

Abstract
Allium species are popular vegetables in China and possess antifungal and antibacterial activities. This study aimed to compare the endophytic bacterial community in the four crucial Allium species in China, Chinese leek (CL), garlic (GA), onion (ON,) and Welsh onion (WO), using sequences of the V3–V4 region of the bacterial 16S rRNA gene. A total of 1,036,637 high-quality sequences and 719 operational taxonomic units (OTUs) were obtained across all libraries. A total of 20 phyla, 50 classes, 80 orders, 134 families, and 234 genera were identified. Among them, 18 OTUs and 19 genera were shared among the four Allium species. Proteobacteria (42.68%) and Bacteroidetes (20.18%) were the dominant phyla in CL, while one unclassified (>70%) was the dominant phyla in the other three Allium species. The alpha-diversity analysis showed the bacterial richness and diversity in CL were significantly higher than those in the other three Allium species. Principal coordinate analysis (PCA) showed endophytic bacterial communities in GA, WO, and ON were more similar than those in CL. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) analysis revealed endophytic bacteria mostly enriched in Membrane Transport, Amino Acid Metabolism and Carbohydrate Metabolism pathway. 17 of the 23 Kyoto Encyclopedia of Genes and Genomes (KEGG) categories and 159 of the 206 lower-level KEGG pathways in CL were significantly higher than those in the other three Allium species. Pearson’s correlation indicated that KEGG pathways with significant differences among the Allium species were closely related to the bacterial genera with significant differences between the Allium species. The findings of our study provided insight into the complex endophytic microbial communities in Allium species.
Subject terms: Symbiosis, Rhizobial symbiosis
Introduction
Healthy plant tissues contain endophytic bacteria that do not cause pathogenic reactions. In recent decades, studies have shown that these diverse and active microbial communities are not merely “passengers” within plants, but instead play an essential role in plant growth, development, and resistance to biotic and abiotic stresses1. Previous studies have shown that endophytic bacteria enhance the growth and development of host plants2,3, improve the efficiency of phytoremediation in heavy metal-degraded soils4–7, inhibit the growth of plant pathogens and effectively reduce the incidence of plant disease8–10, and increase tolerance to salinity stress11,12. Therefore, isolating, identifying, and applying endophytic bacteria have become of interest in research to identify safer biological additives.
Allium species are used worldwide as spices, vegetables, and medicinal plants. Traditionally, they are essential in the daily diet of people in many Asian countries and are used to flavor foods13. They are also an abundant source of endophytic microorganisms. An endophytic bacterium Sphingomonas sp. strain HJY isolated from the leaves of Chinese leek (Allium tuberosum) showed the potential ability to degrade Chlorpyrifos14. The fungal endophyte Clonostachys rosea ICIPE 707 isolated from healthy onion (Allium cepa) colonized onion plants and significantly repressed the feeding punctures, oviposition, and numbers of thrips on plants15. Streptomyces sp.TP-A0569 isolated from the stem of the Welsh onion (Allium fistulosum) and Streptomyces sp.TP-A0595 isolated from Chinese leek produced fistupyrone and 6-prenylindole, respectively, both of which significantly inhibited the infection of Chinese cabbage by Alternaria brassicicola16,17. The endophytic fungus strain Trichoderma brevicompactum 0248 isolated from garlic (Allium sativum) has a marked inhibitory activity on Rhizoctonia solani and Botrytis cinerea18.
Garlic (GA), onion (ON), Welsh onion (WO), and Chinese leek (CL) are typical Allium species that are popular vegetables in China. To some extent, they have similar physicochemical properties. For example, they all are perennial herbs with various kinds of bulbs and used as a flavoring for daily food. They all contain the well-studied sulfur compounds, saponins, flavonoids, nitrogen compounds, and peptides that make Allium species effective against pathogenic microbes19. But so far, very little is known about their endophytes. What is the composition of the endophyte community of the four Allium species? What is the relationship between them? In this study, we comparatively analyzed the endophytic bacterial communities in these four species using Illumina sequencing, to provide valuable information for isolating and utilizing endophytic bacteria with beneficial properties from Allium species.
Results
Sequence characteristics of the four Allium species
After processing, a total of 1,036,637 filtered sequences were obtained from the four Allium species, the numbers of which ranged from 61,541 to 101,135 in these samples (Table 1). Rarefaction curves (Supplementary Fig. S1), together with the estimated coverage values (Table 2), suggested that the libraries were sufficiently large to capture most of the bacterial diversity in the samples. A total of 719 operational taxonomic units (OTUs) were obtained across all libraries at 97% identity. A total of 18 OTUs, accounting for 29.57% of the total abundance, were shared among all samples. There were 396, 64, 24, and 8 OTUs exclusive to CL, GA, ON, and WO, respectively (Fig. 1A). Besides the shared OTUs, 16 bacterial genera were common to all samples. There were 150, 4, 1, and 0 bacterial genus exclusive to CL, GA, ON, and WO, respectively (Fig. 1B).
Table 1.
Statistics of sequences from the four Allium species.
| Sample | Clean sequences | Filter sequences | Percent |
|---|---|---|---|
| CL1 | 104537 | 101,135 | 96.74% |
| CL2 | 101538 | 97,814 | 96.33% |
| CL3 | 75316 | 73,726 | 97.88% |
| WO1 | 80666 | 78,952 | 97.87% |
| WO2 | 63260 | 61,541 | 97.28% |
| WO3 | 103209 | 99,372 | 96.28% |
| GA1 | 101393 | 98,498 | 97.14% |
| GA2 | 90773 | 89,212 | 98.28% |
| GA3 | 84732 | 83,482 | 98.52% |
| ON1 | 90120 | 87,924 | 97.56% |
| ON2 | 83307 | 81,838 | 98.23% |
| ON3 | 84192 | 83,143 | 98.75% |
| Total | 1,063,043 | 1,036,637 | 98.75% |
Note: Clean sequences: the numbers of combinated sequences; Filter sequences: the numbers of sequences after filtration.
Table 2.
Microbial community richness and diversity indices of the four Allium species at a 97% similarity threshold.
| Sample | Nseqs | OTUs | Coverage | Chao | Ace | Shannon | Npshannon |
|---|---|---|---|---|---|---|---|
| CL | 5121.33 ± 1503.40a | 435.33 ± 55.19a | 0.98 ± 0.01a | 484.10 ± 48.97a | 494.02 ± 44.63a | 4.95 ± 0.11a | 5.03 ± 0.10a |
| WO | 1071.67 ± 564.35b | 70.00 ± 35.10b | 0.98 ± 0.01a | 91.70 ± 47.00b | 110.32 ± 51.25c | 2.54 ± 0.51b | 2.62 ± 0.53b |
| GA | 1433.33 ± 117.02b | 113.67 ± 24.10b | 0.97 ± 0.00a | 176.82 ± 28.97b | 243.01 ± 15.87b | 3.07 ± 0.29b | 3.18 ± 0.30b |
| ON | 2093.33 ± 995.73ab | 110.00 ± 26.84b | 0.97 ± 0.01a | 165.59 ± 26.48b | 200.50 ± 35.24bc | 3.04 ± 0.30b | 3.14 ± 0.27b |
Note: Nseqs, number of sequences analyzed; OTU, operational taxonomical unit; ACE, abundance-based coverage estimator. Values are means ± standard error (n = 3). Different letters indicate statistically significant differences at the 0.05 probability level according to Fisher’s least significant difference (LSD) test. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
Figure 1.

Venn diagrams of the endophytic bacteria of the four Allium species. The OTUs (A) and bacterial genera (B) shared between Allium species. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: Onion.
Microbial diversity in CL was higher than that in the other three Allium species
The Alpha diversity of the bacterial communities was calculated using MOTHUR. The numbers of sequences (nseqs) (F = 3.79, P = 0.0584) clustered within OTUs in each of the Allium species were not statistically different, although the OTUs (F = 20.69, P = 0.0004) detected in CL were significantly higher than those in the other three Allium species. The microbial richness parameters such as Chao estimator (F = 19.67, P = 0.0005) and abundance-based coverage estimator (ACE) (F = 17.67, P = 0. 0007) of CL samples were higher than those of GA, ON and WO. The microbial diversity parameters including Shannon (F = 9.94, P = 0.0045) and npshannon (F = 9.7, P = 0.0048) indexes were also statistically higher in CL than those in other three other Allium species. These indicated that the richness and diversity of bacterial communities in CL were significantly higher than those in the other three Allium species (Table 2).
Bacteria in CL was more abundant than those in the other three Allium species at various taxonomic levels
The OTUs were classified into 20 phyla, 50 classes, 81 orders, 134 families, and 234 genera (Supplementary Table S1). Among the bacterial communities with relative amounts more than 1.0% of total abundance, 7 phyla, 11 classes, 14 orders, and 18 families were significantly different (Supplementary Table S2). Proteobacteria, Bacteroidetes, and an unclassified phylum were the three dominant phyla, accounting for 80.31, 94.72, 80.13 and 87.27% of the total bacterial communities in CL, WO, GA, and ON, respectively (Fig. 2). Except for the unclassified phylum, all phyla in CL were significantly more abundant than those in the other three Allium species (Supplementary Table S2). Based on their relative abundances, all the bacterial phyla were clustered into three groups: Proteobacteria was clustered into one independent group, Bacteroidetes and Actinobacteria were clustered into the second group, and the other phyla were gathered into the third group (Fig. 3). The abundance of one unclassified bacterial community in CL was significantly lower than that in the other Allium species at the class (F = 16.84, P = 0.0008), order (F = 16.84, P = 0.0008), and family (F = 16.84, P = 0.0008) levels. while the abundances of other bacterial communities in CL were significantly higher than those in the other three Allium species (Supplementary Table S2).
Figure 2.
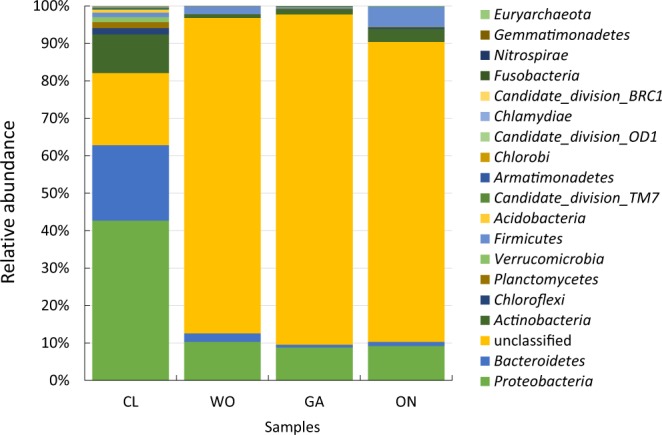
The relative abundance of bacterial phyla in the four Allium species. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
Figure 3.
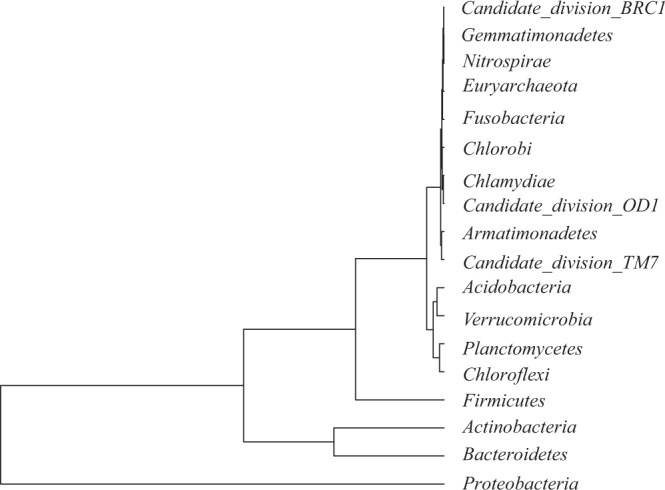
Hierarchical cluster tree of bacterial phyla in the four Allium species.
At the genus level, a total of 234 genera were detected in the four Allium species. Among these, 20 genera with relative amounts larger than 1.0% were selected for further analysis. The selected genera, belonging to 5 phyla, accounted for 62.61, 88.97, 90.97, and 86.04% in CL, WO, GA, and ON, respectively. Among these, 10 genera belonged to Proteobacteria, 4 to Bacteroidetes, 3 to Actinobacteria, 1 to Chloroflexi and 2 to the unclassified phylum. The heatmap showed all the genera were enriched in CL except for the 2 unclassified genera (g_unclassified;p_unclassified and g_unclassified;c_Alphaproteobacteria;p_Proteobacteria) (Fig. 4). The statistical analysis showed 15 of the 20 selected genera were significantly different among the four Allium species. The abundance of one unclassified genus (g_unclassified;p_unclassified) in CL (17.45%) was significantly lower than that in WO (82.12%), GA (70.56%) and ON (76.95%) (F = 16.84, P = 0.0008), while the other 14 genera in CL were significantly higher than those in the other three Allium species (Supplementary Table S2).
Figure 4.

The heatmap displaying the relative abundances of the most dominant genera (top 20, accounting for more than 1.0% total abundance). CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
Bacterial communities in WO, GA, and ON were more similar than those in CL samples
Important distinctions existed in the compositions of bacterial communities among the four Allium species. The LDA Effect Size (LEfSe) (Fig. 5) showed significantly different taxonomic abundances among the four Allium species. Interestingly, at the phylum-to-family level, bacterial communities in CL exhibited relatively higher abundances than those in the other three Allium species. Five phyla (including Chloroflexi and Planctomycetes), 13 classes (including Deltaproteobacteria, Verrucomicrobiae, Acidobacteria), 25 orders (including Micromonosporales, Propionibacteriales, and Pseudonocardiales), and 36 families (including Micromonosporaceae, Nocardioidaceae, and Pseudonocardiaceae) were significantly enriched in the CL samples (Fig. 5A). These abundant taxa could be considered as potential biomarkers (LDA > 3.5, P < 0.05) (Fig. 5B).
Figure 5.

Groups from the phylum-to-genus levels determined to be significant representatives of their sample group, based on LEfSe software analysis. (A) Cladogram representing the taxonomic hierarchical structure of the identified habitat biomarkers generated using LEfSe. Each ring represents a taxonomic level, with phylum, class, order, and family emanating from the center to the periphery. Each circle is a taxonomic unit in the dataset, with circles or nodes shown in color where the taxon represents a significantly more abundant group. (B) Identified biomarkers ranked by their effect size in different samples. The habitat biomarkers were identified as being significantly abundant (p < 0.05) when compared among samples. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
To compare overall microbial communities between the four Allium species, beta-diversity analysis based on a phylogenetic tree (Fig. 6A) and PCA (Fig. 6B) was conducted using weighted UniFrac phylogenetic distances. The phylogenetic tree indicated that the 12 Allium samples were clustered into two groups: group 1 consisted of 3 samples of CL; group 2 consisted of 9 samples of WO, GA, and ON. The highest PCA variations in the bacteria were 68.7% (PC1) and 13.2% (PC2), representing a strong separation of different samples. The samples of CL were clustered together and distinguished from those of the other three Allium species which were overlapped with each other. Furthermore, comparison of within- and between-group distances for all Allium samples revealed that CL tended to be the most homogeneous, while ON was the most heterogenous (Fig. 6C), and that WO, GA, and ON samples were more alike than CL samples (Fig. 6D). B-diversity indicated that bacterial structure and composition of CL was significantly different from those of WO, GA, and ON.
Figure 6.

The endophytic bacterial profile of the four Allium species. Hierarchical cluster tree (A) and principal coordinate analysis (PCA) (B) of different microbiota in different samples based on weighted UniFrac distances. Comparison of within (C) and between (D) group distances for the Allium species. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
The predicted KEGG pathways in CL is more abundant than those in the other three Allium species
Microbiota functions were predicted using the PICRUSt algorithm based on the Greengenes database. A total of 23 relevant KEGG categories were predicted, including Cellular Processes, Environmental Information Processing, Genetic Information Processing and Metabolism. The frequencies ranged from 24 to 3,944,197 (Supplementary Table S3). 17 of these KEGG pathways in CL exhibited significantly higher abundance than those in the other three Allium species. (Supplementary Table S3). Membrane Transport (F = 5.98, P = 0.0193), Amino Acid Metabolism (F = 5.73, P = 0.0216), Carbohydrate Metabolism (F = 5.76, P = 0.0213), Metabolism of Cofactors and Vitamins (F = 5.16, P = 0.0283) and Lipid Metabolism (F = 5.16, P = 0.0282) were the five most enriched KEGG (Fig. 7, Supplementary Table S3).
Figure 7.

KEGG pathways with significant differences among the four Allium species. CL: Chinese leek; WO: Welsh onion; GA: Garlic; ON: onion.
To further identify microbiota function in the four Allium species, we analyzed 206 lower-level KEGG pathways within the 23 KEGG categories the results of which showed that 159 of the lower-level KEGG pathways (accounting for 71.81% of the total abundance) were significantly different among the four Allium species (Supplementary Table S4). Interestingly, all these KEGG pathways in CL, containing Transporters (F = 6.68, P = 0.0143), ABC transporters (F = 6.29, P = 0.0169), Secretion system (F = 4.50, P = 0.0395), Peptidases (F = 5.57, P = 0.0233), Streptomycin biosynthesis (F = 6.62, P = 0.0147), Sulfur metabolism (F = 7.68, P = 0.0097) and Steroid hormone biosynthesis (F = 8.19, P = 0.0080), were also significantly higher than those in the other three Allium species. Otherwise, the 47 KEGG categories were not statistically different among the four Allium species (Supplementary Table S4).
Bacteria were very closely related to genes
To clarify the relationship among the bacterial communities and predicted bacterial functions, the correlation of the 20 bacterial genera (>1.0%) and the 23 KEGG categories were analyzed using the Pearson method. The results showed that the KEGG pathways were firmly related to bacterial genera. Among these relations, 100 pairs comprised of 9 genera and 21 KEGG pathways showed a very significant correlation (r > 0.8, P < 0.01) (Supplementary Table S5, Fig. 8).
Figure 8.

KEGG pathways with significant differences among the four Allium species were highly correlated with bacterial genera with significant differences. *0.01 < p < 0.05, **0.001 < p < 0.01, ***p < 0.001.
Discussion
In the present study, we compared the endophytic bacterial communities between the four Allium species: CL, GA, ON, and WO by using high-throughput sequencing technology. The results showed that the bacterial communities in CL were significantly different from those in the other three Allium species. The diversity and richness of endophytic bacterial communities in CL were more abundant than those in the GA, WO and ON. Also, the predicted gene functions of endophytic bacteria mainly focused on Membrane Transport, Amino Acid Metabolism, and Carbohydrate Metabolism pathway.
Proteobacteria was core phyla in the plant endosphere. In a review, it summarized that Proteobacteria was the dominant phylum, the average abundance of which was approximately 50%1. Other previous studies also supported this conclusion. Endophytic Proteobacteria in apple shoots of different rootstock/scion combinations was as high as 58.4%20, and Proteobacteria comprised approximately 42.40–64.70% of reads across all roots and leaves of the peony tree21. Proteobacteria was also the most abundant phylum (90%) in the leaves of tomato22, and it represented 84% of the total bacteria in sweet clover root23. The results of our present study showed that the endophytic bacterial community was significantly different in the four Allium species. At the phylum level, Proteobacteria was the dominant bacterial community in CL (42.68%), and also the dominant bacterial community in WO (10.31%), GA (8.80%), and ON (9.20%). However, the amount in the three Allium species was significantly lower than that in CL (F = 20.21, P = 0.0004).
Our study revealed that one unclassified phylum was the most dominant bacterial community in WO (82.12%), GA(70.56%) and ON(76.95%) besides Proteobacteria, the abundance of which was significantly higher in the three Allium species than that in CL (17.45%) (F = 16.84, P = 0.0008). The vast abundance of the unclassified phyla was also found in the previous studies24. On the one hand, these unclassified bacterial species reflected the advantages of high-throughput sequencing technology to detect minor, rare, and uncultured bacterial species. On the other hand, it showed that Allium species were abundant in the beneficial bacterial communities25.
The comparative analysis showed significant differences between the four Allium species at the phylum, class, order, family, and genus level. Interestingly, the majority of the different bacterial communities in CL were significantly higher than that in the other Allium species. The alpha diversity indicated higher bacterial richness and diversity in CL than those in the other three Allium species. Meanwhile, the beta-diversity analysis showed that bacterial communities in WO, GA, and ON were more similar than those in CL. This was consistent with the conclusion that the GA, ON, and WO were closer related to each other, and farther away from CL in a genetic relationship26,27. In China, CL was planted once and harvested continuously for 3–5 years, while GA, ON, and WO were planted once in a year and harvested once. That was to say, under this cultivation pattern, the growth period of CL in the soil was 3–5 times more than those of GA, ON and WO. The numbers of sequences (nseqs) and the operational taxonomic units (OTUs) detected in CL were 2.45–4.78 and 3.96–6.22 times higher than those in the other three Allium species. However, the dominant bacteria in the GA, ON and WO was more evident than that in the CL. For example, one unclassified genus was the most dominant bacteria in the four Allium species. The amounts accounted for 82.12, 70.56 and 76.95% in WO, GA and ON, which were 4.04–4.71 times more than that in CL, respectively. These factors, including heredity, cultivation patterns, and dominant bacteria, have contributed to the occurrence of this phenomenon.
Endophytes and host plants established a relationship of mutualistic symbiosis. The growth scale and speed of endophytes in plants were rather crucial for maintaining the relationship. Previous studies have shown that there was a balance point between endophytes and plants. Once the balance was broken, this mutualistic symbiosis would be destroyed. Endophytes obtained a large number of nutrients from plants, degraded the nutrients and cell walls of plants, and changed from mutualistic symbiosis to parasitic relationship. It might even turn into a “pathogen”28–30. This balance was determined by the genetic material of endophytes and plants, and it also was the result of mutual adaptation and natural selection. So the different endophytic bacterial communities in four Allium species might be formed through long-term evolution and were also a necessary condition for maintaining the mutualistic symbiosis relationship between endogenous bacteria and plants.
In the present study, the microbiota function analysis revealed the endophytic bacteria were annotated in 23 KEGG categories. Membrane transport was the richest pathway. It included three lower-level pathway: Transporters, ABC transporters, and secretion system. Transport protein/ABC transporter is vital for bacterial growth and survival for their nutrient absorption and resistance to endogenous and environmental pressures31,32. In plants, it participates in the transport of many substances, such as hormones, lipids, secondary metabolites, and exogenous organisms. It also participates in the interaction between plants and pathogens and the regulation of ion channels33. Secretion system can deliver bacterial effector proteins into host cells to kill or inhibit their growth, and help the bacteria thrive in its host34,35. That is to say, the three pathways are closely related to the survival of endophytic bacteria and host plants. This explained why Transporters, ABC transporters, and secretion system were the three most enriched pathway among the 206 lower-lever pathways in our study.
Amino acids are not only the main components of proteins but also play an essential role in many other physiological processes. They have osmotic effects, regulate ion transport, control stomatal opening, participate in the detoxification of heavy metals, promote redox homeostasis, affect gene expression, and influence the synthesis and activity of some enzymes. Also, amino acids are precursors of many secondary metabolites, which have vital functions such as signal, defense, interaction with other organisms, and photoprotection36. Glycolysis and the citric acid cycle (TCA cycle) are key respiratory pathways in carbohydrate metabolism. They provide plant and bacteria with energy and metabolites for growth and development. Besides, they produce many intermediates for synthesizing other substances37,38. In our study, amino acid metabolism and carbohydrate metabolism pathway were the second and third most enriched pathway in the 23 KEGG categories, which revealed that the majority of the endophytic bacteria of the four Allium species are involved in the two pathways. That may because endophytic bacteria live on the host plants and form a symbiotic relationship with the host. The growth of endophytic bacteria requires various nutrient sources and energy. They participate in the synthesis, degradation, and absorption of amino acids and carbohydrate in the host to provide the nutrient sources and energy for its growth, on the other hand, provide essential amino acids and energy for the host to meet the physiological needs.
Allium plants are known to possess antibacterial and antifungal properties, which are due to their organic sulfides, especially thiosulfinates39. Thiosulfinates decomposed into various sulfur compounds with potent antimicrobial properties19,40. The results showed that endophytic bacteria possess sulfur metabolism function, and it may help the host to regulate the organosulfur compounds.
Besides the well-known sulfur compounds, there exist other kinds of compounds such as saponins, flavonoids, nitrogen compounds, and peptides in Allium species with vigorous inhibitive effects on pathogenic microbes19. Our results showed the endophytic bacteria also involved in other important physiological activities such as peptidases, nitrogen metabolism, terpenoid backbone biosynthesis, streptomycin biosynthesis, tetracycline biosynthesis, biosynthesis of ansamycins, and steroid hormone biosynthesis. Previous studies pointed out that plants and endophytes may have similar or identical pathways for the synthesis of secondary metabolites. Moreover, they can synthesize the same or similar active substances30. Therefore, all these multifunctional endophytic bacteria corporately regulated the metabolism of organosulfur compounds, secondary metabolites, and antibiotics, which demonstrated the antimicrobial properties of Allium species.
PICRUSt can predict the corresponding bacterial metabolic function spectrum through the 16S rRNA gene sequence41. PICRUSt can unscramble potential functions from the microbiome composition data. With PICRUSt, it acts as a bridge between the “composition” and “function” of the microbiome. However, the greengenes reference database tends to study human-related microorganisms, which reduces the accuracy of predicting non-human metagenomes. In our present study, to test the accuracy of the bacterial function prediction, we performed the correlations between bacterial genera and the KEGG pathways, the results revealed they were closely related to each other. For example, Achromobacter spp. were predicted to possess the function of Xenobiotics Biodegradation and Metabolism (r = 0.8346, P = 0.0007). The previous studies reported Achromobacter spp. is efficient in degrading atrazine42; glyphosate43; 2,4-dichlorophenoxyacetic acid (2,4-D) and 2-methyl-4-chlorophenoxy acetic acid (MCPA)44. Enterobacter spp. were estimated to have the function of Biosynthesis of Other Secondary Metabolites (r = 0.836, P = 0.0007) and Amino Acid Metabolism (r = 0.810, P = 0.0014). Previous studies demonstrated Enterobacter spp. produce secondary metabolites such as siderophores and indole-3-acetic acid(IAA)45–47, and enzymes such as pectinase, 1-ami-nocyclopropane-1-carboxylate(ACC) deaminase, and arginine decarboxylase46,48,49. Flavobacterium spp. were estimated to have the function Biosynthesis of Other Secondary Metabolites (r = 0.8445, P = 0.0005) and Glycan Biosynthesis and Metabolism (r = 0.8157, P = 0.0012). The previous studies showed that Flavobacterium spp. synthesized carotenoid zeaxanthin50, produced indolic compounds and biosurfactant51; Flavobacterium spp. also digests alginate oligosaccharides52 and many polysaccharides, including chitin53. So all these previous studies indicated that the predicted function was consistent with the actual function of endophytic bacteria.
Methods
Sample collection
GA, ON, WO, CL were collected from the experimental base of the Qingdao Agricultural University located at Jiaozhuo, Qingdao city, China. The four Allium species were cultured according to conventional agricultural practices, and the underground parts of each plant were used to analyze the endophytic bacterial communities. Individually, the bulbs of garlic and onion, the white pseudostem of Welsh onion, and the rhizome of Chinese leek were analyzed. Three biological replications were sampled; each consisted of five individual plants.
Sample pretreatment was carried out according to references with minor modifications25. The sampled bulbs, pseudostems, and rhizomes were washed using tap water and cut into small pieces of 2-cm long. Then, they were soaked in 70% ethyl alcohol for 2 min, immersed in 2.5% sodium hypochlorite for 5 min, and finally washed with sterile water 4 times. From the fourth wash, 100 μl of the water was added to potato dextrose agar (PDA) plates and incubated at 28 °C for 7 d to determine the disinfection effect. Disinfection grade experiment materials were used to analyze the endophytic bacterial community.
DNA extraction, PCR amplification, and Illumina library generation
DNA of the four Allium species was extracted using TIANamp Bacteria DNA Kit(Tiangen Biotech, Beijing, China) according to manufacturer’s instructions and stored at −20 °C until analysis. The target-specific primers B341F (5′-CCTACGGGNGGCWGCAG-3′) and B785R (5′-GACTACHVGGGTATCTAATCC-3′) were used to amplify the V3-V4 region of the bacterial 16S rRNA gene25,54. PCR reaction referenced to our previous study with minor modifications25. Each PCR reaction was performed using a KAPA HiFi Hotstart PCR Kit (KAPA Biosystems, Wilmington, MA, USA) on a T100TM Thermal Cycler (Bio-Rad, Hercules, CA, USA). Each reaction (25 μL) contained 10 ng of DNA template, 12.5 μL 2 × KAPA HiFi HotStart ReadyMix, and 0.25 μmol L−1 of each primer. The following PCR conditions were used: initial denaturation at 95 °C for 3 min, followed by 25 cycles consisting of denaturation (95 °C for 30 s), annealing (55 °C for 30 s) and extension (72 °C for 30 s) and a final extension step at 72 °C for 5 min. PCR amplicon libraries were purified from a 1.2% agarose gel by QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and quantified using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA, USA), and finally sequenced using the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) at Beijing Ori-Gene Science and Technology (China) to create paired-end reads. The raw reads have been deposited at the Sequence Read Archive (SRA) database of NCBI (SRR8240026 to SRR8240037).
Sequence processing and analysis
FLASH55 was used to merge the paired-end reads. The combined reads were filtered using MOTHUR56 to remove sequences with low quality scores (≤20), to remove sequences with N, to remove sequences with too long homopolymer (>10 bp), to remove sequences with excessive primer mismatch (≥4 bp) and the primer sequence, to remove sequences that are too short (≤200 bp) and too long (≥500 bp). Moreover, using UCHIME57 to remove chimera with Gold dataset as reference58. The filtered sequences were clustered into operational taxonomic units (OTUs) at 97% identity using USEARCH59. Each OTU was systematically classified with the reference of the SILVA database60 using RDP classifiers61. The diversity indices for each species were analyzed using MOTHUR56.
OTUs were aligned to SILVA database60 using PyNAST software62 to construct a phylogenetic tree using FastTree63. UniFrac software64 was used to generate the weighted distance matrix among bacterial communities. Furthermore, we used the LDA Effect Size (LEfSe) to identify differentially abundant families among samples for biomarker discovery65. We also predicted the functional profiling of microbial communities using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) algorithm41. The 16S rRNA sequences were assigned with the Greengenes database to perform closed reference picking, and then the resulting sequences were uploaded onto the PICRUSt website (http://huttenhower.sph.harvard.edu/galaxy/), to generate predicted functional metagenomes using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database66,67 as a functional reference.
Statistical analysis
The data were analyzed using a one-way ANOVA in SAS ver. 8.0 software. Significance differences among the four Allium species were determined using Fisher’s least significant difference test (P ≤ 0.05). Venn diagrams were generated using the “Venn diagram” package, heatmap images were produced using the “pheatmap” package, and Principal coordinate analysis (PCA) figures were created using the “princomp” program in R (v3.1.2). LefSe and PICRUSt were analyzed using LefSe (https://bitbucket.org/biobakery/biobakery/wiki/lefse) and PICRUSt software (http://huttenhower.sph.harvard.edu/galaxy/), respectively. Pearson’s correlations were used to assess the relationships between gene functions and bacterial communities.
Supplementary information
Acknowledgements
I thank Beijing Ori-Gene Science and Technology (China) for providing the Illumina Miseq platform and the assistance of bioinformatical analysis. This work was supported by the National Natural Science Foundation of China (31471864 and 31272151) and the Qingdao Agricultural University High-level Personnel Startup Fund, China (6631115024).
Author contributions
Yonghong Huang contributed to the conception of the study, wrote the manuscript, and performed the experiments and data analyses.
Data availability
Data are available from the Sequence Read Archive (SRA) database of NCBI (SRR8240026 to SRR8240037) https://www.ncbi.nlm.nih.gov/bioproject/PRJNA506734.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-51707-7.
References
- 1.Liu H, et al. Inner Plant Values: Diversity, Colonization and Benefits from Endophytic Bacteria. Frontiers in microbiology. 2017;8:2552. doi: 10.3389/fmicb.2017.02552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan AL, et al. Indole acetic acid and ACC deaminase from endophytic bacteria improves the growth of Solanum lycopersicum. Electronic Journal of Biotechnology. 2016;21:58–64. doi: 10.1016/j.ejbt.2016.02.001. [DOI] [Google Scholar]
- 3.Yaish MW, Antony I, Glick BR. Isolation and characterization of endophytic plant growth-promoting bacteria from date palm tree (Phoenix dactylifera L.) and their potential role in salinity tolerance. Antonie Van Leeuwenhoek. 2015;107:1519–32. doi: 10.1007/s10482-015-0445-z. [DOI] [PubMed] [Google Scholar]
- 4.Khan MU, et al. Cr-resistant rhizo- and endophytic bacteria associated with Prosopis juliflora and their potential as phytoremediation enhancing agents in metal-degraded soils. Frontiers in Plant Science. 2015;5:755. doi: 10.3389/fpls.2014.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y, et al. The hyperaccumulator Sedum plumbizincicola harbors metal-resistant endophytic bacteria that improve its phytoextraction capacity in multi-metal contaminated soil. Journal of Environmental Management. 2015;156:62–9. doi: 10.1016/j.jenvman.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Zhang C, Oliveira RS, Freitas H, Luo Y. Bioaugmentation with Endophytic Bacterium E6S Homologous to Achromobacter piechaudii Enhances Metal Rhizoaccumulation in Host Sedum plumbizincicola. Frontiers in Plant Science. 2016;7:75. doi: 10.3389/fpls.2016.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Syranidou E, et al. Exploitation of Endophytic Bacteria to Enhance the Phytoremediation Potential of the Wetland HelophyteJuncus acutus. Frontiers in microbiology. 2016;7:1016. doi: 10.3389/fmicb.2016.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wicaksono WA, Jones EE, Casonato S, Monk J, Ridgway HJ. Biological control of Pseudomonas syringae pv. actinidiae (Psa), the causal agent of bacterial canker of kiwifruit, using endophytic bacteria recovered from a medicinal plant. Biological control. 2017;116:103–12. doi: 10.1016/j.biocontrol.2017.03.003. [DOI] [Google Scholar]
- 9.D’Alessandro M, et al. Volatiles produced by soil-borne endophytic bacteria increase plant pathogen resistance and affect tritrophic interactions. Plant Cell & Environment. 2014;37:813–26. doi: 10.1111/pce.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glassner Hanoch, Zchori-Fein Einat, Compant Stéphane, Sessitsch Angela, Katzir Nurit, Portnoy Vitaly, Yaron Sima. Characterization of endophytic bacteria from cucurbit fruits with potential benefits to agriculture in melons (Cucumis meloL.) FEMS Microbiology Ecology. 2015;91(7):fiv074. doi: 10.1093/femsec/fiv074. [DOI] [PubMed] [Google Scholar]
- 11.Joe MM, Devaraj S, Benson A, Sa T. Isolation of phosphate solubilizing endophytic bacteria from Phyllanthus amarus Schum & Thonn: Evaluation of plant growth promotion and antioxidant activity under salt stress. Journal of Applied Research on Medicinal & Aromatic Plants. 2016;3:71–7. doi: 10.1016/j.jarmap.2016.02.003. [DOI] [Google Scholar]
- 12.Shahzad R, et al. Inoculation of abscisic acid-producing endophytic bacteria enhances salinity stress tolerance in Oryza sativa. Environmental & Experimental Botany. 2017;136:68–77. doi: 10.1016/j.envexpbot.2017.01.010. [DOI] [Google Scholar]
- 13.Chang TC, et al. A Comparative Study on the Total Antioxidant and Antimicrobial Potentials of Ethanolic Extracts from Various Organ Tissues of Allium spp. Food and Nutrition Sciences. 2013;4:182–90. doi: 10.4236/fns.2013.48A022. [DOI] [Google Scholar]
- 14.Feng F, et al. Isolation, Colonization, and Chlorpyrifos Degradation Mediation of the Endophytic Bacterium Sphingomonas Strain HJY in Chinese Chives (Allium tuberosum) J Agric Food Chem. 2017;65:1131–8. doi: 10.1021/acs.jafc.6b05283. [DOI] [PubMed] [Google Scholar]
- 15.Muvea AM, et al. Colonization of onions by endophytic fungi and their impacts on the biology of Thrips tabaci. PloS one. 2014;9:e108242. doi: 10.1371/journal.pone.0108242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki T, Igarashi Y, Ogawa M, Furumai T. Identification of 6-prenylindole as an antifungal metabolite of Streptomyces sp. TP-A0595 and synthesis and bioactivity of 6-substituted indoles. The Journal of Organic Chemistry. 2003;55:1009–12. doi: 10.7164/antibiotics.55.1009. [DOI] [PubMed] [Google Scholar]
- 17.Igarashi Y, et al. Fistupyrone, a novel inhibitor of the infection of Chinese cabbage by Alternaria brassicicola, from Streptomyces sp TP-A0569. Journal of Antibiotics. 2000;53:1117–22. doi: 10.7164/antibiotics.53.1117. [DOI] [PubMed] [Google Scholar]
- 18.Shentu X, Zhan X, Ma Z, Yu X, Zhang C. Antifungal activity of metabolites of the endophytic fungus Trichoderma brevicompactum from garlic. Brazilian journal of microbiology. 2014;45:248–54. doi: 10.1590/S1517-83822014005000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanzotti V, Bonanomi G, Scala F. What makes Allium species effective against pathogenic microbes? Phytochemistry Reviews. 2013;12:751–72. doi: 10.1007/s11101-013-9295-3. [DOI] [Google Scholar]
- 20.Liu J, et al. Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype-specific influence. Microbiome. 2018;6:18. doi: 10.1186/s40168-018-0403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang R, Liu P, Ye W. Illumina-based analysis of endophytic bacterial diversity of tree peony (Paeonia Sect. Moutan) roots and leaves. Brazilian Journal of Microbiology. 2017;48:695–705. doi: 10.1016/j.bjm.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romero FM, Marina M, Pieckenstain FL. The communities of tomato (Solanum lycopersicum L.) leaf endophytic bacteria, analyzed by 16S‐ribosomal RNA gene pyrosequencing. Fems Microbiology Letters. 2014;351:187–94. doi: 10.1111/1574-6968.12377. [DOI] [PubMed] [Google Scholar]
- 23.Mitter EK, de Freitas JR, Germida JJ. Bacterial Root Microbiome of Plants Growing in Oil Sands Reclamation Covers. Frontiers in microbiology. 2017;8:849. doi: 10.3389/fmicb.2017.00849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Z, et al. Rhizosphere microbial community manipulated by 2 years of consecutive biofertilizer application associated with banana Fusarium wilt disease suppression. Biology & Fertility of Soils. 2015;51:553–62. doi: 10.1007/s00374-015-1002-7. [DOI] [Google Scholar]
- 25.Huang Y. Comparison of rhizosphere and endophytic microbial communities of Chinese leek through high-throughput 16S rRNA gene Illumina sequencing. Journal of Integrative Agriculture. 2018;17:359–67. doi: 10.1016/S2095-3119(17)61731-3. [DOI] [Google Scholar]
- 26.Zhou S, He X, Ge S. trnK-Gene-Based Molecular Phylogeny of Allium. Plants(Lilaceae s.l.) Acta Bot Boreal-Occident Sin. 2006;26:906–14. [Google Scholar]
- 27.He X, Ge S, Xu J, Hong D. PCR-RFLP analysis on phylogeny of Allium. Acta Botanica Sinica. 1998;40:1083–6. [Google Scholar]
- 28.Kogel KH, Franken P, Hückelhoven R. Endophyte or parasite – what decides? Current Opinion in Plant Biology. 2006;9:358–63. doi: 10.1016/j.pbi.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Schulz B, Römmert AK, Dammann U, Aust HJ, Strack D. The endophyte-host interaction: a balanced antagonism? Mycological Research. 1999;103:1275–83. doi: 10.1017/S0953756299008540. [DOI] [Google Scholar]
- 30.Ludwig-Müller J. Plants and endophytes: equal partners in secondary metabolite production? Biotechnology letters. 2015;37:1325–34. doi: 10.1007/s10529-015-1814-4. [DOI] [PubMed] [Google Scholar]
- 31.Piepenbreier H, Fritz G, Gebhard S. Transporters as information processors in bacterial signalling pathways. Molecular Microbiology. 2017;104:1–15. doi: 10.1111/mmi.13633. [DOI] [PubMed] [Google Scholar]
- 32.Garai P, Chandra K, Chakravortty D. Bacterial peptide transporters: Messengers of nutrition to virulence. Virulence. 2017;8:297–309. doi: 10.1080/21505594.2016.1221025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chao-Feng H, Naoki Y, Jian Feng M. Knockout of a bacterial-type ATP-binding cassette transporter gene, AtSTAR1, results in increased aluminum sensitivity in Arabidopsis. Plant physiology. 2010;153:1669–77. doi: 10.1104/pp.110.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mach J. Tracking the Bacterial Type III Secretion System: Visualization of Effector Delivery Using Split Fluorescent Proteins. Plant Cell. 2017;29:1547–8. doi: 10.1105/tpc.17.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho BT, Fu Y, Dong TG, Mekalanos JJ. Vibrio cholerae type 6 secretion system effector trafficking in target bacterial cells. Proc Natl Acad Sci USA. 2017;114:9427–32. doi: 10.1073/pnas.1711219114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Florencio-Ortiz V, Sellés-Marchart S, Zubcoff-Vallejo J, Jander G, Casas JL. Changes in the free amino acid composition of Capsicum annuum (pepper) leaves in response to Myzus persicae (green peach aphid) infestation. A comparison with water stress. PloS one. 2018;13:e0198093. doi: 10.1371/journal.pone.0198093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finger F. Glycolysis Is Dynamic and Relates Closely to Respiration Rate in Stored Sugarbeet Roots. Frontiers in Plant Science. 2017;8:861. doi: 10.3389/fpls.2017.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong M, et al. Effects of exogenous putrescine on glycolysis and Krebs cycle metabolism in cucumber leaves subjected to salt stress. Plant Growth Regulation. 2016;79:319–30. doi: 10.1007/s10725-015-0136-9. [DOI] [Google Scholar]
- 39.Broderick SR, et al. RNA-sequencing reveals early, dynamic transcriptome changes in the corollas of pollinated petunias. BMC plant biology. 2014;14:307. doi: 10.1186/s12870-014-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rose P, Whiteman M, Moore PK, Zhu YZ. Bioactive S-alk(en)yl cysteine sulfoxide metabolites in the genus Allium: the chemistry of potential therapeutic agents. Natural Product Reports. 2005;22:351–68. doi: 10.1039/b417639c. [DOI] [PubMed] [Google Scholar]
- 41.Langille MGI, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology. 2013;31:814. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandes AFT, et al. Degradation of atrazine by Pseudomonas sp. and Achromobacter sp. isolated from Brazilian agricultural soil. International Biodeterioration & Biodegradation. 2018;130:17–22. doi: 10.1016/j.ibiod.2018.03.011. [DOI] [Google Scholar]
- 43.Ermakova IT, et al. Organophosphonates utilization by soil strains of Ochrobactrum anthropi and Achromobacter sp. Archives of Microbiology. 2017;199:665–75. doi: 10.1007/s00203-017-1343-8. [DOI] [PubMed] [Google Scholar]
- 44.Xia ZY, et al. Biodegradation of the Herbicide 2,4-Dichlorophenoxyacetic Acid by a New Isolated Strain of Achromobacter sp. LZ35. Current Microbiology. 2017;74:193–202. doi: 10.1007/s00284-016-1173-y. [DOI] [PubMed] [Google Scholar]
- 45.Lafi FF, et al. Draft Genome Sequence of Enterobacter sp. Sa187, an Endophytic Bacterium Isolated from the Desert Plant Indigofera argentea. Genome Announcements. 2017;5:e01638–16. doi: 10.1128/genomeA.01638-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Wang Q, Wang L, He LY, Sheng XF. Increased growth and root Cu accumulation of Sorghum sudanense by endophytic Enterobacter sp. K3-2: Implications for Sorghum sudanense biomass production and phytostabilization. Ecotoxicology & Environmental Safety. 2016;124:163–8. doi: 10.1016/j.ecoenv.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y, et al. Survival Strategies of the Plant-Associated Bacterium Enterobacter sp. Strain EG16 under Cadmium Stress. Applied and environmental microbiology. 2016;82:1734–44. doi: 10.1128/AEM.03689-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh RP, Jha PN. Mitigation of salt stress in wheat plant (Triticum aestivum) by ACC deaminase bacterium Enterobacter sp. SBP-6 isolated from Sorghum bicolor. Acta Physiologiae Plantarum. 2016;38:1–12. doi: 10.1007/s11738-015-2023-4. [DOI] [Google Scholar]
- 49.Reddy MPC, Saritha KV. Effects of the culture media optimization on pectinase production by Enterobacter sp. PSTB-1. Biotech. 2016;6:207. doi: 10.1007/s13205-016-0502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JH, Jin WK, Lee PC. Complete genome sequence of Flavobacterium kingsejongi WV39, a type species of the genus Flavobacterium and a microbial C40 carotenoid zeaxanthin producer. Journal of Biotechnology. 2018;266:9–13. doi: 10.1016/j.jbiotec.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 51.Sang MK, Kim KD. The volatile-producing Flavobacterium johnsoniae strain GSE09 shows biocontrol activity against Phytophthora capsici in pepper. Journal of applied microbiology. 2012;113:383–98. doi: 10.1111/j.1365-2672.2012.05330.x. [DOI] [PubMed] [Google Scholar]
- 52.Huang L, et al. Characterization of a new alginate lyase from newly isolated Flavobacterium sp. S20. Journal of Industrial Microbiology & Biotechnology. 2013;40:113–22. doi: 10.1007/s10295-012-1210-1. [DOI] [PubMed] [Google Scholar]
- 53.Kharade SS, Mcbride MJ. Flavobacterium johnsoniae PorV is required for secretion of a subset of proteins targeted to the type IX secretion system. Journal of Bacteriology. 2015;197:147–58. doi: 10.1128/JB.02085-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klindworth A, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied & Environmental Microbiology. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edgar RC, Haas BJ, Clemente JC, Christopher Q, Rob K. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haas BJ, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 2013;10:996–8. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 60.Pruesse E, et al. Nucleic Acids Research. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB; pp. 7188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caporaso JG, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–7. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price MN, Dehal PS, Arkin AP. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Molecular Biology & Evolution. 2009;26:1641–50. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Applied & Environmental Microbiology. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanehisa M, Goto S. KEGG: Kyoto Encyclopaedia of Genes and Genomes. Nucleic Acids Research. 2000;ume 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the Sequence Read Archive (SRA) database of NCBI (SRR8240026 to SRR8240037) https://www.ncbi.nlm.nih.gov/bioproject/PRJNA506734.