

Abstract
Relebactam is a small‐molecule β‐lactamase inhibitor developed as a fixed‐dose combination with imipenem/cilastatin. The pharmacokinetics of relebactam and imipenem across 10 clinical studies were analyzed using data from adult healthy volunteers and patients with bacterial infections. Renal function estimated by creatinine clearance significantly affected the clearance of both compounds, whereas weight and health status were of less clinical significance. Simulations were used to calculate probability of joint target attainment (ratio of free drug area under the curve from 0 to 24 hours to minimum inhibitory concentration (MIC) for relebactam and percentage of time the free drug concentration exceeded the MIC for imipenem) for the proposed imipenem/relebactam dose of 500/250 mg, with adjustments for patients with renal impairment, administered as a 30‐minute intravenous infusion four times daily. These dosing regimens provide sufficient antibacterial coverage (MIC ≤ 4 μg/mL) for all renal groups.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ The measure best linked to efficacy for carbapenems is the percentage of time the free drug concentration exceeded the minimum inhibitory concentration, and this value is approximately 6.5% for imipenem. For relebactam, the ratio of free drug area under the curve from 0 to 24 hours to minimum inhibitory concentration required to achieve bacteriostatic effect in the neutropenic mouse thigh model is approximately 5.2.
WHAT QUESTION DID THE STUDY ADDRESS?
☑ This study addressed the following question: What dose of imipenem/relebactam is required to achieve efficacy target criteria for the treatment of bacterial infections in patients with varying degrees of renal function?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Creatinine clearance was the covariate associated with the largest effect on exposure followed by weight and health status. A greater than 90% probability of achieving the imipenem/relebactam efficacy target criteria was calculated for the proposed imipenem/relebactam doses of 500/250 mg in normal patients and 400/200 mg, 300/150 mg, and 200/100 mg for mild, moderate, and severe renal impairment, respectively.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ These data may help to inform imipenem/relebactam recommended dosing for renal‐impaired patients (on the basis of creatinine clearance) if the dosing regimens are approved for use by regulatory authorities.
Relebactam is a small‐molecule β‐lactamase inhibitor active against classes A and C β‐lactamases that is being developed as a fixed‐dose combination with imipenem/cilastatin (PRIMAXIN, Whitehouse Station, NJ).1, 2 Imipenem is an approved carbapenem β‐lactam antibacterial agent that covers many gram‐negative organisms and certain gram‐positive organisms and anaerobes.2 Cilastatin alone has no antibacterial activity, but prevents the metabolism of imipenem by renal dehydropeptidase produced in vivo.
In vitro susceptibility and hollow fiber (HF) time‐kill studies found that relebactam restored the activity of subinhibitory concentrations of imipenem against imipenem‐resistant isolates.3, 4, 5 Animal studies further confirmed the activity of relebactam, and integrated translational pharmacokinetic/pharmacodynamic (PK/PD) modeling suggested that the combination of imipenem/relebactam would be efficacious against the majority of imipenem‐resistant strains at clinically achievable doses and concentrations.6, 7 From these studies, it was established that area under the curve (AUC)/minimum inhibitory concentration (MIC) was the primary driver of relebactam efficacy and that the target value for the free drug AUC from 0 to 24 hours to MIC (fAUC0–24 hours/MIC) ≥ 5.2 was needed to achieve static effect on tissue burden from an in vivo neutropenic mouse thigh model.6 For imipenem and other carbapenem antibiotics, the primary PK/PD driver is the percent of the dosing interval during which the free drug concentration exceeds MIC (fT > MIC).8 Based on the HF data, an imipenem efficacy target criteria of 6.5% fT > MIC was established as needed to achieve 2‐log kill of Pseudomonas aeruginosa and Enterobacteriaceae in the presence of relebactam.5
To inform robust dose selection, the clinical PK data in the targeted patient population need to be compared with preclinical targets to ensure that the majority of patients would achieve target exposure levels at the recommended dose and that any subpopulations requiring a dose adjustment are identified. Therefore, we conducted a population PK analysis of data from adult healthy volunteers and patients with complicated intra‐abdominal infection (cIAI), complicated urinary tract infection (cUTI), or hospital‐acquired/ventilator‐associated bacterial pneumonia (HABP/VABP). We aimed to estimate the population PK parameters of imipenem and relebactam following intravenous (i.v.) infusion and investigate the influence of covariates on drug exposure and resulting target attainment to support dose selection in patients with normal, impaired, and augmented renal function.
Methods
Data analysis
The analysis included data from 10 completed phases I–III clinical studies (Table 1). Plasma concentrations below the limit of quantification (BLQ) in the analytical assay were treated as missing and excluded, as the majority of such samples came from healthy volunteer studies at subclinical doses and at sampling points beyond the end of the clinical dosing interval. Unevaluable PK observations included those without recorded sampling times and those without a dosing event. No missing categorical data were available; therefore, imputations were not required. For the continuous covariates of creatinine clearance (CrCL) and body weight, < 15% of participants had missing information, and imputations with the population median value were implemented.
Table 1.
Summary of studies included in the analysis
| Protocol number | Clinical trial number | Design/objective | Number of participants/patient population | Imipenem doses | Relebactam doses |
|---|---|---|---|---|---|
| PN001 | NA | Phase I, four‐part, double‐blind, randomized, placebo‐controlled, sequential‐panel, rising single dose study, and a sequential panel, multiple‐rising‐dose study to evaluate safety, tolerability and PK of relebactam | 83 healthy males, 18–45 years of age | 500 mg or 500 mg q6h for 7 or 14 days | 25, 50, 125, 250, 500, 1,000, and 1,150 mg or 50, 125, 250, 375, 500, and 625 mg q6h for 7–14 days |
| PN002 | NA | Phase I, randomized, double‐blind, placebo‐controlled, single‐dose study in healthy elderly male, healthy elderly female, and healthy young female participants to evaluate safety, tolerability, and PK of relebactam | 18 healthy participants, 26–75 years of age | 500 mg | 125 mg |
| PN003 | NCT01505634 | Phase II, randomized (1:1:1), double‐blind, comparator‐controlled study to evaluate the efficacy (microbiological response at end of therapy) and safety of relebactam vs. imipenem in adult participants with cUTI | 261 hospitalized adults with cUTI, 18–90 years of age | 500 mg q6h for 4–14 days | 125 or 250 mg q6h for 4–14 days |
| PN004 | NCT01506271 | Phase II, randomized (1:1:1), double‐blind, comparator‐controlled study to evaluate the efficacy and safety of relebactam vs. imipenem in adult participants with cIAI | 313 hospitalized adults with cIAI, 18–88 years of age | 500 mg q6h for 4–14 days | 125 or 250 mg q6h for 4–14 days |
| PN005 | NCT01275170 | Phase I, open‐label, single‐dose study to evaluate safety, tolerability, and PK of relebactam in participants with impaired renal function | 30 participants with renal impairment, 34–75 years of age, 18 healthy matching control participants | 250 mg | 125 mg |
| PN007 | NA | Phase I, open‐label, multiple‐dose study to evaluate the relationship between the intrapulmonary PK of relebactam with respect to the intrapulmonary PK of imipenem in healthy participants | 16 healthy participants, 24–42 years of age | 500 mg q6h for 5 total doses | 250 mg q6h for 5 total doses |
| PN009 | NA | Phase I, single‐dose, double‐blind, randomized, placebo and positive‐controlled, three‐period, balanced crossover study under fasting conditions | 36 healthy participants, 22–55 years of age | 1,150 mg | |
| PN012 | NA | Phase I, randomized, placebo‐controlled, double‐blind, single‐dose and multiple‐dose trial of relebactam and imipenem/cilastatin in healthy Japanese male participants | 16 healthy male Japanese participants, 20–45 years of age | Panels A and B: 500 mg |
Panel A: 125 or 500 mg, or 500 mg q6h for 14 days Panel B: 250 mg or 250 mg q6h for 14 days |
| PN013 | NCT02452047 | Phase III, randomized (2:1), double‐blind, comparator‐controlled study to evaluate the efficacy and safety of relebactam vs. colistin (as CMS) + imipenem in participants with imipenem‐resistant infection | 50 hospitalized adults with HABP/VABP, cUTI, or cIAI, 19–80 years of age |
500/250 mg imipenem + relebactam (FDC) q6h + placebo q12h 300 mg CMS (LD) and 150 mg CMS q12h (MD) + 500 mg imipenem q6h for 5, 7, or 21 days |
|
| PN019 | NA | Phase I, open‐label, randomized, two‐period, crossover study | 14 healthy Japanese participants, 23–55 years of age | 500 mg | 250 mg |
cIAI, complicated intra‐abdominal infection; CMS, colistimethate sodium; cUTI, complicated urinary tract infection; FDC, fixed‐dose combination; HABP, hospital‐associated bacterial pneumonia; LD, loading dose; MD, multiple dose; NA, not available; PK, pharmacokinetics; q12h, every 12 hours; q6h, every 6 hours; VABP, ventilator‐associated bacterial pneumonia.
Data set preparation was performed using SAS (version 9.4; SAS Institute Inc., Cary, NC). Data exploration and data management were conducted using R (version 3.3.1; The R Foundation, Vienna, Austria). The analysis data set included all participants exposed to imipenem and/or relebactam with ≥ 1 measurable postdose concentration.
All study protocols included in the analysis were reviewed and approved by their respective institutional review boards. All studies were conducted in accordance with Good Clinical Practices, the Declaration of Helsinki, and other local regulations.
Modeling approach
The population PK analysis used a nonlinear mixed effects modeling (NONMEM) approach. The optimal structural and stochastic models were obtained using phase I data. The resulting structural (two‐compartment model of disposition with zero‐order i.v. infusion and first‐order linear elimination) and stochastic model (between‐subject variability [BSV] in clearance [CL], apparent volume of the central compartment [V 1], and peripheral compartment [V 2]) were retained, and data from phases II and III were then incorporated.
The population analysis was performed using NONMEM (version 7.3.0; ICON Early Phase, Gaithersburg, MD) executed on an Intel Fortran Compiler (version 12.0.4; Intel, Santa Clara, CA), and Perl‐speaks‐NONMEM (version 4.7.0). Postprocessing of the NONMEM output was conducted within R. Microsoft Excel 2016 (version 16.0; Microsoft, Redmond, WA) was used to output data summaries following the postprocessing of NONMEM output in R.
Covariate analysis
Before formal statistical covariate testing, an exploratory analysis was performed to visualize the relationship between covariate–parameter pairs (Table S1). There were no a priori assumptions regarding body size effects on PK, and a covariate modeling approach of enabling the data to drive model development was preferred. This was considered appropriate based on prior knowledge of the compounds and the large available data set. A stepwise forward selection and backward deletion procedure for covariate selection was implemented using an automated stepwise covariate modeling routine via Perl‐speaks‐NONMEM. Inclusion of covariates was based on statistical significance criteria (P < 0.01; e.g, change in minimum value of the objective function (ΔMVOF) = 6.63 for 1 degree of freedom (df) during the forward inclusion phase, and P < 0.001; e.g, ΔMVOF = 10.83 for 1 df during the backward deletion phase). For categorical covariates, ΔMVOF at the respective P values recognized the df introduced by the selected covariate.
The continuous covariates CrCL and body weight were investigated via power relationships centered on the median value.9 The effect of age on the PK of each compound was initially tested with a linear relationship, with subsequent testing of the power form if merited. Categorical covariates were modeled linearly as a proportional change.
Model qualification
The ability of the final population PK model to describe the observed data was evaluated using standard diagnostic plots and prediction‐corrected visual predictive check (pcVPC).10, 11 Diagnostic plots included observed vs. individual and population predicted concentrations and conditional weighted residuals vs. predicted or time. The shrinkage of the empirical Bayes estimates of the model parameters was evaluated for diagnostic purposes.12
A pcVPC was also performed to assess the model in which a comparison was made between the observed concentrations and model predictions.13 Using the population PK parameter estimates of the final population PK model, time profiles of concentrations in the data set were simulated in 1,000 replicates. For each time point, the 90% prediction interval of simulated concentrations was computed and compared with the observed concentrations. Observations and model predictions were normalized to the median population prediction for each bin to account for multiple covariates. Model performance was acceptable if the observed data were captured appropriately by the 5th, 50th, and 95th percentiles of the simulated data.
A nonparametric bootstrap analysis was performed to evaluate the stability of the model and generate confidence intervals for the model parameters. The bootstrap data sets (n = 1,000) were derived from the original data set through sampling with replacement, and PK parameter estimates from these data sets informed the median and 95% confidence intervals of the population PK parameter estimates.
Probability of target attainment simulations
Probability of target attainment (PTA) simulations employed the final population PK model and included BSV and residual error components. The simulations aimed to confirm the appropriateness of current dosing recommendations by determining the probability of PK/PD target attainment for both relebactam and imipenem (joint PTA). Simulations leveraged demographic data pooled across multiple antibacterial clinical programs (data on file) and relebactam phase II studies to generate a database representative of the intended patient population, which informed the simulation of 10,000,000 virtual participants in R. Virtual participants were randomly selected from this data set to generate populations (n = 1,000) of virtual individuals with normal renal function (CrCL 90 to < 150 mL/minutes); mild, moderate, and severe renal impairment (60 to < 90 mL/minutes, 30 to < 60 mL/minutes, and 15 to < 30 mL/minutes, respectively); end‐stage renal disease (ESRD; < 15 mL/minutes); and varying degrees of augmented renal function (150 to < 180, 180 to < 210, and 210 to < 250 mL/minutes). NONMEM simulation data sets were then created in R.
For selected dosing regimens (Table 2), joint PTA was assessed based on the established imipenem efficacy target criteria of 6.5% fT > MIC and the relebactam efficacy target criteria of fAUC0–24 hours/MIC ≥ 5.2.5, 6 Imipenem and relebactam free fractions in human plasma (0.80 and 0.78, respectively) were used to simulate unbound concentrations.
Table 2.
Doses of imipenem and relebactam chosen for PTA simulations
| Renal impairment category | CrCL range (mL/minutes) a | Dose of imipenem/relebactamb |
|---|---|---|
| Normal and augmented renal function | 90 ≤ CrCL ≤ 250 | 500/250 mg |
| Mild renal impairment | 60 ≤ CrCL < 90 | 400/200 mg |
| Moderate renal impairment | 30 ≤ CrCL < 60 | 300/150 mg |
| Severe renal impairment | 15 ≤ CrCL < 30 | 200/100 mg |
| ESRDc | < 15 | 200/100 mg |
CrCL, creatinine clearance; ESRD, end‐stage renal disease; PTA, probability of target attainment.
aCalculated using the Cockroft‐Gault equation.
bAdministered every 6 hours by intravenous infusion over 30 minutes.
cImipenem/relebactam to be administered only to ESRD patients on hemodialysis, and dose should be administered at intervals timed from the end of that hemodialysis session.
A sensitivity analysis was performed with relebactam fAUC0–24 hours/MIC ≥ 7.5 because this is the fAUC0–24 hours/MIC required to achieve 2‐log kill in an HF model.14 Historically, for imipenem monotherapy, ~ 30% fT > MIC is required to achieve 1‐log kill to 2‐log kill in in vivo animal models.15, 16 Therefore, a sensitivity analysis was also performed with imipenem 30% fT > MIC.
The upper clinical bound of exposure to imipenem was obtained from a steady‐state simulation performed in participants (n = 1,000) with normal renal function receiving 1 g imipenem every 6 hours (q6h), the highest dose per the imipenem/cilastatin label.2 Limits of exposure were derived by taking the 90th percentile PK end point (AUC0–24 hours, maximum plasma drug concentration [Cmax]) of the simulated distribution. The relebactam upper bound exposure was based on clinical experience with a 625 mg dose, the highest dose administered q6h for 7 days to healthy adult volunteers, which was safe and well tolerated.17 Therefore, a 625 mg dose was used for relebactam simulations using the same methodology as described for imipenem. To meet the safety criteria, > 90% of participants were required to remain below the threshold for both compounds. The upper clinical bounds were defined for relebactam (625/250 mg = 2.5) and imipenem (1,000/500 mg = 2.0) based on a ratio of the highest dose and clinical dose in participants with normal renal function.
The lower bound for imipenem was obtained by simulating PK profiles in participants (n = 1,000) at various doses administered q6h over 30 minutes. The lower bound dose equaled the dose at which exactly 90% of participants exceeded the target (at 6.5% fT > MIC, MIC of 4 μg/mL).18 The lower bound dose for relebactam was the dose at which exactly 90% of participants exceeded the AUC0–24 hours of 76.9 μM • h (derived from fAUC0–24 hours/MIC = 5.2 at MIC = 4 μg/mL).18
Current labeling for imipenem/cilastatin recommends either 500 mg q6h or 1 g every 8 hours (q8h) for bacterial infections that are suspected or proven to be susceptible in groups with normal renal function.2 Therefore, imipenem PTA was compared between these two dosing regimens using the methodology described in the previous section.
Results
Data analysis
Quantifiable plasma concentrations from 855 participants (815 imipenem, 649 relebactam) were included in the analysis. Participants provided 4,454 and 4,814 quantifiable plasma concentrations for imipenem and relebactam, respectively. The percentage of samples BLQ within this analysis was considered to be low at 11.8% and 12.5% for relebactam and imipenem, respectively; however, in the trials of phases II and III, BLQ samples accounted for only 1.0% and 1.4% of the relebactam and imipenem samples, respectively. Phase I trials had extensive PK sampling time points up to 24 hours postdose, and a majority of the BLQ samples were from subclinical relebactam and imipenem dose levels. Methods for dealing with BLQ data were therefore considered unnecessary.
Base model
Based on an exploratory data analysis and prior experience modeling imipenem and relebactam data following a phase II study,19 a two‐compartment zero‐order i.v. infusion model with first‐order linear elimination was selected as the structural model. Structural model development was performed using intensively sampled phase I data. Stochastic models to investigate BSV for each PK parameter was assessed in sequence. The most appropriate stochastic model to describe imipenem PK incorporated a log‐normal distribution for BSV in CL, V 1 and V 2. Likewise, the data supported similar BSV for relebactam PK. A proportional error model was selected to describe the residual variability. Goodness‐of‐fit plots confirmed the overall adequacy of the base model to describe the central tendency of the population data and individual data without bias. Next, sparsely sampled data from phase II and phase III data were incorporated into the base model. An evaluation of goodness‐of‐fit plots confirmed the appropriateness of the developed model for describing the patient data.
Covariate model
Study population characteristics are summarized in Table 3. Relebactam is predominantly renally excreted, and CrCL was identified as a significant covariate of relebactam CL (ΔMVOF, −242.22); this was the highest apparent correlation of all covariate–parameter relationships. Subsequently, weight was identified as a significant covariate of relebactam V 1 (ΔMVOF, −37.44). The least significant covariate–parameter pair was health status on relebactam CL, which was dropped during the backward deletion phase (ΔMVOF, −9.11).
Table 3.
Summary of the clinical and demographic data for study participantsa
| Characteristic | Range or N b | % | Medianc |
|---|---|---|---|
| Age (year) | 18–90 | NE | 51 |
| Weight (kg) | 39–180 | NE | 76 |
| CrCL (mL/minutes) | 8–406 | NE | 109 |
| CrCL categoryd | NE | ||
| CrCL < 15 mL/minutes | 5 | 0.6 | |
| CrCL mL/minutes ≥ 15 and < 30 | 9 | 1.1 | |
| CrCL mL/minutes ≥ 30 and < 60 | 70 | 8.2 | |
| CrCL mL/minutes ≥ 60 and < 90 | 199 | 23.4 | |
| CrCL mL/minutes ≥ 90 and < 150 | 439 | 51.5 | |
| CrCL mL/minutes ≥ 150 and < 180 | 103 | 12.1 | |
| CrCL mL/minutes ≥ 180 and < 210 | 18 | 2.1 | |
| CrCL mL/minutes ≥ 210 and < 250 | 4 | 0.5 | |
| CrCL mL/minutes ≥ 250 | 5 | 0.6 | |
| Gender | NE | ||
| Male | 519 | 60.7 | |
| Female | 336 | 39.3 | |
| Health status | NE | ||
| Healthy volunteer | 231 | 27.0 | |
| Patients | 624 | 73.0 | |
| Race | NE | ||
| White | 739 | 86.4 | |
| Black | 32 | 3.7 | |
| Asian | 44 | 5.2 | |
| Other | 40 | 4.7 |
CrCL, creatinine clearance; NE, not evaluated.
a855 participants (815 with imipenem measurements and 649 with relebactam measurements).
bRange (minimum–maximum) is shown for continuous covariates and N, number of participants and %, percentage of total for each category for categorical covariates.
cMedian value is shown for continuous covariates only.
dThree participants had missing CrCL values and therefore calculated statistics are shown for the remaining 852.
For imipenem, CrCL was also the most influential covariate of CL (ΔMVOF, −134.07). Other covariate–parameter relationships identified for imipenem were health status and body weight on V 1 (ΔMVOF, −56.10 and −25.77, respectively) and body weight on CL (ΔMVOF, −18.67). All covariates were retained in the model following backward deletion.
Visual inspection of unexplained BSV random effects vs. covariate plots demonstrated a reduction in unexplained BSV and a tendency toward a normal distribution of BSV centered around zero upon inclusion of the covariate effects (Figure S1 and S2). Meaningful evaluation of race as a covariate was not possible because of an insufficient number of “nonwhite” participants (Table 3); a post hoc analysis showed no obvious trends between unexplained BSV and race. The developed covariate model was further refined to capture the covariance between CL and V 1 in both compounds. The estimated correlation between CL and V 1 was 0.77 and 0.63 for imipenem and relebactam, respectively.
Final model
The final model (Supplementary Material [Link], [Link], [Link]) and bootstrap parameter estimates are presented in Table 4. All parameters in the final model were estimated with relative standard errors < 33%. Diagnostic plots show that the model predictions were scattered randomly around the line of unity, indicating an unbiased model (Figures S3 and S4) and good agreement with their respective population and individual predictions. The condition number (ratio of the largest to the smallest eigenvalue) was 260, indicating that the final model was not overparameterized (condition number < 1,000).
Table 4.
Final imipenem and relebactam model parameter estimates
| Parameter | Imipenem | Relebactam | ||||||
|---|---|---|---|---|---|---|---|---|
| Final model | Bootstrapa | Final model | Bootstrapa | |||||
| Estimate (RSE%)b | 95% CIc | Estimate (RSE%)b | 95% CIc | Estimate (RSE%)b | 95% CIc | Estimate (RSE%)b | 95% CIc | |
| CL (L/hour) | 12.53 (2.0) | 12.04–13.02 | 12.53 (2.0) | 12.08–13.04 | 7.02 (2.0) | 6.75–7.29 | 7.02 (2.0) | 6.74–7.31 |
| V 1 (L) | 15.83 (3.2) | 14.82–16.83 | 15.76 (3.4) | 14.78–16.85 | 11.08 (2.9) | 10.45–11.71 | 11.06 (2.9) | 10.46–11.68 |
| V 2 (L) | 5.84 (4.0) | 5.39–6.29 | 5.86 (3.9) | 5.44–6.32 | 6.41 (3.8) | 5.94–6.89 | 6.43 (3.6) | 5.95–6.89 |
| Q (L/hour) | 11.09 (6.5) | 9.68–12.49 | 11.15 (6.7) | 9.75–12.69 | 10.45 (6.8) | 9.04–11.85 | 10.48 (7.0) | 9.09–11.87 |
| Covariates on CL | ||||||||
| CrCL (power) | 0.46 (8.1) | 0.39–0.53 | 0.46 (8.0) | 0.38–0.53 | 0.75 (8.3) | 0.62–0.87 | 0.75 (8.3) | 0.63–0.86 |
| WT (power) | 0.33 (30.5) | 0.13–0.53 | 0.34 (32.2) | 0.13–0.55 | – | – | – | – |
| Covariates on V 1 | ||||||||
| WT (power) | 0.74 (19.1) | 0.46–1.01 | 0.76 (21.6) | 0.42–1.07 | 0.70 (15.8) | 0.48–0.92 | 0.72 (17.9) | 0.46–0.96 |
| Healthy | −0.29 (9.5) | −0.34 to 0.23 | −0.28 (10.0) | −0.34 to 0.23 | – | – | – | – |
| Random effects BSV | CV% (shrink)d | RSE% | CV%d | RSE% | CV% (shrink)d | RSE% | CV%d | RSE% |
|---|---|---|---|---|---|---|---|---|
| BSV in CL | 51.8 (5.1) | 9.3 | 51.7 | 9.5 | 45.0 (16.4) | 11.6 | 45.0 | 11.6 |
| BSV in V 1 | 74.4 (7.4) | 11.6 | 74.2 | 12.4 | 59.5 (18.8) | 11.9 | 59.4 | 12.5 |
| BSV in V 2 | 35.0 (52.9) | 32.7 | 34.8 | 32.8 | 41.1 (49.8) | 30.3 | 41.0 | 30.9 |
| Corr CL ~ V 1 e | 0.77 | 12.0 | 0.77 | 12.6 | 0.63 | 14.2 | 0.64 | 14.6 |
| Random effects BSV | ||||||||
| Proportional Error | 16.1 (19.0) | 6.7 | 16.1 | 6.7 | 15.3 (14.1) | 5.6 | 15.3 | 5.4 |
BSV, between‐subject variability; CI, confidence interval; Corr, correlation coefficient; CL, clearance; CV, coefficient of variance; NONMEM, nonlinear mixed effects modeling; Q, intercompartmental clearance; RSE, relative standard error; shrink, shrinkage; WT, body weight; V 1, apparent volume of the central compartment; V 2, apparent volume of the peripheral compartment.
aBootstrap is based on n = 1,000 data set replicates.
bMean parameter estimate (RSE% = [standard error/mean] × 100).
c2.5th and 97.5th percentile CIs.
dObtained according to the following equation: CV% = sqrt(ω2) × 100.
eCorrelation between variance parameters calculated as /sqrt().
Model qualification
The maximum likelihood estimate and the appropriateness of the parametric confidence intervals were confirmed by a bootstrap validation of the final model; 109 and 2 runs of 1,000 were discarded because of nonconvergence and parameter estimates near a boundary, respectively. The distributions of the bootstrap estimates for each of the different fixed and random parameters showed a near normal distribution with no difference between the mean estimate of the covariate model and the mean of the bootstrap results. A dose‐normalized pcVPC was also performed using the final model and is presented, stratified by health status, in Figure S5. Overall, the observations were reasonably captured by the simulated median and 5th and 95th prediction intervals.
Assessing clinical relevance of covariates
The magnitude of effect of covariates in the final model on AUC0–24 hours at steady state was assessed through simulations at the clinical doses for imipenem and relebactam of 500 mg and 250 mg, respectively. Figure 1 shows the magnitude of effect for each compound where the fold change in exposure represents a comparison of the mean AUC0–24 hours value of each covariate group to a reference group of participants with normal renal function and body weight of 70–90 kg. The effect of the covariate was judged not clinically relevant if the joint PTA was ≥ 90% and AUC remained within the established clinical upper bound. For imipenem, a lower bound of 0.60 was obtained as the ratio of the lower bound dose (300 mg) that resulted in exactly 90% PTA to the clinical dose (500 mg). For relebactam, the lower clinical bound was 0.4, taken as a ratio of the lower bound dose (100 mg) to a proposed clinical dose (250 mg) in participants with normal renal function.
Figure 1.
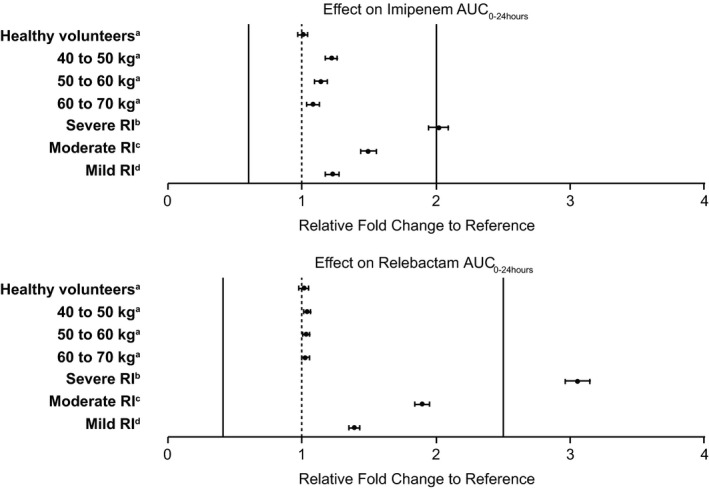
Impact of intrinsic factors on simulated steady‐state AUC 0–24 hours of imipenem and relebactam. aNormal renal function (CrCL 90 to < 150 mL/minutes); bCrCL 15 to < 30 mL/minutes; cCrCL 30 to < 60 mL/minutes; dCrCL 60 to < 90 mL/minutes. Unless otherwise specified, the weight for each group was 70–90 kg. The reference population refers to participants with normal renal function (CrCL 90 to < 150 mL/minutes) and weight 70–90 kg. Lower and upper clinical bounds are shown as solid lines for imipenem (0.6–2.0) and relebactam (0.4–2.5) based on imipenem 500 mg and relebactam 250 mg administered as a 30‐minute intravenous infusion dosed every 6 hours. AUC0–24 hours, area under the plasma concentration‐time curve from 0 to 24 hours; CrCL, creatinine clearance; RI, renal impairment.
The largest covariate effect predicted was based on CrCL in both compounds. The differences in fold exposure of imipenem were 1.22, 1.50, and 2.01 in mild, moderate, and severe renal function groups, respectively, compared with normal renal function. Fold changes in relebactam exposure were 1.38, 1.89, and 3.05 for the mild, moderate, and severe impairment groups, respectively. The magnitude of change in exposure observed due to weight was smaller with differences in fold exposure of imipenem of 1.22 (40–50 kg), 1.14 (50–60 kg), and 1.08 (60–70 kg). The impact of weight on relebactam exposure was not considered to be of significance with predicted fold changes of 1.03 (40–50 kg), 1.02 (50–60 kg), and 1.02 (60–70 kg). Health status did not affect the exposure (AUC0–24 hours) of imipenem or relebactam at steady state.
PTA simulations
The probability of achieving joint PTA was assessed by simulating drug exposures in participants using the proposed dosing shown in Table 2. As described previously, a virtual population database was derived and contained weight and CrCL (the two continuous covariates in the final PK model); pooled data were used to define the variance–covariance matrix between CrCL and body weight. A summary of the percentage of virtual participants achieving these targets are presented alongside the cumulative percentage distribution of isolates from the study for monitoring antimicrobial resistance trends (SMART) surveillance and clinical trials in Table S2. The simulation results are also shown superimposed on the percentage of P. aeruginosa and Enterobacteriaceae‐producing isolates reported in the 2015/2016 SMART global surveillance distribution data and clinical trial data from phases II/III in Figure 2. Simulation results suggest (Table S2) that the proposed dosing regimens would provide participants with sufficient coverage (for imipenem/relebactam MIC ≤ 4 μg/mL) in all renal impairment groups. The simulation‐derived upper exposure threshold limits were as follows: imipenem (AUC0–24 hours: 1959.5 μM • h; Cmax: 364.1 μM) and relebactam (AUC0–24 hours: 1655.2 μM • h; Cmax: 250.8 μM). Greater than 90% of participants remained below these upper thresholds across the renal function categories. The proposed dosing regimen is therefore expected to provide optimal efficacy and safety in renal impairment for the vast majority of patients.
Figure 2.
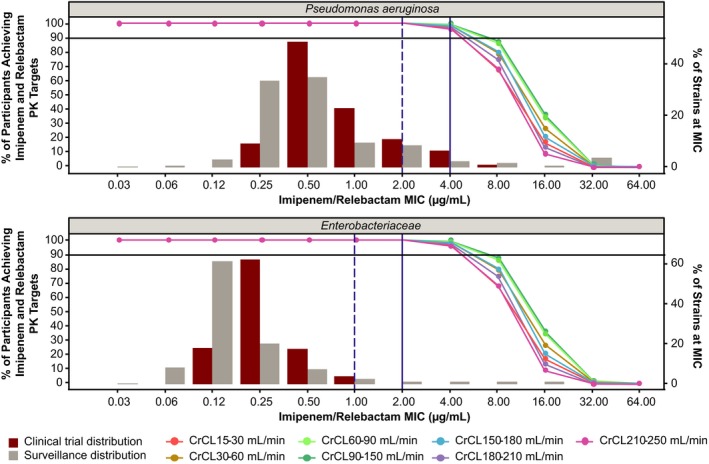
Percentage of participants achieving 6.5% fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam with Pseudomonas aeruginosa (top) and Enterobacteriaceae (bottom) MIC distributions among isolates relevant to proposed indication from clinical phases 2/3 and surveillance data. Simulations used the recommended renal dose adjustments that are detailed in Table 2. Solid and dashed vertical lines represent European Committee on Antimicrobial Susceptibility Testing and Clinical and Laboratory Standards Institute breakpoint MICs, respectively. Solid horizontal line represents 90% probability of target attainment. CrCL, creatinine clearance; fAUC0–24 hours/MIC, ratio of free drug area under the curve from 0 to 24 hours to MIC; fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic.
A sensitivity analysis using a higher PK/PD target for relebactam (fAUC0–24 hours/MIC = 7.5) demonstrated that joint PTA remains > 90% for strains with MICs ≤ 4 μg/mL across all renal function groups (data not shown). The sensitivity analysis based on increased target of 30% fT > MIC for imipenem also showed that joint PTA remained high and at approximately 90% across normal and renal‐impaired groups (Figure S6).
The PTA results comparing imipenem dosing regimens of 1 g q8h and 500 mg q6h are shown in Figure 3. For both regimens, the imipenem PTA was comparable and > 90% when considering 6.5% fT > MIC as the imipenem PK target; PTA for MIC of 4 μg/mL was 99.6% with 500 mg q6h and 99.8% for 1 g q8h, and the PTA for MIC of 2 μg/mL was 100% for both doses. For 30% fT > MIC as the imipenem PK target, the PTA for MIC of 4 μg/mL was 85.8% with 500 mg q6h and 94.6% for 1 g q8h, and the PTA for MIC of 2 μg/mL was 99.0% with 500 mg q6h and 99.6% for 1 g q8h. Considering high PTA, both imipenem regimens should similarly treat bacterial strains with MICs ≤ 4 μg/mL.
Figure 3.
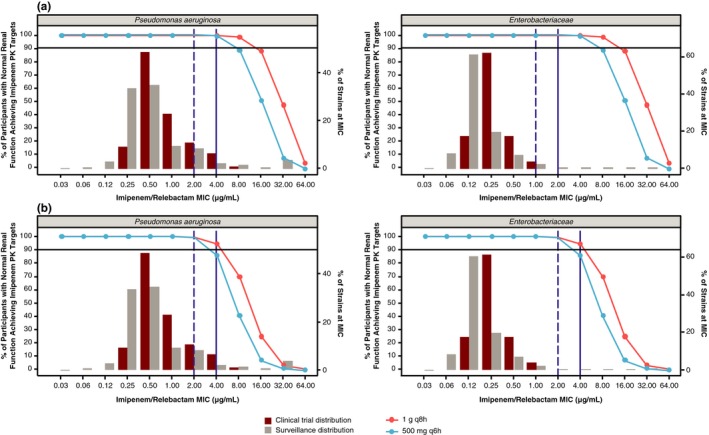
Percentage of participants achieving 6.5% fT > MIC (a) and 30% fT > MIC (b) for imipenem with Pseudomonas aeruginosa (left) and Enterobacteriaceae (right) MIC distributions among isolates relevant to proposed indication from clinical phases 2/3 and surveillance data. Solid and dashed vertical lines represent European Committee on Antimicrobial Susceptibility Testing and Clinical and Laboratory Standards Institute breakpoint MICs, respectively. Solid horizontal line represents 90% probability of target attainment. fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic.
Hemodialysis
In light of current label recommendations that imipenem/cilastatin be administered to ESRD patients undergoing regularly scheduled hemodialysis (HD) at intervals of q6h (after HD)2 and study PN005 results demonstrating high extraction coefficients for imipenem, cilastatin, and relebactam (data on file), simulations in ESRD participants were conducted assuming drug administration after HD. Simulations evaluated exposure on days 1, 2, and 3 of dosing in ESRD participants, consistent with current clinical practice where HD occurs at intervals of 2–3 days. These simulations demonstrated that with 3 days of continuous dosing in ESRD participants, the exposures would help maintain sufficient (> 90%) joint PTA for all days while being within the upper clinical bounds for both imipenem and relebactam (Figure S7). These results support a dosing regimen of 200/100 mg imipenem/relebactam q6h as a 30‐minute infusion after HD and at intervals timed from the end of that HD session.
Discussion
Population PK models were developed that describe imipenem and relebactam concentrations following single‐dose and multiple‐dose administrations to healthy volunteers and patients with cIAI, cUTI, and HABP/VABP. Structural and random model parameters were estimated with adequate precision, and for both compounds the final models (two‐compartment model of disposition with zero‐order i.v. infusion and linear first‐order elimination) were consistent with a priori expectations.19 Although pcVPCs from healthy volunteers showed greater variability (by 95th percentile confidence intervals) than the observed data, the variability was well described in the patient population from phase II and III studies, and simulations of patient data were used to derive dose recommendations.
The most significant covariate identified during the covariate model building procedure was renal function (by CrCL) for the clearance of both compounds. This was consistent with expectations given that a relatively high fraction of drug is renally excreted for relebactam and imipenem. Weight was a significant covariate of imipenem CL, but was accompanied with a smaller ΔMVOF compared with after the addition of CrCL (18.67 units). Given that weight is an input variable in the Cockcroft‐Gault calculation of estimated CrCL,20 this would suggest that there are additional weight effects not accounted for by CrCL alone. The impact of weight on estimated CrCL was also why standard allometric exponents were not applied to weight but were instead estimated directly. An alternative approach could have been to apply an allometric model and subsequently estimate the effect of CrCL. A previously published analysis ruled out the need for imipenem dose adjustments based on weight and concluded that adequate exposures can be achieved with dosing based on renal function, indicating that this effect is not clinically meaningful.19 For imipenem, patients were found to have a higher central volume of distribution (28.6%) than healthy volunteers. This is not an unexpected finding as patients hospitalized for bacterial infections, including those in the intensive care unit, often receive i.v. fluids that increase the volume of distribution.21 Weight was also identified as a statistically significant covariate of the V 1 of both compounds. The impact of these covariates in the final model on steady‐state AUC0–24 hours was assessed through simulations to understand their respective magnitudes of effect on exposure (Figure S4). The largest covariate effect predicted was renal function (CrCL) for imipenem (1.22‐fold to 2.01‐fold) and relebactam (1.38‐fold to 3.05‐fold; Figure 1), and thus was the only covariate that required a dose adjustment.
The final population PK model was used to perform simulations to examine the likelihood of achieving concentrations within a defined therapeutic window for both compounds. In all renal function categories, > 90% of participants were predicted to achieve the target criteria of 6.5% fT > MIC for imipenem and fAUC/MIC = 5.2 for relebactam up to a MIC of 4 μg/mL, the imipenem European Committee on Antimicrobial Susceptibility Testing susceptibility breakpoint for P. aeruginosa.18 Sensitivity analyses using higher PK targets for relebactam and imipenem demonstrated that joint PTA remains high and close to 90% for strains with MICs ≤ 4 μg/mL across normal and all renal impairment groups. In addition, at relevant susceptibility breakpoints of 2–4 μg/mL, little to no difference in PTA was observed between imipenem dosing regimens of 500 mg q6h or 1 g q8h; thus both dosing regimens are expected to provide sufficient coverage for strains with MICs ≤ 4 μg/mL. As additional clinical data are generated with this β‐lactam/β‐lactamase inhibitor combination in patients, it will be important to assess which of the PK/PD targets (e.g., those associated with stasis or 1‐log kill to 2‐log kill) are most closely associated with clinical outcome and thus provide the most relevant value for optimizing dosing. It is also important to consider differences between plasma and target tissue drug levels, such as peritoneal fluid levels for cIAI. For example, imipenem concentrations have been reported to be, on average, 25% lower in peritoneal fluid than in plasma, with considerable variability between patients.22 This demonstrates the challenge of using small studies of tissue PK—how to balance the benefits of understanding concentration at the target site with the need for a population‐level understanding of variability that comes from plasma PK. Alternative approaches may be needed to circumvent the shortcomings of the joint PTA approach, as joint PTA inherently assumes the timing of drug dosing is optimal and concurrent, such that each component achieving its respective exposure target is sufficient and largely ignores the underlying pharmacological interplay between agents. Such alternatives may include the use of a semimechanistic model that more elegantly captures the interaction between the components of the β‐lactam/β‐lactamase inhibitor regimen and could also provide further insight into PK/PD and dose optimization.23, 24 However, such approaches still require a linkage to clinical outcome data to provide full utility.
Our study provides a well‐defined population PK model with covariate effects for imipenem and relebactam. Simulations based on the validated model support imipenem/relebactam dose adjustment recommendations for renal‐impaired subpopulations on the basis of renal function (CrCL) to maintain sufficient joint PTA and maintain exposures below the upper threshold of drug exposure.
Funding
Funding for this research was provided by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Conflict of Interest
M.L., M.X., K.Y., and M.L.R. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. P.B. was a former employee of MSD at the time of study conduct and initial drafting of the manuscript. P.P., K.W., W.C., and P.K. are employees of Certara USA, Inc. or its subsidiaries, which provides consulting services to Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Author Contributions
P.B., P.P., M.L., K.W., W.C., M.X., P.K., K.Y., and M.L.R. wrote the manuscript. M.L.R. and M.L. designed the research. M.L.R. performed the research. P.P., K.W., W.C., and P.K. analyzed the data.
Supporting information
Figure S1. Between‐subject variability (ETA) vs. covariate plots for each covariate: CL parameter pair included in the final covariate model (right) in comparison with the model with no covariate relationships (left). Dashed line denotes smoothing line. Circles are individual observations of the relationship between ETA and covariates of interest. BSV, between‐subject variability; CL, clearance; CrCL, creatinine clearance; ETA, random effects.
Figure S2. Between‐subject variability (ETA) vs. covariate plots for each covariate: V 1 parameter pair included in the final covariate model (right) in comparison with the model with no covariate relationships (left). Dashed line denotes smoothing line. Circles are individual observations of the relationship between ETA and covariates of interest. In box plots, the box denotes the median and 25th and 75th percentiles, and the whiskers denotes the minimum and maximum values. BSV, between‐subject variability; ETA, random effects; HLTH, health status; V 1, apparent volume of the central compartment.
Figure S3. Diagnostic plots from final model: Imipenem. Dashed line denotes smoothing line and the solid line is unity. Circles are individual observations. CWRES, conditioned weighted residuals; h, hour.
Figure S4. Diagnostic plots from final model: Relebactam. Dashed line denotes smoothing line and the solid line is unity. Circles are individual observations. CWRES, conditioned weighted residuals; h, hour.
Figure S5. pcVPC (dose normalized) stratified by health status for imipenem and relebactam (semilogarithmic scale). Solid red lines represent median (50th percentile) observed concentration values. Dashed red lines represent the 5th or 95th percentile values for observations. Red circles = observed concentrations. Solid black lines represent median (50th percentile) model predicted concentration values. Dashed black lines represent the 5th or 95th percentile values for model predictions. Shaded areas represent the 95th CI around the 5th, median and 95th model predictions. CI, confidence interval; h, hour; pcVPC, prediction‐corrected visual predictive check; VPC, visual predictive check.
Figure S6. Percentage of participants achieving 30% fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam with Pseudomonas aeruginosa (left) and Enterobacteriaceae (right) MIC distributions among isolates relevant to proposed indication from clinical phases 2/3 and surveillance data. Solid and dashed vertical lines represent EUCAST and CLSI breakpoint MICs, respectively. Solid horizontal line represents 90% PTA. CLSI, Clinical and Laboratory Standards Institute; CrCL, creatinine clearance; EUCAST, European Committee on Antimicrobial Susceptibility Testing; fAUC0–24 hours/MIC, ratio of free drug area under the curve from 0 to 24 hours to MIC; fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic; PTA, probability of target attainment.
Figure S7. Percentage of participants with ESRD achieving 6.5% of fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam on days 1, 2, and 3 of dosing every 6 hours with 200 mg imipenem and 100 mg relebactam for 3 days. ESRD, end‐stage renal disease; fAUC0–24 hours/MIC, ratio of free drug area under the curve from 0 to 24 hours to MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic.
Table S1. Covariates investigated for their potential impact on the pharmacokinetic parameters of relebactam and imipenem. CL, clearance; CrCL, creatinine clearance (Cockcroft‐Gault); HLTH, health status (healthy or with infection); V 1, apparent volume of the central compartment; WT, body weight.
Table S2. Distribution of isolate data with PTA for 6.5% fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam. aFor the surveillance data, MIC of 32 μg/mL represents MIC values ≥ 32 μg/mL. ESBL, extended‐spectrum β‐lactamase; ESRD, end‐stage renal disease; fAUC/MIC, ratio of free drug area under the curve to minimum inhibitory concentration; fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PTA, probability of target attainment.
Supplementary Material S1. Model definitions.
Supplementary Material S2. Model code.
Supplementary Material S3. Model data set.
Acknowledgments
Medical writing and/or editorial assistance was provided by Robert Schupp, PharmD, CMPP, of The Lockwood Group, Stamford, CT, USA. This assistance was funded by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
References
- 1. Blizzard, T.A. et al Discovery of MK‐7655, a beta‐lactamase inhibitor for combination with Primaxin® . Bioorg. Med. Chem. Lett. 24, 780–785 (2014). [DOI] [PubMed] [Google Scholar]
- 2. PRIMAXIN® (imipenem and cilastatin) [prescribing information] . (Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Inc.,Whitehouse Station, NJ, 2018). [Google Scholar]
- 3. Hirsch, E.B. , Ledesma, K.R. , Chang, K.T. , Schwartz, M.S. , Motyl, M.R. & Tam, V.H. In vitro activity of MK‐7655, a novel beta‐lactamase inhibitor, in combination with imipenem against carbapenem‐resistant Gram‐negative bacteria. Antimicrob. Agents Chemother. 56, 3753–3757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Livermore, D.M. , Warner, M. & Mushtaq, S. Activity of MK‐7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 68, 2286–2290 (2013). [DOI] [PubMed] [Google Scholar]
- 5. Wu, J. et al Exploring the pharmacokinetic/pharmacodynamic relationship of relebactam (MK‐7655) in combination with imipenem in a hollow‐fiber infection model. Antimicrob. Agents Chemother. 62, e02323‐17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mavridou, E. , Melchers, R.J. , van Mil, A.C. , Mangin, E. , Motyl, M.R. & Mouton, J.W. Pharmacodynamics of imipenem in combination with beta‐lactamase inhibitor MK7655 in a murine thigh model. Antimicrob. Agents Chemother. 59, 790–795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rizk, M.L. When mechanism matters: impacting antibacterial drug development through application of mechanistic models. American Conference on Pharmacometrics 2017, Fort Lauderdale, FL, October 14–18, 2017.
- 8. Neuner, E.A. & Gallagher, J.C. Pharmacodynamic and pharmacokinetic considerations in the treatment of critically Ill patients infected with carbapenem‐resistant Enterobacteriaceae . Virulence 8, 440–452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holford, N.H. A size standard for pharmacokinetics. Clin. Pharmacokinet. 30, 329–332 (1996). [DOI] [PubMed] [Google Scholar]
- 10. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Guideline on reporting the results of population pharmacokinetic analyses (Doc. Ref. CHMP/EWP/185990/06). <https://www.ema.europa.eu/documents/scientific-guideline/guideline-reporting-results-population-pharmacokinetic-analyses_en.pdf> (2007). Accessed November 29, 2018.
- 11. United States Department of Health and Human Services, Food and Drug Administration . Population pharmacokinetics guidance for industry. Draft guidance. <https://www.fda.gov/media/128793/download> (2019). Accessed August 29, 2019.
- 12. Karlsson, M.O. & Savic, R.M. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 82, 17–20 (2007). [DOI] [PubMed] [Google Scholar]
- 13. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhagunde, P et al A translational pharmacokinetic/pharmacodynamic model to characterize bacterial kill in the presence of imipenem‐relebactam. Int. J. Infect. Dis. 10.1016/j.ijid.2019.08.026. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 15. Andes, D. & Craig, W.A. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19, 261–268 (2002). [DOI] [PubMed] [Google Scholar]
- 16. Drusano, G.L. Prevention of resistance: a goal for dose selection for antimicrobial agents. Clin. Infect. Dis. 36, S42–S50 (2003). [DOI] [PubMed] [Google Scholar]
- 17. Rhee, E.G. et al Pharmacokinetics, safety, and tolerability of single and multiple doses of relebactam, a beta‐lactamase inhibitor, in combination with imipenem and cilastatin in healthy participants. Antimicrob. Agents Chemother. 62 (2018). 10.1128/aac.00280-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The European Committee on Antimicrobial Susceptibility Testing . Breakpoint tables for interpretation of MICs and zone diameters. Version 9.0. <http://www.eucast.org> (2019). Accessed May 15, 2019.
- 19. Lala, M. , Brown, M. , Kantesaria, F. , Walker, B. , Paschke, A. & Rizk, M.L. Simplification of imipenem dosing by removal of weight‐based adjustments. J. Clin. Pharmacol. 59, 646–653 (2019). [DOI] [PubMed] [Google Scholar]
- 20. Cockcroft, D.W. & Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 16, 31–41 (1976). [DOI] [PubMed] [Google Scholar]
- 21. Blot, S.I. , Pea, F. & Lipman, J. The effect of pathophysiology on pharmacokinetics in the critically ill patient–concepts appraised by the example of antimicrobial agents. Adv. Drug Deliv. Rev. 77, 3–11 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Dahyot‐Fizelier, C. et al Kinetics of imipenem distribution into the peritoneal fluid of patients with severe peritonitis studied by microdialysis. Clin. Pharmacokinet. 49, 323–334 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Bulitta, J.B. et al Generating robust and informative nonclinical in vitro and in vivo bacterial infection model efficacy data to support translation to humans. Antimicrob. Agents Chemother. 63 (2019). 10.1128/aac.02307-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sy, S. , Zhuang, L. , Xia, H. , Beaudoin, M.E. , Schuck, V.J. & Derendorf, H. Prediction of in vivo and in vitro infection model results using a semimechanistic model of avibactam and aztreonam combination against multidrug resistant organisms. CPT. Pharmacometrics. Syst. Pharmacol. 6, 197–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Between‐subject variability (ETA) vs. covariate plots for each covariate: CL parameter pair included in the final covariate model (right) in comparison with the model with no covariate relationships (left). Dashed line denotes smoothing line. Circles are individual observations of the relationship between ETA and covariates of interest. BSV, between‐subject variability; CL, clearance; CrCL, creatinine clearance; ETA, random effects.
Figure S2. Between‐subject variability (ETA) vs. covariate plots for each covariate: V 1 parameter pair included in the final covariate model (right) in comparison with the model with no covariate relationships (left). Dashed line denotes smoothing line. Circles are individual observations of the relationship between ETA and covariates of interest. In box plots, the box denotes the median and 25th and 75th percentiles, and the whiskers denotes the minimum and maximum values. BSV, between‐subject variability; ETA, random effects; HLTH, health status; V 1, apparent volume of the central compartment.
Figure S3. Diagnostic plots from final model: Imipenem. Dashed line denotes smoothing line and the solid line is unity. Circles are individual observations. CWRES, conditioned weighted residuals; h, hour.
Figure S4. Diagnostic plots from final model: Relebactam. Dashed line denotes smoothing line and the solid line is unity. Circles are individual observations. CWRES, conditioned weighted residuals; h, hour.
Figure S5. pcVPC (dose normalized) stratified by health status for imipenem and relebactam (semilogarithmic scale). Solid red lines represent median (50th percentile) observed concentration values. Dashed red lines represent the 5th or 95th percentile values for observations. Red circles = observed concentrations. Solid black lines represent median (50th percentile) model predicted concentration values. Dashed black lines represent the 5th or 95th percentile values for model predictions. Shaded areas represent the 95th CI around the 5th, median and 95th model predictions. CI, confidence interval; h, hour; pcVPC, prediction‐corrected visual predictive check; VPC, visual predictive check.
Figure S6. Percentage of participants achieving 30% fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam with Pseudomonas aeruginosa (left) and Enterobacteriaceae (right) MIC distributions among isolates relevant to proposed indication from clinical phases 2/3 and surveillance data. Solid and dashed vertical lines represent EUCAST and CLSI breakpoint MICs, respectively. Solid horizontal line represents 90% PTA. CLSI, Clinical and Laboratory Standards Institute; CrCL, creatinine clearance; EUCAST, European Committee on Antimicrobial Susceptibility Testing; fAUC0–24 hours/MIC, ratio of free drug area under the curve from 0 to 24 hours to MIC; fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic; PTA, probability of target attainment.
Figure S7. Percentage of participants with ESRD achieving 6.5% of fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam on days 1, 2, and 3 of dosing every 6 hours with 200 mg imipenem and 100 mg relebactam for 3 days. ESRD, end‐stage renal disease; fAUC0–24 hours/MIC, ratio of free drug area under the curve from 0 to 24 hours to MIC; MIC, minimum inhibitory concentration; PK, pharmacokinetic.
Table S1. Covariates investigated for their potential impact on the pharmacokinetic parameters of relebactam and imipenem. CL, clearance; CrCL, creatinine clearance (Cockcroft‐Gault); HLTH, health status (healthy or with infection); V 1, apparent volume of the central compartment; WT, body weight.
Table S2. Distribution of isolate data with PTA for 6.5% fT > MIC for imipenem and fAUC0–24 hours/MIC = 5.2 for relebactam. aFor the surveillance data, MIC of 32 μg/mL represents MIC values ≥ 32 μg/mL. ESBL, extended‐spectrum β‐lactamase; ESRD, end‐stage renal disease; fAUC/MIC, ratio of free drug area under the curve to minimum inhibitory concentration; fT > MIC, time the free drug concentration exceeds the MIC; MIC, minimum inhibitory concentration; PTA, probability of target attainment.
Supplementary Material S1. Model definitions.
Supplementary Material S2. Model code.
Supplementary Material S3. Model data set.