

Abstract
Objective
Genome wide association studies identified novel loci in phospholipid phosphatase 3 (PLPP3) that associate with coronary artery disease (CAD) risk independently of traditional risk factors. PLPP3 encodes lipid phosphate phosphatase 3 (LPP3), a cell surface enzyme that can regulate the availability of bioactive lysophopsholipids including lysophosphatidic acid (LPA). The protective allele of PLPP3 increases LPP3 expression during cell exposure to oxidized lipids, however, the role of LPP3 in atherosclerosis remains unclear.
Approach and Results
In this study, we sought to validate LPP3 as a determinate of the development of atherosclerosis. In experimental models of atherosclerosis, LPP3 is upregulated and co-localizes with endothelial, smooth muscle cell (SMC), and CD68-positive cell markers. Global post-natal reductions in Plpp3 expression in mice substantially increase atherosclerosis, plaque-associated LPA, and inflammation. Although LPP3 expression increases during ox-LDL-induced phenotypic modulation of bone marrow-derived macrophages, myeloid Plpp3 does not appear to regulate lesion formation. Rather, SMC LPP3 expression is a critical regulator of atherosclerosis and LPA content in lesions. Moreover, mice with inherited deficiency in LPA receptor signaling are protected from experimental atherosclerosis.
Conclusions
Our results identify a novel lipid signaling pathway that regulates inflammation in the context of atherosclerosis and is not related to traditional risk factors. Pharmacological targeting of bioactive LPP3 substrates, including LPA, may offer an orthogonal approach to lipid lowering drugs for mitigation of CAD risk.
Keywords: Atherosclerosis, Coronary Artery Disease, Lysophospholipids, Lipid Phosphate Phosphatase, Lysophosphatidic Acid
Introduction
Coronary artery disease (CAD) is a major component of cardiovascular disease and a leading cause of death worldwide. Due to the complexity of the disease, contributors to CAD risk can be divided into several factors, but more generally they include both heritable and non-heritable risk factors. Genome wide association studies identified heritable single nucleotide polymorphisms (SNPs) in phospholipid phosphatase 3 (PLPP3), previously referred to as PPAP2B, as novel loci associated with risk of CAD and its complications independent of traditional risk factors1–3. Lipid phosphate phosphatase 3 (LPP3), encoded by the gene PLPP3, is a cell surface enzyme that can regulate the availability and hence the signaling actions of bioactive lipids including lysophosphatidic acid (LPA). The risk-associated haplotype block is in the final intron of PLPP3 and contains a CCAAT enhancer binding protein beta (CEBPβ) motif. The CAD risk allele disrupts transcriptional enhancer activity induced by CEBPβ binding4. CEBPβ binding increases transcription from the PLPP3 promoter, and polymorphisms that disrupt this interaction are associated with lower PLPP3 gene expression in monocytes exposed to oxidized LDL4. In addition to identifying a local expression quantitative trait loci (eQTL) effect in monocytes, we have demonstrated that exposure of human monocytes to oxidized low-density lipoprotein (oxLDL) increases LPP3 expression and activity, and that the CAD risk allele in PLPP3 is associated with lower LPP3 expression4, 5. Others have reported similar findings in other vascular cells6.
We have previously demonstrated a protective role for LPP3 and its biological target LPA in vascular injury models in a tissue-specific manner7–9. In fact, the loss of LPP3 in vascular smooth muscle cells (vSMCs) exacerbates local inflammation and proliferation following carotid artery ligation that leads to increased neointimal formation likely via heightened ERK activity and Rho activation7. We also demonstrated vSMC LPP3 expression increases following injury and vSMC proliferation increases with exposure to LPA7. In addition, endothelial, but not hematopoietic, deletion of Plpp3 increases vascular permeability in healthy mice and enhances sensitivity following inflammation-induced vascular leak8. Both pharmacological and genetic inhibition of LPA production attenuates the increased vascular leak observed in this model8. With respect to CAD, LPP3 expression in vascular endothelial cells is critical in maintaining an anti-inflammatory phenotype and vascular barrier integrity in regions of athero-protective blood flow10. In addition, LPP3 expressed in the liver of mice limits atherosclerosis progression and modulates systemic LPA levels11. Finally, treating mice susceptible to atherosclerosis, either through diet or peritoneal injections, with unsaturated LPA species prone to oxidation accelerates lesion formation compared to mice fed saturated LPA species12, 13. Cumulatively these data demonstrate LPP3 harbors a protective role against injury in the vasculature and that its regulation of bioavailable LPA may be a crucial mechanism.
Together these observations suggest a link between polymorphisms in PLPP3 and CAD and, specifically, that upregulation of LPP3 following injury may protect against the development and complications of CAD in humans. To functionally validate these observations, we sought to determine whether LPP3 expressed globally, or in a tissue-specific manner, influences the development of experimental atherosclerosis. Herein we present that the loss of Plpp3 globally or in vSMCs alone accelerates the progression of experimental atherosclerosis possibly in part via LPA signaling through the LPA receptor 4 (LPA4).
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All procedures conformed to the recommendations of Guide for the Care and Use of Laboratory Animals14 and were approved by the Institutional Animal Care and Use Committee. The production, initial characterization, and backcrossing of mice has previously been described15. In order to generate mice susceptible to atherosclerosis, Plpp3fl/fl animals (fl/fl) with or without the MX1-Cre or SMC-22 Cre (referred to as SM22-Δ) transgene were crossed to Ldlr−/− mice (The Jackson Laboratory; Bar Harbor, ME). Littermate pups with and without the MX-1 Cre transgene were treated with polyinosinic-polycytidylic acid (pI:pC; Sigma) between post-natal days 3 to 5 to activate the MX1 promoter (referred to as MX1-Δ). To generate Ldlr−/− mice lacking Plpp3 in myeloid cells, lethally irradiated animals (900rads) were reconstituted with bone marrow from fl/fl mice without or with LysM-Cre transgene (referred to as LysM-Δ). To simulate LDL receptor deficiency, mice lacking Lpar416 were backcrossed to the C57BL/6J background, and then bred to generate Lpar4Y/+ and Lpar4Y/− mice, which were injected with PCSK9D377Y.AAV (1×1011 genomic copies/mouse; provided by University of Pennsylvania, Penn Vector Core) before feeding Western diet. Mice were housed in cages with HEPA-filtered air in rooms on 12-hour light cycles, and fed Western diet (Research Diets, New Brunswick, NJ, D12079B) ad libitum for 12 – 20 weeks. Both male and female mice were used for the MX1 and Sm22 models of Plpp3 ablation and their data combined as no sex-specific differences were observed (Data not shown). Due to a lack of differences in these models, only male mice were used for the LysM bone marrow transplant model of Plpp3 ablation. Finally, because Lpar4 is on the X-chromosome, both male and female mice were used and reported separately. For procedural anesthesia, 2% isoflurane was delivered using the Harvard Apparatus Anesthetic Vaporizer. For euthanasia, mice were anesthetized with 5% isoflurane followed by cervical dislocation. This study was designed in adherence to the guidelines for experimental atherosclerosis studies as recommended in the AHA statement, which includes the use of our mouse models (including proper use of sample size and use of both sexes where appropriate), quantification of atherosclerosis, characterizing plasma lipids, statistical analyses and presentation of data17.
Quantification of Atherosclerosis and Immunohistochemistry
Atherosclerosis was quantified along the proximal aorta by en face technique and also by measuring lesion size at the aortic root by oil-red-O staining of tissue sections. Aortas were cleaned of the adventitia and dissected from the aortic root to the iliac bifurcation. Aortas were cut open longitudinally, exposing the intimal surface, and atherosclerosis was quantified on the intimal surface of the aorta by an en face technique and reported as proportion of total area. Aortic roots were serially sectioned (10 μm) starting ≈3mm from the base of the aortic root as described previously18. Sections were stained with filtered oil red-O (0.25% in 2-propanol), counterstained with hematoxylin, and oil red-O area was quantified in sections using ImageJ software19 and reported as lesion area.
Immunohistochemistry
Primary antibodies were LPP320, smooth muscle alpha-actin (2μg/mL; Abcam AB5694), CD68 (10μg/mL; Abcam AB53444), PECAM1 (5μg/mL; BD Biosciences 553370), and podocalyxin (2μg/mL; R&D Systems AF1556). In situ hybridization and proximity ligation assays were performed as previously described21. Imaging was performed using a Nikon A1R confocal microscope with a spectral detector (Nikon, Melville, NY). For lipid measurements, aortic roots were homogenized in 0.5mL of 0.1M HCl and extracted and GC-MS/MS performed with internal standards as described previously22. Using this method, S1P was undetectable in the proximal aortic tissue.
Gene Expression Analysis of Proximal Aortas
Proximal aortas were homogenized and treated with the Qiagen RNeasy Fibrous Tissue Mini Kit (Qiagen, Germantown, MD, ID:74704), and RNA (1 μg) was used for the generation of a cDNA library using MultiScribe™ Reverse Transcriptase (Life Technologies, Waltham, MA,4311235). cDNA was diluted 1:10 and used in a reaction with the TaqMan® Fast Universal PCR Master Mix (2X) (Life Technologies, Waltham, MA, 4364103) with VIC/FAM primers from Life Technologies designed for specific gene targets.
Plasma Analysis
Whole blood was collected into CTAD tubes supplemented with EDTA, and plasma separated by centrifugation (12,000 rpm x 4 min). Total plasma cholesterol was measured using the Wako Diagnostics Cholesterol E Assay (Wako Diagnostics, Richmond, VA, 999–02601) and plasma triglycerides were measured using the Wako Diagnostics L-type TG M Assay with the Multi-Calibrator Lipid Standard (Wako Diagnostics, Richmond, VA, 464–01601) according to the manufacturer’s protocol. Plasma lipoproteins were separated by fast-performance liquid chromatography on to a Superose 6 column (Sigma-Aldrich, St. Louis, MO) and was performed on individual mouse plasma samples (N=4/genotype). Cholesterol concentration in fractions was determined by enzymatic colorimetric assay (Wako Diagnostics, Richmond, VA).
Lysophosphatidic Acid Levels
LPA was measured in plasma and tissue by LC-MS/MS as previously described by our group23.
Isolation and Culture of Bone Marrow Derived Macrophages
To isolate bone marrow cells, femur and tibia were isolated and flushed with DMEM through a 25- gauge needle. Media-cell suspension was filtered through 70-μm nylon mesh and centrifuged at 1000×g for 5 minutes. Cells were plated at a concentration of 1 million cells per well in DMEM Glutamax containing 10% FBS, 1%PenStrep and 10% L929 media. Media was changed every 2 to 3 days to generate bone marrow-derived macrophages (BMDM). To isolate SMC, aorta was separated from surrounding tissue starting from the arch to the renal arteries. The aorta was digested and SMC cultured as previously described9. Primary human coronary artery SMC (Clonetics™, Lonza, Allendale, NJ) were treated with methyl-β-cyclodextrin (MβCD) cholesterol (20μg/mL) or vehicle for 48 hours for the generation of SMC-derived foam cells. 12-well transwell (Sigma, St Louis MO) plates with 8.0μm pore size were used for migration assays.
Statistical Analysis
All data are expressed as mean ± SEM unless otherwise noted. Data was classified as parametric if it passed Shapiro-Wilk normality test and Brown-Forsythe equal variance tests or nonparametric if either test failed. Statistical analysis was performed using an unpaired, two-tailed Student t-test unless otherwise noted. P-value <0.05 is regarded as significant. Two-way ANOVA and the Holm-Sidak post hoc test was used when multiple comparisons were made. Statistical analysis was performed using SigmaPlot 14.0 (Systat Inc., San Jose, CA). In cases where small sample sizes were used a power analysis was performed to determine the sample size required to detect differences of 30%. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
LPP3 is dynamically upregulated during atherosclerosis
The association of PLPP3 alleles with human CAD risk and their correlation with gene and LPP3 protein expression in vascular cells suggests that regulation of LPP3 levels could influence the development of atherosclerotic vascular disease. LPP3 expression is readily detected in human atheromas4. To enable a detailed histological comparison of LPP3 expression in healthy and atherosclerotic coronary arteries, we took advantage of experimental pig models in which we could compare normal coronary arteries and well-characterized coronary atherosclerosis resembling lesions in humans from normocholesterolemic or heterozygous familial hypercholesterolemic pigs, respectively24. Healthy porcine coronary arteries displayed relatively low levels of LPP3 expression (Supplemental Fig 1A, left). Coronary arteries from hypercholesterolemic animals fed a high fat diet exhibited more prominent LPP3 immunostaining that co-localized with SMC makers and that was present in other cells within the vessel wall (Supplemental Fig 1A, right). Next, we profiled LPP3 expression in two different murine models of atherosclerosis and observed prominent LPP3 expression in aortic root from both Apoe−/− mice (Supplemental Fig 1B) and in Ldlr−/− mice fed a Western diet in comparison to wild-type (WT) controls (Supplemental Fig 1C and D). In the atherosclerotic aortic root lesions, LPP3 expression colocalized with the endothelial markers PECAM1 (Supplemental Fig 1C and 1D) and podocalyxin (Supplemental Fig 1E), with CD68 (Supplemental Fig 1F), and with SMC marker SM α-actin staining (Supplemental Fig 1G). Upregulation of LPP3 protein, which is variably glycosylated as previously reported25, was detected by immunoblot analysis and was restricted to the atherosclerotic-prone proximal region of the aorta but not the descending thoracic aorta (Supplemental Fig 1H). Together, these findings confirm that LPP3 is dynamically upregulated during atherosclerosis.
Post-natal reduction in Plpp3 promotes atherosclerosis
Having established that LPP3 levels are increased in several vascular cell types in atherosclerotic arteries, we investigated the consequences of global reduction of LPP3 expression in experimental models of murine atherosclerosis. Disruption of Plpp3 in mice results in embryonic lethality due to defects in patterning and failure of the development of the extra-embryonic vasculature26, 27. Therefore, we used a post-natal targeting strategy to broadly reduce Plpp3 expression. Mice carrying a previously described floxed Plpp3 allele and Cre recombinase under the control of the Mx1 promoter were generated on the Ldlr−/− background8. Control (fl/fl) and Mx1-Cre expressing neonates were treated with pI-pC, to activate Mx1 directed recombination (MX1-Δ). Sustained reduction in LPP3 expression was confirmed by analysis of bone marrow cells from adult mice (Supplemental Fig 2A) with some attenuation of overall protein expression in proximal aorta (Supplemental Fig 2B). After consuming Western diet for 12 weeks both fl/fl and MX1-Δ mice displayed hypercholesterolemia (Supplemental Fig 2C) without differences in plasma lipoprotein-associated cholesterol profiles (Supplemental Fig 2D and 2E) or plasma triglyceride levels (Supplemental Fig 2F). We observed no differences in complete blood counts between fl/fl and MX1-Δ mice after 12 weeks on Western diet (Supplemental Figure 3A). En face examination of proximal aortas revealed a 30% higher atherosclerotic burden in the MX1-Δ mice (Fig 1A; P = 0.009). Similarly, oil red-O staining of the aortic sinus revealed a similar increase in lesion area in MX1-Δ mice (Fig 1B; p=0.028). Reduced LPP3 was accompanied by a nearly two-fold increase in CD68-positive lesion area (P<0.001; Fig 1C) and an accompanying increase in inflammatory markers ICAM (Fig 1D) and CD68 mRNA levels (P<0.05; Fig 1E) in proximal aorta of the MX1-Δ mice. Global reduction of LPP3 increased plasma LPA, with increases in both saturated and unsaturated LPA species (Fig 1F). The global loss of LPP3 also led to an approximately 2-fold increase in total LPA content in the proximal aorta (Fig 1G). Together, these findings support a role for LPP3 in attenuating inflammatory responses and atherosclerotic plaque development. However, the broad activity of the MX-1 promoter does not provide information about cell types contributing the observed protective effect of LPP3 against experimental atherosclerosis. Thus, we employed cell-type specific approaches to attenuate LPP3 expression.
Figure 1. Global reduction in postnatal Plpp3 expression accelerates atherosclerosis.
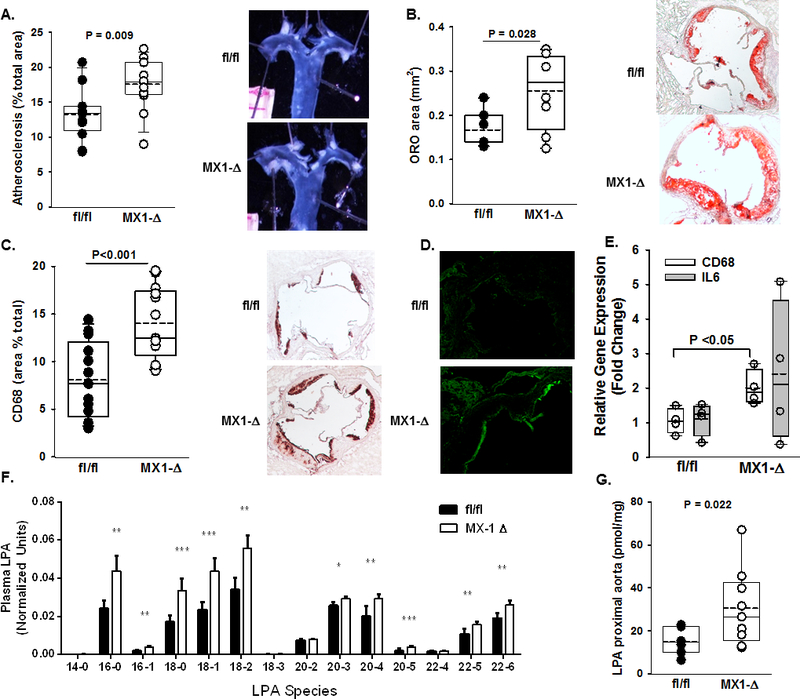
A) Left Panel: En face analysis of atherosclerosis lesion area (% of arch area) in proximal aortas (n=12–14/group). Right Panel: Representative proximal aortas from fl/fl and MX1-Δ mice on the Ldlr−/− background after 12 weeks on Western diet. B) Oil red-O staining of aortic sinus lesions reported as % area of aortic sinus (n=5–7/group). C) Left Panel: Immunohistological quantification of CD68 from sections of aortic roots from fl/fl and MX1-Δ mice on the Ldlr−/− background after 12 weeks on Western diet (n=12–14/group). Right Panel: Representative images of CD68 staining in the aortic sinus. D) Immunofluorescence staining of ICAM-1 in proximal aortic roots from fl/fl (top) and MX1-Δ (bottom) mice on the Ldlr−/− background after 12 weeks on Western diet. E) Cd68 and Il-6 gene expression (fold change; mean ± SEM) determined by qRT-PCR from proximal aortas of fl/fl and MX1-Δ mice on the Ldlr−/− background after 12 weeks on Western diet (n=4–5/group). F) Plasma LPA species quantification from fl/fl (black bars) and MX1-Δ (white bars) mice on the Ldlr−/− background after 12 weeks on Western diet (n=15/group). *P<0.05, **P<0.01, ***P<0.001. G) Total LPA content (pmol/mg tissue) in proximal aortas from fl/fl and MX1-Δ mice on the Ldlr−/− background 12 weeks after Western diet (n=7–9/group).
Myeloid LPP3 may not be a major regulator of atherosclerosis
We previously reported that levels of LPP3 in human monocyte/macrophages was extremely low and substantially increased in response to atherogenic oxidized low-density lipoprotein (ox-LDL) in a manner that is governed by CAD risk alleles4. However, the functional consequences of LPP3 expression in macrophages is presently unknown. We therefore investigated whether monocyte / macrophage expression of LPP3 regulates the extent of atherosclerosis in mice. As observed with peripheral blood monocytes from humans, Plpp3 expression in bone-marrow derived macrophages (BMDM) from control (fl/fl) mice is low and increased significantly following exposure to ox-LDL but not LDL (P<0.001; Fig 2A). BMDM isolated from mice with LysM-Cre driven deletion of Plpp3 (LysM-Δ) lacked LPP3 upregulation during ox-LDL stimulated foam cell formation. To determine if LPP3 regulates ox-LDL stimulated gene expression, responses of fl/fl control and LysM-Δ cells treated without or with oxLDL were compared. No difference in Cebpb expression occurred under any condition. Fos, transcriptional factor A mitochondrial (Tfam), and Cd36 expression were substantially upregulated in LysM-Δ as compared to fl/fl BMDM at baseline, and Cd36 was further amplified by environmental exposure to ox-LDL (Fig 2B). Having established dynamic regulation of LPP3 in murine BMDM and effects of LPP3 deficiency on ox-LDL stimulated gene expression in these cells, the role of myeloid LPP3 in experimental atherosclerosis was investigated by performing bone marrow transplant using fl/fl and LysM-Δ bone marrow cells into lethally irradiated Ldlr−/− mice that were subsequently fed a Western diet. After 12 weeks on Western diet, animals displayed characteristic hyperlipidemia with no differences in total plasma cholesterol or plasma LPA between the groups (Fig 2C and D). Development of atherosclerosis was also similar, without significant differences in the extent of atherosclerotic area by en face (Fig 2E) or by aortic root analysis (Fig 2F). These results indicate that although Plpp3 expression is dynamically regulated as macrophages assume a foam cell-like phenotype in vitro, myeloid LPP3 expression may not be a primary influencer of the development of experimental atherosclerosis.
Figure 2. Myeloid deficiency of Plpp3 does not alter atherosclerosis.
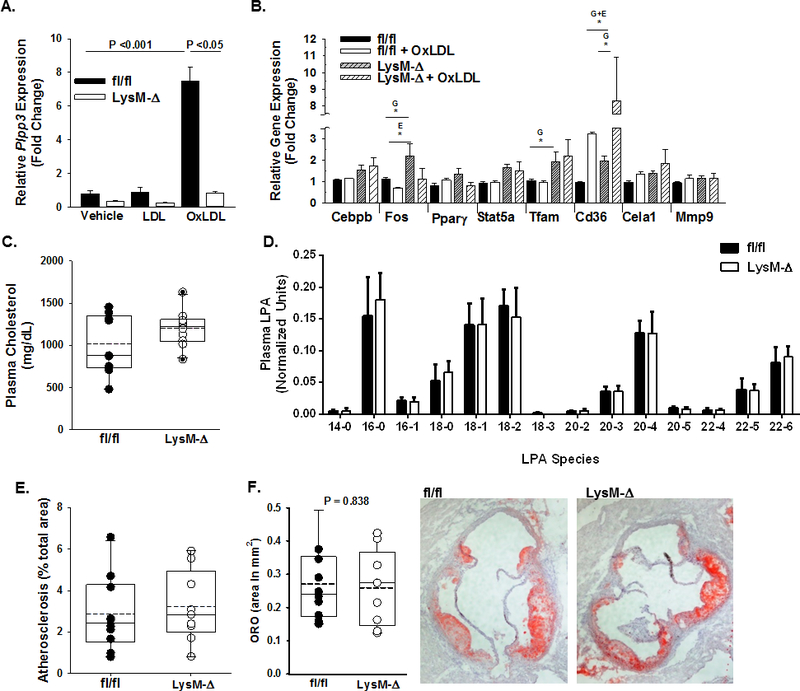
A) Plpp3 gene expression (fold change; mean ± SEM) in bone marrow-derived macrophages (BMDMs) from fl/fl and LysM-Δ mice (n = 3 – 5 per group) treated with human LDL (50 μg/mL), human oxLDL (50 μg/mL) or vehicle for 18 hours. Two-way ANOVA with Holm-Sidak pairwise multiple comparisons test was used to determine differences between treatment groups and genotype. B) Gene expression was profiled in BMDMs from fl/fl or LysM-Δ mice (n= 3 – 5/group) exposed to vehicle or human oxLDL. Two-way ANOVA with Holm-Sidak pairwise multiple comparisons test was used to analyze effects of genotype (G) and ox-LDL as an environmental stimulus for foam cell formation (E). C) Plasma cholesterol (mg/dL) in chimeric Ldlr−/− mice injected with bone marrow cells harvested from fl/fl (closed circles) or LysM-Δ (open circles) mice fed western diet for 12 weeks beginning 4 weeks after irradiation and reconstitution (n=10/group). D) Plasma LPA species quantification from fl/fl (black bars) and LysM-Δ (white bars) mice on the Ldlr−/− background after 12 weeks on Western diet (n=10/group). E) En face analysis of atherosclerosis (% of arch area) in Western-diet fed, chimeric Ldlr−/− mice with fl/fl (closed circles) or LysM-Δ (open circles) bone marrow cells (n=10/group). F) Oil red-O staining of aortic sinus lesions reported as % area of aortic sinus (n=10/group). *P<0.05
Loss of SMC LPP3 increases atherosclerosis
In both murine and porcine vessels, LPP3 was prominently upregulated in cells expressing SM α-actin in atherosclerotic lesions. Our previous work established a role for LPP3 in regulating SMC responses to the bioactive lysolipid LPA, in particular, LPP3 attenuates LPA-promoted migration and RhoA and ERK activation in isolated SMC, likely due to LPP3-mediated degradation of LPA7. The absence of SMC LPP3 in mice promotes the development of intimal hyperplasia in normolipidemic mice, suggesting that the enzyme normally attenuates events associated with SMC migration and proliferation after injury7. Based on this evidence implicating LPP3 in SMC function, we examined the impact of loss of SMC-derived LPP3 (SM22-Δ) on the development of atherosclerosis in Ldlr−/− mice fed a Western diet. Measurement of LPP3 expression in primary cultures of SMC from fl/fl and SM22-Δ Ldlr−/− aortas confirmed efficiency of the knock-down strategy (Fig 3A). After 12 weeks on Western diet, no differences in plasma cholesterol levels were observed in fl/fl and SM22-Δ mice (Fig 3B). Unlike our MX1 mediated model of reduced Plpp3, no change in plasma LPA levels were detected between SM22-Δ and fl/fl mice (Fig 3C). However, SMC-specific deletion of LPP3 resulted in an approximate doubling of the extent of atherosclerosis in SM22-Δ mice as compared to the fl/fl control mice measured by en face (P = 0.0002; Fig 3D) and confirmed in measurements of lesion areas in the aortic sinus by oil red-O staining (P=0.012; Fig 3E). SM22-Δ mice also displayed a significant increase in CD68 lesion area compared to fl/fl controls (P=0.006; Fig 3F). We observed a 3-fold higher expression of the inflammatory marker Il6 in SM22-Δ proximal aorta (P <0.05; Fig 3G). In keeping with the loss of local lipid degrading activity, aortic LPA content increased approximately 2-fold in the SM22-Δ tissue (P=0.011; Fig 3H).
Figure 3. SMC deficiency of Plpp3 accelerates atherosclerosis.
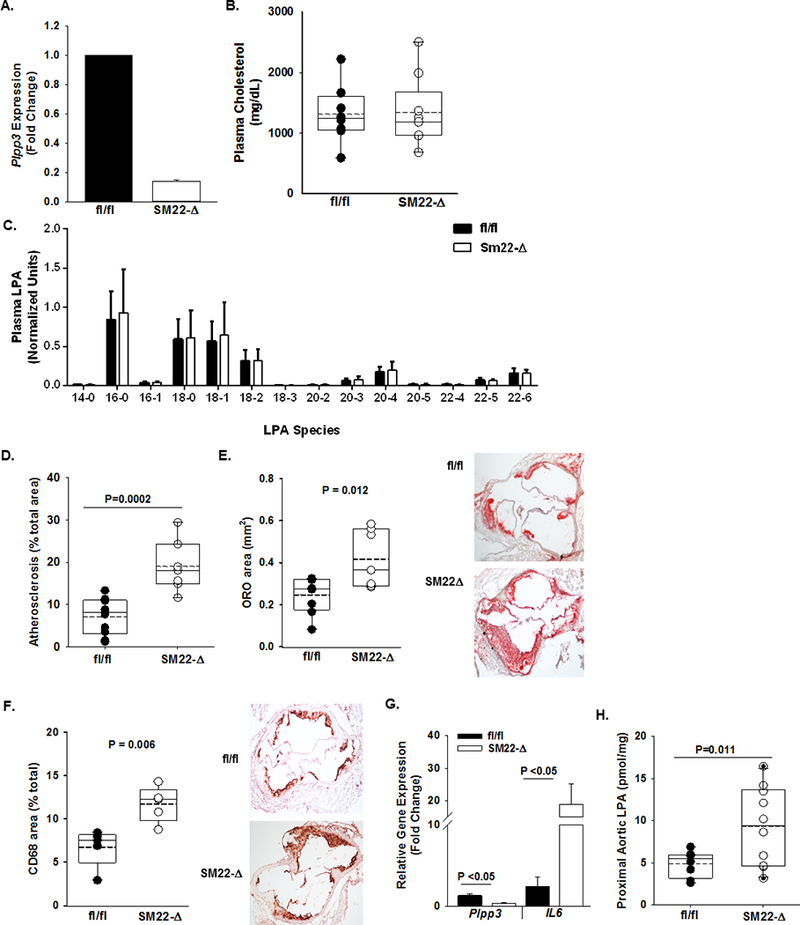
A) Plpp3 expression (fold change; mean ± SEM) from SMC isolated from fl/fl (dark) and SM22-Δ (open) mice on the Ldlr−/− background (n=3/group). B) At 6 – 8 weeks of age, fl/fl (dark circles) and SM22-Δ (open circles) mice on the Ldlr−/− background were placed on Western diet for 12 weeks and total plasma cholesterol (mg/dL) measured (n=7/group). C) Plasma LPA species quantification from fl/fl (black bars) and Sm22-Δ (white bars) mice on the Ldlr−/− background after 12 weeks on Western diet (n=9–11/group). D) En face analysis of atherosclerosis (% of arch area) (n=7–9/group). E) Oil red-O staining of aortic sinus lesions reported as % area of aortic sinus (n=5–6/group). F) Left Panel: Immunohistological quantification of CD68 from sections of aortic roots from fl/fl and MX1-Δ mice on the Ldlr−/− background after 12 weeks on Western diet (n=5–6/group). Right Panel: Representative images of CD68 staining in the aortic sinus. G) Plpp3 and Il-6 expression (fold change; mean ± SEM) in proximal aortas from fl/fl and SM22-Δ (n=5/group). H) Total LPA content (pmol/mg) in proximal aortas of fl/fl and SM22-Δ (n=7–9/group).
The results of tissue-specific targeting implicate SMC-derived, but not myeloid-derived, LPP3 as an intrinsic negative regulator of the development of atherosclerosis. Our earlier work suggests that the polymorphisms in humans associated with CAD risk influence upregulation of LPP3 in foam cells derived from monocytes. In particular, the protective allele results in higher LPP3 expression in macrophage foam cells. Cultured SMCs can assume a foam cell-like phenotype upon loading with cholesterol, and intimal SMCs contribute to a substantial proportion of the foam cell population in advanced murine atherosclerosis28–30. Based on our findings in myeloid-derived foam cells, we sought to determine whether LPP3 expression was dynamically regulated during phenotypic modulation of SMC after cholesterol uptake. Exposure of human aortic SMCs to cholesterol reduced the SMC marker ACTA2 and increased CD68 consistent with downregulation of SMC markers and the assumption of an inflammatory phenotype (P<0.05; Fig 4A). Additionally, SMCs exposed to cholesterol displayed a 2-fold increased expression of PLPP3 and more than a10-fold increase in LPA receptor 4 (LPAR4) (P<0.05; Fig 4A). Cholesterol-loading also significantly reduced cell migration in response to either serum or LPA (Fig 4B). SMC isolated from fl/fl and SM22-Δ mice respond to cholesterol loading by upregulating Plpp3, Cd68 and Lpar4 similarly to human aortic SMCs (P<0.05; Fig 4C). LPP3 deficiency attenuated upregulation of Cd68 and Lpar4 (Fig 4D and E). These findings suggest that LPP3 may influence the phenotype of SMC-derived foam cells. To determine if LPP3 expression regulated the ability of intimal SMC to assume properties of foam cells within atherosclerotic plaque, in situ hybridization and proximity ligation assays (ISH-PLA) were employed to visualize in tissue sections the H3K4dime marker of the myosin heavy chain 11(MYH11) locus that is restricted to the SMC lineage21. Approximately 40% of the CD68+ staining cells within atherosclerotic lesions of control mice displayed the H3K4dime mark, which persists in phenotypically modulated SMC (Fig 4F). No difference was observed in sections from SM22-Δ mice (Fig 4F). These results indicate that the absence of LPP3 does not fundamentally alter the phenotype transition of SMC to foam cells, as assessed by CD68 staining. Instead, LPP3 may regulate functional response of the cells to bioactive lipids, such as LPA.
Figure 4. PLPP3 expression is dynamically regulated during SMC phenotypic modulation.
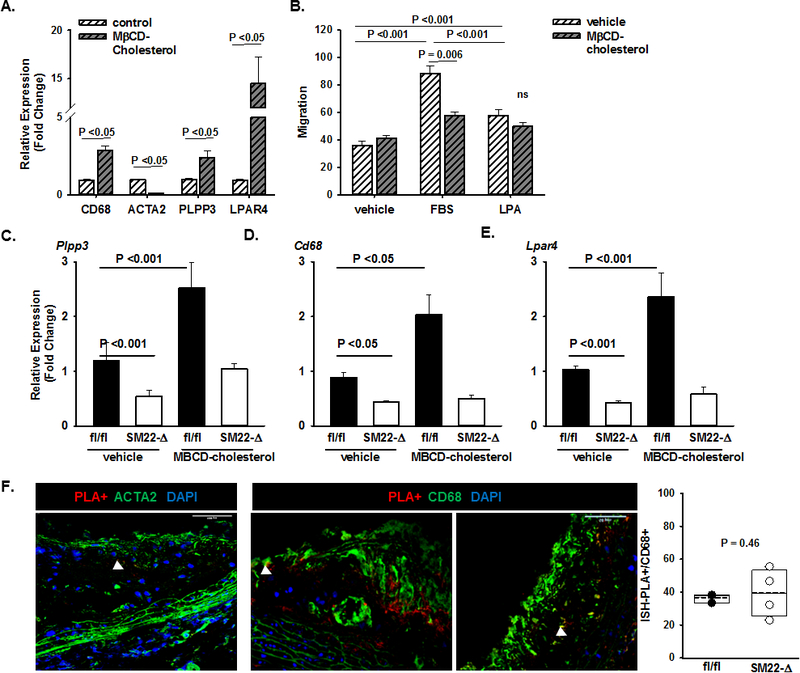
A) Expression of the indicated genes in primary human coronary artery SMC treated with vehicle control or methyl-β-cyclodextrin (MβCD) complexed cholesterol (20μg/mL) for 48 hours to induce modulation towards a foam-cell like phenotype. Relative gene expression (fold change; mean ± SEM) was determined. B) Cell migration to vehicle (DMEM with 0.01% FBS), FBS (DMEM with 10% FBS) or LPA (DMEM with 10μM 18:1 LPA) for 24 hours (right panel; mean numbers of migrated cells ± SEM). Two-way ANOVA was used to determine differences between treatment groups and chemotaxis agent. Relative gene expression (fold change; mean ± SEM) of C) Plpp3, D) Cd68 and E) Lpar4 in primary SMCs from fl/fl and SM22-Δ on the Ldlr−/− background treated with vehicle or MβCD-cholesterol (20μg/mL) for 48 hours. Two-way ANOVA was used to determine differences between treatment groups and genotypes. F) H3K4me2 marker of the MYH11 promoter (red) visualized by ISH-PLA in aortic root sections stained with ACTA (green) and DAPI (blue). Cells positive for the H3K4me2 marker as a percentage of total CD68+ cells (cells/field quantified) in aortic root sections from fl/fl and SM22Δ animals on the Ldlr−/− background fed Western diet for 12 weeks (n=2–4/group).
LPAR4 in involved in the development of atherosclerosis
Based on the findings observed in SMC, we next focused on the role LPAR4 in the development of atherosclerosis. Control or Lpar4-deficient mice were injected with PCSK9D377Y.AAV to lower LDLR expression and phenotypically mimic the Ldlr−/− background, and then placed on Western diet to elicit hypercholesterolemia31. Lpar4 is carried on the X chromosome in mice, so analysis of both sexes was performed. After 16 weeks on Western diet, total plasma cholesterol was similar regardless of genotype (Fig 5A), although overall lower levels were detected in female mice injected with PCSK9 virus. Despite similar levels of hyperlipidemia after PCSK9D377Y.AAV injection, both Lpar4−/− male (P=0.019) and female (P=0.011) mice were significantly protected from the development of atherosclerosis as measured by en face analysis (Fig 5B). Oil red-O staining of aortic sinus lesions revealed female Lpar4−/− mice were significantly protected from atherosclerosis compared to WT mice (P=0.008), and while male Lpar4−/− mice appeared to be protected from lesion formation the difference did not reach statistical significance (P=0.05; Fig 5C). The area of CD68 staining was statistically lower in aortic sinus lesions of male (P=0.046), but not female (P=0.11), Lpar4−/− mice compared to WT controls (Fig 5D) with a corresponding increase in the area of SMC α-actin staining (P=0.038; Fig 5E).
Figure 5. LPAR4 regulates experimental atherosclerosis.
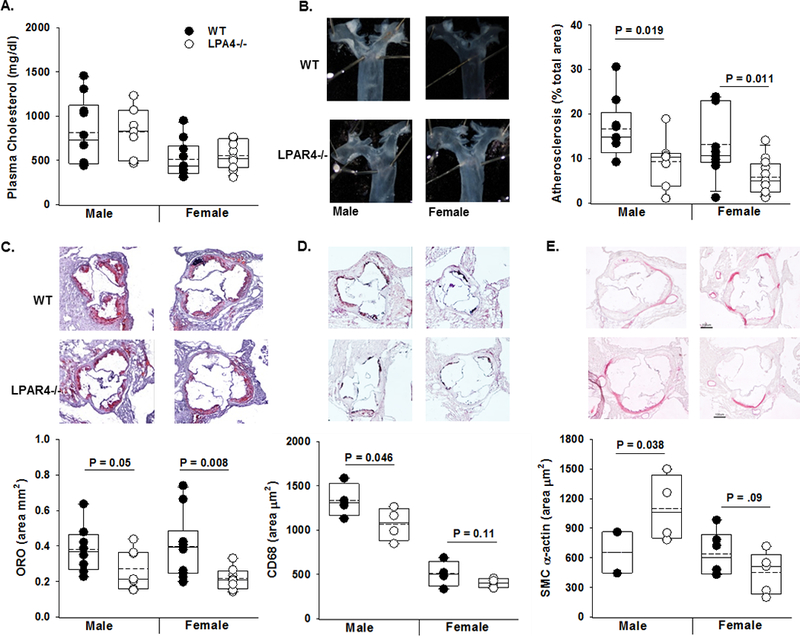
A) Six - 8-week-old wild-type (WT; closed circle) control or Lpar4−/− (open circles) mice were infected with PCSK9 adeno-associated virus. One week later, they were placed on Western diet for 16- weeks and total plasma cholesterol (mg/dL) (n=7–9/group). B) Left Panel: Representative images of proximal aorta. Right Panel: En face analysis of atherosclerosis (% of arch area) in male and female WT (closed circle) and Lpar4−/− (open circles) mice infected with PCSK9 adeno-associated virus and Western diet fed for 16- weeks (n=7–9/group). C) Oil red-O staining and lesion area (% total aorta in root section; n=7–9/group). D) CD68 immunostaining (n=4/group) and E) SM α-actin staining from aortic root sections of male mice (n=2–5/group).
Discussion
Heritable variant loci in PLPP3 that associate with lower LPP3 expression are strong predictors of CAD risk1. We previously reported both a local and distant eQTL effect in monocyte/macrophages and found that higher expression of LPP3 was a characteristic of oxLDL-induced foam cells from individuals with the protective allele4. In this report, we describe dynamic upregulation of LPP3 during the development of atherosclerosis in porcine and murine models of atherosclerosis. Herein, we report that LPP3 serves as an intrinsic negative regulator of plaque formation in experimental models of murine atherosclerosis and its loss either globally or in SMCs coincide with increased plaque macrophage cellularity, increased inflammatory cytokine expression and elevated LPA levels in atheromas. Importantly, Lpar4 expression is modulated by cholesterol loading in both human and murine SMCs and Lpar4−/− mice prone to atherosclerosis are protected against plaque formation suggesting that elevated LPA signaling may directly modulate CAD in mice. These findings provide functional validation and support a causal relationship of observations linking PLPP3 alleles with LPP3 expression and risk of CAD.
Global reductions in LPP3 expression using the Mx1-Cre system substantially increase atherosclerosis and are accompanied by elevations in plaque-associated LPA and inflammation. Interestingly, although LPP3 expression increases during oxLDL-induced phenotypic modulation of BMDM, myeloid Plpp3 does not appear to regulate the development of atherosclerosis. Instead, SMC expression of LPP3 is a major determinant of the extent of both atherosclerosis and LPA content in lesions. Importantly, the pro-atherosclerotic effects of targeting LPP3 with either Mx1-Cre or Sm22-Cre are independent of changes in circulating lipoprotein or plasma triglyceride levels.
We previously reported that SMC LPP3 is a negative regulator of neointimal formation in the absence of hyperlipidemia, in part due to an ability to attenuate inflammatory cell accumulation and SMC migration22. In SMCs, LPP3 regulates LPA-induced ERK activity, Rho activation, and migration, and lentiviral expression of human or mouse LPP3 (but not catalytically inactive variants) attenuates these effects22. These results imply that LPP3 may function within the vasculature to regulate responses to bioactive lipids, such as LPA. Herein, we expand these findings to demonstrate that LPP3 expression is upregulated in both human and murine primary SMCs as they assume a foam-cell like phenotype characterized by upregulation of CD68 and down regulation of SMC marker ACTA2 in response to cholesterol uptake. In this context, upregulation of LPP3 is associated with decreased FBS-stimulated SMC migration. We also demonstrate that LPP3 regulates LPA levels in atherosclerotic vessels, in that tissue LPA levels are higher from mice with reduced LPP3. Thus, the effect of LPP3 on inflammation may be a consequence of broad effects on extracellular bioactive lipid levels and hence signaling responses of multiple cell types during the development of atherosclerosis.
While LPA is not the only substrate of LPP3, in support of a role for LPA-mediated pro-atherosclerotic effects, we demonstrate that global deletion of Lpar4 in hyperlipidemic mice significantly reduced the development of atherosclerosis in the absence of an effect on plasma cholesterol. As to be expected with decreased atheroma size, we observed significantly less macrophage content in male Lpar4−/− mice; however, this effect was not observed in female mice. Additionally, we characterized SMC cell content in these knockout mice and observed a significant increase in SMC area in male Lpar4−/− lesions that also was not present in females. These data suggest that loss of Lpar4−/− may affect SMC proliferation and migration during atherosclerosis progression differently in males than females. Regardless of LPAR4’s role in SMC modulation during atherosclerosis, the overall result was protection from atherosclerotic lesion formation. LPA signaling can promote CD14 and SR-A1 expression in these cells which may influence the mechanisms linking oxLDL uptake and foam cell formation. Indeed, previous studies have reported an atherogenic role for LPA signaling via inducing endothelial expression of the monocyte adhesion molecule CXCL112. Zhou et al. report that treatment of mice with unsaturated but not saturated LPA species increases atherogenesis and macrophage accumulation in aortic sinus lesions that can be attenuated via treatment with an inhibitor to LPA receptors 1 and 312. These data are supported by another report wherein intestinally derived unsaturated LPA species increased atherosclerotic lesion size in mice relative to normal rodent diet and saturated LPA fed mice13. It is important to note that our MX1 model of Plpp3 reduction did increase circulating LPA content; however, there were no species-specific modulation of LPA by global loss of LPP3. Additionally, the loss of SMC-derived LPP3 accelerated atherosclerosis progression independently of changes in circulating LPA. Our findings reported here propose a novel LPA signaling pathway through LPAR4 that modulates atherosclerosis progression.
While our work focuses attention on SMC LPP3 and LPAR4 as regulators of atherosclerosis, expression of LPP3 in other cell-types may also attenuate development of atherosclerotic lesions. We also observed increases in endothelial LPP3 in atherosclerotic lesions. We and others have demonstrated that LPP3 controls endothelial barrier function8, 31 perhaps by spatial restriction of LPA receptor signaling32. LPP3 also modulates mechanotransduction in endothelial cells in an anti-inflammatory manner10, 33; thus, endothelial LPP3 expression may also protect against atherosclerosis progression. A recent report implicates hepatocyte LPP3 as a regulator of plasma LPA levels and experimental atherosclerosis11. These findings are intriguing because they suggest that LPP3 expression in the liver could have a systemic effect, likely by regulating levels of key bioactive lipids that stimulate atherosclerosis. However, in the models reported here, inactivation of mouse Plpp3 was not associated with significant differences in plasma lipid or lipoprotein profiles or, as observed in the SM22-model, circulating levels of LPA.
It is important to note that Sm22 expression has been observed in cells other than SMC, such as in monocytes and neutrophils in mice 34. Therefore, it is possible that the results observed are not the result of the lack of LPP3 in SMC. However, loss of Plpp3 in myeloid derived cells achieved with the LysM promoter had no impact on atherogenesis, suggesting that the primary effect of SM22-mediated knockdown of Plpp3 is unlikely to be mediated by loss of expression in granulocytes. Importantly, due to the fact that SM22-mediated Plpp3 deletion is constitutive, we cannot rule out that loss of SMC-derived LPP3 during development did not impact susceptibility of these mice to the development of atherosclerosis. Finally, it is worth mentioning that we did not directly test whether activation of the MX1-Cre recombinase alone modulated atherogenesis. While this cannot be excluded as a possibility, our model utilizing MX1-Cre mediated deletion of Plpp3 demonstrates increased plasma and atherosclerotic plaque LPA, the former of which has been previously reported as a mechanism driving atherogenesis11, 13.
Allelic variation of loci within the final intron of PLPP3 associates with inter individual differences in CAD risk. Functional genomic studies identify this as a dynamically regulated locus containing regulatory elements for C/EBP and AP1 family transcription factors. While these loci could act in trans to modulate expression of distal genes, our finding that Plpp3 deficiency exacerbates experimental atherosclerosis is consistent with the concept that cis actions of these loci on activity of the PLPP3 promoter account for or contribute to the CAD risk effect. Taken together, our results identify and validate the PLPP3 gene as an associated biological pathway that regulates CAD risk but is not obviously related to traditional risk factors, in particular those involving elevations in circulating cholesterol and triglycerides. While strategies to elevate PLPP3 expression to protect against atherosclerosis may not be feasible, our findings suggest that pharmacological targeting of LPA synthesis and signaling might offer an orthogonal approach to lipid lowering drugs for mitigation of CAD risk.
Supplementary Material
{kind=link}
Highlights.
LPP3 is augmented during atherosclerosis.
PLPP3 deficiency diminishes experimental atherosclerosis.
Deficiency of SMC LPP3 increases atherosclerosis.
Acknowledgements
PM: designed and conducted experiments, performed analysis, drafted manuscript.
LY: designed and conducted experiments, performed analysis, critically reviewed manuscript.
GM: designed and conducted experiments, performed analysis, critically reviewed manuscript.
MU: designed and conducted experiments, performed analysis.
JB: designed and conducted experiments, performed analysis, critically reviewed manuscript.
JV: designed and conducted experiments, performed analysis, critically reviewed manuscript.
TN: provided essential reagents, critically reviewed manuscript.
DEA: provided essential reagents, critically reviewed manuscript.
AJM: designed and analyzed experiments, drafted and reviewed manuscript.
SSS: designed and analyzed experiments, drafted and reviewed manuscript.
Sources of Funding
This project was supported by a grant from the Heart Lung and Blood Institute (R01HL120507), the National Center for Research Resources (P20RR021954) and by an IDeA award from the National Institute of General Medical Sciences (P20GM103527) of the National Institutes of Health. This material is the result of work supported in part with resources and the use of facilities at the Lexington VA Medical Center. This work was supported in part by VA merit Award # BX002769 (ATX) and BX001984 (SSS) from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program. PM was supported in part by a grant from the Heart Lung and Blood Institute (T32HL072743) and National Center for Advancing Translational Sciences (TL1TR000115).
Non-standard Abbreviations and Acronyms
- PLPP3
Phospholipid phosphatase 3
- CAD
Coronary artery disease
- LPP3
Lipid phosphate phosphatase 3
- LPA
Lysophosphatidic acid
- SMC
Smooth muscle cell
- SNPs
Single nucleotide polymorphisms
- CEBPβ
CCAAT enhancer binding protein beta
- LPPs
Lipid phosphate phosphatases
- ATX
Autotaxin
- LPC
Lysophosphatidylcholine
- pI:pC
Polyinosinic-polycytidylic acid
- ox-LDL
Oxidized low-density lipoprotein
- BMDM
Bone-marrow derived macrophages
- Tfam
Transcriptional factor A mitochondrial
Footnotes
Disclosures
The authors have no financial disclosures to report.
References
- 1.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M and al. e. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nature Genet. 2011:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun YXGC, Lu Y, Fu X, Jia JG, Zhao YJ, Li LD, Dui HZ, Zhang XY, Li ZY, Lei L, Zhang WF, Yuan YQ. Association between PPAP2B gene polymorphisms and coronary heart disease susceptibility in Chinese Han males and females. Oncotarget. 2017;8:13166–13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wirtwein MMO, Sjőgren M, Hoffmann M, Narkiewicz K, Gruchala M, Sobiczewski W. Relationship between selected DNA polymorphisms and coronary artery disease complications. Int J Cardiol. 2017;228:814–820. [DOI] [PubMed] [Google Scholar]
- 4.Reschen ME, Gaulton KJ, Lin D, Soilleux EJ, Morris AJ, Smyth SS and O’Callaghan CA. Lipid-induced epigenomic changes in human macrophages identify a coronary artery disease-associated variant that regulates PPAP2B Expression through Altered C/EBP-beta binding. PLoS Genet. 2015;11:e1005061. doi: 10.1371/journal.pgen.1005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth SSMP, Yang F, Brandon JA, Morris AJ. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erbilgin ACM, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ. Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res. 2013;54:1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panchatcharam M, Miriyala S, Salous A, Wheeler J, Dong A, Mueller P, Sunkara M, Escalante-Alcalde D, Morris AJ and Smyth SS. Lipid phosphate phosphatase 3 negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia. Arterioscler Thromb Vasc Biol. 2013;33:52–59. doi: 10.1161/atvbaha.112.300527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panchatcharam M, Salous AK, Brandon J, Miriyala S, Wheeler J, Patil P, Sunkara M, Morris AJ, Escalante-Alcalde D and Smyth SS. Mice with targeted inactivation of ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and permeability. Arterioscler Thromb Vasc Biol. 2014;34:837–845. doi: 10.1161/atvbaha.113.302335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panchatcharam M, Miriyala S, Yang F, Rojas M, End C, Vallant C, Dong A, Lynch K, Chun J, Morris AJ and Smyth SS. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. 2008;103:662–670. doi: 10.1161/circresaha.108.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu C, Huang RT, Kuo CH, Kumar S, Kim CW, Lin YC, Chen YJ, Birukova A, Birukov KG, Dulin NO, Civelek M, Lusis AJ, Loyer X, Tedgui A, Dai G, Jo H and Fang Y. Mechanosensitive PPAP2B Regulates Endothelial Responses to Atherorelevant Hemodynamic Forces. Circ Res. 2015;117:e41–e53. doi: 10.1161/circresaha.117.306457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Busnelli M, Manzini S, Hilvo M, Parolini C, Ganzetti GS, Dellera F, Ekroos K, Janis M, Escalante-Alcalde D, Sirtori CR, Laaksonen R and Chiesa G. Liver-specific deletion of the Plpp3 gene alters plasma lipid composition and worsens atherosclerosis in apoE(−/−) mice. Sci Rep. 2017;7:44503. doi: 10.1038/srep44503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J, Saulnier-Blache Jean S, Reinholz M, van Zandvoort M, Weber C and Schober A. Lipoprotein-Derived Lysophosphatidic Acid Promotes Atherosclerosis by Releasing CXCL1 from the Endothelium. Cell Metabolism. 2011;13:592–600. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Navab M, Chattopadhyay A, Hough G, Meriwether D, Fogelman SI, Wagner AC, Grijalva V, Su F, Anantharamaiah GM, Hwang LH, Faull KF, Reddy ST and Fogelman AM. Source and role of intestinally derived lysophosphatidic acid in dyslipidemia and atherosclerosis. J Lipid Res. 2015;56:871–887. doi: 10.1194/jlr.M056614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guide for the Care and Use of Laboratory Animals: Eighth Edition. Washington, DC: The National Academies Press; 2011. [PubMed] [Google Scholar]
- 15.Escalante-Alcalde D, Sanchez-Sanchez R and Stewart CL. Generation of a conditional Ppap2b/Lpp3 null allele. Genesis. 2007;45:465–469. doi: 10.1002/dvg.20314. [DOI] [PubMed] [Google Scholar]
- 16.Lee Z, Cheng CT, Zhang H, Subler MA, Wu J, Mukherjee A, Windle JJ, Chen CK and Fang X. Role of LPA4/p2y9/GPR23 in negative regulation of cell motility. Mol Biol Cell. 2008;19:5435–5445. doi: 10.1091/mbc.E08-03-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME and Virmani R. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/atv.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 18.Daugherty A and Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods. 2005;36:129–138. doi: 10.1016/j.ymeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Rasband WS. ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA: Retreived from https://imagej.nih.gov/ij/.1997-2016. [Google Scholar]
- 20.Sciorra VA and Morris AJ. Sequential actions of phospholipase D and phosphatidic acid phosphohydrolase 2b generate diglyceride in mammalian cells. Mol Biol Cell. 1999;10:3863–3876. PMC25685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG and Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/circresaha.114.302937. [DOI] [PubMed] [Google Scholar]
- 22.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM and Fisher EA. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/atvbaha.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraemer MP, Halder S, Smyth SS and Morris AJ. Measurement of Lysophosphatidic Acid and Sphingosine-1-Phosphate by Liquid Chromatography-Coupled Electrospray Ionization Tandem Mass Spectrometry. Methods Mol Biol. 2018;1697:31–42. doi: 10.1007/7651_2017_55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nichols TC, Merricks EP, Bellinger DA, Raymer RA, Yu J, Lam D, Koch GG, Busby WH Jr. and Clemmons DR. Oxidized LDL and Fructosamine Associated with Severity of Coronary Artery Atherosclerosis in Insulin Resistant Pigs Fed a High Fat/High NaCl Diet. PLoS One. 2015;10:e0132302. doi: 10.1371/journal.pone.0132302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smyth SS, Sciorra VA, Sigal YJ, Pamuklar Z, Wang Z, Xu Y, Prestwich GD and Morris AJ. Lipid phosphate phosphatases regulate lysophosphatidic acid production and signaling in platelets: studies using chemical inhibitors of lipid phosphate phosphatase activity. J Biol Chem. 2003;278:43214–43223. doi: 10.1074/jbc.M306709200M306709200 [pii]. [DOI] [PubMed] [Google Scholar]
- 26.Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN and Lynch KR. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J. 2009;419:611–618. doi: 10.1042/bj20081888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escalante-Alcalde D, Hernandez L, Le Stunff H, Maeda R, Lee HS Jr, Gang C, Sciorra VA, Daar I, Spiegel S, Morris AJ and Stewart CL. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development. 2003;130:4623–4637. doi: 10.1242/dev.00635. [DOI] [PubMed] [Google Scholar]
- 28.Bennett MR, Sinha S and Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/circresaha.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez D, Shankman LS, Nguyen AT and Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177. doi: 10.1038/nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Erh Jaw J, Sin DD, Seidman MA, Leeper NJ and Francis GA. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol. 2019:Atvbaha119312434. doi: 10.1161/atvbaha.119.312434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterjee I, Baruah J, Lurie EE and Wary KK. Endothelial lipid phosphate phosphatase-3 deficiency that disrupts the endothelial barrier function is a modifier of cardiovascular development. Cardiovasc Res. 2016;111:105–118. doi: 10.1093/cvr/cvw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yukiura H, Hama K, Nakanaga K, Tanaka M, Asaoka Y, Okudaira S, Arima N, Inoue A, Hashimoto T, Arai H, Kawahara A, Nishina H and Aoki J. Autotaxin regulates vascular development via multiple lysophosphatidic acid (LPA) receptors in zebrafish. J Biol Chem. 2011;286:43972–43983. doi: 10.1074/jbc.M111.301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Touat-Hamici Z, Weidmann H, Blum Y, Proust C, Durand H, Iannacci F, Codoni V, Gaignard P, Therond P, Civelek M, Karabina SA, Lusis AJ, Cambien F and Ninio E. Role of lipid phosphate phosphatase 3 in human aortic endothelial cell function. Cardiovasc Res. 2016;112:702–713. doi: 10.1093/cvr/cvw217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Z, Li C, Frieler RA, Gerasimova AS, Lee SJ, Wu J, Wang MM, Lumeng CN, Brosius FC 3rd, Duan SZ and Mortensen RM. Smooth muscle protein 22 alpha-Cre is expressed in myeloid cells in mice. Biochemical and biophysical research communications. 2012;422:639–642. doi: 10.1016/j.bbrc.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.