

Abstract
Purpose
Very high-risk prostate cancer (PC) is associated with poor response to local and systemic treatments; however, few cases have been molecularly profiled. We studied clinical outcomes and molecular profiles of patients with clinically localized primary Gleason pattern 5 PC.
Patients and Methods
Clinicopathologic features, targeted somatic and germline sequencing, and PTEN, TP53, and ERG status by immunohistochemistry were assessed in patients undergoing surgery from 2005 to 2015; 60 consecutive patients were identified with Gleason score 5 + 4 = 9 or 5 + 5 = 10 PC after radical prostatectomy with available tissue and clinical follow-up. Clinicopathologic and genomic parameters were correlated with biochemical relapse, metastasis-free survival, time to castration resistance, and overall survival using Cox proportional hazards models.
Results
Of patients with somatic sequencing data and clinical follow-up, 34% had DNA repair gene mutations, including 22% (11 of 49) with homologous recombination and 12% (six of 49) with mismatch repair gene alterations. Homologous recombination mutations were germline in 82% (nine of 11) of patients. In addition, 33% (16 of 49) had TP53 mutation, and 51% (29 of 57) had PTEN loss. Overall, 43% developed metastasis, with a time to castration resistance of 12 months. On multivariable analysis of clinicopathologic variables, only ductal/intraductal histology (hazard ratio, 4.43; 95% CI, 1.76 to 11.15; P = .002) and seminal vesicle invasion (hazard ratio, 5.14; 95% CI, 1.83 to 14.47; P = .002) were associated with metastasis. Among genomic alterations, only TP53 mutation and PTEN loss were associated with metastasis on univariable analysis, and neither remained significant in multivariable analyses. These data are retrospective and hypothesis generating.
Conclusion
Potentially actionable homologous recombination and mismatch repair alterations are observed in a significant proportion of patients with very high-risk PC at the time of radical prostatectomy. These findings could inform the design of prospective trials in this patient population.
CONTEXT
Key Objective
We performed an integrated clinical and genomic analysis of prostate tumors with primary Gleason pattern 5 at radical prostatectomy to identify potentially actionable alterations.
Knowledge Generated
In this very high-risk patient subset, adverse pathologic findings and common molecular alterations, such as TP53 mutation, PTEN loss, and ERG expression, were associated with poor outcomes. Importantly, one third of patients had pathogenic mutations in DNA repair genes, including in the homologous repair and mismatch repair pathways.
Relevance
These data support the notion that aggressive subsets of primary prostate cancer are enriched for actionable genomic alterations and could inform the design of prospective clinical trials in this population.
INTRODUCTION
The National Comprehensive Cancer Network very high-risk prostate cancer (PC) subset includes patients with T3b to T4 disease or more than four biopsy cores with Gleason score 8 to 10 (grade group 4/5) or those presenting with Gleason score 5 + 5 = 10 or 5 + 4 = 9 (primary Gleason pattern 5 [PG5]; grade group 5).1 These patients have the poorest oncologic outcomes, with higher relapse rates, frequent development of distant metastasis, poor response to systemic treatments, and short survival.2-4 Previous studies have suggested that the proportion of Gleason pattern 5 tumor on biopsy or surgical specimen defines a subset with independent prognostic implications and distinct outcomes after local and systemic treatments.5-8 Although we have a wealth of data substantiating the poor prognosis of patients with predominant or PG5, we know surprisingly little about the molecular alterations in these poorly differentiated tumors. Among the 333 primary prostate tumors profiled for The Cancer Genome Atlas (TCGA), only eight patient cases (2%) had PG5.9 Because the prevalence of targetable alterations is relatively increased in other aggressive histologic subtypes of PC,10-12 it is critically important to fill in these gaps in our molecular characterization of this disease.
Evolving data on metastatic castration-resistant PC (mCRPC) have demonstrated that druggable genomic alterations are not uncommon. Patients with homologous recombination defects, accounting for 20% to 25%13,14 of patients with mCRPC, benefit from the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib.15 In addition, between 4% and 8%16,17 of patients with mCRPC have mismatch repair (MMR) –deficient tumors, which are associated with high mutational burden and striking responses to programmed death 1 (PD-1) inhibitors.18 Yet how these results in mCRPC might translate into treatment for clinically localized disease remains unclear. Several clinical trials evaluating perioperative therapies in patients with high-risk localized PC, including androgen-deprivation therapy (ADT),19 antiandrogens,20 and docetaxel,21 have failed to demonstrate relevant benefit. However, the utility of neoadjuvant and/or adjuvant PARP and PD-1 inhibitors has not yet been studied, in large part because of the relative rarity of these alterations in primary PC cohorts.
Here, we assembled a consecutive cohort of clinically localized prostate tumors with PG5 and long-term clinical follow-up after radical prostatectomy (RP). In the largest such cohort with molecular characterization to date to our knowledge, we find that PG5 is highly enriched with potentially targetable alterations in DNA repair pathway genes, providing a rationale for the design of adjuvant clinical trials in this population.
PATIENTS AND METHODS
Patient Cohort
The Johns Hopkins University Institutional Review Board approved this retrospective study. In the overall Johns Hopkins RP cohort from 1975 to 2018, PG5 (5 + 5 = 10 or 5 + 4 = 9) comprised 0.8% (197 of 24,917) of all patient cases. To maximize duration of oncologic follow-up with the most recent pathologic specimens possible to preserve DNA quality, we queried the pathology database for consecutive RPs with PG5 from 2005 to 2015 and identified 60 patient cases with available tissue and clinical follow-up (Appendix Fig A1).
Immunohistochemistry
Tissue microarrays were constructed as previously reported.11 Immunostaining for ERG, PTEN, and p53 status was performed using previously reported and genetically validated rabbit monoclonal antibodies and staining and scoring protocols.22-26
Macrodissection of Tumor Tissue for DNA Isolation
Formalin-fixed paraffin-embedded tissue blocks from the dominant tumor nodule were used to obtain 5 × 0.6 mm punches from regions with the highest percentage of Gleason pattern 5 tumor. Tumor was extracted from the dominant acinar carcinoma, whereas areas with a minor component of ductal or intraductal carcinoma (n = 9) were not intentionally sampled. DNA was extracted from formalin-fixed paraffin-embedded material as previously described.11
Next-Generation Tumor Sequencing
Targeted next-generation deep sequencing to evaluate 262 cancer-related genes at a 500× average depth was performed using UW-OncoPlex (University of Washington, Seattle, WA) as previously described.27 Reported alterations were limited to those deemed pathogenic or likely pathogenic in the ClinVar database. Germline mutations were inferred by expert molecular pathologist review through cross-referencing against the ClinVar database and by variant allele fraction in the context of tumor content, ploidy, and loss of heterozygosity status.28,29 Two patients (two of 49) did not have tumor DNA available for UW-OncoPlex but had prior somatic sequencing performed using a clinical platform for clinical care purposes (PGDx; Foundation Medicine, Cambridge, MA, and Baltimore, MD) and were included in the analysis. Patient cases in which tumor purity and/or DNA quality was not high enough to exclude false-negative results are listed in the Data Supplement.
Germline Sequencing
Altogether, 55% patient cases (27 of 49) underwent germline sequencing (Data Supplement). Of these, five underwent clinical-grade germline sequencing (Invitae Genetics, San Francisco, CA; Color Genomics, Burlingame, CA) from saliva samples, and the remaining 22 had undergone sequencing of benign seminal vesicle or leukocyte DNA performed as part of previously described studies.30
Clinical Outcome Analysis
Clinical end points were time to biochemical recurrence (BCR), defined by prostate-specific antigen (PSA) less than 0.2 ng/mL measured 6 to 13 weeks after RP, followed by a confirmatory test,31 and metastasis-free survival (MFS), defined as the time from RP to the first detection of distant metastasis on imaging or death resulting from any cause, whichever occurred first. Time to castration resistance (TCR), defined as the time from ADT to two consecutive elevations in PSA at least 4 weeks apart, and overall survival (OS), defined as the time from RP to death, were also assessed. Kaplan-Meier curves were used to visualize time-to-event data. Univariable and multivariable Cox proportional hazards models were used to estimate hazard ratios (HRs) and corresponding 95% CIs and test for the association of the variables with clinical outcomes. All statistical tests were two sided, with statistical significance set at P ≤ .05. Because this study was hypothesis generating, we did not perform corrections for multiple comparisons.
RESULTS
Baseline Characteristics
Demographic, clinical, and pathologic characteristics of the PG5 cohort (N = 60) at RP are listed in Table 1. Two patients received neoadjuvant ADT before surgery. At surgery, most patients had pathologic evidence of locally advanced disease, including non–organ-confined (pT3/T4) tumors (54 [90%] of 60), seminal vesicle invasion (SVI) (35 [58%] of 60), positive lymph nodes (16 [27%] of 60), and positive surgical margins (28 [47%] of 60). In addition, 15% of patients had a minor component of tumor with histologic features of ductal or intraductal carcinoma. Of the 37 patients who received either salvage or adjuvant radiation therapy, 27 (73%) also received adjuvant ADT. Also, of the 16 patients who had positive lymph nodes (pN+), 11 (69%) received adjuvant ADT.
TABLE 1.
Baseline Demographic, Clinical, and Pathologic Characteristics of Patient Cohort (N = 60)

Prevalence of Gene Mutations
Overall, 81.7% (49 of 60) of patients had available sequencing data on somatic genomic alterations (Data Supplement), and 35% (17 of 49) of these had at least one pathogenic mutation in a gene involved in DNA repair (Table 2; Data Supplement). The most common alterations were homologous recombination alterations, found in 22% (n = 11) of patients and including mutations in ATM (n = 4), BRCA2 (n = 3), CHEK2 (n = 2), BRCA1 (n = 1), and PALB2 (n = 1). Of these alterations, 82% (nine of 11) were inferred to be likely germline mutations. Overall, 55% (27 of 49) had germline sequencing results available, including six patients with presumed germline homologous recombination alterations on the basis of somatic sequencing, all of which were confirmed on germline sequencing (BRCA2, n = 3; BRCA1, CHEK2, and PALB2, n = 1 each). In addition to these alterations in homologous recombination genes, MMR deficiency was found in 12% (six of 49) of patients (all in MSH2 and included in our previous immunohistochemistry [IHC] study11).
TABLE 2.
Frequency of Genomic Alterations in Cohort

Other mutations common in PC were also identified (Data Supplement). Mutations in TP53 were identified in 33% of patients (16 of 49), with good correlation with p53 IHC results (Data Supplement). ERG and PTEN status were also generally concordant by sequencing and IHC in patient cases with high DNA quality and/or tumor purity (Data Supplement). ERG expression by IHC, indicating an underlying ERG gene rearrangement, was seen in 39% of patient cases (22 of 57). Of 57 patients who had tumors evaluated by IHC, 51% (29 of 57) had PTEN protein loss. FOXA1 mutations were seen in 12% (six of 49) of patients, and SPOP mutations were seen in 8% (four of 49) of patients. WNT pathway mutations were seen in 8% of patients, including three activating mutations in CTNNB1 and one inactivating mutation in APC. PIK3CA-activating mutations (p.H1047R) were seen in 6% (three of 49) of patients.
BCR
The median time to BCR in our cohort was 17.9 months (95% CI, 8.93 to 74.78 months). Several pathologic characteristics were associated with increased risk of BCR on univariable analysis, including SVI (Appendix Table A1; Appendix Fig A2A) and presence of ductal or intraductal histology (Appendix Table A1; Appendix Fig A2B). Multivariable analysis including all clinicopathologic variables corroborated these findings for SVI (HR, 4.41; 95% CI, 2.03 to 9.58; P < .01) and ductal or intraductal histology (HR, 4.58; 95% CI, 1.90 to 11.01; P < .001).
Among genomic alterations on univariable analysis, patients with TP53 mutations had a statistically significant higher risk of BCR compared with patients who had wild-type TP53 (HR, 2.61; 95% CI, 1.29 to 5.29; P = .008). Patients with PTEN loss (HR, 2.47; 95% CI, 1.25 to 4.86; P = .009) or ERG expression (HR, 2.25; 95% CI, 1.18 to 4.31; P = .014) by IHC also had a higher risk of BCR (Appendix Figs A2C and A2D; Table A2). No statistically significant differences were seen for other categories of genomic alterations, including homologous recombination and MMR defects. Multivariable models were fit separately for each genomic alteration, adjusting for ductal or intraductal histology and SVI; however, none of the genomic alterations remained significantly associated with BCR (Appendix Table A2).
MFS
Overall, 43% (26 of 60) of the cohort developed metastasis, with a median MFS of 86.4 months (95% CI, 40.7 months to not reached). Bone and lymph nodes were the most common sites of metastatic disease, seen in 77% and 73% of patients with metastasis, respectively. Visceral disease was seen in 31% of patients who developed metastasis, with liver (23%) and lung (19%) the most common sites. Among clinicopathologic factors on univariable analysis, presence of SVI (Table 3; Fig 1A) and ductal or intraductal histology (Table 3; Fig 1B), along with patient age, PSA, and nodal status, were both associated with increased risk of metastasis (Fig 1B). On multivariable analysis, including all clinicopathologic factors, only SVI (HR, 5.14; 95% CI, 1.83 to 14.47; P = .002) and ductal or intraductal histology (HR, 4.43; 95% CI, 1.76 to 11.15; P = .002) remained independently associated with risk of MFS, in addition to patient age when treated as a continuous variable (HR, 0.93 for each year of age; 95% CI, 0.88 to 0.98; P = .008; Table 3).
TABLE 3.
Association of Patient Characteristics With MFS (N = 60)

FIG 1.

Kaplan-Meier analysis of metastasis-free survival in primary Gleason pattern 5 cohort by (A) seminal vesicle invasion (SVI) status, (B) ductal or intraductal (DOI) histology status, (C) TP53 mutation status, and (D) PTEN protein status using immunohistochemistry. NR, not reached.
Among genomic alterations, presence of deleterious TP53 mutation (HR, 3.06; 95% CI, 1.26 to 7.43; P = .013) and PTEN loss (HR, 3.97; 95% CI, 1.65 to 9.53; P = .002) were associated with worse MFS on univariable analysis (Table 4; Figs 1C and 1D). However, no genomic alterations remained significant when multivariable models were fit separately for each genomic alteration adjusting for age, ductal or intraductal histology, and SVI (Table 4).
TABLE 4.
Association of Genomic Alternations With MFS (n = 49)

TCR
Thirty-four patients (56%) received ADT, with a median TCR of 12.0 months (95% CI, 9.59 to 30.98 months). Among pathologic variables, presence of positive surgical margins was the only variable statistically associated with shorter TCR on multivariable analysis (HR, 6.91; 95% CI, 2.3 to 20.77; P < .001). The only genomic alteration associated with TCR was presence of TP53 mutation (HR, 3.08; 95% CI, 1.2 to 7.91; P = .019), although this did not remain significant on multivariable analysis after adjusting for positive surgical margins (Appendix Table A3 and A4).
OS
The median OS of the whole cohort was 111.1 months (95% CI, 93.9 months to not reached). SVI (HR, 3.8; 95% CI, 1.06 to 13.64; P = .04) and positive surgical margins (HR, 3.91; 95% CI, 1.22 to 12.53; P = .04) were associated with OS on multivariable analysis. Presence of TP53 mutation was also statistically associated with worse OS (HR, 3.86; 95% CI, 1.25 to 11.9; P = .019) on univariable analysis, although this did not remain significant after adjusting for SVI and positive surgical margins on multivariable analysis (Tables 5 and 6).
TABLE 5.
Association of Patient Characteristics With OS (N = 60)

TABLE 6.
Association of Genomic Alterations With OS (n = 49)

DISCUSSION
To our knowledge, this is the first study to characterize, clinically and molecularly, a large cohort of patients with PG5 PC at RP. The primary objective of this study was to search for biomarkers that could enhance our ability to predict outcomes and inform choices of management strategies. We chose a subset of patients with established unfavorable clinical outcomes who were diagnosed before evidence of metastatic disease and received the same initial local treatment to minimize potential biases associated with extent of disease. Our findings show that adverse surgical pathologic features are commonly present at the time of initial diagnosis, suggesting a high biologic potential for early metastasis. Almost all patients had non–organ-confined disease, more than half had SVI, and a quarter had node-positive disease, demonstrating the aggressive behavior of these poorly differentiated tumors.
The prevalence of genomic abnormalities in the DNA repair pathway in clinically localized PG5 seems to be comparable to or perhaps even higher than that in patients with mCRPC. Pritchard et al13 showed that 12% of patients with mCRPC harbor germline mutations in genes responsible for DNA repair, and more than 20% of patients at the same stage have somatic mutations in these genes.14 Our study shows that more than one third of patients with PG5 have DNA repair pathogenic mutations, including 22% with mutations in homologous recombination genes and 12% in MMR genes. These results corroborate observations that pathogenic homologous recombination gene defects are enriched among men with higher-grade disease within the TCGA PC cohort.10 The rate of homologous recombination gene defects in PG5 PC in our study is notably higher than that within all of grade group 5 tumors in TCGA, likely because of the small number (2%) of PG5 tumors profiled within the TCGA cohort. Abnormalities in homologous recombination and MMR genes could have potential therapeutic implications for the treatment of all stages of PC, including the design of adjuvant and neoadjuvant trials in the PG5 population. In addition, our findings have important implications for the families of patients with very high-risk disease, given that 82% of patients with homologous recombination mutations in this study had apparent underlying germline mutations. These findings clearly support the recent National Comprehensive Cancer Network guidelines, which recommend genetic testing in all patients with high-risk or very high-risk PC.
Our results show that there are pathologic and molecular features that are useful for risk stratification after RP. Patients with SVI or ductal or intraductal histology have more than four times greater risk of metastasis, and TP53 mutation, PTEN loss, and ERG expression were associated with increased risk of biochemical failure and metastasis. Notably, our study validates current clinicopathologic variables used for risk stratification and suggests that pathologic findings after RP may be more significant for risk prediction than molecular alterations in multivariable models. However, where full pathologic data are not available (eg, in patients who do not undergo RP), molecular alterations could be of added value. Future studies should evaluate prospectively the utility of TP53 mutation and PTEN loss in well-designed biomarker-driven clinical trials cohorts, particularly in the biopsy setting. In addition, although we believe that our data are unique and informative, these results should be viewed as preliminary because of relatively the small numbers and multiplicity of clinical, pathologic, and molecular data points, which limit the power of the observations with regard to outcomes. It is likely that a prospective evaluation with additional numbers and follow-up time will allow for a more precise study of the prognostic significance of clinicopathologic and molecular parameters in this group of patients.
The pivotal role of ADT in PC with Gleason scores 9 to 10 has been called into question in recent years.8 A recent large retrospective study of patients undergoing external-beam radiation therapy in combination with ADT compared outcomes of patients with Gleason score 8 versus 9 to 10, suggesting that those in the higher Gleason group may derive less benefit than patients with Gleason score 8.8 Recent studies have shown that the addition of either docetaxel21,32 or abiraterone33,34 improves OS in the locally advanced or metastatic hormone-sensitive setting, with higher benefit among high-risk patients, including those with high disease burden,32 T3 to T4 disease,21,34 PSA greater than 40 ng/mL,21,34 and Gleason score 8 to 10.21,33,34 Our data suggest that compared with lower-grade tumors, PG5 tumors may be less dependent on androgens and potentially more dependent on alternative oncogenic drivers, including defects in DNA repair pathways.
Limitations of the study include the fact that it was retrospective, and clinical follow-up and DNA sequencing were available only for a subset of patients, although the clinicopathologic parameters of patients with and without follow-up and sequencing were not significantly different (Appendix Fig A1). Also, the quality of the archival DNA and tumor purity were not uniformly high, and false negatives at sequencing could not be excluded in all patient cases, suggesting that our results may actually underestimate the prevalence of DNA repair alterations among PG5 tumors. A final limitation is that we had germline DNA available only in approximately half of the total cohort, although it is notable we found no errors in inferred germline status based on somatic DNA sequencing.
In conclusion, we performed the first integrated clinical and genomic analysis to our knowledge of PG5 prostate tumors at RP, a subset that has not been well represented in other sequencing efforts. Clinical outcomes and response to conventional treatment are worse compared with lower-risk groups, and adverse pathologic findings and common molecular alterations such as TP53 mutation, PTEN loss, and ERG expression are associated with poor outcomes. The relatively high incidence of HRD and MMR deficiency observed herein could inform the design of multimodal clinical trials employing PARP inhibitors or PD-1/PD-1 ligand monoclonal antibodies in very high-risk patients with clinically localized disease. Prospective studies are necessary to further define the prognostic and predictive roles of molecular alterations in this patient population.
Appendix
TABLE A1.
Association of Patient Characteristics With BCR (N = 60)

TABLE A2.
Association of Genomic Alterations With BCR (n = 49)

TABLE A3.
Association of Patient Characteristics With Castration-Resistant Disease (n = 26)

TABLE A4.
Association of Genomic Alterations With Castration-Resistant Disease (n = 49)

FIG A1.
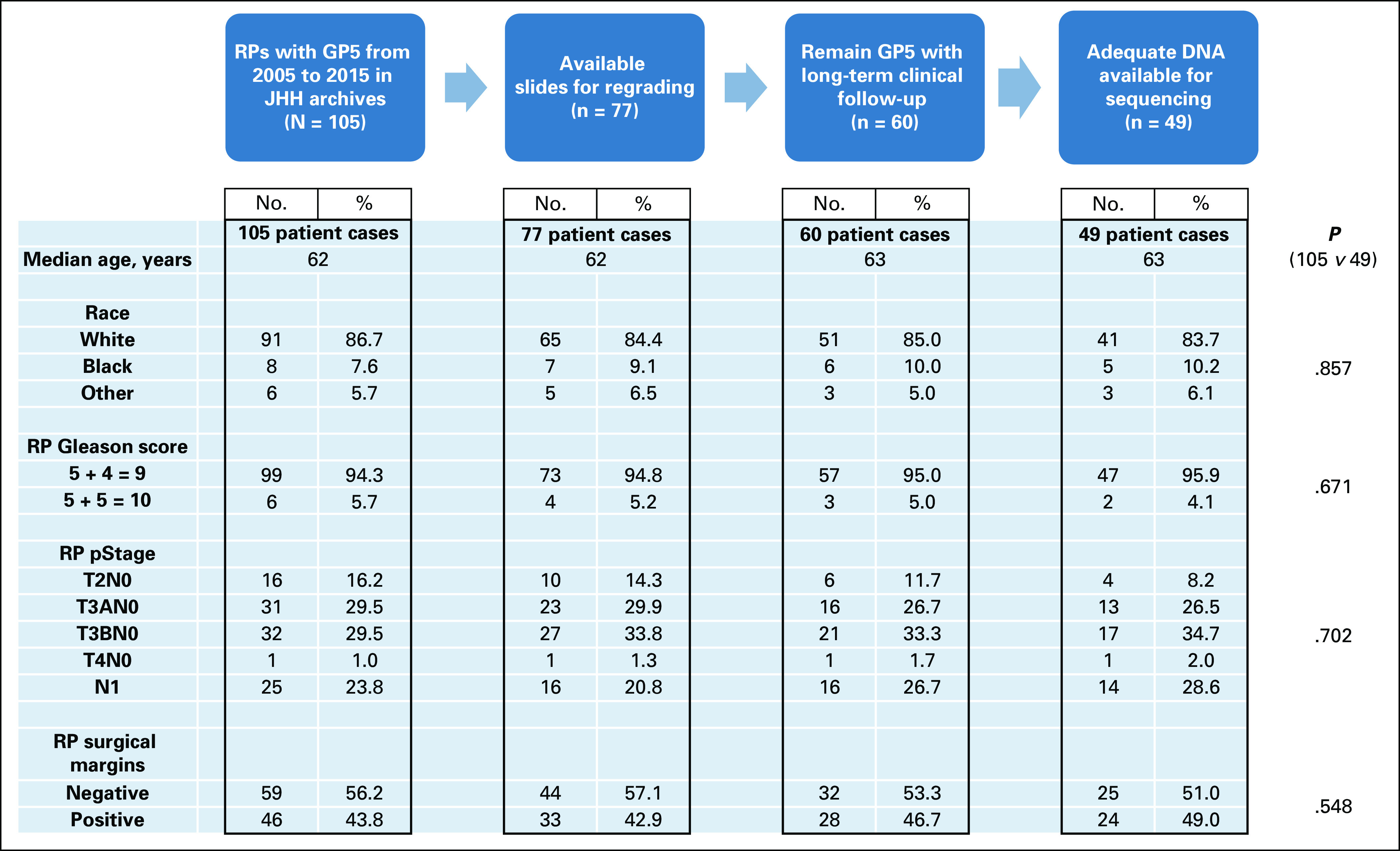
Pathology database query for consecutive radical prostatectomies (RPs) with primary Gleason score 5 (PG5) from 2005 to 2015, where 60 patient cases were identified with available tissue and clinical follow-up. JHH, Johns Hopkins Hospital.
FIG A2.

Kaplan-Meier analysis of biochemical recurrence–free survival in primary Gleason pattern 5 cohort by (A) seminal vesicle invasion (SVI) status, (B) ductal or intraductal (DOI) histology status, (C) p53 mutation status, and (D) PTEN protein status.
Footnotes
Supported by the Patrick Walsh Research Fund; by National Cancer Institute (NCI), National Institutes of Health, Prostate Specialized Program of Research Excellence Grant No. P50CA58236; and by NCI Cancer Center Support Grant No. 5P30CA006973-52.
AUTHOR CONTRIBUTIONS
Conception and design: Pedro Isaacsson Velho, Mark C. Markowski, Emmanuel S. Antonarakis, Mario A. Eisenberger, Tamara L. Lotan
Financial support: Colin C. Pritchard, Tamara L. Lotan
Administrative support: Tamara L. Lotan
Provision of study material or patients: Michael A. Carducci, Samuel R. Denmeade, Emmanuel S. Antonarakis
Collection and assembly of data: Pedro Isaacsson Velho, Jong Chul Park, Harsimar B. Kaur, Fawaz Almutairi, Samuel R. Denmeade, Mark C. Markowski, William B. Isaacs, Emmanuel S. Antonarakis, Colin C. Pritchard, Mario A. Eisenberger, Tamara L. Lotan
Data analysis and interpretation: Pedro Isaacsson Velho, David Lim, Hao Wang, Michael A. Carducci, Mark C. Markowski, Emmanuel S. Antonarakis, Colin C. Pritchard, Mario A. Eisenberger, Tamara L. Lotan
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Pedro Isaacsson Velho
Honoraria: Bayer HealthCare Pharmaceuticals
Speakers’ Bureau: AstraZeneca, Pfizer, Bristol-Myers Squibb
Research Funding: Bristol-Myers Squibb, Pfizer (Inst)
Expert Testimony: Bayer HealthCare Pharmaceuticals
Travel, Accommodations, Expenses: AstraZeneca, Astellas Pharma, Pfizer, Merck Serono, Merck
Michael A. Carducci
Consulting or Advisory Role: Astellas Pharma, AbbVie, Roche/Genentech, Pfizer, Foundation Medicine
Research Funding: Bristol-Myers Squibb (Inst), Pfizer (Inst), AstraZeneca (Inst), Gilead Sciences (Inst), EMD Serono (Inst), eFFECTOR Therapeutics (Inst)
Samuel R. Denmeade
Stock and Other Ownership Interests: Sophiris Bio
Consulting or Advisory Role: Sophiris Bio
Travel, Accommodations, Expenses: Sophiris Bio
Mark C. Markowski
Honoraria: Clovis, Exelixis
William B. Isaacs
Honoraria: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Emmanuel S. Antonarakis
Honoraria: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma, Merck, AstraZeneca, Clovis Oncology
Consulting or Advisory Role: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma, Merck, AstraZeneca, Clovis Oncology
Research Funding: Janssen Biotech (Inst), Johnson & Johnson (Inst), Sanofi (Inst), Dendreon (Inst), Aragon Pharmaceuticals (Inst), Exelixis (Inst), Millennium Pharmaceuticals (Inst), Genentech (Inst), Novartis (Inst), Astellas Pharma (Inst), Tokai Pharmaceuticals (Inst), Merck (Inst), AstraZeneca (Inst), Clovis Oncology (Inst), Constellation Pharmaceuticals (Inst)
Patents, Royalties, Other Intellectual Property: Coinventor of biomarker technology licensed to Qiagen
Travel, Accommodations, Expenses: Sanofi, Dendreon, Medivation
Mario A. Eisenberger
Leadership: Veru
Stock and Other Ownership Interests: Veru
Honoraria: Sanofi, Pfizer
Consulting or Advisory Role: Astellas Pharma, Ipsen, Bayer HealthCare Pharmaceuticals, Sanofi, Pfizer
Research Funding: Sanofi, Tokai Pharmaceuticals, Genentech
Travel, Accommodations, Expenses: Bayer HealthCare Pharmaceuticals, Astellas Pharma, Sanofi, Pfizer, Veru
Tamara L. Lotan
Consulting or Advisory Role: Janssen Oncology
Research Funding: Ventana Medical Systems
No other potential conflicts of interest were reported.
REFERENCES
- 1. Mohler JL, Lee RJ, Antonarakis ES, et al: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. Prostate Cancer 2018:151.
- 2.Epstein JI, Zelefsky MJ, Sjoberg DD, et al. A contemporary prostate cancer grading system: A validated alternative to the Gleason score. Eur Urol. 2016;69:428–435. doi: 10.1016/j.eururo.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. doi: 10.1002/cncr.31833. Sundi D, Tosoian JJ, Nyame YA, et al: Outcomes of very high-risk prostate cancer after radical prostatectomy: Validation study from 3 centers. Cancer 125:391-397, 2019. [DOI] [PubMed] [Google Scholar]
- 4.Sundi D, Wang VM, Pierorazio PM, et al. Very-high-risk localized prostate cancer: Definition and outcomes. Prostate Cancer Prostatic Dis. 2014;17:57–63. doi: 10.1038/pcan.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis CL, Partin AW, Han M, et al. Adenocarcinoma of the prostate with Gleason score 9-10 on core biopsy: Correlation with findings at radical prostatectomy and prognosis. J Urol. 2013;190:2068–2073. doi: 10.1016/j.juro.2013.05.056. [DOI] [PubMed] [Google Scholar]
- 6.Cheng L, Davidson DD, Lin H, et al. Percentage of Gleason pattern 4 and 5 predicts survival after radical prostatectomy. Cancer. 2007;110:1967–1972. doi: 10.1002/cncr.23004. [DOI] [PubMed] [Google Scholar]
- 7. doi: 10.1001/jamaoncol.2018.4836. Tilki D, Chen MH, Wu J, et al: Surgery vs radiotherapy in the management of biopsy Gleason score 9-10 prostate cancer and the risk of mortality. JAMA Oncol doi:10.1001/jamaoncol.2018.4836 [epub ahead of print on November 15, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. doi: 10.1016/j.eururo.2018.08.033. Yang DD, Mahal BA, Muralidhar V, et al: Androgen deprivation therapy and overall survival for Gleason 8 versus Gleason 9-10 prostate cancer. Eur Urol 75:35-41, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. doi: 10.1038/s41391-018-0086-1. Marshall CH, Fu W, Wang H, et al: Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: Association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis 22:59-65, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guedes LB, Antonarakis ES, Schweizer MT, et al. MSH2 loss in primary prostate cancer. Clin Cancer Res. 2017;23:6863–6874. doi: 10.1158/1078-0432.CCR-17-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isaacsson Velho P, Silberstein JL, Markowski MC, et al. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate. 2018;78:401–407. doi: 10.1002/pros.23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. doi: 10.1016/j.cell.2015.05.001. Robinson D, Van Allen EM, Wu YM, et al: Integrative clinical genomics of advanced prostate cancer. Cell 161:1215-1228, 2015 [Erratum: Cell 162:454, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. doi: 10.1016/j.eururo.2018.10.009. Antonarakis ES, Shaukat F, Isaacsson Velho P, et al: Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur Urol 75:378-382, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nava Rodrigues D, Rescigno P, Liu D, et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J Clin Invest. 2018;128:4441–4453. doi: 10.1172/JCI121924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powell IJ, Tangen CM, Miller GJ, et al. Neoadjuvant therapy before radical prostatectomy for clinical T3/T4 carcinoma of the prostate: 5-year followup, phase II Southwest Oncology Group Study 9109. J Urol. 2002;168:2016–2019. doi: 10.1016/S0022-5347(05)64285-1. [DOI] [PubMed] [Google Scholar]
- 20.Iversen P, McLeod DG, See WA, et al. Antiandrogen monotherapy in patients with localized or locally advanced prostate cancer: Final results from the bicalutamide early prostate cancer programme at a median follow-up of 9.7 years. BJU Int. 2010;105:1074–1081. doi: 10.1111/j.1464-410X.2010.09319.x. [DOI] [PubMed] [Google Scholar]
- 21.Vale CL, Burdett S, Rydzewska LHM, et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: A systematic review and meta-analyses of aggregate data. Lancet Oncol. 2016;17:243–256. doi: 10.1016/S1470-2045(15)00489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tosoian JJ, Almutairi F, Morais CL, et al. Prevalence and prognostic significance of PTEN loss in African-American and European-American men undergoing radical prostatectomy. Eur Urol. 2017;71:697–700. doi: 10.1016/j.eururo.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lotan TL, Wei W, Ludkovski O, et al. Analytic validation of a clinical-grade PTEN immunohistochemistry assay in prostate cancer by comparison with PTEN FISH. Mod Pathol. 2016;29:904–914. doi: 10.1038/modpathol.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaux A, Albadine R, Toubaji A, et al. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol. 2011;35:1014–1020. doi: 10.1097/PAS.0b013e31821e8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guedes LB, Almutairi F, Haffner MC, et al. Analytic, preanalytic, and clinical validation of p53 IHC for detection of TP53 missense mutation in prostate cancer. Clin Cancer Res. 2017;23:4693–4703. doi: 10.1158/1078-0432.CCR-17-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maughan BL, Guedes LB, Boucher K, et al. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018;21:260–268. doi: 10.1038/s41391-017-0027-4. [DOI] [PubMed] [Google Scholar]
- 27.Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirts BH, Casadei S, Jacobson AL, et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016;18:974–981. doi: 10.1038/gim.2015.212. [DOI] [PubMed] [Google Scholar]
- 30.Na R, Zheng SL, Han M, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–747. doi: 10.1016/j.eururo.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cookson MS, Aus G, Burnett AL, et al. Variation in the definition of biochemical recurrence in patients treated for localized prostate cancer: The American Urological Association Prostate Guidelines for Localized Prostate Cancer Update Panel report and recommendations for a standard in the reporting of surgical outcomes. J Urol. 2007;177:540–545. doi: 10.1016/j.juro.2006.10.097. [DOI] [PubMed] [Google Scholar]
- 32.Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373:737–746. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. 2017;377:352–360. doi: 10.1056/NEJMoa1704174. [DOI] [PubMed] [Google Scholar]
- 34.James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–351. doi: 10.1056/NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]