

Abstract
Palmitic acid is a saturated fatty acid whose blood concentration is elevated in obese patients. This causes inflammatory responses, where toll-like receptors (TLR), TLR2 and TLR4, play an important role. Nevertheless, palmitic acid is not only a TLR agonist. In the cell, this fatty acid is converted into phospholipids, diacylglycerol and ceramides. They trigger the activation of various signaling pathways that are common for LPS-mediated TLR4 activation. In particular, metabolic products of palmitic acid affect the activation of various PKCs, ER stress and cause an increase in ROS generation. Thanks to this, palmitic acid also strengthens the TLR4-induced signaling. In this review, we discuss the mechanisms of inflammatory response induced by palmitic acid. In particular, we focus on describing its effect on ER stress and IRE1α, and the mechanisms of NF-κB activation. We also present the mechanisms of inflammasome NLRP3 activation and the effect of palmitic acid on enhanced inflammatory response by increasing the expression of FABP4/aP2. Finally, we focus on the consequences of inflammatory responses, in particular, the effect of TNF-α, IL-1β and IL-6 on insulin resistance. Due to the high importance of macrophages and the production of proinflammatory cytokines by them, this work mainly focuses on these cells.
Keywords: Palmitic acid, Saturated fatty acid, Obesity, Inflammation, Insulin resistance, Macrophage
Introduction
In developed countries, an overweight and obesity is a growing epidemiological problem. It is estimated that in the North America and Europe nearly 60% of the population is overweight and 15% are obese [1–3]. This percentage is increasing steadily for over 30 years.
Obese patients have significantly increased free fatty acid (FFA) levels in the blood [4–8]. The FFA includes palmitic acid (PA) and other fatty acids such as stearic acid, monounsaturated fatty acids (MUFA) (oleic acid) and polyunsaturated fatty acids (PUFA) (linoleic acid) [8, 9]. Consuming large amounts of saturated fatty acids (SFA), in particular PA, and metabolism disorders, increases the concentration of these fatty acids in the blood. This leads to inflammatory responses, which are an important factor in the development of diseases associated with obesity, for instance, insulin resistance [10].
PA induces inflammatory responses; however, it does so by activating different signaling pathways. Some pathways may interact, while others may only occur in specific cells. This work focuses mainly on macrophages, due to their high importance and role in proinflammatory cytokines production.
The impact of the palmitic acid on the cell functions
When PA gets into the cell, it is metabolized to saturated phospholipids (mainly to lysophosphatidylcholine) [11–14], diacylglycerol (DAG) [11, 15–18] and ceramides [17, 19–23].
In general, fatty acids such as MUFA are metabolized and then accumulated in the form of low-toxic triacylglycerol (TAG). However, a large amount of PA inhibits the TAG synthesis at the DAG stage, which is then accumulated in the cell. The exact mechanism of this process is still poorly understood. Probably the diglyceride acyltransferase (DGAT), an enzyme involved in the synthesis of TAG from DAG and acyl-CoA, has less activity when the substrates are saturated DAG and saturated acyl-CoA [20, 24, 25]. Another possible explanation is that PA induces the production of reactive oxygen species (ROS), which inhibits the DGAT2 activity [26].
Palmitate is mainly incorporated into DAG because its incorporation into TAG is reduced by the fall in the expression of DGAT2. It was shown that after 12 h incubation with 150 μM of PA, the DGAT2 mRNA expression was reduced in the murine proximal tubular epithelial cell model [27] and after 16 h of incubation with 500 μM PA in murine C2C12 myoblasts [28]. However, after 24-h incubation with 300–900 μM PA, the expression of DGAT1 and DGAT2 increased in anserine primary hepatocytes [29]. In turn, when they treated those hepatocytes with even higher concentration of PA, DGAT1 and DGAT2 expression started to decrease to the control level.
In addition to DGAT1 and DGAT2, PA does not change the activity of diacylglycerol kinases involved in DAG metabolism, which was shown in vascular smooth-muscle cells [30]. Further, DAG activates protein kinases C (PKC), which is important in TLR2 and TLR4 activation, as well as in the activation of nuclear factor κB (NF-κB).
It is known that DAG is the activator of conventional PKC (cPKC) and novel PKC (nPKC). However, different DAGs may activate different PKCs [31]. For example, PKCδ is poorly activated by 16:0/16:0-DAG, but strongly by other DAGs containing unsaturated fatty acids in its structure. In turn, PKCε is poorly activated by all DAGs. PKCθ, on the other hand, is strongly activated by all types of DAGs. In addition to the activation, also palmitoyl-CoA can cause acylation of PKC, which increases the activity of the kinases of this group [32]. However, DAG does not activate PKCζ [33]. Activators for this kinase are ceramides [34, 35]. Nevertheless, the importance of individual PKCs depends on the level of its expression in the tissue. PKCζ and PKCδ expression occurs in all tissues [36]. The expression of PKCε occurs mostly in the brain, kidneys and heart, and in other tissues, it is very low [36, 37]. In contrast, PKCθ expression is specific for muscle but is not expressed in adipocytes, macrophages or liver cells [38].
The PA, along with PKC activation, can also affect the endoplasmic reticulum (ER) stress (Fig. 1). PA is metabolised to phospholipids (mainly to lysophosphatidylcholine) and to the DAG. Both of these substances contain saturated hydrocarbon chains. High concentration of PA leads to saturated DAG and saturated lysophosphatidylcholine accumulation in the ER [14, 18, 39], which causes destructive changes in its structure. These changes are detected by transmembrane domain of inositol-requiring enzyme 1α (IRE1α) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) but not by activating transcription factor (ATF)6 [40–42]. This leads to the activation of ER stress sensors; however, under the influence of saturated lipids, there is no formation of large cluster with IRE1α, but only dimerization of this ER stress sensor. IRE1α is a kinase and endonuclease that catalyzes the excision of an intron from X-box-binding protein-1 (XBP-1) mRNA to form splice XBP-1 (XBP-1s) [14, 43].
Fig. 1.
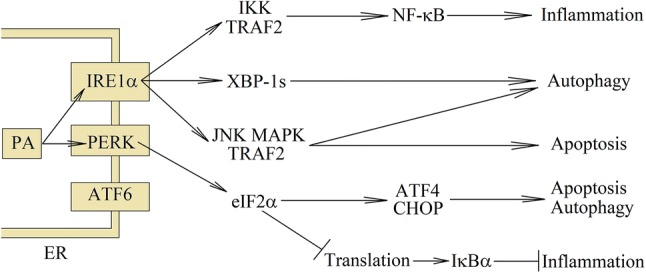
The consequences of PA-induced ER stress. At high concentrations, PA is converted to saturated lysophosphatidylcholine and DAG, which are incorporated into the ER. This causes ER stress and activation of ER stress sensors: IRE1α and PERK. The same pathways are activated during the detection of unfolded proteins. In particular, eIF2α phosphorylation, represses the translation of many genes with the exception of few, such as CHOP or ATF4. Then NF-κB is activated, which leads to apoptosis suppression and induction of inflammatory reactions. Activation of ER stress sensors is involved in increasing the capacity to maintain autophagy in stressed cells; however, severe ER stress leads to cell apoptosis
PERK, on the other hand, is a kinase that phosphorylates eukaryotic initiation factor 2α (eIF2α), what causes the repression of translation. Nevertheless, PERK activation by ER stress increases translation of C/EBP homologous protein (CHOP) and ATF4 and then CHOP- and ATF4-dependent autophagy genes, which are involved in increasing the capacity to maintain autophagy in stressed cells [14, 43–47]. Autophagy also depends on c-Jun NH2-terminal kinase (JNK)–mitogen-activated protein kinase (MAPK). Autophagy is a key mechanism to protect the cell against lipotoxicity. Nevertheless, the prolonged exposure of the cells to the lipotoxic environment causes the mammalian target of rapamycin (mTOR)-dependent autophagy inhibition [48] and leads to cell apoptosis. CHOP is involved in apoptosis induction in hepatocytes [49, 50]. Although activation of JNK–MAPK by glycogen synthase kinase-3β (GSK-3β) is also important in the induction of apoptosis [12, 49, 51, 52], the exact mechanism of GSK-3β activation through PA is still unclear. It can not only be activated by the ER stress, but also independently of the ER. Despite apoptosis induction, ER stress is important in the induction of inflammatory response [53, 54]. It activates NF-κB and NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome, which results in increased production of proinflammatory cytokines.
Various functions of mitochondria are strongly affected by PA treatment. Normally, ROS generation is relatively low, but in the presence of PA it increases significantly. This increase is mostly due to the partial inhibition of complexes I and complexes III of the respiratory chain [55–58]. Another effect of PA is the inhibition of the mitochondrial adenine nucleotide translocator activity, which causes accumulation of ATP in these organelles and increased production of ROS [59, 60]. ROS functions as specific second messenger that participates in the induction of inflammatory response, for example, it may trigger the activation of NF-κB.
The other lipids synthesized from PA are ceramides. Their increased amount causes an up-regulation in the expression and increased activity of neutral sphingomyelinase (nSMase) and serine palmitoyltransferase (SPT) [23]. Probably, it happens through the activation of NF-κB and increased expression of its downstream genes [61] or through the ER stress, which activates the IRE1α => XBP-1s pathway [62]. This results in an increased de novo synthesis of ceramides as well as increased release of sphingosine from the cell membrane, which is transformed into ceramides. Also, incubation of the cells with PA causes the accumulation of palmitoyl-CoA, which is a substrate for ceramide production. It seems that the production of de novo ceramides under the influence of PA is important for enhancing the signal transduction through TLR4 [21, 22].
Macrophages in obesity
One of the molecular symptoms of obesity is the occurrence of chronic low-grade inflammation. Macrophages play an important role in these processes in adipose tissue [63–65] and to a lesser extent in the liver [66]. This immune system cells can be tissue-resident macrophages such as microglia in the brain and Kupffer cells in the liver or be recruited into the tissues from the blood monocytes. In this process, monocytes differentiate and then polarize into specific macrophage phenotypes, depending on the factors acting on these cells [67]. There are two major macrophage sub-populations with different functions: inflammatory M1 and anti-inflammatory M2 macrophages. Functionally, the M1 macrophages produce pro-inflammatory cytokines and participate in the removal of pathogens and cancer cells. In turn, M2 macrophages produce anti-inflammatory cytokines, e.g., interleukin (IL)-10 and participate in the remodeling of the tissue during wound healing, regulation of the immune system and dampening of inflammation.
In lean animals and healthy people, resident macrophages in adipose tissue are polarized toward anti-inflammatory M2 state [64, 68–70]. Nevertheless, in obesity, adipocytes and these adipose tissue-resident macrophages produce chemokines that cause the recruitment of pro-inflammatory macrophages [64, 69–72]. This is a C–C motif chemokine receptor (CCR)2, CCR5, CCR7 and C–X3–C motif receptor 1 (CX3CR1) chemokine-dependent process. These pro-inflammatory macrophages localize in the environment of necrotic adipocytes, forming so-called crown-like structures [64, 71, 73] and start to produce pro-inflammatory cytokines [68]. Nevertheless, in humans, these macrophages have mixed phenotype [74]. The surface marker expression (integrin αvβ5, CD163, CD200, CD206, CD209, CD1b and CD1c) on these cells is similar to that on M2-polarized macrophages. At the same time, they show very high basal production of pro-inflammatory cytokines, even higher than in M1 macrophages. This mixed phenotype is associated with various factors that affect these macrophages. In adipose tissue, cell debris and free lipid droplets from necrotic adipocytes enhance inflammatory reactions and polarization of M1 macrophages [71, 73, 75]. Also in obesity, adipose tissue hypoxia induces pro-inflammatory M1 activation [76, 77]. Other pro-inflammatory factors affecting macrophages in advanced obesity are hyperglycemia [78, 79] and hyperinsulinemia [80, 81]. Nevertheless, in the described macrophages, there is a high expression of the peroxisome proliferator-activated receptor (PPAR)γ, which limits inflammation [82, 83].
Palmitic acid is a toll-like receptor agonist
In many studies, where specific inhibitors and siRNAs have been used, the results showed that TLR2 and TLR4 are activated by SFA, such as PA [61, 84–92] and lauric acid [93, 94]. The effect was especially visible in cells incubated for more than 12 h with a given SFA.
PA induces the activation and dimerization of TLR2 with TLR1, TLR2 with TLR6 or TLR4 [88, 90]. After TLR4 or TLR2 activation, the receptor dimerization and recruitment to lipid rafts take place. This is followed by signal transduction through MyD88 and NADPH oxidase activation [93–95]. The signal is transmitted through two pathways, the myeloid differentiation factor 88 (MyD88) => IL-1 receptor-associated kinase (IRAK) => TNF receptor-associated factor (TRAF)6 and the phosphatidylinositol 3-kinase (PI3K) => protein kinase B (PKB)/Akt pathway [93, 94]. As a consequence, it activates NF-κB. However, also the SFA-mediated TLR4 activation may initiate the MyD88-independent signaling pathway: TLR4 => IFNβ-mediated transcription factor (TRIF) => interferon regulator factor (IRF)3 (Fig. 2) [94]. The signaling mechanism induced by SFAs is mediated by TLR4, but activation of TLR4 requires complex formation with an accessory protein called myeloid differentiation protein 2 (MD2). PA, as well as natural TLR4 agonist, LPS, associates with the hydrophobic binding pocket of this TLR4 adaptor protein MD2, which activates signal transduction [96, 97].
Fig. 2.
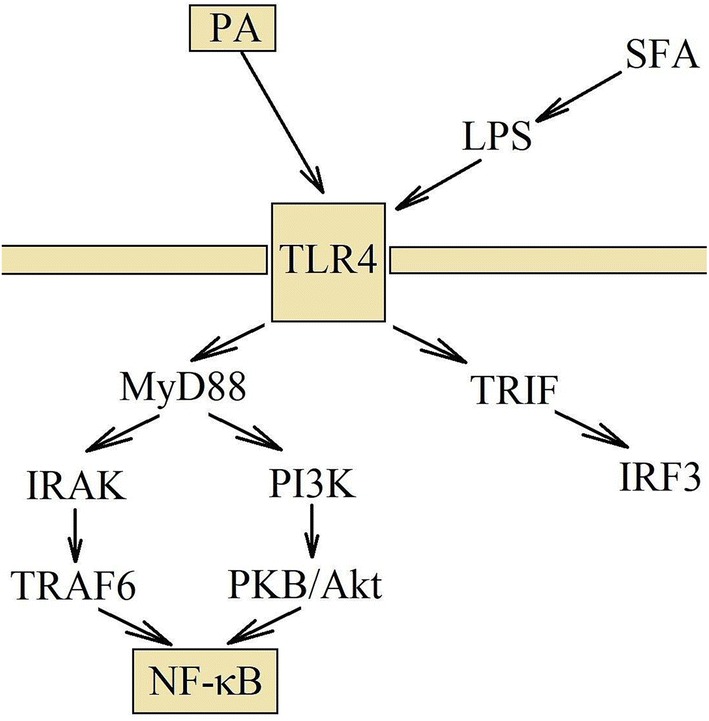
TLR4 activation via PA and signal transduction. PA activates TLR4 directly, but it can also activate this receptor indirectly. Consuming large amounts of fats causes disorder in intestinal functions, which leads to increased amount of LPS in the blood. After TLR4 activation, the signal transduction takes place via the IRAK and PI3K =>PKB/Akt pathways. They lead to the activation of NF-κB and the induction of inflammatory reactions
Notably, a number of groups have independently proposed that SFAs, including PA may also indirectly stimulate TLR-dependent signaling [98–100], especially after a very short exposure time of the cells to a given SFA [98]. Moreover, PA treatment at either time point induces only 8% of the genes induced by LPS [54]. Molecular simulation of PA interactions with TLR4-MD2 also questions whether PA is an agonist of this receptor [100]. In turn, some papers showed the TLR4-dependent effect of PA on inflammatory responses, but at a different time point than the effect of its agonist, LPS alone [101, 102]. This may indicate the indirect PA activation of TLR4, by increasing the production of some noncanonical TLR4 activators. The explanation may also be that PA enhances the signal transduction, or that TLR4 forms complexes with other receptors, e.g., with cluster of differentiation 36 (CD36), through which TLR4 may be activated by PA [103]. Nevertheless, it is possible that PA may directly activate TLR4, as well as indirectly by inducing the same signaling pathways causing ER stress and generation of DAGs and ceramides in the cell.
Palmitic acids produce activators for toll-like receptors
In addition to LPS, other substances can also activate TLR4. For example, extracellular ceramides in electronegative LDL or extracellular histones. Electronegative LDL is the LDL fraction whose blood levels are elevated in obese people [104]. Electronegative LDL presents a PLC-like activity [105], which is related to high ceramide content. This is important in inducing inflammatory reactions through CD14 and TLR4 in macrophages and monocytes [106–108]. Nevertheless, the incubation of hepatocytes with PA causes the release of very similar particles: extracellular vesicles, which contain ceramides. The hepatocyte exposure to PA causes the ER stress. It activates the IRE1α => XBP-1s pathway and thus increases the expression of SPT1. This results in increased de novo ceramide production, which is secreted in extracellular vesicles outside the cell [62]. The formation of the extracellular vesicles also depends on IRE1α activation. Extracellular vesicles, which contain ceramides, may be pro-inflammatory; however, this hypothesis requires confirmation and careful research.
Another possible mechanism for activating TLR by PA is the increase of extracellular histone release. It was already shown that PA induces the release of histone H3 from activated macrophage RAW 264.7 cell line [109], but this process is not related to cell death. PA induces the release of histone H3 from macrophages, in part, through the ROS generation and the JNK–MAPK signaling pathway [109]. Extracellular histones directly bind and activate TLR2 [110–113], TLR4/MD2 [110–113] and TLR9 [114]. Although histones in the complex with DNA activate TLR much better than histones alone [111], this TLR activation causes MyD88-dependent activation of NF-κB and thus increases the production of proinflammatory cytokines. Extracellular histones induce the expression of adhesion molecules, intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), in endothelial cells [109]. This results in the trafficking of monocytes and macrophages across the vessel wall and recruitment of these cells to the tissues. However, it is postulated that extracellular histones are actually a component derived from neutrophil extracellular traps (NET) [115]. NET is the process by which immune cells, mainly neutrophils, defend the organism against pathogens. It involves the release of the cell nucleus content or mitochondrial DNA, outside of the cell. Due to the fact that histones are proteins associated with DNA, in this process, they are removed out of the cell as well.
Indirect effect of palmitic acid on toll-like receptor 4 activation: increase in lipopolysaccharide levels
PA may indirectly act on TLR4. In particular, it may help to activate this receptor by increasing the amount of LPS in the blood. High lipid concentration in the intestines causes impairment in intestinal barrier function [116, 117]. This facilitates the passage of bacteria and LPS through the intestinal wall. LPS goes to chylomicrons and through the portal vein enters the bloodstream [118]. At the same time, in obese and diabetic people, there are changes in the composition of gut microbiota, which affect the function of the intestines and the amount of LPS entering the blood [116, 119, 120]. Hence, consuming food with a large amount of SFA, and with a small amount of fiber and 3-n PUFA, causes an increase of LPS concentration in the blood [121–124]. Also people with obesity, atherosclerosis or type 2 diabetes mellitus (T2DM) have increased concentration of LPS in their blood [121, 125–128]. Consumption of large amounts of PA results in increased levels of PA and LPS in the blood. Due to the fact that LPS is a TLR4 agonist, large amounts of PA may indirectly activate TLR4.
Indirect effect of palmitic acid on toll-like receptors activation
In the cells, PA can act in different ways, activating many TLR-dependent signaling pathways. As a consequence, TLR can be repeatedly activated during multi-day treatment of the cells with this SFA [87]. PA may increase the mRNA expression and protein levels of TLR4, which enhances the signal transduction of this receptor [129–131]. It can also support the activation of TLR4. After activation by LPS, TLR4 is translocated into the lipid raft to assemble the complex involved in signal transduction. At this first stage, most important is the composition of lipids in the cell membrane, in particular, endogenous cholesterol synthesis. Thanks to the fact that PA, the same as LSP, increases the expression of the fatty acid synthase (FAS), and thus increases production of substrates for the production of cholesterol and facilitates the activation of TLR4 [132, 133].
Role of protein kinase C in toll-like receptor-mediated signaling pathway
After LPS-mediated TLR4 activation, a signal transduction occurs. This results in the activation of NF-κB and an increased production of proinflammatory cytokines. PKC is involved in the transmission of this signal. PKCζ is important in the translocation of TLR4 into the lipid rafts and NF-κB activation, which was proofed in the myometrial cell model [134], THP-1 macrophages [135] and human peripheral blood monocytes and macrophages [136]. After the activation of TLR4, PKCζ is activated by RhoA. Next, the PKCζ activates the transforming growth factor beta-activated kinase (TAK)1, which is then involved in the activation of NF-κB. Identical signal transduction occurs via TLR2 [137]. However, experiments on RAW264.7 macrophages showed that TLR2 and TLR4 activate PKCδ which then binds to Toll–Interleukin 1 Receptor Domain Containing Adaptor Protein (TIRAP)/Mal. This is important in the p38 MAPK and NF-κB activation [138]. Also in the same macrophages, activation of TLR4 (through the MyD88-depending pathway) causes binding and phosphorylate of PKCε, which is important in the activation of NF-κB [139]. Because PA increases the concentration of PKC activators: DAG and ceramides, it can enhance, by synergy effect, the TLR4- and TLR2-mediated signaling pathways [22, 140, 141].
Significance of endoplasmic reticulum stress in toll-like receptor-mediated signaling pathway
One of the ER stress sensors activated by TLR is IRE1α. Interestingly, TLR does not activate other ER stress sensors, such as ATF6 nor PERK [142]. TRAF6 plays a key role in TLR-mediated IRE1α activation by catalyzing IRE1α ubiquitination and blocking the recruitment of protein phosphatase 2A (PP2A) (Fig. 3) [143]. Phosphorylation of IRE1α and thereby increased activation of this protein result in the XBP-1 mRNA splicing, which results in the creation of XBP-1s. Activation of this pathway does not cause the expression of the ER stress proteins, or even decrease the activation of ATF6 and PERK. The consequence of TLR-induced IRE1α activation is the production of proinflammatory cytokines [53, 142, 143], in particular IL-1β and CC motif chemokine ligand (CCL)5/regulated on activation, normal T cell expressed and secreted (RANTES) and, in part, tumor necrosis factor-α (TNF-α).
Fig. 3.
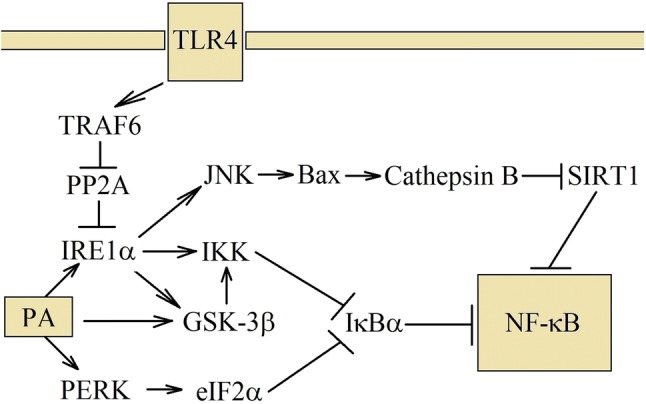
Mechanism of NF-κB activation by PA. PA and TLR4 share some of the signaling pathways. Both activates IRE1α, but in a different way. TLR4 activates this ER stress sensor via TRAF6. In turn, activation through PA depends on the damage of ER membranes and incorporation into them. Then IRE1α activates JNK–MAPK pathway, which destabilizes the lysosomes. Cathepsin B is released, which is involved in the NF-κB activation. IRE1α also activates IKK, which participates in the canonical activation of NF-κB. PA can also cause activation of PERK, which inhibits translation and thereby reduces the level of IκBα that leads, as well, to the NF-κB activation
SFA treatment also displays ER stress and activates the ER stress sensor, IRE1α. This, in turn, activates NF-κB and increases IL-1β production [53, 54]. However, in this model, TNF-α expression is, in part, dependent on the IRE1α =>XBP-1s pathway.
Unlike TLR, PERK can be activated by SFA, which may be an argument that SFAs do not act as TLR agonist, but they induce signaling pathways with the same effect as TLR activation [54, 142, 143], although, in the hepatocyte HepG2 cell line, activation of IRE1α by PA is TLR4 dependent [144]. Most probably, PA participates in two processes simultaneously. First, it integrates with the ER, causing the activation of IRE1α and PERK, and second, induces TLR4-mediated IRE1α activation.
Activation of nuclear factor κB and NOD-like receptor pyrin domain containing 3 inflammasome
Two factors play an important role in increasing IL-1β production: increased expression of pre-IL-1β, and activation of inflammasome. In the latter, the pre-IL-1β proteolysis to IL-1β occurs.
ER stress and activation of IRE1α are responsible for the production of IL-1β in macrophages under the PA treatment (Fig. 4) [53, 54]. NF-κB activation is responsible for the increase of pre-IL-1β expression. During the ER stress, activated IRE1α forms a complex with inhibitor of NF-kB (IκB) kinase (IKK) and TRAF2, which results in the activation of IKK and subsequent activation of NF-κB [145]. In turn, PERK phosphorylates eIF2α, which represses the translation of certain genes including IκBα [146]. IκBα is a protein with a short half-life. With repressed translation, the amount of IκBα is reduced and hence the activation of NF-κB. In the ER stress, activation of GSK-3β results in increased production of IL-1β [53]. Nevertheless, the role of GSK-3β in inflammatory responses induced by PA is still unclear. Probably this kinase phosphorylates IKKγ/NEMO, which causes activation of NF-κB [147].
Fig. 4.
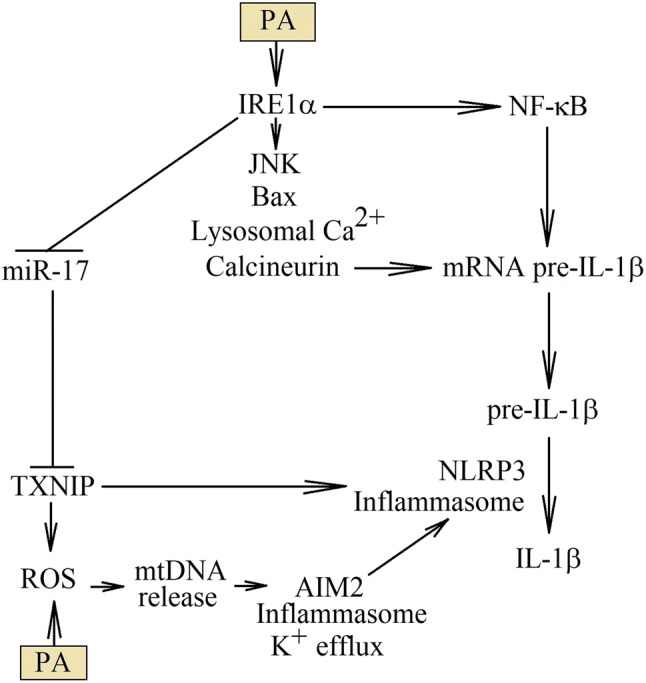
PA results in increased production of IL-1β. PA increases the production of IL-1β at various levels of this cytokine synthesis. First, PA activates NF-κB, which increases the expression levels of pre-IL-1β mRNA. Second, PA can increase the stability of this transcript by destabilizing lysosomes, releasing from them the Ca2+ ions and thereby activating calcineurin. Finally, PA activates NLRP3 inflammasome, which is associated with increased TXNIP expression, or released mitochondrial DNA to the cytoplasm
The ER stress induced by PA causes inflammatory responses through the destabilization of lysosomes. This happens by translocating Bax to lysosomes and destabilizing the lysosomal membrane [148–150]. It may depend on the JNK–MAPK activation [49, 151]. However, the mechanism of JNK–MAPK activation by PA is still unknown. It may also depend on IRE1α [152] or be activated by GSK-3β, independently of the ER stress [12]. Without a doubt, the process of lysosomes destabilization is independent of ceramides and cathepsin B. Destabilization of the lysosome membrane releases cathepsins into the cytoplasm. When this proteases, including cathepsin B, can perform the sirtuin (SIRT)1 proteolysis and the activity of this NF-κB deacetylase is decreased. As a consequence, the acetylation increases and thus the activation of p65 NF-κB [153]. This pathway is also known to be important in the activation of TLR4-mediated NF-κB signaling.
As shown by the experiments on high-fat choline-deficient food-fed mice, the activation of NLRP3 inflammasome alone may not depend on cathepsin B [153]. Even more, in macrophages, activating NLRP3 inflammasome can destabilize lysosomes [154]. Nevertheless, mainly it is possible to activate NLRP3 inflammasome by cathepsin B, as demonstrated by experiments on microvascular endothelial cells treated with PA [155]. In addition to cathepsins, also the Ca2+ ions are released from lysosomes, which causes the activation of calcineurin, increases the stability of pre-IL-1β mRNA and increases the expression of this polypeptide [156].
However, the PA may trigger the activation of NLRP3 inflammasome through other mechanisms. For example, PA damages the mitochondria, by reducing membrane potential and increasing ROS production. This results in mitochondrial DNA release into the cytoplasm [157], which leads to an activation of AIM2 inflammasome [158]. As a consequence, there is a cell membrane perforation, efflux of K+ ions and activation of NLRP3 inflammasome.
Another way to activate NLRP3 inflammasome is the PA-induced ER stress, leading to thioredoxin interacting protein (TXNIP) protein expression [159, 160] Activation of IRE1α reduces the expression of miR-17 which normally destabilizes TXNIP mRNA. Thanks to this, PA increases the expression of TXNIP protein by inducing ER stress. In the mitochondria, TXNIP binds and disturbs the action of thioredoxin H-type 1 (TRX1), and increases the generation of ROS. ROS is involved in the activation of NLRP3 inflammasome [161]. Moreover, TXNIP can directly bind to the NLRP3 inflammasome, which results in the activation of this inflammasome and the formation of IL-1β [161].
Fatty acid-binding protein 4/aP2 and peroxisome proliferator-activated receptor γ
Another important role of PA-induced inflammatory response is the increase of fatty acid-binding protein 4 (FABP4)/aP2 expression (Fig. 5). In macrophages, the PA increases the expression of FABP4/aP2 protein, but not FABP4/aP2 mRNA [162, 163] and this process is related to the ER stress induction. In turn, LPS increases mRNA and protein levels of FABP4/aP2 found in macrophages, which shows that PA does not affect the expression of this protein via TLR4, only through the ER stress induction [164]. The FABP4/aP2, another name for adipocyte fatty acid-binding protein (A-FABP), is a fatty acid-binding protein. Nevertheless, unlike other FABPs, FABP4/aP2 has the same affinities to PA as to oleic acid and docosahexaenoic acid (DHA) [165]. As a result, the concentration of free MUFA and PUFA in the cells is reduced by FABP4/aP2 [166].
Fig. 5.
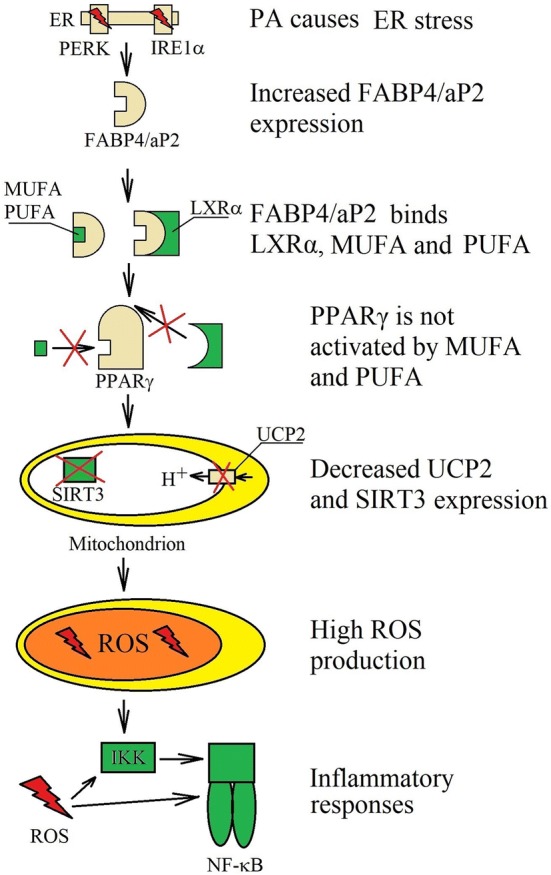
Role of FABP4/aP2 in PA activity. PA-induced ER stress increase the expression of FABP4/aP2. It is a protein that binds MUFA and PUFA that prevents the activation of PPARγ. FABP4/aP2 also binds LXRα, disrupting the expression of PPARγ-dependent genes. This reduces the expression of SIRT3 and UCP2, which in turn results in increased generation of ROS that is involved in inflammatory responses. Moreover, PPARγ inhibits NF-κB activation; therefore, functional disorders in PPARγ, results in increased activation of NF-κB
Increased FABP4/aP2 expression by PA, results in the lower expression of many proteins, including SIRT3, uncoupling protein 2 (UCP2) and peroxisome proliferator receptor-γ coactivator 1 (PGC-1)α, which interferes with mitochondrial function [163, 166–168]. This leads to increased ROS generation and increased inflammatory responses. This has been confirmed in C2C12 skeletal muscle cells where palmitate reduces PGC-1α expression through a mechanism involving NF-κB activation [169].
FABP4/aP2 binds LXRα, MUFA and PUFA which results in the reduction of the expression of LXRα-dependent and PPARγ-dependent genes [170]. PPARγ is a transcription factor and a nuclear receptor which is activated by MUFA and PUFA. Inhibiting the function of PPARγ reduces the expression of ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1), proteins involved in the clearance of cholesterol from macrophages [162, 171]. Also, the stearoyl-coenzyme A desaturase (SCD) expression, which is SFA processing desaturase, protecting from negative effects of PA, is inhibited. Inhibition of PPARγ enhances the activity of IKK and NF-κB [170] and reduces the expression of CD36 [170, 172]. However, it should be noted that PA increases the expression of CD36 by inducing the ER stress [43, 83, 103, 173].
The experiments on macrophages showed that the expression of ABCA1 and ABCG1 is increased in the cells treated with PA [83, 174]. This is related to the other effect caused by PA, in particular, the ER stress and increased PPARγ expression. Probably, increased PPARγ expression depends on the ER stress activation of the IRE1α =>XBP-1s pathway [175].
FABP4/aP2 can also decrease the expression of UCP2. It is an uncoupling protein that reduces the generation of ROS in the mitochondria. However, increasing ROS production by PA treatment results in increased expression of UCP2 [45, 168, 176–178]. Nevertheless, increased expression of FABP4/aP2 in macrophages abolishes this effect, or even reduces the expression of UCP2, which enhances the generation of ROS and induces inflammatory responses [166, 178]. The reduced expression of UCP2 is due to reduced PPARγ activation [168].
Another protein whose expression in macrophages is reduced under the influence of FABP4/aP2 is SIRT3 [166]. It is a mitochondrial protein that causes deacetylation of superoxide dismutase (SOD)2, which increases the activity of this antioxidant enzyme and thus reduces the concentration of ROS [179]. Reduction in SIRT3 expression results in decreased activity of SOD2 which leads to increased generation of ROS in the mitochondria.
Free fatty acid receptor 1/G protein-coupled receptor 40 plays a role as a receptor for palmitic acid in inflammatory responses
In addition to the presented way of action, PA may also activate its own free fatty acid receptor 1 (FFA1)/G protein-coupled receptor 40 (GPR40) receptor, which enhances inflammatory response (Fig. 6).
Fig. 6.

Activation of FFA1/GPR40 receptor. FFA1/GPR40 is the PA receptor whose activation enhances inflammatory reactions in neutrophils (a). This receptor causes signal transduction through PLC and PI3K, which activates IKK and consequently NF-κB. This transcription factor is involved in inflammatory reactions; however, activation of FFA1/GPR40 in pancreatic β-cells results in the release of insulin (b). Activation of PLC and PKC causes the release of Ca2+ from the ER to the cytoplasm. Higher cytoplasmic concentration of Ca2+ leads then to insulin release by pancreatic β-cells. However, continuous activation of FFA1/GPR40 results in Ca2+ depletion from ER and consequently the ER stress and pancreatic β-cells apoptosis
The highest expression of FFA1/GPR40 occurs in the brain, pancreas and monocytes, least in muscles, liver and adipose tissue [180]. Activation of FFA1/GPR40 causes signal transduction through the PI3K =>PKB/Akt pathway, and the PLC =>PKC => p38/ERK–MAPK pathway, which causes phosphorylation and degradation of IκBα [181]. This leads to the NF-κB activation. As a consequence, the expression of IL-8/C–X–C motif chemokine ligand (CXCL)8 and cyclooxygenase-2 (COX-2) increases in neutrophils [181] and IL-6 increases in human cardiac microvascular endothelial cells [140].
Consequences of the proinflammatory action of palmitic acid
Palmitic acid increases proinflammatory cytokine production
PA can activate TLR4-mediated proinflammatory signaling pathways through the MyD88-dependent [92, 182] and MyD88-independent [88] activation of NF-κB. As a consequence, in macrophages and monocytes, there is an increased expression of cytokines such as IL-1β [85], TNF-α [86, 88], CCL2/monocyte chemoattractant protein-1 (MCP-1) [91, 130, 182], CCL4/macrophage inflammatory protein 1β (MIP-1β) [183] and increase in COX-2 [84, 136] and matrix metallopeptidase 9 (MMP-9) expression [85, 92]. PA also increases the LPS effect on the IL-1β [130], CXCL2 [131] and TNF-α production [130, 131]. However, PA can also induce TNF-α expression independently of TLR4 activation [54].
Nevertheless, in some cells, PA can increase the expression of proinflammatory cytokines. For example, CCL2/MCP-1 is secreted in adipocytes [86, 184]. In contrast, in TLR4-dependent manner, PA causes the increased expression of CCL2/MCP-1 and CXCL1 in pancreatic β-cells [185]. Also in the experiments on C2C12 myoblasts, PA causes TLR4-dependent increase in the IL-6 and TNF-α expression [102]. However, this effect is significant only after 6 h of incubation with PA. After 24 h, the importance of TLR4 is negligible.
In hepatocytes, PA causes IRE1α and JNK MAPK-dependent increase in the production of extracellular vesicles [62, 186]. These extracellular vesicles contain sphingosine-1-phosphate (S1P) [62, 187]. Also PA by the activation of the mixed lineage kinase 3 (MLK3) =>MAPK kinase (MKK)3/6 =>p38 MAPK =>signal transducer and activator of transcription 1 (STAT1) pathway increases the expression of CXCL10/IP-10 in extracellular vesicles [186, 188]. Activation of MLK3 may depend on DAG and PKC [189]. Other signaling molecule produced by hepatocytes under the influence of PA is tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [190]. It is a ligand for death receptor 5 (DR5)/TNFRSF10B that activates macrophages and NF-κB, which increases the production of IL-1β and IL-6 by these cells.
The increased concentration of PA causes the production of chemokines by the cells. In particular, hepatocytes produce CXCL10/IP-10 [186, 188] and S1P [62, 187], adipocytes produce CCL2/MCP-1 [86, 184], and pancreatic β-cells the CCL2/MCP-1 [185]. All these chemokines cause recruitment of macrophages in the environment of these cells [64, 185, 191, 192]. Macrophages begin to form so-called crown-like structures in adipose tissue [64, 71, 73] and start to produce the proinflammatory cytokines involved in inflammatory responses and in insulin resistance [185, 191–193], such as TNF-α, IL-1β and IL-6 (Fig. 7).
Fig. 7.
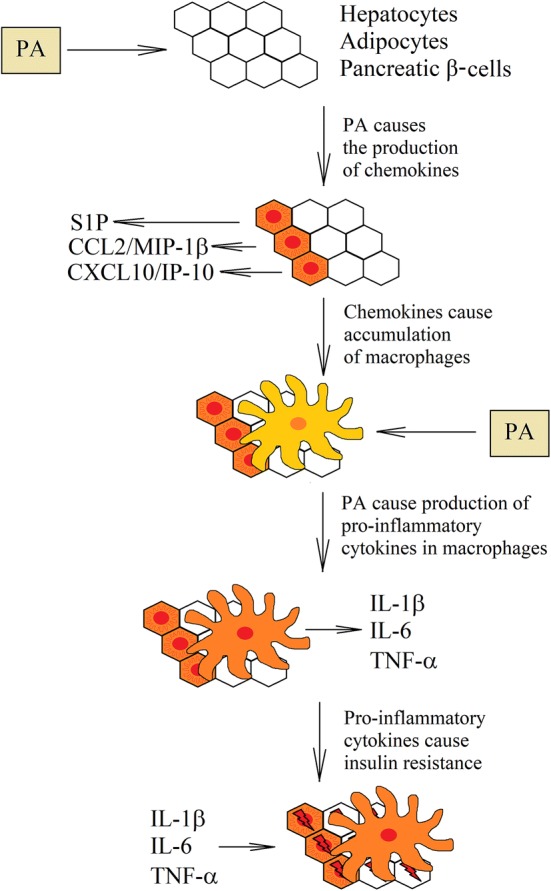
Role of macrophages in PA-induced insulin resistance. Healthy tissues contain a very small number of macrophages. However, under the influence of PA, hepatocytes, pancreatic β-cells and adipocytes begin to produce chamokines. This causes recruitment of macrophages to the liver, pancreas and adipose tissue. In turn, macrophages in these tissues begin to accumulate PA that causes inflammatory reactions. Increased production of proinflammatory cytokines results in insulin resistance in cells that are near the activated macrophages
Consequences of high palmitic acid levels in the blood: insulin resistance
Under the influence of PA, macrophages accumulated in the tissues start to produce proinflammatory cytokines, which leads to insulin resistance (Tab. 1) [10]. The most important cytokines that cause insulin resistance are TNF-α [193, 194], IL-1β [195, 196] and IL-6 [190, 197].
Table 1.
Mechanisms of PA-induced insulin resistance caused by the most important proinflammatory cytokines produced by macrophages
| Cytokines | Cellular mechanisms for insulin resistance |
|---|---|
| IL-1β |
Reduction of the IRS-1, IRS-2 and GLUT4 expression Apoptosis of pancreatic β-cells |
| IL-6 |
PKCδ-dependent phosphorylation of IRS-1 Increase of SOCS-3 expression |
| TNF-α |
JNK–MAPK-dependent phosphorylation of IRS-1 Increase of PTP1B expression Increase of SOCS-3 expression IKK2-dependent increase in the S6K1 expression IKK-dependent phosphorylation of IRS-1 |
TNF-α causes insulin resistance in different ways. In hepatocytes, it causes the activation of JNK–MAPK signaling pathway, which phosphorylates insulin receptor substrate (IRS)-1 [198]. Also in myocytes [199] and adipocytes [200], there is a higher expression of protein tyrosine phosphatase 1B (PTP1B), which inhibits the effect of insulin receptor (IR). In adipocytes, the TNF-α leads to the increase of suppressor of cytokine signaling 3 (SOCS-3) expression [201]. This protein suppresses the action of cytokines, but also binds to the IR, disrupting the function of this receptor. SOCS-3 may also cause the proteolytic degradation of IRS-1 [202]. Furthermore, TNF-α causes an IKK2-dependent increase in the ribosomal protein S6 kinase (S6K1) expression in adipocytes and hepatocytes [203]. S6K1 phosphorylates IRS-1 causing disruption in IR signal transduction. However, IKK itself can also phosphorylate IRS-1, which causes insulin resistance in muscles [204].
Another important cytokine involved in PA-induced insulin resistance is IL-1β [195]. This cytokine reduces in adipocytes the expression of IRS-1, IRS-2 and glucose transporter 4 (GLUT4) [196, 205]. IL-1β is cytotoxic to pancreatic β-cells, causing apoptosis of these cells [195, 206]. However, it should not be forgotten that PA alone acts cytotoxically on pancreatic β-cells as well. It activates its FFA1/GPR40 receptor, which causes the release of Ca2+ ions from the ER [207]. This is the signal to start releasing insulin. However, the chronic activation of FFA1/GPR40 by PA causes the ER Ca2+ depletion, and consequently the ER stress and apoptosis of pancreatic β-cells [208].
In turn, IL-6 [77, 190, 197] causes the PKCδ-dependent phosphorylation of IRS-1 in muscles, which leads to insulin resistance [209]. Nevertheless, this effect is tissue specific. In hepatocytes, IL-6 works by inducing the expression of SOCS-3 [210].
It should be remembered that the production of proinflammatory cytokines by macrophages is only one of the possible mechanisms of PA-induced insulin resistance. Very important are also signaling pathways which are directly induced by PA. In particular, an increased amount of DAG causes phosphorylation of IRS-1 by PKCδ [211, 212], PKCε [213] and PKCθ [214, 215]. PKCθ and PKCε activate IKK, which also phosphorylates IRS-1 [213]. Another way leading to PA-induced insulin resistance is the increased amount of ceramides in the cell. This causes activation of PP2A and dephosphorylation of PKB/Akt [19, 216, 217]. Ceramides can also activate PKCζ, which binds and phosphorylates PKB/Akt [218, 219]. Beside its influence on PKC, PA leads to ER stress and activation of JNK–MAPK pathway, which phosphorylate IRS-1. This makes PA a very important factor contributing to insulin resistance [213, 220, 221].
Conclusion
In obesity, a high concentration of PA causes insulin resistance which leads to diabetes. This state is called “diabesity”. There are many known PA-induced insulin resistance mechanisms. For instance, PA may induce signaling pathways that interfere with IR signal transduction. PA may also indirectly lead to insulin resistance by causing inflammatory reactions in macrophages. This results in increased proinflammatory cytokine production that causes insulin resistance in the cells closely located to activated macrophages. It seems that an important factor inducing the PA-mediated inflammatory response is the activation of TLR2- and TLR4-mediated signaling pathway.
However, in the cell, PA is metabolized to saturated DAG, ceramides and lysophosphatidylcholine. They can cause various effects, leading to inflammatory reactions. In particular, a high concentration of PA in macrophages leads to ER stress. Also, PKC activation by DAG and ceramides strengthens inflammatory reactions. Another way to induce inflammatory reactions by PA is to increase the ROS generation, which contributes to the disruption of the mitochondrial function.
Because PA can induce and enhance the inflammatory reactions through many mechanisms, it is very difficult to interfere with these reactions in people with obesity, mainly because of the fact that changes in one path are balanced by other proinflammatory pathways. Therefore, the best way to reduce the inflammatory response in obese patients is to reduce the free blood FA concentration. To do this, you should apply the appropriate diet or drugs that cause normalization of lipid metabolism.
Abbreviations
- ATF
Activating transcription factor
- CHOP
C/EBP homologous protein
- CCL
CC motif chemokine ligand
- JNK
c-Jun NH2-terminal kinase
- CD36
Cluster of differentiation 36
- DAG
Diacylglycerol
- DGAT
Diglyceride acyltransferase
- ER
Endoplasmic reticulum
- eIF2α
Eukaryotic initiation factor 2α
- FABP4
Fatty acid-binding protein 4
- GSK-3β
Glycogen synthase kinase-3β
- IκB
Inhibitor of NF-kB
- IKK
Inhibitor of NF-kB kinase
- IRE1α
Inositol-requiring enzyme 1α
- IRS
Insulin receptor substrate
- IL
Interleukin
- LPS
Lipopolysaccharide
- MAPK
Mitogen-activated protein kinase
- MUFA
Monounsaturated fatty acids
- MyD88
Myeloid differentiation factor 88
- NLRP3
NOD-like receptor pyrin domain containing 3
- NF-κB
Nuclear factor κB
- PPAR
Peroxisome proliferator-activated receptor
- PI3K
Phosphatidylinositol 3-kinase
- PUFA
Polyunsaturated fatty acids
- PKB
Protein kinase B
- PERK
Protein kinase RNA-like endoplasmic reticulum kinase
- PKC
Protein kinases C
- PP2A
Protein phosphatase 2A
- ROS
Reactive oxygen species
- RANTES
Regulated on activation, normal T cell expressed and secreted
- SFA
Saturated fatty acid
- SPT
Serine palmitoyltransferase
- SIRT
Sirtuin
- XBP-1s
Spliced X-box-binding protein-1
- TXNIP
Thioredoxin-interacting protein
- TRAF
TNF receptor-associated factor
- TLR
Toll-like receptor
- TAG
Triacylglycerol
- TNF-α
Tumor necrosis factor-α
- UCP2
Uncoupling protein 2
Author contributions
JK: manuscript concept, literature search and review, and writing the manuscript, KB-R: participated in writing the manuscript and translation of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the statutory budget of the Department of Molecular Biology, School of Medicine in Katowice, Medical University of Silesia, Poland.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jan Korbecki, Phone: (+48) 32 208 83 94, Email: jan.korbecki@onet.eu.
Karolina Bajdak-Rusinek, Email: kbajdak-rusinek@sum.edu.pl.
References
- 1.Pasco JA, Nicholson GC, Brennan SL, Kotowicz MA. Prevalence of obesity and the relationship between the body mass index and body fat: cross-sectional, population-based data. PLoS One. 2012 doi: 10.1371/journal.pone.0029580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hruby A, Hu FB. The epidemiology of obesity: a big picture. Pharmacoeconomics. 2015 doi: 10.1007/s40273-014-0243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism. 2019 doi: 10.1016/j.metabol.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Mook S, Halkes CJ, Bilecen S, Cabezas MC. In vivo regulation of plasma free fatty acids in insulin resistance. Metabolism. 2004;53:1197–2001. doi: 10.1016/j.metabol.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 5.Pankow JS, Duncan BB, Schmidt MI, Ballantyne CM, Couper DJ, Hoogeveen RC, Golden SH, Atherosclerosis Risk in Communities Study Fasting plasma free fatty acids and risk of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes Care. 2004;27:77–82. doi: 10.2337/diacare.27.1.77. [DOI] [PubMed] [Google Scholar]
- 6.Soriguer F, García-Serrano S, García-Almeida JM, Garrido-Sánchez L, García-Arnés J, Tinahones FJ, Cardona I, Rivas-Marín J, Gallego-Perales JL, García-Fuentes E. Changes in the serum composition of free-fatty acids during an intravenous glucose tolerance test. Obesity (Silver Spring). 2009 doi: 10.1038/oby.2008.475. [DOI] [PubMed] [Google Scholar]
- 7.Kim JY, Park JY, Kim OY, Ham BM, Kim HJ, Kwon DY, Jang Y, Lee JH. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS) J Proteome Res. 2010 doi: 10.1021/pr100101p. [DOI] [PubMed] [Google Scholar]
- 8.Feng R, Luo C, Li C, Du S, Okekunle AP, Li Y, Chen Y, Zi T, Niu Y. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: a case–control study. Lipids Health Dis. 2017 doi: 10.1186/s12944-017-0551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodríguez-Carrio J, Alperi-López M, López P, Ballina-García FJ, Suárez A. Non-esterified fatty acids profiling in rheumatoid arthritis: associations with clinical features and th1 response. PLoS One. 2016 doi: 10.1371/journal.pone.0159573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl Res. 2016 doi: 10.1016/j.trsl.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 11.Peng G, Li L, Liu Y, Pu J, Zhang S, Yu J, Zhao J, Liu P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology. 2011 doi: 10.1210/en.2010-1369. [DOI] [PubMed] [Google Scholar]
- 12.Kakisaka Keisuke, Cazanave Sophie C., Fingas Christian D., Guicciardi Maria E., Bronk Steven F., Werneburg Nathan W., Mott Justin L., Gores Gregory J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2012;302(1):G77–G84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leamy AK, Egnatchik RA, Shiota M, Ivanova PT, Myers DS, Brown HA, Young JD. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J Lipid Res. 2014 doi: 10.1194/jlr.M050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SK, Oh E, Yun M, Lee SB, Chae GT. Palmitate induces cisternal ER expansion via the activation of XBP-1/CCTα-mediated phospholipid accumulation in RAW 264.7 cells. Lipids Health Dis. 2015 doi: 10.1186/s12944-015-0077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys. 2003;419:101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 17.Lee JS, Pinnamaneni SK, Eo SJ, Cho IH, Pyo JH, Kim CK, Sinclair AJ, Febbraio MA, Watt MJ. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: role of intramuscular accumulation of lipid metabolites. J Appl Physiol. 2006;100:1467–1474. doi: 10.1152/japplphysiol.01438.2005. [DOI] [PubMed] [Google Scholar]
- 18.Akoumi A, Haffar T, Mousterji M, Kiss RS, Bousette N. Palmitate mediated diacylglycerol accumulation causes endoplasmic reticulum stress, Plin2 degradation, and cell death in H9C2 cardiomyoblasts. Exp Cell Res. 2017 doi: 10.1016/j.yexcr.2017.03.032. [DOI] [PubMed] [Google Scholar]
- 19.Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 20.Lee JY, Cho HK, Kwon YH. Palmitate induces insulin resistance without significant intracellular triglyceride accumulation in HepG2 cells. Metabolism. 2010 doi: 10.1016/j.metabol.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Jin J, Zhang X, Lu Z, Perry DM, Li Y, Russo SB, Cowart LA, Hannun YA, Huang Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am J Physiol Endocrinol Metab. 2013 doi: 10.1152/ajpendo.00251.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schilling JD, Machkovech HM, He L, Sidhu R, Fujiwara H, Weber K, Ory DS, Schaffer JE. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J Biol Chem. 2013 doi: 10.1074/jbc.M112.419978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma MK, Yateesh AN, Neelima K, Pawar N, Sandhya K, Poornima J, Lakshmi MN, Yogeshwari S, Pallavi PM, Oommen AM, Somesh BP, Jagannath MR. Inhibition of neutral sphingomyelinases in skeletal muscle attenuates fatty-acid induced defects in metabolism and stress. Springerplus. 2014 doi: 10.1186/2193-1801-3-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman R, Bell RM. Triacylglycerol synthesis in isolated fat cells. Studies on the microsomal diacylglycerol acyltransferase activity using ethanol-dispersed diacylglycerols. J Biol Chem. 1976;251:4537–4543. [PubMed] [Google Scholar]
- 25.Pinnamaneni SK, Southgate RJ, Febbraio MA, Watt MJ. Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia. 2006;49:3027–3037. doi: 10.1007/s00125-006-0427-9. [DOI] [PubMed] [Google Scholar]
- 26.Jung S, Choi M, Choi K, Kwon EB, Kang M, Kim DE, Jeong H, Kim J, Kim JH, Kim MO, Han SB, Cho S. Inactivation of human DGAT2 by oxidative stress on cysteine residues. PLoS One. 2017 doi: 10.1371/journal.pone.0181076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soumura M, Kume S, Isshiki K, Takeda N, Araki S, Tanaka Y, Sugimoto T, Chin-Kanasaki M, Nishio Y, Haneda M, Koya D, Kashiwagi A, Maegawa H, Uzu T. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem Biophys Res Commun. 2010 doi: 10.1016/j.bbrc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Coll T, Eyre E, Rodríguez-Calvo R, Palomer X, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem. 2008 doi: 10.1074/jbc.M708700200. [DOI] [PubMed] [Google Scholar]
- 29.Pan Z, Wang J, Tang H, Li L, Lv J, Xia L, Han C, Xu F, He H, Xu H, Kang B. Effects of palmitic acid on lipid metabolism homeostasis and apoptosis in goose primary hepatocytes. Mol Cell Biochem. 2011 doi: 10.1007/s11010-010-0680-6. [DOI] [PubMed] [Google Scholar]
- 30.Du X, Jiang Y, Qian W, Lu X, Walsh JP. Fatty acids inhibit growth-factor-induced diacylglycerol kinase alpha activation in vascular smooth-muscle cells. Biochem J. 2001;357:275–282. doi: 10.1042/0264-6021:3570275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamiya Y, Mizuno S, Komenoi S, Sakai H, Sakane F. Activation of conventional and novel protein kinase C isozymes by different diacylglycerol molecular species. Biochem Biophys Rep. 2016 doi: 10.1016/j.bbrep.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ford DA, Horner CC, Gross RW. Protein kinase C acylation by palmitoyl coenzyme A facilitates its translocation to membranes. Biochemistry. 1998;37:11953–11961. doi: 10.1021/bi980565w. [DOI] [PubMed] [Google Scholar]
- 33.Ways DK, Cook PP, Webster C, Parker PJ. Effect of phorbol esters on protein kinase C-zeta. J Biol Chem. 1992;267:4799–4805. [PubMed] [Google Scholar]
- 34.Wang G, Silva J, Krishnamurthy K, Tran E, Condie BG, Bieberich E. Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J Biol Chem. 2005;280:26415–26424. doi: 10.1074/jbc.M501492200. [DOI] [PubMed] [Google Scholar]
- 35.Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem. 2007;282:12450–12457. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- 36.Wetsel WC, Khan WA, Merchenthaler I, Rivera H, Halpern AE, Phung HM, Negro-Vilar A, Hannun YA. Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J Cell Biol. 1992;117:121–133. doi: 10.1083/jcb.117.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogoyevitch MA, Parker PJ, Sugden PH. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ Res. 1993;72:757–767. doi: 10.1161/01.res.72.4.757. [DOI] [PubMed] [Google Scholar]
- 38.Meller N, Altman A, Isakov N. New perspectives on PKCtheta, a member of the novel subfamily of protein kinase C. Stem Cells. 1998;16:178–192. doi: 10.1002/stem.160178. [DOI] [PubMed] [Google Scholar]
- 39.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells. 2013 doi: 10.1111/gtc.12074. [DOI] [PubMed] [Google Scholar]
- 41.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013 doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kono N, Amin-Wetzel N, Ron D. Generic membrane-spanning features endow IRE1α with responsiveness to membrane aberrancy. Mol Biol Cell. 2017 doi: 10.1091/mbc.E17-03-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DH, Cho YM, Lee KH, Jeong SW, Kwon OJ. Oleate protects macrophages from palmitate-induced apoptosis through the downregulation of CD36 expression. Biochem Biophys Res Commun. 2017 doi: 10.1016/j.bbrc.2017.05.066. [DOI] [PubMed] [Google Scholar]
- 44.Go DH, Lee YG, Lee DH, Kim JA, Jo IH, Han YS, Jo YH, Kim KY, Seo YK, Moon JH, Jung CH, Jeon TI. 3-Decylcatechol induces autophagy-mediated cell death through the IRE1α/JNK/p62 in hepatocellular carcinoma cells. Oncotarget. 2017;8:58790–58800. doi: 10.18632/oncotarget.17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zezina E, Snodgrass RG, Schreiber Y, Zukunft S, Schürmann C, Heringdorf DMZ, Geisslinger G, Fleming I, Brandes RP, Brüne B, Namgaladze D. Mitochondrial fragmentation in human macrophages attenuates palmitate-induced inflammatory responses. Biochim Biophys Acta Mol Cell Biol Lipids. 2018 doi: 10.1016/j.bbalip.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 46.B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin J, Wang Y, Gu L, Fan N, Ma Y, Peng Y. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: implications for apoptosis and inflammation. Int J Mol Med. 2015 doi: 10.3892/ijmm.2015.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin J, Gu L, Wang Y, Fan N, Ma Y, Peng Y. Rapamycin improves palmitate-induced ER stress/NF κ B pathways associated with stimulating autophagy in adipocytes. Mediat Inflamm. 2015 doi: 10.1155/2015/272313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, Gores GJ. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009 doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010 doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, Billadeau DD, Gores GJ. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011 doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao J, Feng XX, Yao L, Ning B, Yang ZX, Fang DL, Shen W. Saturated free fatty acid sodium palmitate-induced lipoapoptosis by targeting glycogen synthase kinase-3β activation in human liver cells. Dig Dis Sci. 2014 doi: 10.1007/s10620-013-2896-2. [DOI] [PubMed] [Google Scholar]
- 53.Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, Ryter SW, Surh YJ, Chung HT. Endoplasmic reticulum stress-induced IRE1α activation mediates cross-talk of GSK-3β and XBP-1 to regulate inflammatory cytokine production. J Immunol. 2015 doi: 10.4049/jimmunol.1401399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, Volmer R, Iwawaki T, Koliwad SK. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cocco T, Di Paola M, Papa S, Lorusso M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic Biol Med. 1999;27:51–59. doi: 10.1016/s0891-5849(99)00034-9. [DOI] [PubMed] [Google Scholar]
- 56.Loskovich MV, Grivennikova VG, Cecchini G, Vinogradov AD. Inhibitory effect of palmitate on the mitochondrial NADH:ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem J. 2005;387:677–683. doi: 10.1042/BJ20041703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schönfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 58.Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010 doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ventura FV, Ruiter J, Ijlst L, de Almeida IT, Wanders RJ. Differential inhibitory effect of long-chain acyl-CoA esters on succinate and glutamate transport into rat liver mitochondria and its possible implications for long-chain fatty acid oxidation defects. Mol Genet Metab. 2005;86:344–352. doi: 10.1016/j.ymgme.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 60.Ciapaite J, Bakker SJ, Diamant M, van Eikenhorst G, Heine RJ, Westerhoff HV, Krab K. Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-CoA effects Implications for a mechanism linking obesity and type 2 diabetes. FEBS J. 2006;273:5288–5302. doi: 10.1111/j.1742-4658.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- 61.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011 doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner. J Lipid Res. 2016 doi: 10.1194/jlr.M063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, Harrison LC. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010 doi: 10.2337/db09-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitz J, Evers N, Awazawa M, Nicholls HT, Brönneke HS, Dietrich A, Mauer J, Blüher M, Brüning JC. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol Metab. 2016 doi: 10.1016/j.molmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ, Chung H, Bogner-Strauss JG, Evans RM, Olefsky JM, Oh DY. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes. 2015 doi: 10.2337/db14-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018 doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 68.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeyda M, Gollinger K, Kriehuber E, Kiefer FW, Neuhofer A, Stulnig TM. Newly identified adipose tissue macrophage populations in obesity with distinct chemokine and chemokine receptor expression. Int J Obes (Lond) 2010 doi: 10.1038/ijo.2010.103. [DOI] [PubMed] [Google Scholar]
- 70.Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabetes. 2012 doi: 10.2337/db11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008 doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto K, Ginsberg HN, Mukaida N, Kaneko S, Ota T. CCR22 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012 doi: 10.2337/db11-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Zeyda M, Farmer D, Todoric J, Aszmann O, Speiser M, Györi G, Zlabinger GJ, Stulnig TM. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes (Lond) 2007;31:1420–1428. doi: 10.1038/sj.ijo.0803632. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki T, Gao J, Ishigaki Y, Kondo K, Sawada S, Izumi T, Uno K, Kaneko K, Tsukita S, Takahashi K, Asao A, Ishii N, Imai J, Yamada T, Oyadomari S, Katagiri H. ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.01.076. [DOI] [PubMed] [Google Scholar]
- 76.Fujisaka S, Usui I, Ikutani M, Aminuddin A, Takikawa A, Tsuneyama K, Mahmood A, Goda N, Nagai Y, Takatsu K, Tobe K. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia. 2013 doi: 10.1007/s00125-013-2885-1. [DOI] [PubMed] [Google Scholar]
- 77.Snodgrass RG, Boß M, Zezina E, Weigert A, Dehne N, Fleming I, Brüne B, Namgaladze D. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J Biol Chem. 2016 doi: 10.1074/jbc.M115.686709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moganti K, Li F, Schmuttermaier C, Riemann S, Klüter H, Gratchev A, Harmsen MC, Kzhyshkowska J. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology. 2017 doi: 10.1016/j.imbio.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 79.Pavlou S, Lindsay J, Ingram R, Xu H, Chen M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018 doi: 10.1186/s12865-018-0261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Blüher M, Kolanus W, Kahn CR, Brüning JC. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010 doi: 10.1371/journal.pgen.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manowsky J, Camargo RG, Kipp AP, Henkel J, Püschel GP. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am J Physiol Endocrinol Metab. 2016 doi: 10.1152/ajpendo.00427.2015. [DOI] [PubMed] [Google Scholar]
- 82.Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet-induced obesity in mice. Diabetes. 2010 doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014 doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 85.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 86.Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S, Kamei Y, Ogawa Y. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27:84–91. doi: 10.1161/01.ATV.0000251608.09329.9a. [DOI] [PubMed] [Google Scholar]
- 87.Lee SM, Choi SE, Lee JH, Lee JJ, Jung IR, Lee SJ, Lee KW, Kang Y. Involvement of the TLR4 (Toll-like receptor4) signaling pathway in palmitate-induced INS-1 beta cell death. Mol Cell Biochem. 2011 doi: 10.1007/s11010-011-0820-7. [DOI] [PubMed] [Google Scholar]
- 88.Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, Adams SH, Hwang DH. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012 doi: 10.1194/jlr.D029546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Z, Liu D, Wang F, Liu S, Zhao S, Ling EA, Hao A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling. Br J Nutr. 2012 doi: 10.1017/S0007114511002868. [DOI] [PubMed] [Google Scholar]
- 90.Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. 2013 doi: 10.4049/jimmunol.1300298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cullberg KB, Larsen JØ, Pedersen SB, Richelsen B. Effects of LPS and dietary free fatty acids on MCP-1 in 3T3-L1 adipocytes and macrophages in vitro. Nutr Diabetes. 2014 doi: 10.1038/nutd.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sindhu S, Al-Roub A, Koshy M, Thomas R, Ahmad R. Palmitate-induced MMP-9 expression in the human monocytic cells is mediated through the TLR4-MyD88 dependent mechanism. Cell Physiol Biochem. 2016 doi: 10.1159/000447798. [DOI] [PubMed] [Google Scholar]
- 93.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- 94.Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- 95.Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009 doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicholas DA, Zhang K, Hung C, Glasgow S, Aruni AW, Unternaehrer J, Payne KJ, Langridge WHR, De Leon M. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1β. PLoS One. 2017 doi: 10.1371/journal.pone.0176793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Y, Qian Y, Fang Q, Zhong P, Li W, Wang L, Fu W, Zhang Y, Xu Z, Li X, Liang G. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat Commun. 2017 doi: 10.1038/ncomms13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009 doi: 10.1161/ATVBAHA.109.194050. [DOI] [PubMed] [Google Scholar]
- 99.Anderson EK, Hill AA, Hasty AH. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler Thromb Vasc Biol. 2012 doi: 10.1161/ATVBAHA.112.250142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, Weir J, Mellett NA, Pernes G, Conway JRW, Lee MKS, Timpson P, Murphy AJ, Masters SL, Gerondakis S, Bartonicek N, Kaczorowski DC, Dinger ME, Meikle PJ, Bond PJ, Febbraio MA. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018 doi: 10.1016/j.cmet.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 101.Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, Handa P, Schwartz MW, Kim F. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol. 2009 doi: 10.1161/ATVBAHA.109.188813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perry BD, Rahnert JA, Xie Y, Zheng B, Woodworth-Hobbs ME, Price SR. Palmitate-induced ER stress and inhibition of protein synthesis in cultured myotubes does not require Toll-like receptor 4. PLoS One. 2018 doi: 10.1371/journal.pone.0191313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu Z, Li Y, Brinson CW, Kirkwood KL, Lopes-Virella MF, Huang Y. CD36 is upregulated in mice with periodontitis and metabolic syndrome and involved in macrophage gene upregulation by palmitate. Oral Dis. 2017 doi: 10.1111/odi.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Freitas MCP, Fernandez DGE, Cohen D, Figueiredo-Neto AM, Maranhão RC, Damasceno NRT. Oxidized and electronegative low-density lipoprotein as potential biomarkers of cardiovascular risk in obese adolescents. Clinics (Sao Paulo) 2018 doi: 10.6061/clinics/2018/e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Estruch M, Sanchez-Quesada JL, Beloki L, Ordoñez-Llanos J, Benitez S. The induction of cytokine release in monocytes by electronegative low-density lipoprotein (LDL) is related to its higher ceramide content than native LDL. Int J Mol Sci. 2013 doi: 10.3390/ijms14022601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Estruch M, Bancells C, Beloki L, Sanchez-Quesada JL, Ordóñez-Llanos J, Benitez S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis. 2013 doi: 10.1016/j.atherosclerosis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 107.Estruch M, Sánchez-Quesada JL, Ordóñez-Llanos J, Benítez S. Ceramide-enriched LDL induces cytokine release through TLR4 and CD14 in monocytes. Similarities with electronegative LDL. Clin Investig Arterioscler. 2014 doi: 10.1016/j.arteri.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 108.Ligi D, Benitez S, Croce L, Rivas-Urbina A, Puig N, Ordóñez-Llanos J, Mannello F, Sanchez-Quesada JL. Electronegative LDL induces MMP-9 and TIMP-1 release in monocytes through CD14 activation: inhibitory effect of glycosaminoglycan sulodexide. Biochim Biophys Acta Mol Basis Dis. 2018 doi: 10.1016/j.bbadis.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 109.Shrestha C, Ito T, Kawahara K, Shrestha B, Yamakuchi M, Hashiguchi T, Maruyama I. Saturated fatty acid palmitate induces extracellular release of histone H3: a possible mechanistic basis for high-fat diet-induced inflammation and thrombosis. Biochem Biophys Res Commun. 2013 doi: 10.1016/j.bbrc.2013.06.117. [DOI] [PubMed] [Google Scholar]
- 110.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011 doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011 doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, Anders HJ. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012 doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res. 2016 doi: 10.1016/j.thromres.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 114.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT, Tsung A. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology. 2011 doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rosin DL, Okusa MD. Dying cells and extracellular histones in AKI: beyond a NET effect? J Am Soc Nephrol. 2012 doi: 10.1681/ASN.2012060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012 doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 117.Genser L, Aguanno D, Soula HA, Dong L, Trystram L, Assmann K, Salem JE, Vaillant JC, Oppert JM, Laugerette F, Michalski MC, Wind P, Rousset M, Brot-Laroche E, Leturque A, Clément K, Thenet S, Poitou C. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J Pathol. 2018 doi: 10.1002/path.5134. [DOI] [PubMed] [Google Scholar]
- 118.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009 doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- 119.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009 doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Szulińska M, Łoniewski I, van Hemert S, Sobieska M, Bogdański P. Dose-dependent effects of multispecies probiotic supplementation on the lipopolysaccharide (LPS) level and cardiometabolic profile in obese postmenopausal women: a 12-week randomized clinical trial. Nutrients. 2018 doi: 10.3390/nu10060773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, Al-Attas OS, Sabico S, O’Hare JP, Ceriello A, Saravanan P, Kumar S, McTernan PG. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. 2012 doi: 10.2337/dc11-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mani V, Hollis JH, Gabler NK. Dietary oil composition differentially modulates intestinal endotoxin transport and postprandial endotoxemia. Nutr Metab (Lond). 2013 doi: 10.1186/1743-7075-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lyte JM, Gabler NK, Hollis JH. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016;15:186. doi: 10.1186/s12944-016-0357-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ahola AJ, Lassenius MI, Forsblom C, Harjutsalo V, Lehto M, Groop PH. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci Rep. 2017 doi: 10.1038/s41598-017-06885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, Pörsti I, Rissanen A, Kaprio J, Mustonen J, Groop PH, Lehto M, FinnDiane Study Group Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011 doi: 10.2337/dc10-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Al-Disi DA, Al-Daghri NM, Khan N, Alfadda AA, Sallam RM, Alsaif M, Sabico S, Tripathi G, McTernan PG. Postprandial effect of a high-fat meal on endotoxemia in Arab women with and without insulin-resistance-related diseases. Nutrients. 2015 doi: 10.3390/nu7085290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lassenius MI, Ahola AJ, Harjutsalo V, Forsblom C, Groop PH, Lehto M. Endotoxins are associated with visceral fat mass in type 1 diabetes. Sci Rep. 2016 doi: 10.1038/srep38887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carnevale R, Nocella C, Petrozza V, Cammisotto V, Pacini L, Sorrentino V, Martinelli O, Irace L, Sciarretta S, Frati G, Pastori D, Violi F. Localization of lipopolysaccharide from Escherichia coli into human atherosclerotic plaque. Sci Rep. 2018 doi: 10.1038/s41598-018-22076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu SP, Li XY, Li Z, He LN, Xiao Y, Yan K, Zhou ZG. Octanoylated ghrelin inhibits the activation of the palmitic acid-induced TLR4/NF-κB signaling pathway in THP-1 macrophages. ISRN Endocrinol. 2012 doi: 10.5402/2012/237613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tashiro H, Takahashi K, Sadamatsu H, Kato G, Kurata K, Kimura S, Sueoka-Aragane N. Saturated fatty acid increases lung macrophages and augments house dust mite-induced airway inflammation in mice fed with high-fat diet. Inflammation. 2017 doi: 10.1007/s10753-017-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ann SJ, Kim KK, Cheon EJ, Noh HM, Hwang I, Yu JW, Park S, Kang SM, Manabe I, Miller YI, Kim S, Lee SH. Palmitate and minimally-modified low-density lipoprotein cooperatively promote inflammatory responses in macrophages. PLoS One. 2018 doi: 10.1371/journal.pone.0193649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, Semenkovich CF. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016 doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carroll RG, Zasłona Z, Galván-Peña S, Koppe EL, Sévin DC, Angiari S, Triantafilou M, Triantafilou K, Modis LK, O’Neill LA. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J Biol Chem. 2018 doi: 10.1074/jbc.RA118.001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dallot E, Méhats C, Oger S, Leroy MJ, Breuiller-Fouché M. A role for PKCzeta in the LPS-induced translocation NF-kappaB p65 subunit in cultured myometrial cells. Biochimie. 2005;87:513–521. doi: 10.1016/j.biochi.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 135.Cuschieri J, Umanskiy K, Solomkin J. PKC-zeta is essential for endotoxin-induced macrophage activation. J Surg Res. 2004;121:76–83. doi: 10.1016/j.jss.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 136.Huang X, Chen LY, Doerner AM, Pan WW, Smith L, Huang S, Papadimos TJ, Pan ZK. An atypical protein kinase C (PKC zeta) plays a critical role in lipopolysaccharide-activated NF-kappa B in human peripheral blood monocytes and macrophages. J Immunol. 2009 doi: 10.4049/jimmunol.0804073. [DOI] [PubMed] [Google Scholar]
- 137.Teusch N, Lombardo E, Eddleston J, Knaus UG. The low molecular weight GTPase RhoA and atypical protein kinase Czeta are required for TLR2-mediated gene transcription. J Immunol. 2004;173:507–514. doi: 10.4049/jimmunol.173.1.507. [DOI] [PubMed] [Google Scholar]
- 138.Kubo-Murai M, Hazeki K, Sukenobu N, Yoshikawa K, Nigorikawa K, Inoue K, Yamamoto T, Matsumoto M, Seya T, Inoue N, Hazeki O. Protein kinase Cdelta binds TIRAP/Mal to participate in TLR signaling. Mol Immunol. 2007;44:2257–2264. doi: 10.1016/j.molimm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 139.Faisal A, Saurin A, Gregory B, Foxwell B, Parker PJ. The scaffold MyD88 acts to couple protein kinase Cepsilon to Toll-like receptors. J Biol Chem. 2008 doi: 10.1074/jbc.M710330200. [DOI] [PubMed] [Google Scholar]
- 140.Lu Z, Li Y, Jin J, Zhang X, Hannun YA, Huang Y. GPR40/FFA1 and neutral sphingomyelinase are involved in palmitate-boosted inflammatory response of microvascular endothelial cells to LPS. Atherosclerosis. 2015 doi: 10.1016/j.atherosclerosis.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jin J, Lu Z, Li Y, Cowart LA, Lopes-Virella MF, Huang Y. Docosahexaenoic acid antagonizes the boosting effect of palmitic acid on LPS inflammatory signaling by inhibiting gene transcription and ceramide synthesis. PLoS One. 2018 doi: 10.1371/journal.pone.0193343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010 doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, Zhang T, Kong Q, Li H, Chen S, Chen A, Kaufman RJ, Yang WL, Lin HK, Zhang D, Perlman H, Thorp E, Zhang K, Fang D. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013 doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shen C, Ma W, Ding L, Li S, Dou X, Song Z. The TLR4-IRE1α pathway activation contributes to palmitate-elicited lipotoxicity in hepatocytes. J Cell Mol Med. 2018 doi: 10.1111/jcmm.13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Medunjanin S, Schleithoff L, Fiegehenn C, Weinert S, Zuschratter W, Braun-Dullaeus RC. GSK-3β controls NF-kappaB activity via IKKγ/NEMO. Sci Rep. 2016 doi: 10.1038/srep38553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 149.Kågedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, Jürgensmeier JM, Ollinger K. Lysosomal membrane permeabilization during apoptosis–involvement of Bax? Int J Exp Pathol. 2005;86:309–321. doi: 10.1111/j.0959-9673.2005.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 152.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 153.de Mingo Á, de Gregorio E, Moles A, Tarrats N, Tutusaus A, Colell A, Fernandez-Checa JC, Morales A, Marí M. Cysteine cathepsins control hepatic NF-κB-dependent inflammation via sirtuin-1 regulation. Cell Death Dis. 2016 doi: 10.1038/cddis.2016.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013 doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 2016 doi: 10.18632/oncotarget.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem. 2014 doi: 10.1074/jbc.M113.531202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, He K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 2018 doi: 10.1016/j.cellimm.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 158.Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, Flavell RA, Zamboni DS. AIM2 engages active but unprocessed caspase-1 to induce noncanonical activation of the NLRP3 inflammasome. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.06.086. [DOI] [PubMed] [Google Scholar]
- 159.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012 doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Szpigel A, Hainault I, Carlier A, Venteclef N, Batto AF, Hajduch E, Bernard C, Ktorza A, Gautier JF, Ferré P, Bourron O, Foufelle F. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia. 2018 doi: 10.1007/s00125-017-4462-5. [DOI] [PubMed] [Google Scholar]
- 161.Xu L, Lin X, Guan M, Zeng Y, Liu Y. Verapamil attenuated prediabetic neuropathy in high-fat diet-fed mice through inhibiting TXNIP-mediated apoptosis and inflammation. Oxid Med Cell Longev. 2019 doi: 10.1155/2019/1896041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, Hotamisligil GS. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009 doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li H, Xiao Y, Tang L, Zhong F, Huang G, Xu JM, Xu AM, Dai RP, Zhou ZG. Adipocyte fatty acid-binding protein promotes palmitate-induced mitochondrial dysfunction and apoptosis in macrophages. Front Immunol. 2018 doi: 10.3389/fimmu.2018.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR. Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by toll-like receptor agonists. Arterioscler Thromb Vasc Biol. 2005;25:1220–1224. doi: 10.1161/01.ATV.0000159163.52632.1b. [DOI] [PubMed] [Google Scholar]
- 165.Richieri GV, Ogata RT, Zimmerman AW, Veerkamp JH, Kleinfeld AM. Fatty acid binding proteins from different tissues show distinct patterns of fatty acid interactions. Biochemistry. 2000;39:7197–7204. doi: 10.1021/bi000314z. [DOI] [PubMed] [Google Scholar]
- 166.Xu H, Hertzel AV, Steen KA, Bernlohr DA. Loss of fatty acid binding protein 4/aP2 reduces macrophage inflammation through activation of SIRT3. Mol Endocrinol. 2016 doi: 10.1210/me.2015-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gan L, Liu Z, Cao W, Zhang Z, Sun C. FABP4 reversed the regulation of leptin on mitochondrial fatty acid oxidation in mice adipocytes. Sci Rep. 2015 doi: 10.1038/srep13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xu H, Hertzel AV, Steen KA, Wang Q, Suttles J, Bernlohr DA. Uncoupling lipid metabolism from inflammation through fatty acid binding protein-dependent expression of UCP2. Mol Cell Biol. 2015 doi: 10.1128/MCB.01122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Coll T, Jové M, Rodríguez-Calvo R, Eyre E, Palomer X, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes. 2006;55:2779–2787. doi: 10.2337/db05-1494. [DOI] [PubMed] [Google Scholar]
- 170.Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J Biol Chem. 2005;280:12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ikeda J, Ichiki T, Takahara Y, Kojima H, Sankoda C, Kitamoto S, Tokunou T, Sunagawa K. PPARγ agonists attenuate palmitate-induced ER stress through up-regulation of SCD-1 in macrophages. PLoS One. 2015 doi: 10.1371/journal.pone.0128546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 173.Hua Y, Kandadi MR, Zhu M, Ren J, Sreejayan N. Tauroursodeoxycholic acid attenuates lipid accumulation in endoplasmic reticulum-stressed macrophages. J Cardiovasc Pharmacol. 2010 doi: 10.1097/FJC.0b013e3181c37d86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mauerer R, Ebert S, Langmann T. High glucose, unsaturated and saturated fatty acids differentially regulate expression of ATP-binding cassette transporters ABCA1 and ABCG1 in human macrophages. Exp Mol Med. 2009;41:126–132. doi: 10.3858/emm.2009.41.2.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cho YM, Kim DH, Lee KH, Jeong SW, Kwon OJ. The IRE1α-XBP1s pathway promotes insulin-stimulated glucose uptake in adipocytes by increasing PPARγ activity. Exp Mol Med. 2018 doi: 10.1038/s12276-018-0131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.McCarty MF. High mitochondrial redox potential may promote induction and activation of UCP2 in hepatocytes during hepatothermic therapy. Med Hypotheses. 2005;64:1216–1219. doi: 10.1016/j.mehy.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 177.Lou J, Wang Y, Wang X, Jiang Y. Uncoupling protein 2 regulates palmitic acid-induced hepatoma cell autophagy. Biomed Res Int. 2014 doi: 10.1155/2014/810401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Steen KA, Xu H, Bernlohr DA. FABP4/aP2 regulates macrophage redox signaling and inflammasome activation via control of UCP2. Mol Cell Biol. 2017 doi: 10.1128/MCB.00282-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Traba J, Geiger SS, Kwarteng-Siaw M, Han K, Ra OH, Siegel RM, Gius D, Sack MN. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J Biol Chem. 2017 doi: 10.1074/jbc.M117.791715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 181.Mena SJ, Manosalva C, Carretta MD, Teuber S, Olmo I, Burgos RA, Hidalgo MA. Differential free fatty acid receptor-1 (FFAR1/GPR40) signalling is associated with gene expression or gelatinase granule release in bovine neutrophils. Innate Immun. 2016 doi: 10.1177/1753425916656765. [DOI] [PubMed] [Google Scholar]
- 182.Ahmad R, Al-Roub A, Kochumon S, Akther N, Thomas R, Kumari M, Koshy MS, Tiss A, Hannun YA, Tuomilehto J, Sindhu S, Rosen ED. The synergy between palmitate and TNF-α for CCL2 production is dependent on the TRIF/IRF3 pathway: implications for metabolic inflammation. J Immunol. 2018 doi: 10.4049/jimmunol.1701552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kochumon S, Wilson A, Chandy B, Shenouda S, Tuomilehto J, Sindhu S, Ahmad R. Palmitate activates CCL4 expression in human monocytic cells via TLR4/MyD88 dependent activation of NF-κB/MAPK/ PI3K signaling systems. Cell Physiol Biochem. 2018 doi: 10.1159/000488824. [DOI] [PubMed] [Google Scholar]
- 184.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, Tobe K, Arai H, Kadowaki T, Nagai R. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012 doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 186.Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, Goodfellow VS, Malhi H, Gores GJ. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016 doi: 10.1002/hep.28252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liao CY, Song MJ, Gao Y, Mauer AS, Revzin A, Malhi H. Hepatocyte-derived lipotoxic extracellular vesicle sphingosine 1-phosphate induces macrophage chemotaxis. Front Immunol. 2018 doi: 10.3389/fimmu.2018.02980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tomita K, Kabashima A, Freeman BL, Bronk SF, Hirsova P, Ibrahim SH. Mixed lineage kinase 3 mediates the induction of CXCL10 by a STAT1-dependent mechanism during hepatocyte lipotoxicity. J Cell Biochem. 2017 doi: 10.1002/jcb.25973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, Charlton MR, Shah VH, Malhi H, Gores GJ. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016 doi: 10.1053/j.gastro.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008 doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li P, Lu M, Nguyen MT, Bae EJ, Chapman J, Feng D, Hawkins M, Pessin JE, Sears DD, Nguyen AK, Amidi A, Watkins SM, Nguyen U, Olefsky JM. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J Biol Chem. 2010 doi: 10.1074/jbc.M110.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Talbot NA, Wheeler-Jones CP, Cleasby ME. Palmitoleic acid prevents palmitic acid-induced macrophage activation and consequent p38 MAPK-mediated skeletal muscle insulin resistance. Mol Cell Endocrinol. 2014 doi: 10.1016/j.mce.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Haghani K, Pashaei S, Vakili S, Taheripak G, Bakhtiyari S. TNF-α knockdown alleviates palmitate-induced insulin resistance in C2C12 skeletal muscle cells. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2015.03.137. [DOI] [PubMed] [Google Scholar]
- 195.Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology. 2008 doi: 10.1210/en.2007-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gao D, Madi M, Ding C, Fok M, Steele T, Ford C, Hunter L, Bing C. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Endocrinol Metab. 2014 doi: 10.1152/ajpendo.00430.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kern L, Mittenbühler MJ, Vesting AJ, Ostermann AL, Wunderlich CM, Wunderlich FT. Obesity-induced TNFα and IL-6 signaling: the missing link between obesity and inflammation-driven liver and colorectal cancers. Cancers (Basel). 2018 doi: 10.3390/cancers11010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li L, He Q, Huang X, Man Y, Zhou Y, Wang S, Wang J, Li J. NOX3-derived reactive oxygen species promote TNF-alpha-induced reductions in hepatocyte glycogen levels via a JNK pathway. FEBS Lett. 2010;584:995–1000. doi: 10.1016/j.febslet.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 199.Nieto-Vazquez I, Fernández-Veledo S, de Alvaro C, Rondinone CM, Valverde AM, Lorenzo M. Protein-tyrosine phosphatase 1B-deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-alpha-induced insulin resistance. Diabetes. 2007;56:404–413. doi: 10.2337/db06-0989. [DOI] [PubMed] [Google Scholar]
- 200.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008 doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, Hotamisligil GS, Van Obberghen E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem. 2001;276:47944–47949. doi: 10.1074/jbc.M104602200. [DOI] [PubMed] [Google Scholar]
- 202.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 203.Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem. 2008 doi: 10.1074/jbc.M806480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.de Alvaro C, Teruel T, Hernandez R, Lorenzo M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J Biol Chem. 2004;279:17070–17078. doi: 10.1074/jbc.M312021200. [DOI] [PubMed] [Google Scholar]
- 205.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148:241–251. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, Laybutt DR. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes. 2008 doi: 10.2337/db07-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schnell S, Schaefer M, Schöfl C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol Cell Endocrinol. 2007;263:173–180. doi: 10.1016/j.mce.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 208.Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009 doi: 10.1152/ajpendo.90525.2008. [DOI] [PubMed] [Google Scholar]
- 209.Weigert C, Hennige AM, Lehmann R, Brodbeck K, Baumgartner F, Schaüble M, Häring HU, Schleicher ED. Direct cross-talk of interleukin-6 and insulin signal transduction via insulin receptor substrate-1 in skeletal muscle cells. J Biol Chem. 2006;281:7060–7067. doi: 10.1074/jbc.M509782200. [DOI] [PubMed] [Google Scholar]
- 210.Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278:13740–13746. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 211.Greene MW, Morrice N, Garofalo RS, Roth RA. Modulation of human insulin receptor substrate-1 tyrosine phosphorylation by protein kinase Cdelta. Biochem J. 2004;378:105–116. doi: 10.1042/BJ20031493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dey D, Mukherjee M, Basu D, Datta M, Roy SS, Bandyopadhyay A, Bhattacharya S. Inhibition of insulin receptor gene expression and insulin signaling by fatty acid: interplay of PKC isoforms therein. Cell Physiol Biochem. 2005;16:217–228. doi: 10.1159/000089847. [DOI] [PubMed] [Google Scholar]
- 213.Kewalramani G, Fink LN, Asadi F, Klip A. Palmitate-activated macrophages confer insulin resistance to muscle cells by a mechanism involving protein kinase C θ and ε. PLoS One. 2011 doi: 10.1371/journal.pone.0026947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 215.Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) J Biol Chem. 2004;279:45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 216.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 217.Galbo T, Olsen GS, Quistorff B, Nishimura E. Free fatty acid-induced PP2A hyperactivity selectively impairs hepatic insulin action on glucose metabolism. PLoS One. 2011 doi: 10.1371/journal.pone.0027424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. 2004;382:619–629. doi: 10.1042/BJ20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mahfouz R, Khoury R, Blachnio-Zabielska A, Turban S, Loiseau N, Lipina C, Stretton C, Bourron O, Ferré P, Foufelle F, Hundal HS, Hajduch E. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: a mechanistic insight. PLoS One. 2014 doi: 10.1371/journal.pone.0101865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 221.Chen YY, Sun LQ, Wang BA, Zou XM, Mu YM, Lu JM. Palmitate induces autophagy in pancreatic β-cells via endoplasmic reticulum stress and its downstream JNK pathway. Int J Mol Med. 2013 doi: 10.3892/ijmm.2013.1530. [DOI] [PubMed] [Google Scholar]