

Abstract
Although highly active antiretroviral therapy (HAART) can reduce HIV-1 viremia to levels that are below the limit of detection of clinical assays, the virus persists in reservoirs and trace levels of free virions can be found in the plasma. Whether this residual viremia represents ongoing cycles of replication continuing despite HAART or simply the release of virus from stable reservoirs has been controversial. Here we summarize the evidence that HAART can stop ongoing cycles of replication. The evidence comes from a detailed analysis of the residual viremia, which shows it to be archival and non-evolving in character. In addition, new pharmacodynamic measures incorporating a previous ignored slope parameter have provided the first real indication of how well HAART actually suppresses viral replication in vivo. Together, these results argue that the ultimate theoretical potential of HAART to control viral replication has already been reached. Progress towards eradication of the infection will require novel approaches to target the stable reservoirs that persist even when viral replication is completely halted.
Keywords: HAART, latency, reservoir, antiretroviral drug, residual viremia, slope, eradication
Introduction
Remarkable progress has been made in the treatment of HIV-1 infection. There are now 22 licensed antiretroviral drugs which, when used in combinations of three or more, can drive plasma virus levels down below the limit of detection of clinical assays (50 copies of HIV-1 RNA/ml of plasma) in the majority of patients1–3. In those who remain adherent, long term suppression of viremia appears possible.
Key insights into the ability of HAART to suppress viral replication came from classic quantitative studies of the initial response to HAART by Ho, Perelson, Shaw, Haase, and others3–8. These studies showed that there is a rapid, biphasic decay in the level of viremia following the initiation of therapy. The first phase involves the rapid decay of the cells that produce the vast majority of the plasma virus4,5,9. These appear to be activated CD4+ T cells9. In the productively infected state, these cells have a very short half-life, surviving only about 1 day before dying from viral cytopathic effects or host cytolytic effector mechanisms. When most of these cells have died, it is possible to observe a 2nd slower phase of decay3. This reflects the turnover of a minor population of virus-producing cells. These may be infected macrophages, which are more resistant to the cytopathic effects of infection10, or CD4+ T cells that are in a state of partial activation3,9. These cells turn over with a half-life of about 2 weeks. Importantly, this classic analysis of viral dynamics assumes that HAART stops all new rounds of infection, revealing the intrinsic decay rates of the different populations of cells infected before therapy was initiated.
The striking discovery that the vast majority of the plasma virus is produced by cells with relatively short half-lives led to the notion that eradication of HIV-1 infection with HAART might be possible. Perelson and colleagues pointed out that eradication would likely be dictated not simply by the size of an infected cell population but also by its half-life3. Using the measured half-life of the 2nd phase of decay, they predicted that eradication might be possible after 2–3 years of continuous treatment3. However, this optimistic assessment was followed shortly by the demonstration that HIV-1 could persist in a latent form in resting CD4+ T cells even in patients who had an undetectable viral load on HAART11–15. These latently infected cells constitute a viral reservoir, defined as a cell type or anatomical site in which a replication-competent form of the virus persists with more stable kinetics than the main pool of actively replicating virus. During the past decade, a great deal has been learned about this reservoir (for reviews see references 16–18), and evidence is accumulating for additional reservoirs including cells in the monocyte-macrophage lineage and free virus bound to follicular dendritic cells19–21. A further blow to the eradication hypothesis came from the discovery that low levels of free virus were present in the plasma even in patients on HAART who had consistent suppression of viremia to <50 copies/ml22. More recently, this residual viremia (RV) has been quantified down to 1 copy/ml using extremely sensitive RT-PCR assays23, 24, and it now appears that HAART reduces viremia to a new steady state level that is on the order of 1–5 copies/ml in most patients.
Understanding the nature of this RV is a major problem in HIV-1 research. Although the presence of RV was originally taken as evidence for ongoing cycles of replication in the setting of suppressive HAART22, an alternative hypothesis deserves consideration (Figure 1). We have hypothesized that the RV reflects production of virus from stable reservoirs, such as the latent reservoir in resting CD4+ T cells, without additional cycles of replication. The virus produced by latently infected cells that become activated is detectable in the plasma as HIV-1 RNA, but is prevented from completing new rounds of infection because of the inhibitory effects of the drugs. Distinguishing between these two hypotheses is important for several reasons. If RV reflects ongoing cycles of replication, then intensification of treatment may be needed to prevent the evolution of resistance and the reseeding of viral reservoirs. If, on the other hand, RV simply reflects the production of virus from stable reservoirs, then intensification of treatment is likely to be of little clinical benefit and progress towards eradication will be dependent upon finding ways to target viral reservoirs. In this article, we review the experimental evidence supporting the hypothesis that RV reflects the production of virus from stable reservoirs, including studies of the composition of the RV as well as striking new pharmacodynamic measures of the ability of antiretroviral drugs to inhibit viral replication. We conclude with an analysis of the treatment implications of these findings
Figure 1.
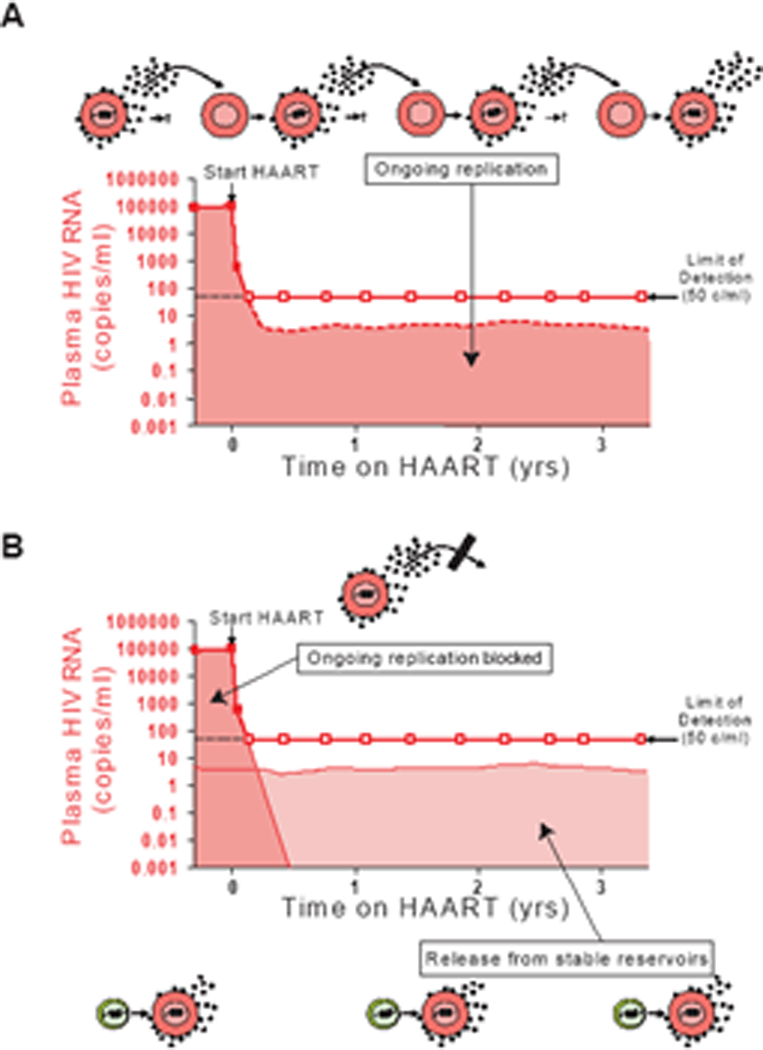
Two theories to RV in patients on HAART. (A) RV represents ongoing cycles of replication that continue at a lower level because of the suppressive effects of the drugs. (B) HAART stops all ongoing cycles replication, and the RV reflects release of virus from stable reservoirs such as the latent reservoir in resting CD4+ T cells.
Genetic analysis of RV
Our hypothesis predicts that the genetic composition of the RV should mirror that of the stable reservoirs that give rise to the RV. Direct sequence analysis of RV is challenging because of the extremely low amount of template viral RNA in the plasma of patients who are responding well to HAART. By ultracentrifugation of patient plasma and careful RT-PCR amplification of the pelleted virion RNA, it is possible to obtain sequences of plasma virus from patients who have <50 copies/ml of HIV-1 RNA20, 25–28. Special care must be taken to avoid PCR error and PCR resampling20. When this is done, a clear picture of the RV emerges. Overall, it is archival in character, and lacks new drug resistance mutations. In patients who were treatment naïve when starting a suppressive HAART regimen, the RV is composed of viruses without drug resistance mutations25,26. This is true even in patients who have been on HAART for several years, and even for patients whose regimens include drugs with a very low genetic barrier to resistance. Interestingly, the recommended initial HAART regimen consisting of tenofovir, emtricitabine (FTC), and efavirenz (EFV), a regimen which has never been beaten in head to head clinical trials, consists of two drugs (FTC and EFV) for which a single amino acid substitution can give high level resistance29,30. Yet in patients on this regimen as their initial treatment, the RV is devoid of resistance mutations. In patients who have developed drug resistance as a result of prior non-suppressive therapy and then gone on a suppressive salvage regimen, the RV consists of wild type viruses and viruses with archival drug resistance mutations, but no new resistance mutations25,27. Longitudinal studies of RV have shown a striking lack of evolutionary change20,27. The RV does not have a temporal structure consistent with ongoing evolution27. Thus, the RV is archival in character, non-evolving, and sensitive to drugs in the current regimen.
The same properties apply to the viruses present during the occasional “blips” that many patients experience28. These are transient elevations of viremia into the detectable range with a subsequent spontaneous return to levels below 50 copies/ml. The viruses present during these blips do not contain new drug resistance mutations28. In fact, most blips are not reproducible when the same blood sample is tested in independent approved laboratories. Rather, isolated blips of <200 copies/ml appear to represent normal biological and statistical fluctuation in the level of RV. Consistent with this notion is the work of Palmer and colleagues who quantified RV in large numbers of patients starting HAART. It now appears that the decline in viremia occurs in four phases31. After the two initial phases described above, a slow third phase brings the level of viremia down to a final fourth phase, a steady state level which does not show any measurable decay. Typical levels of steady state RV are 1–5 copies/ml with considerable patient to patient variation. This variation is correlated with pre-HAART viral loads, suggesting that the RV originates from long-lived viral reservoirs which are larger in patients who had higher levels of viral replication before starting HAART31. Palmer and colleagues have also shown that the level of RV is similar in patients on HAART regimens with different expected levels of antiviral activity, suggesting that all effective HAART regimens will suppress RV to the same average level, a level that is determined not by the regimen but by the size of the viral reservoirs that give rise to the RV24. In patients whose viral reservoirs are large, the steady state level of RV may be closer to 50 copies/ml, and as a result of random fluctuations, these patients experience more frequent blips while still maintaining durable control of viremia.
Phylogenetic studies suggest that the RV is in general similar to the viruses detected in resting CD4+ T cells with one exception mentioned below. Plasma and reservoir viruses are phylogenetically intermingled, and in some cases the same exact viral sequence can be found in the RV and in the latent reservoir in resting CD4+ T cells20. Of course, sequence similarity or identity cannot be used to prove the origin of the RV, but these results and the static and archival nature of the RV suggest that at least some of the RV is derived from latently infected cells that become activated.
Predominant Plasma Clones
Although the detection of viruses with identical sequences in both plasma and resting CD4+ T cells is consistent with the hypothesis that at least some of the RV is derived from latently infected cells, detailed phylogenetic analysis of sequences from some patients has provided evidence that another cell type also contributes to RV20. In ~50% of the patients studied, the majority of plasma sequences were identical to one another, representing a single branch of a complex phylogenetic tree of patient specific sequences (Figure 2). Interestingly, the predominant plasma sequence was profoundly underrepresented in resting CD4+ T cells. Because identical sequences were obtained by limiting dilution PCR from many separate blood samples, it is certain that these sequences represent a common sequence found in the plasma in vivo and not PCR resampling. Linkage between predominant pol and env sequences established that these predominant plasma sequences in fact represent predominant clonal species of virus (termed predominant plasma clones, PPC). In some patients, these PPCs are the most common sequence in the plasma. PPCs persist without sequence change for periods ranging from months to as long as 3 years. The cellular source of the PPCs remains unclear, but because PPC sequences are not found in resting CD4+ cells, the source may be another cell type. PPCs have not been previously described because few studies have examined the sequences of plasma virus in patients with plasma HIV-1 RNA levels below 50 copies/ml20,25–28. Interestingly, in a careful study of pediatric patients whose viral loads had risen transiently into the detectable range, Tobin et al. noted that some patients had multiple identical sequences in the analysis of both the RT and env genes32.
Figure 2.
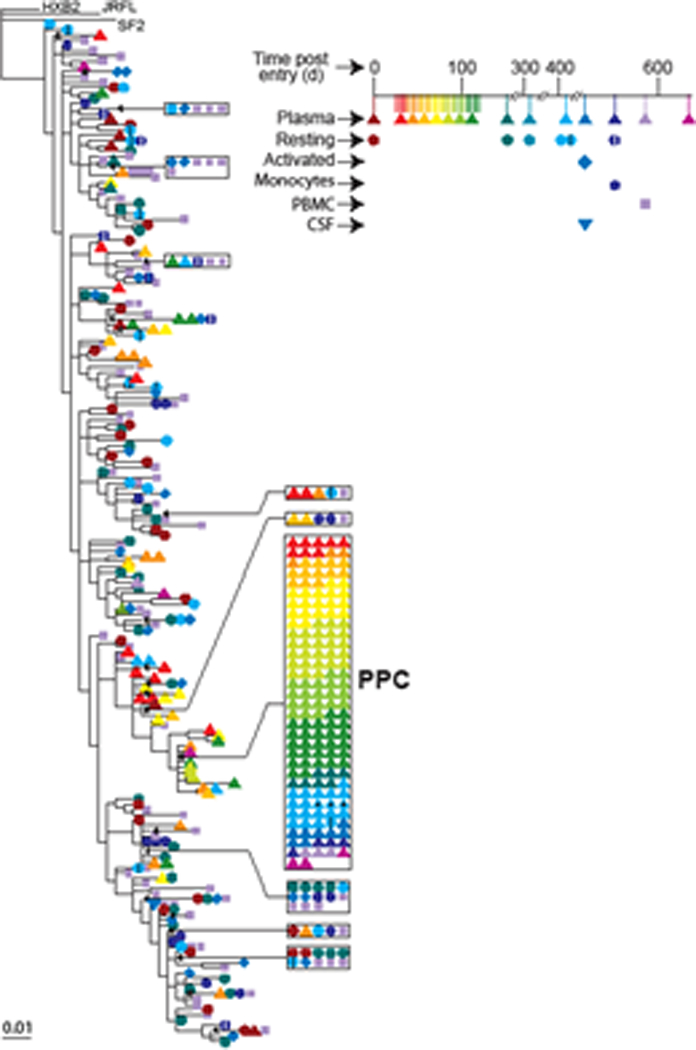
Phylogenetic analysis of plasma and reservoir sequences from a patient on HAART illustrating the presence of a predominant plasma clone (PPC). The figure shows a phylogenetic tree of pol gene sequences from free virus in the plasma (triangles) and CSF (inverted triangles) and from provirus in resting CD4+ T cells (circles), activated CD4+ T cells (diamonds), monocytes (hexagons), and unfractionated PBMC (squares). Colors indicate sampling times. Note the repeated isolation of a PPC sequence over a 2 year period. This same sequence was profoundly underrepresented among resting CD4+ T cells. For details, see reference 20.
We have advanced a hypothesis to explain the nature of the PPC. We suggest that a rare infection event establishes an integrated viral genome in a cell that has proliferative capacity, for example a stem cell in the monocyte-macrophage lineage. This infected cell proliferates, copying the viral genome without introducing errors and generating an expanded set of differentiated progeny cells that release virus. According to this hypothesis, the relevant cell type must be capable of both clonal expansion after infection and of low level virus production. The nature of the PPC suggests that infection of this cell type is inefficient and/or that only rarely does clonal expansion of this cell type reach the level where virus produced by the cell and its progeny predominate in the plasma.
Evidence for ongoing replication
Despite the above evidence that the RV is archival, non-evolving and in some cases oligoclonal, many investigators believe that ongoing cycles of viral replication continue even in patients who have suppression of viremia to <50 copies/ml. It is therefore worth reviewing the evidence supporting ongoing cycles of replication. Unfortunately, most of the assays used to analyze this question do not distinguish between continuing cycles of replication and continuous production of virus from stable reservoirs. For example, the mere detection of free virus in the plasma or of HIV-1 RNA positive cells in the tissues does not prove that ongoing cycles of replication are occurring. These findings can be equally well explained by the frequent activation of latently infected cells. In this situation, the cells may have been infected prior to the initiation of HAART. Following activation, they begin to transcribe the integrated provirus and produce virus particles. The viruses that are produced by these cells may not initiate new rounds of replication due to inhibition by the drugs, but they are nevertheless detectable as plasma HIV-1 RNA. Other proposed methods for detecting ongoing cycles of replication are equally problematic. The detection of 2LTR circles has been suggested as a marker for ongoing replication33,34, but we and others have shown that these circles are stable and do not necessarily indicate recent infection. Perhaps the most definitive way to demonstrate ongoing cycles of replication in patients on HAART is to document viral evolution. Studies of viral evolution in the setting of HAART have given variable results, with evolution detected in some but not other patients27,32,35,36. Unless extensive sampling is done, it is difficult to exclude the possibility that any newly observed variant was actually present previously and missed due to inadequate sampling. In addition, because the effectiveness of HAART varies among patients, this result is not surprising. The important point is that at one end of the spectrum there are always patients who show no evidence of evolution, suggesting that HAART can stop most or all of the ongoing cycles of replication, at least in some cases.
In summary, the extent to which viral replication continues in patients on HAART who have suppression of viremia to <50 copies/ml remains unclear. The existence of a stable latent reservoir means that there will always be RV even in the setting of a 100% effective HAART regimen that prevents any new rounds of infection. This reflects production of virus from cells that were infected before HAART was started, for example latently infected cells that become activated. We believe that the bulk of evidence supports the conclusion that ongoing cycles of viral replication are not the source of the bulk of the RV. Whether some extremely low level of ongoing replication persists in patients on HAART remains to be established.
Stability of the latent reservoir
The discovery that RV is dominated by PPCs in some patients on HAART also allowed us to address a long standing controversy regarding the stability of the latent reservoir in resting CD4+ T cells. Our initial longitudinal studies showed that the pool of latently infected cells harboring replication-competent HIV-1 has an extremely long half-life (44 months)37,38. We attributed this to the intrinsic stability of memory T cells, the cells which harbor latent HIV-1. However, it has been argued that the stability of the latent pool is the result of continual replenishment by ongoing cycles of new infection that continue despite HAART39. The presence of a PPC that is rare in resting CD4+ T cells allowed us to evaluate the rate at which new viral species enter the latent reservoir in patients on HAART. We are able to do this because the PPC has a unique sequence which serves as a label for viruses that are dominant in the plasma at a particular time point. It is then possible to ask whether these sequences appear in the latent reservoir at later time points as would be expected if the reservoir is continuously turning over. In general, we did not see the PPC entering the latent reservoir at later time points. These findings constrain the rate at which the viruses that comprise the RV can enter the reservoir. Using standard mathematical models for labeling of T cell compartments, we find that no more than 100 cells can be entering the reservoir per day in patients on HAART40. This presents less that 0.0001% of the total reservoir12. This low rate of entry argues against the idea that the reservoir is being continuously replenished by ongoing replication.
Pharmacodynamic explanation for the effectiveness of HAART
The arguments presented above suggest that in adherent patients, HAART suppresses all ongoing cycles of replication without affecting stable viral reservoirs. How is it that HAART can suppress replication so completely? Standard HAART regimens consist of three drugs, typically two nucleoside/nucleotide analogue reverse transcriptase inhibitors (NRTIs) and a third drug which may be either a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a protease inhibitor (PI)30,41. To understand how well HAART suppresses viral replication in vivo, it is important to quantitate how well individual antiretroviral drugs or drug combinations suppress viral replication at clinically relevant concentrations through in vitro infectivity assays. However, there is no widely accepted system for comparing the antiviral activity of different drugs at clinically relevant concentrations. A major advance in the field has been the development of single round infectivity assays42–45 which provide a more accurate measure of the instantaneous inhibition caused by a drug than do conventional multi-round assays46. A further improvement is the use of primary CD4+ T lymphoblasts as target cells for infection. These cells mimic the normal target cells for HIV-1 in vivo much more closely than the transformed cell lines used in many studies47.
Quantitative analysis of the activity of antiviral drugs also requires a mathematical model of the dose-effect relationship and a parameter to describe antiviral activity. One of the classic models is the median effect model developed by Chou48, which is based on the law of mass action and is closely related to the widely used sigmoidal Emax model49. The median effect model is given by the equations:
where fa is the fraction of infection events inhibited by a drug, fu is the fraction that are not inhibited (1- fa), D is the concentration of the drug, IC50 is the drug concentration that inhibits the maximum effect by 50%, and m is a slope parameter. This parameter is mathematically analogous to the Hill coefficient50, which represents the steepness of the dose-response curve. It is clear from the model that the degree of inhibition of infection by a given concentration of an antiretroviral drug is dependent on three factors: the drug concentration (D), the IC50, and the slope parameter m. IC50 is the most widely used parameter to compare drug potency. Another commonly used measure is the inhibitory quotient (IQ), the ratio between a clinically achievable concentration of the drug and its IC5051–53. Both of these indices neglect the slope parameter m. However, in a recent study47 using the single round infectivity assay, we showed that slope values are distinct for different classes of anti-HIV drugs and appear to be an intrinsic characteristic for each drug class. NRTIs and integrase inhibitors have slope close to 1, characteristic of non-cooperative reactions, whereas NNRTIs, PIs and fusion inhibitors surprisingly have slopes >1. The importance of this observation is that the slope parameter dramatically influences antiviral activity at clinically relevant concentrations. Thus it represents a critical missing dimension in the measurement of antiviral activity
To illustrate the importance of the slope parameter, we developed a new index, instantaneous inhibitory potential (IIP), to describe antiviral activity at clinically relevant drug concentrations (Figure 3). IIP equals to the log reduction in single round infectivity at clinically relevant concentrations. IIP is equivalent to log (1/fu) and can be calculated by the median effect equation. We found that the IIP values of approved antiretroviral drugs at clinically relevant concentrations varied in a class-dependent manner by over 9 logs! IIP values are strongly influenced by slope. Inhibition of single-round infectivity by more than 4 logs is observed only for drug classes that have high slope values. Thus the slope parameter sets intrinsic limitations on antiviral activity for drug classes. As a result of higher slope values, the NNRTI EFV and a subset of PIs produce >5 logs of inhibition at Cave. Most impressively, the PIs darunavir, indinavir and saquinavir, produce >9 logs of inhibition at Cmax. IIP values for NNRTIs and PIs are several logs greater than those observed for NRTIs, fusion inhibitors, or integrase inhibitors (Figure 3), which partially explains why the most effective initial HAART regimens invariably include an NNRTI or PI30. As a result of their long half lives, the NNRTI efavirenz and the PI darunavir can maintain IIP values above 5 logs even with several missed doses, which is consistent with their unrivaled success in clinical trials30.
Figure 3.
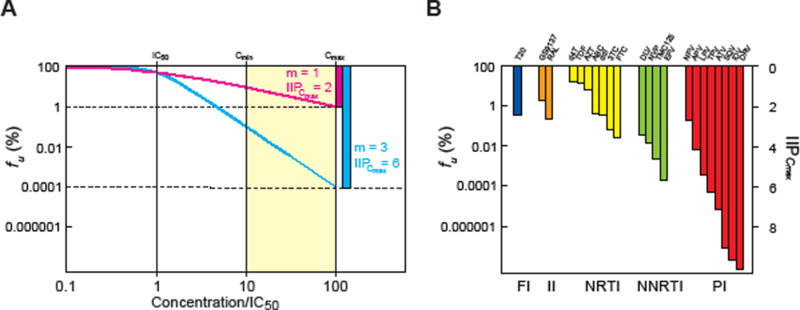
Clinical concentrations of some antiretroviral drugs cause profound inhibition of viral replication as a result of high slope values. (A) Hypothetical dose-response curves for two antiviral drugs with the same IC50 but different slope values. The response is plotted as fu, the percent of infection events that are unaffected by a given concentration of drug. For drugs with an m value of 1, the dose-response curves are shallow, and the degree of inhibition achieved in the clinical concentration range (shaded area, 10–100 times the IC50) is limited. For a drug with an m value of 3 and the same IC50, 10,000 fold greater inhibition is achieved at Cmax. (B) IIP of current antiretroviral drugs at Cmax plotted on the same scale as in (A). Note that for some PIs, IIP is on the order of 10 logs, a 10,000,000,000 fold inhibition of replication.
The discovery that the slope parameter is a crucial determinant of antiviral activity at clinical relevant concentrations provide fundamental new insights into how well HAART works and why effective HAART regimens tends to include a PI or NNRTI. These results further support the hypothesis that optimal HAART regimens stop all ongoing cycles of viral replication.
Treatment Implications
The studies described above suggest that we have may reached the theoretical limit of antiretroviral therapy – the complete arrest of ongoing cycles of replication. The RV that is observed in patients on HAART may be due in large part to the production of virus from stable reservoirs including the latent reservoir in resting CD4+ T cells and a second reservoir responsible for releasing the PPCs. We cannot exclude the possibility that some ongoing cycles of replication also contribute to the RV, although sequence evolution is not observed. If a substantial portion of the RV is due to virus production from stable reservoirs, then eradication of HIV-1 infection cannot be achieved by intensification of therapy. Rather, all of the stable reservoirs must be identified and methods for targeting each of them must be developed. Before this goal is achieved, we can at least offer patients the reassurance that modern HAART regimens, if taken correctly, can maintain suppression of viral replication indefinitely.
Acknowledgements
This work was supported by NIH grant 51178 and by the Howard Hughes Medical Institute.
Abbreviations:
- D
drug concentration
- DRV
darunavir
- EFV
efavirenz
- FTC
emtricitabine
- HAART
highly active antiretroviral therapy
- IC50
inhibitory concentration 50%
- IIP
instantaneous inhibitory potential
- IQ
inhibitory quotient
- LTR
long terminal repeat
- m
slope parameter
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- NRTI
nucleoside analogue reverse transcriptase inhibitor
- PCR
polymerase chain reaction
- PI
protease inhibitor
- PPC
predominant plasma clone
- RT
reverse transcriptase
- RV
residual viremia
References
- 1.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS clinical trials group 320 study team. N Engl J Med 1997. September 11;337(11):725–33. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med 1997. September 11;337(11):734–9. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997. May 8;387(6629):188–91. [DOI] [PubMed] [Google Scholar]
- 4.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995. January 12;373(6510):123–6. [DOI] [PubMed] [Google Scholar]
- 5.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995. January 12;373(6510):117–22. [DOI] [PubMed] [Google Scholar]
- 6.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996. March 15;271(5255):1582–6. [DOI] [PubMed] [Google Scholar]
- 7.Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 1996. November 8;274(5289):985–9. [DOI] [PubMed] [Google Scholar]
- 8.Cavert W, Notermans DW, Staskus K, Wietgrefe SW, Zupancic M, Gebhard K, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 1997. May 9;276(5314):960–4. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 1999. November 12;286(5443):1353–7. [DOI] [PubMed] [Google Scholar]
- 10.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 1986. July 11;233(4760):215–9. [DOI] [PubMed] [Google Scholar]
- 11.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat Med 1995. December;1(12):1284–90. [DOI] [PubMed] [Google Scholar]
- 12.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997. May 8;387(6629):183–8. [DOI] [PubMed] [Google Scholar]
- 13.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997. November 14;278(5341):1295–300. [DOI] [PubMed] [Google Scholar]
- 14.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997. November 14;278(5341):1291–5. [DOI] [PubMed] [Google Scholar]
- 15.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 1997. November 25;94(24):13193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. The multifactorial nature of HIV-1 latency. Trends Mol Med 2004. November;10(11):525–31. [DOI] [PubMed] [Google Scholar]
- 17.Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol 2007. February;5(2):95–106. [DOI] [PubMed] [Google Scholar]
- 18.Williams SA, Greene WC. Regulation of HIV-1 latency by T-cell activation. Cytokine 2007. July;39(1):63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, et al. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J Virol 2002. January;76(2):707–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol 2006. July;80(13):6441–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, et al. Characterization of the follicular dendritic cell reservoir of HIV-1. J Virol 2008. April 2. [DOI] [PMC free article] [PubMed]
- 22.Dornadula G, Zhang H, VanUitert B, Stern J, Livornese L Jr, Ingerman MJ, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999. November 3;282(17):1627–32. [DOI] [PubMed] [Google Scholar]
- 23.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 2003. October;41(10):4531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog 2007. April;3(4):e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermankova M, Ray SC, Ruff C, Powell-Davis M, Ingersoll R, D’Aquila RT, et al. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 2001. July 11;286(2):196–207. [DOI] [PubMed] [Google Scholar]
- 26.Persaud D, Siberry GK, Ahonkhai A, Kajdas J, Monie D, Hutton N, et al. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J Virol 2004. January;78(2):968–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, et al. Genotypic analysis of HIV-1 drug resistance at the limit of detection: Virus production without evolution in treated adults with undetectable HIV loads. J Infect Dis 2004. April 15;189(8):1452–65. [DOI] [PubMed] [Google Scholar]
- 28.Nettles RE, Kieffer TL, Kwon P, Monie D, Han Y, Parsons T, et al. Intermittent HIV-1 viremia (blips) and drug resistance in patients receiving HAART. JAMA 2005. February 16;293(7):817–29. [DOI] [PubMed] [Google Scholar]
- 29.Bartlett JA, Fath MJ, Demasi R, Hermes A, Quinn J, Mondou E, et al. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults. AIDS 2006. October 24;20(16):2051–64. [DOI] [PubMed] [Google Scholar]
- 30.Bartlett JG, Lane HC. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents . 2007. Available from: http://aidsinfo.nih.gov/contentfiles/AdultandAdolescentGL.pdf. [PubMed]
- 31.Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 2008. March 11 [cited 5/2/2008];105(10):3879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tobin NH, Learn GH, Holte SE, Wang Y, Melvin AJ, McKernan JL, et al. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: Expression of archival virus and replication of virus. J Virol 2005. August;79(15):9625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharkey ME, Teo I, Greenough T, Sharova N, Luzuriaga K, Sullivan JL, et al. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat Med 2000. January;6(1):76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharkey M, Triques K, Kuritzkes DR, Stevenson M. In vivo evidence for instability of episomal human immunodeficiency virus type 1 cDNA. J Virol 2005. April;79(8):5203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med 1999. May 27;340(21):1605–13. [DOI] [PubMed] [Google Scholar]
- 36.Frenkel LM, Wang Y, Learn GH, McKernan JL, Ellis GM, Mohan KM, et al. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J Virol 2003. May;77(10):5721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 1999. May;5(5):512–7. [DOI] [PubMed] [Google Scholar]
- 38.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 2003. June;9(6):727–8. [DOI] [PubMed] [Google Scholar]
- 39.Ramratnam B, Mittler JE, Zhang L, Boden D, Hurley A, Fang F, et al. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat Med 2000. January;6(1):82–5. [DOI] [PubMed] [Google Scholar]
- 40.Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog 2007. August 31;3(8):e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammer SM, Saag MS, Schechter M, Montaner JS, Schooley RT, Jacobsen DM, et al. Treatment for adult HIV infection: 2006 recommendations of the international AIDS society-USA panel. JAMA 2006. August 16;296(7):827–43. [DOI] [PubMed] [Google Scholar]
- 42.Kellam P, Larder BA. Recombinant virus assay: A rapid, phenotypic assay for assessment of drug susceptibility of human immunodeficiency virus type 1 isolates. Antimicrob Agents Chemother 1994. January;38(1):23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hertogs K, de Bethune MP, Miller V, Ivens T, Schel P, Van Cauwenberge A, et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob Agents Chemother 1998. February;42(2):269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob Agents Chemother 2000. April [cited 7/17/2007];44(4):920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol 2004. February;78(4):1718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferguson NM, Fraser C, Anderson RM. Viral dynamics and anti-viral pharmacodynamics: Rethinking in vitro measures of drug potency. Trends Pharmacol Sci 2001. February;22(2):97–100. [DOI] [PubMed] [Google Scholar]
- 47.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med 2008;In press. [DOI] [PMC free article] [PubMed]
- 48.Chou TC. Derivation and properties of michaelis-menten type and hill type equations for reference ligands. J Theor Biol 1976. July 7;59(2):253–76. [DOI] [PubMed] [Google Scholar]
- 49.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006. September;58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 50.Hill AV. The possible effects of the aggregation of the molecules of huemoglobin on its dissociation curves. J Physiol (London) 1910;40:iv–vii. [Google Scholar]
- 51.Ellner PD, Neu HC. The inhibitory quotient. A method for interpreting minimum inhibitory concentration data. JAMA 1981. October 2;246(14):1575–8. [DOI] [PubMed] [Google Scholar]
- 52.Neu HC, Ellner PD. The inhibitory quotient. Bull N Y Acad Med 1983. June;59(5):430–42. [PMC free article] [PubMed] [Google Scholar]
- 53.Morse GD, Catanzaro LM, Acosta EP. Clinical pharmacodynamics of HIV-1 protease inhibitors: Use of inhibitory quotients to optimise pharmacotherapy. Lancet Infect Dis 2006. April [cited 2/7/2008];6(4):215–25. [DOI] [PubMed] [Google Scholar]