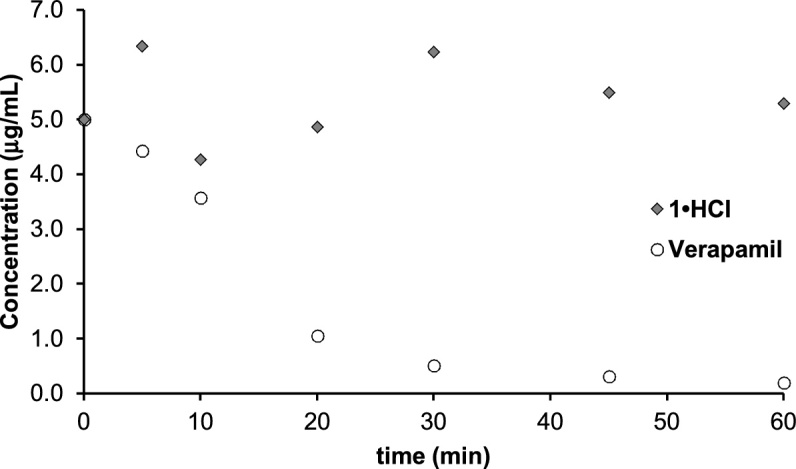
Microsomal stability of 1.
DPAGT1 assay
The enzymatic substrate, UDP-Glucosamine-C6-FITC was chemically synthesized according to the reported procedures [10]. DPAGT1 was expressed in suspended Expi293 cells for 36 h. The cells were lysed by drawing through a 26 g needle (10 times) and membrane protein was extracted using buffer containing 1% DM (decyl β-d-maltopyranoside) detergent. DPAGT1 was purified using HA (hemagglutinin)-agarose resin and a superdex 200 size exclusion column (Fig. 5).
Fig. 5.

DPAGT1-catalyzed reactions.
UDP-Glucosamine-C6-FITC (2 mM stock solution, 0.56 μL), MgCl2 (0.5 M, 4 μL), β-mercaptoethanol (50 mM, 5 μL), CHAPS (20%, 2.5 μL), Tris buffer (pH 8.0, 50 mM), C55-dolichyl phosphate (4 mM, 1.68 μL), and 1•HCl (0–50 μg/mL in Tris buffer) were place in a 500 μL Eppendorf tube. To a stirred reaction mixture, DPAGT1 solution (10 μL) was added (total volume of reaction mixture: 50 μL adjust with Tris buffer). The reaction mixture was incubated for 4 h at 37 °C and quenched with n-butanol (150 μL). Two phases were mixed via vortex and centrifuged at 10,000 × g for 3 min. The upper organic phase was assayed via reverse-phase HPLC. The organic phase (30 μL) was injected into HPLC (solvent: gradient elution of 85:15–95:5 MeOH/0.05 M aq. NH4HCO3 over 20 min; UV: 485 nm; flow rate: 0.5 mL/min; column: Kinetex 5 μm C8, 100 Å, 150 × 4.60 mm), and the area of the peak for C55-P-P-glucosamine-C6-FITC was quantified to obtain the IC50 value. The IC50 values were calculated from plots of the percentage product inhibition versus the inhibitor concentration (Fig. 6).
Fig. 6.

IC50 curve for APPB (1).
Acknowledgments
The National Institutes of Health is greatly acknowledged for financial support of this work (Grant GM114611). MK also thank University of Tennessee Health Science Center for generous financial support (CORNET award). NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.mex.2019.09.031.
Contributor Information
Katsuhiko Mitachi, Email: kmitachi@uthsc.edu.
Shou M. Kurosu, Email: kurosu@wisc.edu.
Cody D. Gillman, Email: gillmanc@caltech.edu.
Hyun Gi Yun, Email: hyun@caltech.edu.
William M. Clemons, Jr., Email: clemons@caltech.edu.
Michio Kurosu, Email: mkurosu@uthsc.edu.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- 1.Mitachi K., Aleiwi B.A., Schneider C.M., Siricilla S., Kurosu M. Stereocontrolled total synthesis of muraymycin D1 having a dual mode of action against Mycobacterium tuberculosis. J. Am. Chem. Soc. 2016;138:12975–12980. doi: 10.1021/jacs.6b07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitachi K., Yun H.-G., Kurosu S.M., Eslamimehr S., Lemieux M.R., Klaić L., Clemons W.M., Kurosu M. Novel FR-900493 analogs that inhibit outgrowth of Clostridium difficile spores. ACS Omega. 2018;3:1726–1739. doi: 10.1021/acsomega.7b01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y., Kurosu M. A new protecting group and linker for uridine ureido nitrogen. Tetrahedron. 2012;68:4797–4804. doi: 10.1016/j.tet.2012.03.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Wang Q., Wang Y., Kurosu M. A new oxyma derivative for nonracemizable amide-forming reactions in water. Org. Lett. 2012;14:3372–3375. doi: 10.1021/ol3013556. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y., Aleiwi B.A., Wang Q., Kurosu M. Selective esterifications of primary alcohols in a water-containing solvent. Org. Lett. 2012;14:4910–4913. doi: 10.1021/ol3022337. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aleiwi B.A., Mitahi K., Kurosu M. Mild and convenient N-formylation protocol in water-containing solvents. Tetrahedron Lett. 2013;54:2077–2081. doi: 10.1016/j.tetlet.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang S., Kim R.Y., Seo M.J., Lee S., Kim Y.M., Seo M., Seo J.J., Ko Y., Choi I., Jang J., Nam J., Park S., Kang H., Kim H.J., Kim J., Ahn S., Pethe K., Nam K., No Z., Kim J. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014;57:5293–5305. doi: 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- 7.Box K.J., Völgyi G., Baka E., Stuart M., Takács-Novák K., Comer J.E.A. Equilibrium versus kinetic measurements of aqueous solubility, and the ability of compounds to supersaturate in solution—a validation study. J. Pharm. Sci. 2006;95:1298–1307. doi: 10.1002/jps.20613. [DOI] [PubMed] [Google Scholar]
- 8.Baka E., Comer J.E.A., Takács-Novák K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 2008;46:335–341. doi: 10.1016/j.jpba.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 9.Völgyi G., Baka E., Box K.J., Comer J.E.A., Takács-Novák K. Study of pH-dependent solubility of organic bases. Revisit of Henderson-Hasselbalch relationship. Anal. Chim. Acta. 2010;673:40–46. doi: 10.1016/j.aca.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Mitachi K., Siricilla S., Yang D., Kong Y., Skorupinska-Tudek K., Swiezewska E., Kurosu M. Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of a new WecA inhibitor that kills non-replicating Mycobacterium tuberculosis. Anal. Biochem. 2016;512:78–90. doi: 10.1016/j.ab.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.