

Abstract
The ability to use soluble organic amine bases in Pd-catalyzed C–N cross-coupling reactions has provided a long-awaited solution to the many issues associated with employing traditional, heterogeneous reaction conditions. However, little is known about the precise function of these bases in the catalytic cycle and about the effect of variations in base structure on catalyst reactivity. We used 19F NMR to analyze the kinetic behavior of C–N coupling reactions facilitated by different organic bases. In the case of aniline coupling reactions employing DBU, the resting state was a DBU-bound oxidative addition complex, LPd(DBU)(Ar)X, and the reaction was found to be inhibited by base. In general, however, depending on the binding properties of the chosen organic base, increased concentration of the base can have a positive or negative influence on the reaction rate. Furthermore, the electronic nature of the aryl triflate employed in the reaction directly affects the reaction rate. The fastest reaction rates were observed with electronically neutral aryl triflates, while the slowest were observed with highly electron-rich and –deficient substrates. We propose a model in which the turnover-limiting step of the catalytic cycle depends on the relative nucleophilicity of the base compared to that of the amine. This hypothesis guided the discovery of new reaction conditions for the coupling of weakly binding amines, including secondary aryl amines, which were unreactive nucleophiles in our original protocol.
Graphical Abstract
INTRODUCTION
The capability to study and understand organometallic transformations through mechanistic analysis has enabled the development of efficient, practical, and robust synthetic methodologies.1 , 2 In particular, investigation of palladium-catalyzed carbon-nitrogen (C–N) bond forming reactions3 has produced more active catalysts and reliable protocols4 that have been widely applied in both academic and industrial settings.5 Many of these studies have focused on systems reliant on partially or almost completely insoluble inorganic bases, including metal alkoxide and carbonate bases, to facilitate the deprotonation step (Figure 1). As a result of this heterogeneity, it is difficult to accurately determine the base concentration and to quantify its effect on the reaction rate.6,7 In contrast, recently developed homogeneous systems employing soluble organic bases, including phosphazines, amidines, and guanidines,8 feature single-phase reaction mixtures that are potentially amenable to precise quantification of dissolved base. Specifically, our laboratory recently reported the use of the soluble organic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in the coupling of aryl amines, amides, and primary aliphatic amines with a variety of aryl (pseudo)halides.9 While the soluble organic bases have mitigated the operational issues and functional group incompatibilities associated with inorganic bases, little is known about their mechanistic role at the molecular level. Therefore, better understanding of these catalytic systems would provide fundamental information on this emerging area of homogenous catalysis.
Figure 1.
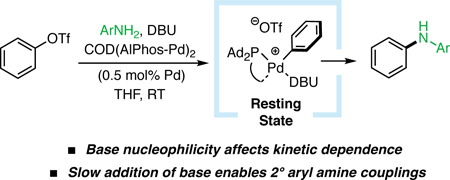
(A) The general catalytic cycle for the Pd-catalyzed coupling of aryl (pseudo)halides with amines: I, monoligated Pd(0) catalyst; II , oxidative addition (OA) complex; III, amine-bound OA complex; IV, amido complex. (B) Potential intermediates involving coordination of a base: V , base-bound OA complex; VI, base-bound/amine-bound OA complex.
The incorporation of nitrogen-containing bases in Pd-catalyzed cross-coupling reactions introduces many potential mechanisms for these Lewis-basic species to influence catalytic intermediates, depending on the inherent nucleophilicity and steric properties of these compounds 10 , 11 Previously, in alternative catalytic systems, the potential non-innocent nature of bases in C–N coupling has been proposed. For example, in a computational study of the coupling of bromobenzene and morpholine, Norrby and coworkers showed that DBU is able to stabilize catalytic intermediates (Figure 1, VI) by binding to the Pd center. 12 Mechanistic studies by Hartwig have uncovered relationships between the identity of differently substituted alkoxide bases, their concentration, and their effect on overall reaction rate in the coupling of aryl chlorides with N-methylbenzylamine.13 The ability to understand the effect that amine bases have on C–N cross-coupling would provide valuable fundamental mechanistic information that would also serve as a practical guide for reaction optimizations and rational determination of which base to employ. Herein, we report the first experimental mechanistic investigations into the effect of organic amine bases in Pd-catalyzed C–N coupling. Based on these findings, we are able to overcome certain limitations of our previously reported protocol by developing modified conditions that permit the coupling of secondary aryl amines with aryl triflates, a transformation that has not yet been facilitated by a weak, inexpensive organic amine base.14
RESULTS AND DISCUSSION
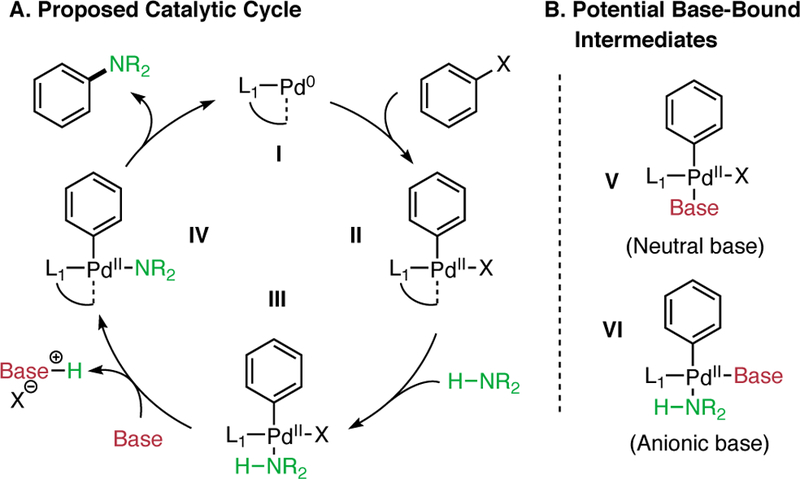
To probe the effect of organic bases in coupling reactions facilitated by an AlPhos-supported palladium catalyst, 19F NMR spectroscopy was used to monitor the rate of cross-coupling between p-tolyl triflate (1) with 4-fluoroaniline (2) (Figure 2). The kinetic orders in [Pd], [DBU], [1], and [2] were determined by measuring the rate of formation of 3 over time in THF solutions. As the concentration of 2 was varied between 0.5 and 1.5 M at room temperature, the rate of product formation increased. A plot of ln(Rate) vs ln([2]) fit well with a line with slope +1.02 (Figure 2, A), a result that is consistent with a positive first order dependence on 2 . In contrast, when the concentration of DBU was varied, the rate of the reaction decreased. The plot of ln(Rate) vs ln([DBU]) fit a line with slope −1.16, indicating that there was a negative first order dependence on DBU (Figure 2, B) . These results suggest that both the amine and the base are involved prior to, or during, the rate-limiting step(s) (RLS) of the reaction. Varying the concentration of 1 did not influence the reaction rate (zeroth order), and positive, linear effect (first order dependence) was observed when increasing the total [Pd] from 0.5 to 1.5 mol% Pd (see the Supporting Information for kinetic data and a description of the studies carried out). As we expected, these results are consistent with a monomeric active Pd species. Moreover, because there is no dependence on [aryl triflate], we believe that the oxidative addition (Figure 1, II) sequence is facile under these reaction conditions. These kinetic data show that the base inhibits (negative order) the coupling reaction of aryl triflates and aryl amines, suggesting that this reagent does more than facilitate the removal of a proton from a palladium-bound amine intermediate or act as a simple acid scavenger.
Figure 2.
Orders in reactants for the coupling of aryl triflate (1) with aniline (2) in the presence of DBU. Reaction rates were measured in triplicate by 19F NMR and represent six time points per reaction (18 time points total). The average rate is plotted for reactions performed at 0.5, 1.0, and 1.5 M relative to the varied reagent. Error bars represent the logarithmic standard deviation of the three reaction rates. (A) Logarithmic plot showing a first order dependence on [2]. (B) Logarithmic plot showing a negative first order dependence on [DBU].
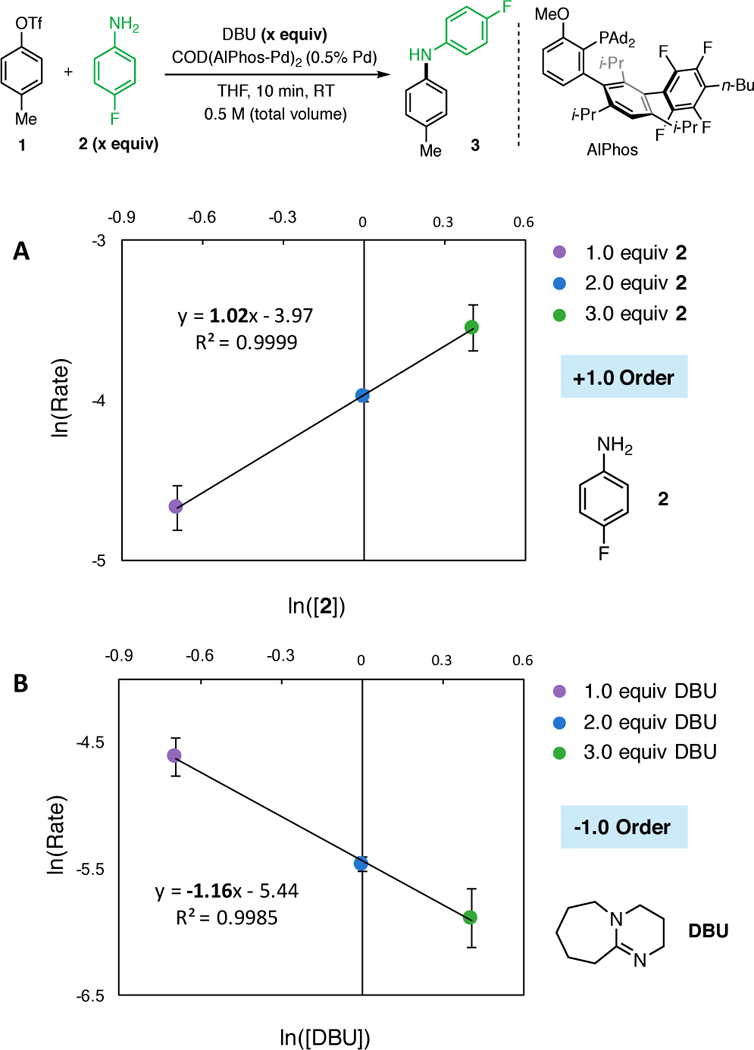
To interpret the effect of [2] and [DBU] on the reaction rate, we used 31P NMR to determine the likely resting state of the catalytic cycle. First, the relative binding affinity of Pd for 2 and DBU was studied via sequential addition of each reaction component to an NMR tube. The addition of a substoichiometric amount (4 mol% Pd) of precatalyst (COD(Pd-AlPhos)2) to a solution of 1 in THF (Figure 3, A) showed only one signal (118.1 ppm) consistent with the oxidative addition (OA) complex (Figure 1, II), which has been previously isolated and reported.9 Next, addition of aniline to this solution resulted in the complete consumption of the OA complex and the formation of a species exhibiting a broad resonance at 77 ppm (Figure 3, B), which we believe to be the aniline-bound OA complex (Figure 1, III) based on previous experiments involving aliphatic amines.9 Addition of DBU to this solution initiated the catalytic reaction and resulted in the formation of a new, sharp resonance at 70.1 ppm (Figure 3, C) . This spectrum exhibits a chemical shift equivalent to that of a separately prepared DBU-bound OA complex (Figure 3, D), providing evidence that this base-bound Pd species is likely the resting state of the catalyst during the catalytic reaction.15 While the involvement of the base in the resting state is generally uncommon, Hartwig has shown that Pd-catalyzed cross-coupling of weakly binding fluoroalkylamines facilitated by phenoxide bases proceeds through a Pd(Ar)OPh intermediate, demonstrating the ability for the base to bind to Pd.16 Taking into account the resting state of our reaction, the kinetic orders that we have determined suggest that the RLS of the reaction involves the substitution of DBU by p-fluoroaniline on the DBU-bound OA complex. This process would be expected to produce a negative order in DBU and a positive order in 2. Other possible rate-limiting steps, such as deprotonation of the amine-bound OA complex (Figure 4, VII) and reductive elimination of the amido complex (Figure 4, VIII), would likely exhibit zeroth order dependence on base in disagreement with our experimental results. 17 The identity of the resting state and the kinetic orders implicate an amine–DBU exchange process at palladium as the turnover-limiting step. The general mechanism is shown in Figure 4. The rate-limiting exchange can take place through either a dissociation-association (D/A) sequence, in which reversible coordination of an amine takes place before DBU dissociation (Figure 5, A), or through an association-dissociation (A/D) sequence (Figure 5, B), in which reversible detachment of DBU takes place before amine association. Due to the observed negative kinetic order in DBU as well as the known reluctance of the highly hindered AlPhos-supported arylpalladium(II) to accept two amine ligands, (see Supporting Information for comparisons and further discussion), we propose the D/A mechanism to be operative. To determine which step within this manifold limits the rate of the reaction, we conducted a Hammett analysis. 18 The rates of catalytic reactions of 2 with aryl triflates bearing various para-substituents were measured using 19F NMR spectroscopy. These rates, normalized to that of phenyl triflate, were plotted against previously determined Hammett constants19 associated with the substituents (Figure 6). For electron-withdrawing substituents (σ > 0), such as p-chloro, phenylazo, and nitro, a negative Hammett correlation was observed (ρ = −1.97), indicating a deceleration effect for electron-poor triflates. In contrast, for electron-releasing substituents (σ < 0), such as p-methyl, methoxy, and N,N-dimethylamino, a positive Hammett correlation was observed (ρ = +1.27), indicating a deceleration effect for electron-rich triflates. This class of downward curving Hammett plots20 is indicative of a shift in rate-limiting step within this two-step sequence.21
Figure 3.
Analysis of reaction intermediates by 31P NMR: (A) OA complex. (B) aniline-bound OA complex. (C) DBU-bound OA complex resting state. (D) DBU-bound OA complex. Pd Precat. denotes COD(AlPhos-Pd)2.
Figure 4.
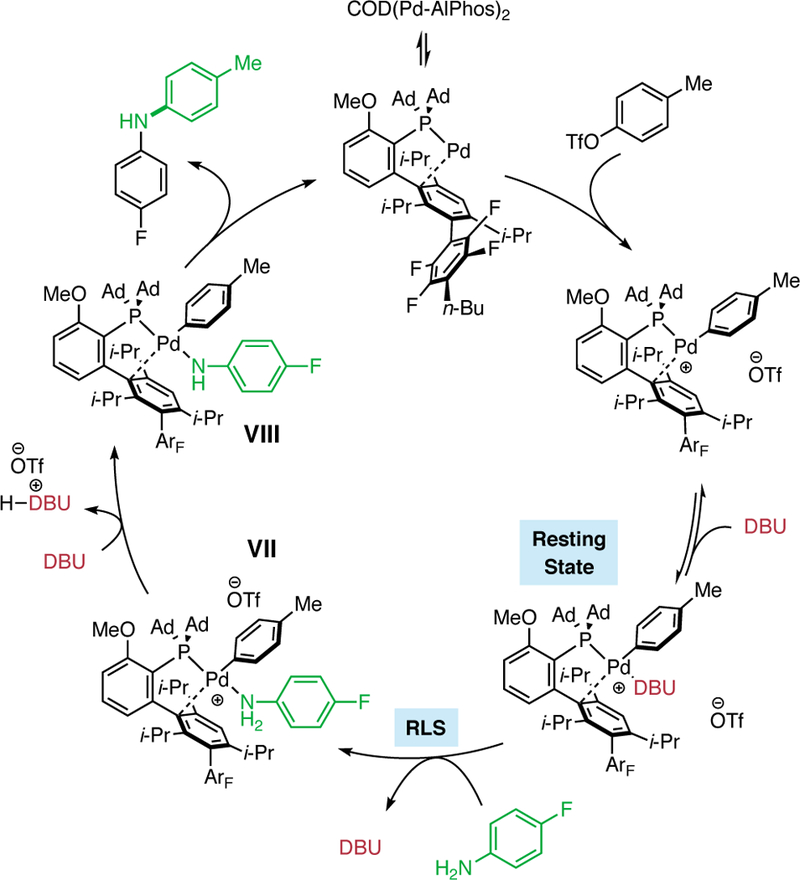
Proposed catalytic cycle of the coupling of aryl triflates and aryl amines in the presence of DBU.
Figure 5.
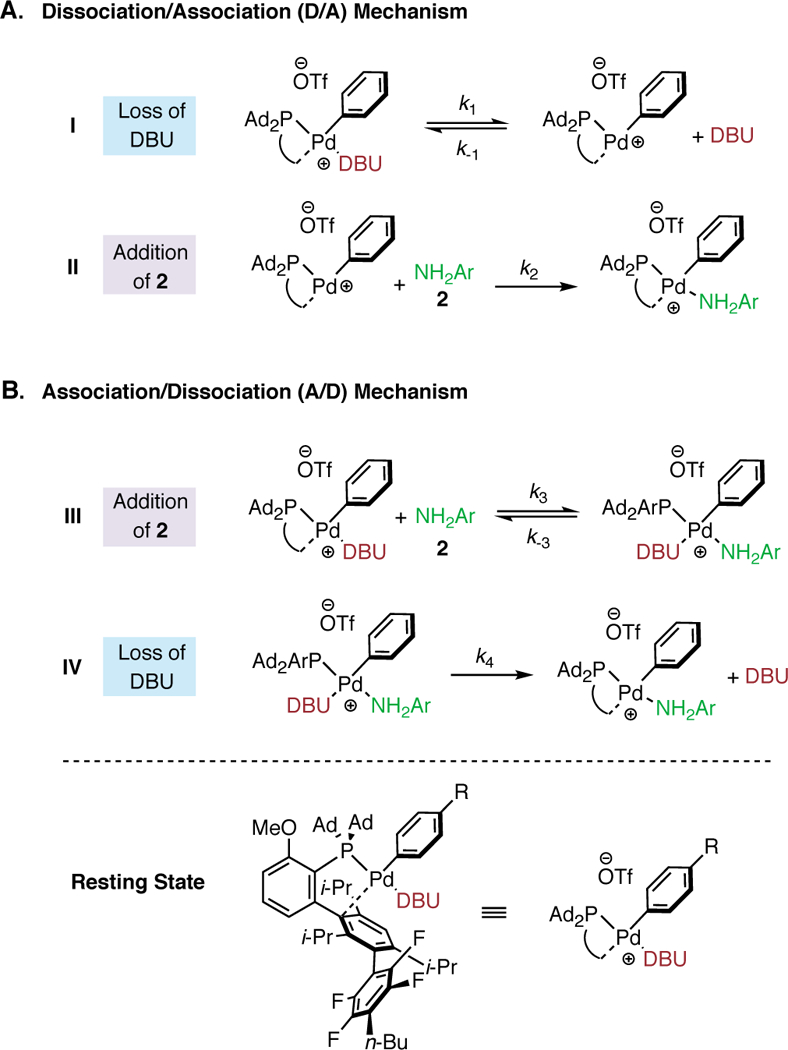
Elementary steps of the two possible rate-limiting steps. (A) Dissociation/Association mechanism; I, DBU-dissociation event; II, amine binding event. (B) Association/Dissociation mechanism; III, amine binding event; IV, DBU-dissociation event.
Figure 6.
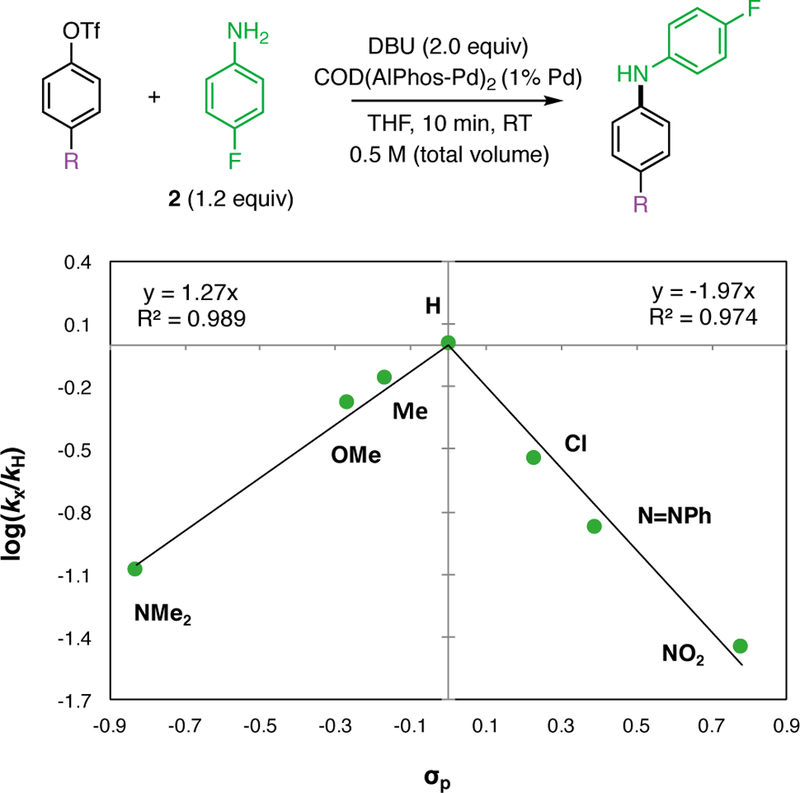
Hammett plot of the coupling of 4-substituted aryl triflates and 2 in the presence of DBU. Data point labels indicate para substituent. Reaction rates are plotted as the average of two runs. Positive and negative trend line intercepts are fit to the plot origin.
We can explain this behavior more precisely by examining the steady-state rate law of the D/A mechanism (equation 1, see Supporting Information for derivation).22 We can assume that step II (Figure 5, A) is effectively irreversible because of fast and irreversible subsequent steps: deprotonation by DBU and reductive elimination, respectively.23 Kinetically, we observed a negative dependence on DBU, which supports our assumption regarding deprotonation. If this step were rate limiting, one would expect to observe zero dependence on this reagent. Thus, the observed rate (kobs) is proportional to a function of two constants (equation 2). Accordingly, the logarithm of the rate constant is the sum of two terms (plus neglected constants, equation 3). The first term, log(k2), can be predicted to have a small, positive dependence on the Hammett parameter of the aryl substituent, since the positive charge at palladium decreases in the transition state of step II. The second term can be analyzed by examining its behavior at the extremes. For K >> [DBU], this term approaches zero. Meanwhile, for K << [DBU], the term approaches log(K/[DBU]), which can be predicted to have a large, negative dependence on the Hammett parameter of the aryl substituent, since the positive charge at palladium increases during dissociation of DBU in step I. We note that this negative dependence, which results from an equilibrium (ground state) effect in step I, is likely larger in magnitude than the positive dependence of the first term, which results from a kinetic (transition state) effect in step II. A summary of predicted behavior of each of these terms is shown in Figure 7. The composite log(kobs) is shown in red as the sum of the two terms of equation (3), which are separately shown in blue and green. The red curve exhibits the same qualitative behavior as is observed in experiment. Intuitively, the non-linear rate relationship indicates the combination of negative ρ for step I, positive ρ for step II, and a shift from step II rate-limiting to step I rate-limiting with increasing ρ. Finally, while this proposed mechanistic scheme provides good agreement with our kinetically-derived mathematical model and understanding of inductive effects, these experiments do not exclusively rule out other deviations from the D/A mechanism. 24
Figure 7.
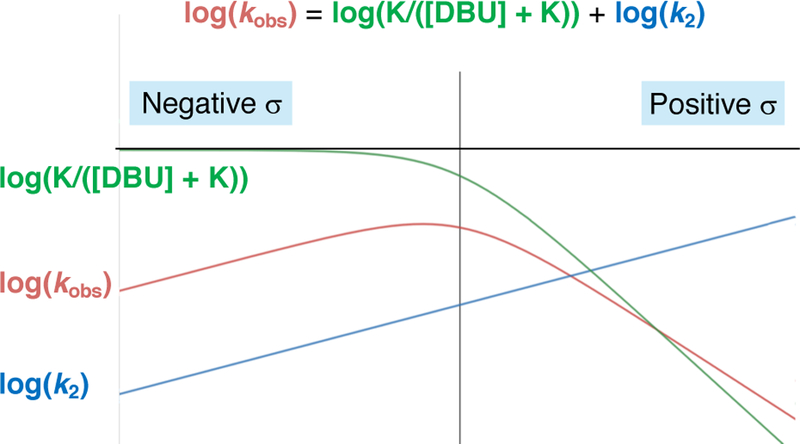
Mathematical model of log(Kobs) as a function of Hammett parameter. The green curve and blue curve show the individual effects of σ on the separate terms of the equation, while the red curve shows the sum of these effects.
Based on the intricate dependence of the reaction rate on electronic effects, it is clear that the mechanism of this transformation is sensitive to subtle changes in substrate structure. Accordingly, we considered whether structural variations to the base would have large effects on the kinetic behavior of the reaction. If so, the ability to perturb the system to circumvent inhibition by DBU might lead to a catalytic system manifesting greater reactivity. To study this, we varied both the steric properties and overall concentration of the base and measured the effects of these changes on the reaction rate. We expected that the use of a more hindered base should disfavor the formation of a base-bound Pd complex due to steric interactions. We examined N,N-diisopropylethylamine (DIPEA) and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD), both soluble, organic bases with Lewis-basic nitrogen atoms that are in sterically hindered environments.
| (1) |
| (2) |
| (3) |
The kinetic orders of the reaction of 1 and 2 in the presence of either DIPEA or MTBD were determined by measuring the rate of formation of 3 over time, in an analogous manner to the above-describe kinetic experiments. As the concentration of DIPEA was varied between 0.5 and 1.5 M, a positive linear relationship was observed. Plotting ln(Rate) vs ln([DIPEA]) provided a straight line with a slope of +0.79 (Figure 8), which indicates a positive, partial first order dependence on [DIPEA]. To investigate the origin of the partial order, we determined the resting state of the catalyst during the reaction catalytic process using 31P NMR. We observed multiple broad signals which we believe correspond to both the DIPEA-bound OA complex and 2-bound OA complex (See the Supporting Information for copies of the spectra). This result demonstrates that there is an equilibrium between the base-bound and amine-bound OA complexes and suggests that more hindered bases bind to the Pd center less efficiently than DBU. It is likely that the presence of multiple resting states of the catalytic reaction results in non-integral order reaction kinetics. For example, if the deprotonation of the amine-bound OA complex, which is predicted to exhibit +1 order in DIPEA, and the dissociation of the DIPEA from the OA complex, which is predicted to exhibit −1 order in DIPEA, are each partially rate-limiting, a non-integral order could result.
Figure 8.
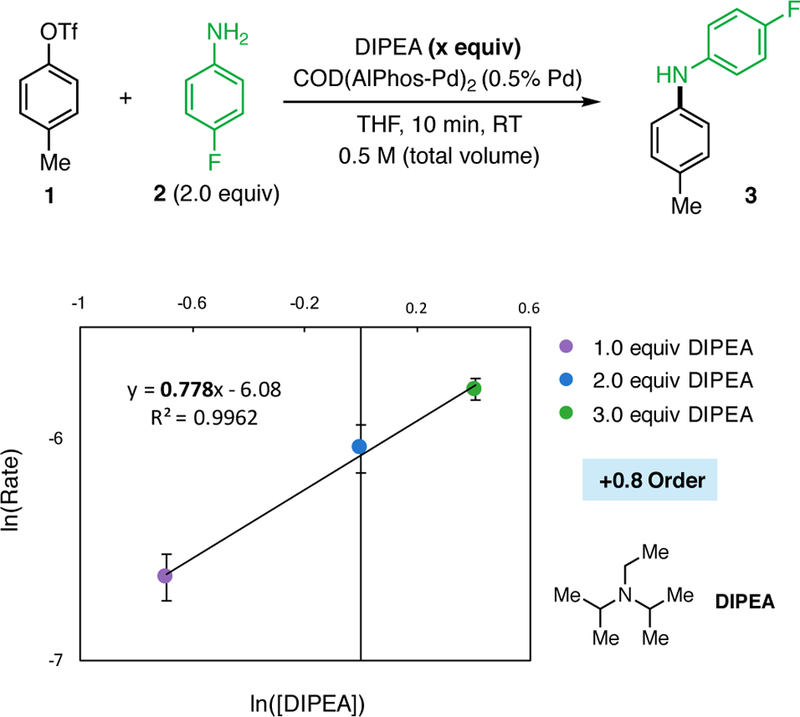
Logarithmic plot showing a positive, partial order dependence on DIPEA in the coupling of 1 and 2. The average rate is plotted for reactions performed at 0.5, 1.0, and 1.5 M relative to DIPEA. Error bars represent the logarithmic standard deviation of the three reaction rates that were determined from six time points per reaction (18 time points total).
For coupling reactions carried out using MTBD as the base (cf. Figure 9), the rate of product formation decreased as the concentration of base increased. The logarithmic plot of Rate vs [MTBD] showed a slope of −0.51, indicating a negative partial order in MTBD concentration (Figure 9). As in the case of DIPEA, we believe that the steric environment of the key basic nitrogen in MTBD decrease the efficiency of its binding to the Pd center. In this case the resting state in the catalytic process is likely a mixture of MTBD-bound and 2-bound oxidative addition complex.25 Comparing these results with those employing DBU (vide supra) suggests that the resting state of the catalytic reaction depends on the relative binding ability of the base and the aniline substrate. In coupling reactions where the base outcompetes the aniline for binding to the Pd center of the OA complex, both a base-bound resting state and a negative order dependence on base are observed. In contrast, if the amine is more nucleophilic and/or less hindered than the base, then it may outcompete the base in binding to the Pd center. In this case, both an amine-bound OA complex resting state and positive order dependence on amine are observed.
Figure 9.
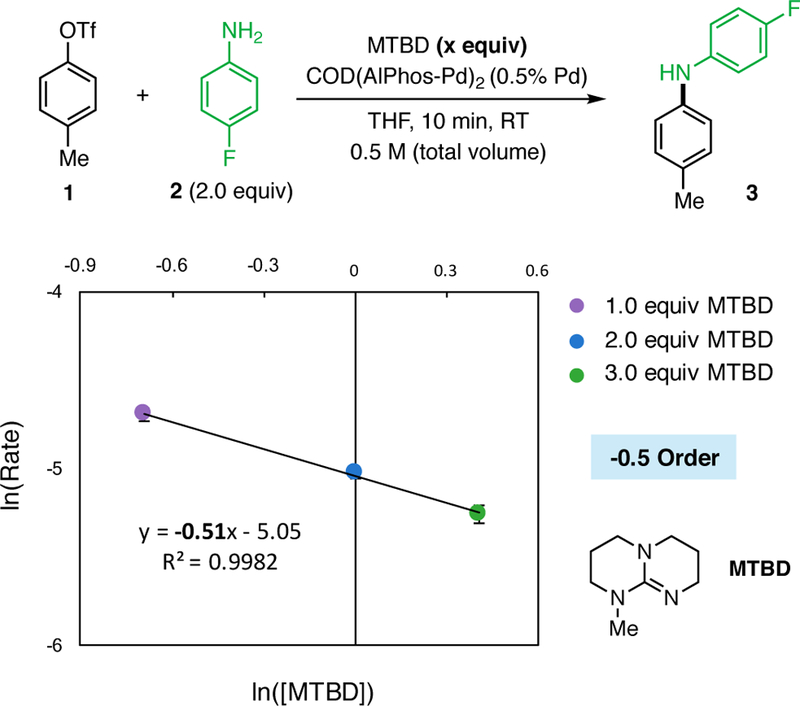
Logarithmic plot showing a −0.5 order dependence on MTBD in the coupling of 1 and 2. The average rate is plotted for reactions performed at 0.5, 1.0, and 1.5 M relative to MTBD. Error bars represent the logarithmic standard deviation of the three reaction rates that were determined from six time points per reaction (18 time points total).
While the steric properties of the bases had a predictable effect on the reaction order, the overall reaction rates depend on a combination of less predictable factors, including the kinetic and thermodynamic basicity of the chosen base. In the case of reactions performed at low base concentrations (1.0 M), the reactions facilitated by DBU and MTBD were found to be faster than the reaction with DIPEA (Figure 10, A) despite their inhibitory behavior. While DIPEA was found to have a positive dependence on the reaction (+0.8), this base is much weaker than DBU and MTBD, which may affect the overall rate of the reaction due to a slow deprotonation event. In the case of high base concentrations (3.0 M), the rate of reactions facilitated by DBU and MTBD decreased significantly (Figure 10, B) as expected based on their order dependence. The rate of the reaction facilitated by DIPEA increased under these conditions such that it surpassed the rate of the reaction facilitated by DBU. These results demonstrate that both the base structure and concentration affect the reaction rate and should be taken into consideration when employing a soluble base in a Pd-catalyzed C–N cross-coupling. From these observations, we conceived two approaches to improve difficult coupling reactions involving weakly binding amines and potentially nucleophilic bases. First, in reactions that employ bases that slow the rate of the reaction, such as DBU, decreasing the concentration of base should favor formation of an aniline-bound OA complex, increasing the rate of the reaction. We chose to test this hypothesis through slow (syringe-pump) addition of DBU in the coupling reaction of aryl triflates with secondary anilines and aromatic amines. While a number of methods that rely on inorganic bases can efficiently couple these nucleophiles, methods using soluble organic bases rely on expensive phosphazene bases such as P2Et ($40/mmol) instead of economical bases like DBU ($0.02/mmol). As a result, improving these reaction conditions would have a direct impact on the utility of these methodologies.
Figure 10.
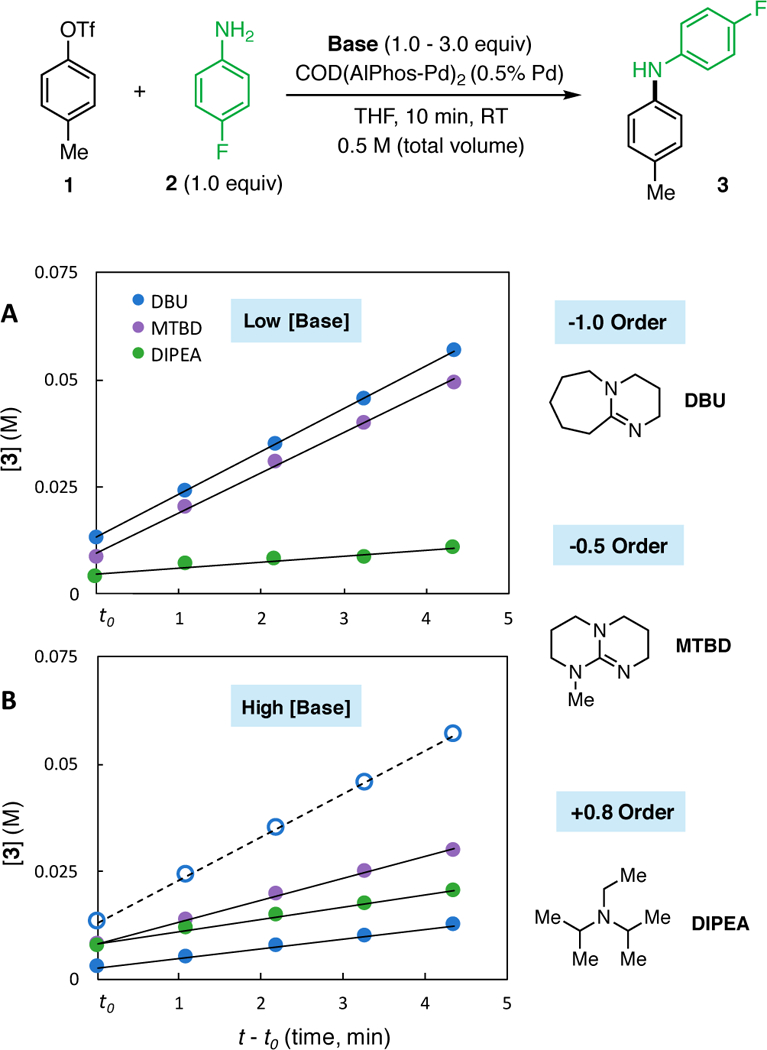
Comparison of the order in [base] and the initial rates of the reactions facilitated by MTBD, DBU, and DIPEA at low and high base concentrations. Data points are the average of three runs. (A) Reactions performed at a low base concentration (1.0 M). (B) reactions performed at a high base concentration (3.0 M). The dotted reference line shows the original rate under low [DBU] concentrations.
Under our previously reported reaction conditions using soluble amine bases,9 the coupling reactions of secondary amines with aryl halides or triflates resulted in low or no formation of the desired product. For example, the reaction of 1 and indoline resulted in a 10% yield of the desired product (Table 1, 1a) when heated to 60 °C in 2-methyl tetrahydrofuran (2-MeTHF) in the presence of an AlPhos-supported Pd catalyst and DBU. However, when 2.0 equivalents of DBU were added slowly (0.167 mmol/h) via syringe pump, the desired product was obtained in nearly quantitative isolated yield (Table 1, 1a). Simply employing lower loadings of base resulted in incomplete conversion of the starting material. 26 A number of substituted aryl triflate electrophiles and weakly binding amines were examined, and the yields of the reaction employing the slow addition of base (red yields) were compared to those obtained using the previously reported reaction conditions (black yields). More complex electrophiles, including a Flumazenil derivative (5a) could be efficiently transformed to products under these reaction conditions when slightly elevated temperatures were used (80 °C). Notably, a benzoxadiazole electrophile (8a) was found to undergo the desired C-N coupling in modest yield despite the possibility for these heteroatom-heteroatom containing substrates to undergo undesired reactions with Pd.8c Other weakly coordinating amines, including acyclic secondary amines such as N-methyl aniline derivatives are suitable substrates (2a, 6a, and 8a). Aromatic amines, including a pyrrole (3a ), an indazole (4a ), and an imidazole (9a) were found to be reactive coupling partners under the slow addition reaction conditions. In the case of the indazole and imidazole couplings, N1 selectivity was observed and there was no detectable formation of the N2-regioisomer.27 In a few cases, limiting the DBU concentration has no effect on the reaction yield, such as with substrate 7a and indole (result not shown). In these cases, the desired product is still obtained in high yields. Currently, the reactions of sterically demanding secondary aryl amine nucleophiles, including N-benzyl aniline, exhibit low or no reactivity under these conditions.
Table 1:
Coupling of heterocyclic amines and secondary aryl amines.a
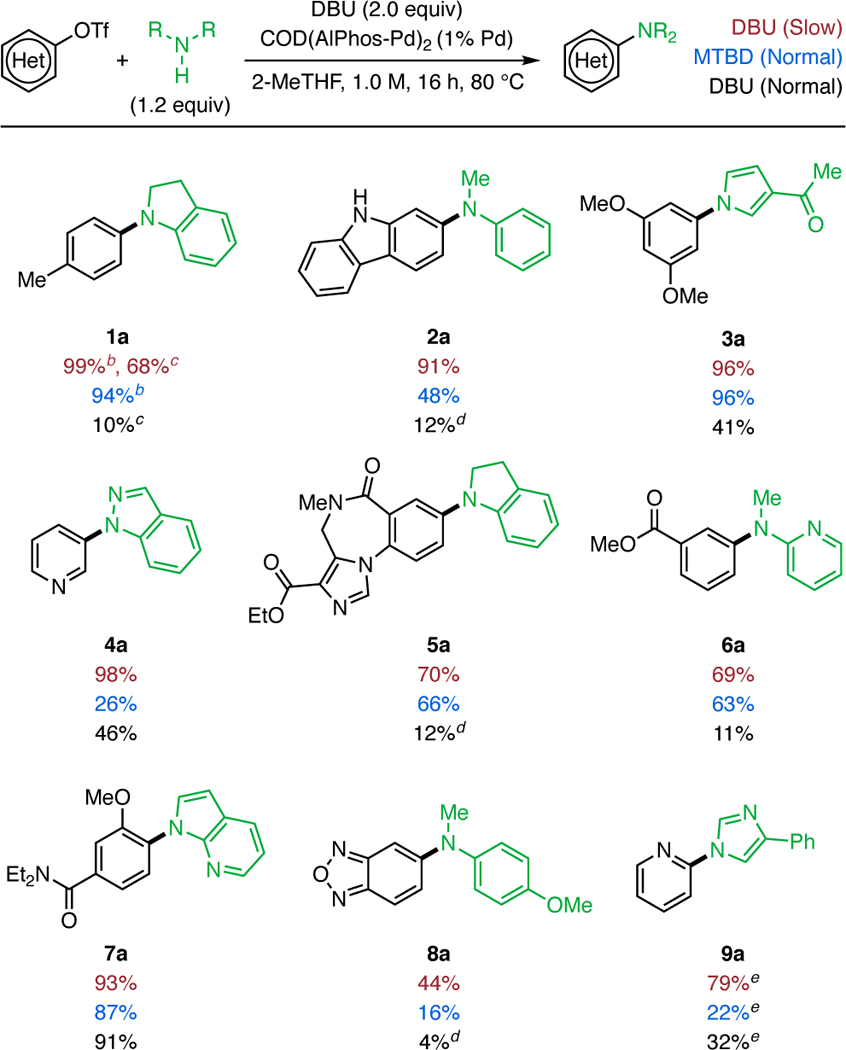 |
Red yields indicate the average isolated yield of two reactions performed with a slow addition of DBU (syringe pump, 12 h, 0.167 mmol/h); blue isolated yields indicate the normal addition of MTBD and are denoted as a single isolated yield. black yields indicate a normal addition of DBU and are denoted as a single isolated or NMR yield. Standard reaction conditions: aryl triflate (0.50 mmol), nucleophile (0.60 mmol), base (1.0 mmol), COD(AlPhos-Pd)2 (2.5 μmol, 1% Pd) 2-methyltetrahydrofuran (2-MeTHF, 1.0 M), 80 °C for 16 h.
Reaction performed at 60 °C.
1H NMR yield of reaction performed at 60 °C with 1.2 equiv of DBU.
1H NMR yield.
Reaction performed with 2% Pd.
In a second approach to overcome the inhibiting effect of DBU, MTBD, a considerably more expensive base than DBU, was found to be a suitable alternative in the coupling reactions of some weakly binding amines for cases in which syringe pump addition of DBU is considered undesirable. Because MTBD is less inhibiting than DBU (vide supra), it could be employed as base without requiring the use of syringe pump addition. The yields of coupling reactions facilitated by MTBD (Table 1, blue yields) were similar to those observed with the slow addition protocol using DBU in coupling reactions of many N-alkyl aryl amines ( 1a, 5a, and 6a). However, in some cases, the desired product was still observed in modest yields (e.g., 2a,b and 4a). The ability to use either DBU with syringe pump addition or MTBD to facilitate these coupling reactions provides two complementary approaches for the synthetic chemist to choose from depending on whether cost-efficiency or operational simplicity is more important to them.
CONCLUSION
In summary, we have demonstrated that organic amine bases play an important but complicated role in the coupling of aryl triflates with aryl amines. We have determined that the nucleophilicity of the base, relative to that of the amine substrate, dictates the resting state of the reaction, as well as the rate limiting step. For example, it was determined that for the Pd-catalyzed coupling of an aryl triflate and an aniline, the reaction displayed a negative first order in DBU when this amidine was used as the base. This and other mechanistic insights were used to develop modified reaction conditions that enabled the coupling of weakly binding heterocyclic amines and secondary aryl amines with aryl triflates. The slow addition of DBU was found to offer a solution for these couplings, while the normal, single-batch addition of MTBD provided an operationally simpler but costlier alternative procedure. Finally, while this study focuses on one catalyst system, we believe that these conclusions provide key considerations in developing and optimizing future cross-coupling methodologies that employ nitrogen-containing bases.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (Grant Nos. GM122483 and GM58160), the NSF Graduate Research Fellowship Program under Grant No. 1122374 (J.M.D.), and an NIH Postdoctoral Fellowship under Grant No. 1F32GM120847–01 (N.A.W.). Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF or NIH. The authors thank Dr. Jorge Garcia Fortanet for synthesizing the aryl triflate used to prepare product 5a. We also thank Dr. Scott McCann (MIT) and Dr. Andy Thomas (MIT) for insightful conversations relating to the interpretation of this data and for assistance in preparing this manuscript. We acknowledge Dr. Christine Nguyen (MIT) for editing this manuscript. We gratefully acknowledge Robert Pollice (ETH Zürich) for alerting us to an error in our rate law derivation prior to publication. Finally, we recognize Prof. Mu-Hyun Baik (KAIST), Seoung-Tae Kim (KAIST) and Dr. Bimal Pudaisaini (KAIST) for their computational support and valuable conversations about the mechanism of this transformation.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, raw kinetic data, and spectral data.
Notes
MIT has filed patents on ligands and precatalysts that are described in this manuscript, from which S.L.B and former coworkers receive royalty payments.
REFERENCES
- (1).Kochi JK Organometallic Mechanisms and Catalysis; Academic Press: New York, 1978. [Google Scholar]
- (2).For a perspective on this topic, see: Blum SA; Tan KL; Bergman RG Application of Physical Organic Methods to the Investigation of Organometallic Reaction Mechanisms. J. Org. Chem 2003, 68, 4127–4137. [DOI] [PubMed] [Google Scholar]
- (3) (a).Shekhar S; Ryberg P; Hartwig J; Mathew JS; Blackmond DG; Strieter ER; Buchwald SL Reevaluation of the Mechanism of the Amination of Aryl Halides Catalyzed by BINAP-Ligated Palladium Complexes. J. Am. Chem. Soc 2006, 128, 3584–3591. [DOI] [PubMed] [Google Scholar]; (b) Arrechea PL; Buchwald SL; Biaryl Phosphine Based Pd(II) Amido Complexes: The Effect of Ligand Structure on Reductive Elimination. J. Am. Chem. Soc 2016, 138, 12486–12493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Klingensmith LM; Strieter ER; Barder T; Buchwald SL New Insights into Xantphos/Pd-Catalyzed C−N Bond Forming Reactions: A Structural and Kinetic Study. Organometallics 2006, 25, 82–91. [Google Scholar]; (d) Barder TE; Biscoe MR; Buchwald SL Structural Insights into Active Catalyst Structures and Oxidative Addition to (Biaryl)phosphine−Palladium Complexes via Density Functional Theory and Experimental Studies. Organometallics 2007, 26, 2183–2192. [Google Scholar]; (e) Klinkenberg JL; Hartwig JF Slow Reductive Elimination from Arylpalladium Parent Amido Complexes. J. Am. Chem. Soc 2010, 132, 11830–11833. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shekhar S; Ryberg P; Hartwig JF; Oxidative Addition of Phenyl Bromide to Pd(BINAP) vs Pd(BINAP)(amine). Evidence for Addition to Pd(BINAP). Org. Lett 2006, 8, 851–854. [DOI] [PubMed] [Google Scholar]; (g) Hooper MW; Utsunomiya N; Hartwig JF; J. Org. Chem 2003, 68, 2861–2873. [DOI] [PubMed] [Google Scholar]; (h) Alcazar-Roman LM; Hartwig JF Mechanistic Studies on Oxidative Addition of Aryl Halides and Triflates to Pd(BINAP)2 and Structural Characterization of the Product from Aryl Triflate Addition in the Presence of Amine. Organometallics 2002, 21, 491–502. [Google Scholar]; (i) Alcazar-Roman LM; Hartwig JH Mechanism of Aryl Chloride Amination: Base-Induced Oxidative Addition. J. Am. Chem. Soc 2001, 123, 12905–12906. [DOI] [PubMed] [Google Scholar]; (j) Hamann BC; Hartwig JF Systematic Variation of Bidentate Ligands Used in Aryl Halide Amination. Unexpected Effects of Steric, Electronic, and Geometric Perturbations. J. Am. Chem. Soc 1998, 120, 3694–3703. [Google Scholar]; (k) Hartwig JF; Richards S; Barañano D; Paul F Influences on the Relative Rates for C−N Bond Forming Reductive Elimination and the β-Hydrogen Elimination of Amides. A Case Study on the Origins of Competing Reduction in the Palladium-Catalyzed Amination of Aryl Halides. J. Am. Chem. Soc 1996, 118, 3626–3633. [Google Scholar]; (l) Louie J; Paul F; Hartwig JF Catalysis with Platinum-Group Alkylamido Complexes. The Active Palladium Amide in Catalytic Aryl Halide Aminations as Deduced from Kinetic Data and Independent Generation. Organometallics, 1996, 15, 2794–2805. [Google Scholar]; (m) Driver MS; Hartwig JF A Rare, Low-Valent Alkylamide Complex, a Diphenyl Amido Complex, and Their Reductive Elimination of Amines by Three-Coordinate Intermediates. J. Am. Chem. Soc 1995, 117, 4708–4709. [Google Scholar]; (n) Paul F; Patt J; Hartwig JF Palladium-Catalyzed Formation of Carbon-Nitrogen Bonds. Reaction Intermediates and Catalyst Improvements in the Hetero Cross-Coupling of Aryl Halides and Tin Amides. J. Am. Chem. Soc 1994, 116, 5969–5970. [Google Scholar]; (o) Hoi KH; Çalimsiz S; Froese RDJ; Hopkinson AC; Organ MG Amination with Pd–NHC Complexes: Rate and Computational Studies on the Effects of the Oxidative Addition Partner. Chem. Eur. J 2011, 17, 3086–3090. [DOI] [PubMed] [Google Scholar]; (p) Hoi KH; Çalimsiz S; Froese RDJ; Hopkinson AC; Organ MG Amination with Pd−NHC Complexes: Rate and Computational Studies Involving Substituted Aniline Substrates. Chem. Eur. J 2012, 18, 145–151. [DOI] [PubMed] [Google Scholar]; (q) Organ MG; Abdel-Hadi M; Avola S; Dubovyk I; Hadei N; Kantchev EAB; O’Brien CJ; Sayah M; Valente C Pd-Catalyzed Aryl Amination Mediated by Well Defined, N-Heterocyclic Carbene (NHC)–Pd Precatalysts, PEPPSI. Chem. Eur. J 2008, 14, 2443–2452. [DOI] [PubMed] [Google Scholar]; (r) Strotman N,A; Soumeillant MC; Zhu K; Markwalter CE; Wei CS; Hsiao Y; Eastgate MD Effects of Multiple Catalyst Deactivation Pathways and Continuous Ligand Recycling on the Kinetics of Pd-Catalyzed C-N Coupling Reactions. J. Org. Chem 2019. 8b02214. [DOI] [PubMed]
- (4) (a).For examples of mechanistic studies related to ligand discovery, see: Ruiz-Castillo P; Blackmond DG; Buchwald SL Rational Ligand Design for the Arylation of Hindered Primary Amines Guided by Reaction Progress Kinetic Analysis. J. Am. Chem. Soc 2015, 137, 3085–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Olsen EPK; Arrechea PL; Buchwald SL Mechanistic Insight Leads to a Ligand Which Facilitates the Palladium-Catalyzed Formation of 2-(Hetero)Arylaminooxazoles and 4-(Hetero)Arylaminothiazoles. Angew. Chem., Int. Ed 2017, 56, 10569–10572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hicks JD Hyde AM; Martinez Cuezva A; Buchwald SL Pd-Catalyzed N-Arylation of Secondary Acyclic Amides: Catalyst Development, Scope, and Computational Study. J. Am. Chem. Soc 2009, 131, 16720–16734. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Biscoe MR; Barder TE; Buchwald SL Angew. Chem., Int. Ed. Electronic Effects on the Selectivity of Pd-Catalyzed C–N Bond-Forming Reactions Using Biarylphosphine Ligands: The Competitive Roles of Amine Binding and Acidity. Angew. Chem., Int. Ed 2007, 46, 7232–7235. [DOI] [PubMed] [Google Scholar]; (e) Lundgren RJ; Stradiotto M Addressing Challenges in Palladium-Catalyzed Cross-Coupling Reactions Through Ligand Design. Chem. Eur. J 2012, 18, 9758–9769. [DOI] [PubMed] [Google Scholar]; (f) Crawford SM; Wheaton CA; Mishra V; Stradiotto M Probing the Effect of Donor-Fragment Substitution in Mor-DalPhos on Palladium-Catalyzed C−N and C−C Cross-Coupling Reactivity. Can. J. Chem 2018, 96, 578–586. [Google Scholar]
- (5).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For a discussion of this effect in the context of Cu-catalyzed C–N coupling facilitated by soluble bases, see: Sung S; Sale D; Braddock DC; Armstrong A; Brennan C; Davies RP Mechanistic Studies on the Copper-Catalyzed N-Arylation of Alkylamines Promoted by Organic Soluble Ionic Bases. ACS Catal 2016, 6, 3965–3974. [Google Scholar]
- (7) (a).Meyers C; Maes BUW; Loones KTJ; Bal G; Lemière GLF; Dommisse RA Study of a New Rate Increasing “Base Effect” in the Palladium-Catalyzed Amination of Aryl Iodides. J. Org. Chem 2004, 69, 6010–6017. [DOI] [PubMed] [Google Scholar]
- (8) (a).Buitrago Santanilla A; Christensen M; Campeau L-C; Davies IW; Dreher SD P2Et Phosphazene: A Mild, Functional Group Tolerant Base for Soluble, Room Temperature Pd-Catalyzed C–N, C–O, and C–C Cross-Coupling Reactions. Org. Lett 2015, 17, 3370–3373. [DOI] [PubMed] [Google Scholar]; (b) Buitrago Santanilla A; Regalado EL; Pereira T; Shevlin M; Bateman K; Campeau L-C; Schneeweis J; Berritt S; Shi Z-C; Nantermet P; Liu Y; Helmy R; Welch CJ; Vachal P; Davies IW; Cernak T; Dreher SD Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53. [DOI] [PubMed] [Google Scholar]; (c) Ahneman DT; Estrada JE; Lin S; Dreher SD; Doyle AG Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning. Science 2018, 360, 186–190. [DOI] [PubMed] [Google Scholar]; (d) Uehling MR; King RP; Krska SW; Cernak T; Buchwald SL Pharmaceutical Diversification Via Palladium Oxidative Addition Complexes. Science 2019, 363, 405–408. [DOI] [PubMed] [Google Scholar]
- (9).Dennis JM White NA; Liu RY; Buchwald SL Breaking the Base Barrier: An Electron-Deficient Palladium Catalyst Enables the Use of a Common Soluble Base in C–N Coupling. J. Am. Chem. Soc 2018, 140, 4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Widenhoefer RA; Buchwald SL; Halide and Amine Influence in the Equilibrium Formation of Palladium Tris(o-tolyl)phosphine Mono(amine) Complexes from Palladium Aryl Halide Dimers. Organometallics, 1996, 15, 2755–2763. [Google Scholar]
- (11). For computational investigations into the binding of amines to dialkyl biarylmonophosphine ligated Pd complexes, see: Barder TE; Buchwald SL Insights into Amine Binding to Biaryl Phosphine Palladium Oxidative Addition Complexes and Reductive Elimination from Biaryl Phosphine Arylpalladium Amido Complexes via Density Functional Theory. J. Am. Chem. Soc 2007, 129, 12003–12010. [DOI] [PubMed] [Google Scholar]
- (12).Sunesson Y; Limé E; Nilsson Lill SO; Meadows RE; Norrby P-O Role of the Base in Buchwald–Hartwig Amination. J. Org. Chem 2014, 79, 11961–11969 [DOI] [PubMed] [Google Scholar]
- (13).Shekhar S; Hartwig JF Effects of Bases and Halides on the Amination of Chloroarenes Catalyzed by Pd(PtBu3)2. Organometallics 2007, 26, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14). For an example of a secondary aryl amine coupling facilitated by Ni/Ir photoredox catalysis and 1,4-Diazabicyclo[2.2.2]octane (DABCO) as the base, see: Corcoran EB; Pirnot MT; Lin S; Dreher SD; DiRocco DA; Davies IW; Buchwald SL; MacMillan DWC Aryl Amination Using Ligand-Free Ni(II) Salts and Photoredox Catalysis. Science 2016, 353, 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).This resonance was also observed when a solution containing both 2 and DBU were added to a pre-formed OA complex, again demonstrating that the base preferentially binds to the Pd center, likely due to inherent nucleophilicities. Quantitatively, Mayr reactivity parameters have shown that DBU (N = 15.29, MeCN) is more nucleophilic than aniline (N = 12.64, MeCN). See: (a) Baidya M; Mayr H Nucleophilicities and carbon basicities of DBU and DBN. Chem. Commun 2008, 0, 1792–1794. [DOI] [PubMed] [Google Scholar]; (b) Brotzel F; Chu YC; Mayr H Nucleophilicities of Primary and Secondary Amines in Water. J. Org. Chem 2007, 72, 3679–3688. [DOI] [PubMed] [Google Scholar]
- (16).Brusoe AT; Hartwig JF Palladium-Catalyzed Arylation of Fluoroalkylamines. J. Am. Chem. Soc 2015, 137, 8460–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). These predicted orders take into account that the resting state of the reaction is a DBU-bound OA complex. If either deprotonation or reductive elimination were rate limiting, the base would need to first dissociate from the Pd center (−1 order) and then deprotonate the amine-bound OA complex (+1 order) prior to (or during) the rate limiting step. These combined events result in a net 0th order dependence.
- (18).For examples of other Hammett analyses in the context of Pd-catalyzed C–N Coupling, see: (a) Driver MS; Hartwig JF Carbon–Nitrogen-Bond-Forming Reductive Elimination of Arylamines from Palladium(II) Phosphine Complexes. J. Am. Chem. Soc 1997, 119, 8232–8245. [Google Scholar]; (b) Yamashita M; Cuevas Vicario JV; Hartwig JF; Trans Influence on the Rate of Reductive Elimination. Reductive Elimination of Amines from Isomeric Arylpalladium Amides with Unsymmetrical Coordination Spheres. J. Am. Chem. Soc 2003, 125, 16347–16360. [DOI] [PubMed] [Google Scholar]
- (19).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent Constants and Resonance and Field Parameters Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (20).For a discussion on concave and convex Hammett plots, see: Schreck JO Nonlinear Hammett Relationships J. Chem. Educ 1971, 48, 103–107. [Google Scholar]
- (21).For a discussion on the shape of Hammett plots in relation to changes in mechanism and rate-limiting step, see: Um I-T; Han H-J Ahn J-A; Kang S; Buncel E Reinterpretation of Curved Hammett Plots in Reaction of Nucleophiles with Aryl Benzoates: Change in Rate-Determining Step or Mechanism versus Ground-State Stabilization. J. Org. Chem 2002, 67, 8475–8480. [DOI] [PubMed] [Google Scholar]
- (22). We observed the same resting state (DBU-bound OA complex, see Supporting Information) and kinetic dependences (positive order in aniline, negative order in DBU) for the coupling of p-nitro phenyl triflate. Because the relative nucleophilicities of DBU and 4-fluoroaniline do not change, the resting state of the reaction is presumed be a DBU-bound OA complex no matter the electrophilicity of the Pd center. In the coupling of p-N,N-dimethylamine phenyl triflate with aniline, we observed that the use of excess DBU (3.0 vs 1.0 equivalents) resulted in lower reaction yields. This result is consistent with a negative dependence on base for the reactions of electron-rich substrates, which is consistent with our other data. As a result of these experiments, we believe that the derived rate law applies to substrates with both electron donating, neutral, and withdrawing substituents. Moreover, these observed orders and resting states suggest that there is no kinetic dependence on aryl triflate.
- (23). If the base is not strong enough to rapidly deprotonate the amine-bound OA complex, then the amine may dissociate to reform the OA complex. In this case, there is an equilibrium between the two complexes (Figure 5A, II). We believe that our assumption holds when DBU or bases of a greater strength are employed.
- (24). Other theoretical rate limiting steps include deprotonation of the amine-bound OA complex and reductive elimination of the amido complex. Because the kinetics showed a clear inverse dependence on DBU for the coupling p-tolyl triflate, a slightly electron-rich substrate, it is unlikely that deprotonation is rate limiting. Moreover, for coupling reactions facilitated by dialkylbiaryl monophosphine ligands, the reductive elimination step is rarely rate-limiting. For particularly large ligands such as AlPhos, this is also thought to be true. In two special cases, the reductive elimination of diaryl amines and fluoroalkyl amines have been rate-limiting. See references 3b and 16 respectively.
- (25). Attempts to study the resting state by 31P NMR did not yield a definitive result. Both 2 and MTBD independently bind the OA complex, but the combination of these reagents did not yield any readily identifiable species, which we attribute to rapid exchange of the aniline and MTBD at the Pd(II) center.
- (26). Optimizing the addition rate of DBU may enable the cross-coupling with less than 2.0 equivalents of base. However, this may be substrate dependent given the easily influenced reaction rate. For the purpose of this study, 2.0 equivalents added at 0.167 mmol/h provided the product in high yields for substrates with diverse substitution patterns and electronic characteristics.
- (27).For other examples of N1-selective arylations of imidazoles, see: Ueda S; Su M; Buchwald SL Completely N1-Selective Palladium-Catalyzed Arylation of Unsymmetric Imidazoles: Applications to the Synthesis of Nilotinib. J. Am. Chem. Soc 2012, 134, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.