

Abstract
Rapid proliferation of cancer cells assisted by endothelial cell-mediated angiogenesis and acquired inflammation at the tumor microenvironment (TME) lowers the success rate of chemotherapeutic regimens. Therefore, targeting these processes using localized delivery of a minimally toxic drug combination may be a promising strategy. Here, we present engineering of a biocompatible self-assembled lithocholic acid-dipeptide derived hydrogel (TRI-Gel) that can maintain sustained delivery of antiproliferating doxorubicin, antiangiogenic combretastatin-A4 and anti-inflammatory dexamethasone. Application of TRI-Gel therapy to a murine tumor model promotes enhanced apoptosis with a concurrent reduction in angiogenesis and inflammation, leading to effective abrogation of tumor proliferation and increased median survival with reduced drug resistance. In-depth RNA-sequencing analysis showed that TRI-Gel therapy induced transcriptome-wide alternative splicing of many genes responsible for oncogenic transformation including sphingolipid genes. We demonstrate that TRI-Gel therapy targets the reversal of a unique intron retention event in β-glucocerebrosidase 1 (Gba1), thereby increasing the availability of functional Gba1 protein. An enhanced Gba1 activity elevates ceramide levels responsible for apoptosis and decreases glucosylceramides to overcome drug resistance. Therefore, TRI-Gel therapy provides a unique system that affects the TME via post-transcriptional modulations of sphingolipid metabolic genes, thereby opening a new and rational approach to cancer therapy.
Short abstract
Hydrogel-mediated delivery of antiproliferating, antiangiogenic, and anti-inflammatory drugs aid in tumor regression by altering the sphingolipid metabolism through alternative splicing.
Introduction
Tumor microenvironment (TME) consists of rapidly proliferating cancer cells infiltrated by different host cell types like vascular endothelial cells, macrophages, tumor-associated fibroblasts, and other immune cells.1 Intercellular communications occurring through a network of cytokines, chemokines, growth factors, and matrix remodeling enzymes generate a conducive environment for cancer cells to proliferate, invade, metastasize, and develop drug resistance.2 Angiogenesis and inflammation induced by directed migration of endothelial cells and immune cells trigger an immunosuppressive and pro-proliferative niche at the tumor site.2 Such a programmed neoplastic transformation reduces the efficacy of chemotherapeutic drugs and allows the tumor to develop drug resistance.3 Therefore, angiogenesis and inflammation along with uncontrolled tumor cell proliferation are critical therapeutic targets to achieve effective tumor regression and patient survival and for combating drug resistance.4
Recent advances in lipid research endorse the role of sphingolipids in the pathophysiology of tumor progression including cell proliferation, inflammation, angiogenesis, and drug resistance.5−7 Many enzymes of sphingolipid metabolism have emerged as key therapeutic targets for cancer therapy, like activation of ceramide synthases can cause apoptosis, downregulation of glucosylceramide synthase can diminish drug resistance, and inhibition of sphingosine kinase 1 can reduce proliferation as well as angiogenesis.5−7 Chemotherapeutic drugs can regulate the sphingolipid metabolism by activating different enzymes of the biosynthetic pathway,8,9 like ceramide synthases activated by doxorubicin and staurosporine can catalyze an increase in production of specific ceramides responsible for apoptosis.8 Interestingly, chemotherapy-stimulated induction of ceramides can also mediate post-transcriptional regulation of apoptotic proteins by alternative splicing (AS).10 However, delivery of these chemotherapeutic drugs is usually associated with high toxicity due to their uncontrolled release, nonspecific interactions with blood cells, and accumulation in undesired organs that kill healthy cells.11
Hydrogels are self-assembled supramolecular organization of small molecules or polymers, where electrostatic, hydrophobic, and H-bonding interactions help in packing of these molecules.12,13 Low molecular weight hydrogels (LMWHs) based on self-assembled amphiphilic molecules are a suitable alternative to polymeric hydrogels on account of their high-water content and tunable mechanical properties.14 Recent studies have shown that LMWHs can encapsulate and maintain a sustained release of drugs at localized sites.15,16 However, weak electrostatic and hydrophobic interactions in LMWHs do not allow the encapsulation of multiple drugs with distinct hydrophobicity. Therefore, existing LMWHs find it challenging to encapsulate and maintain sustained release of a combination of drugs in desired manner required for cancer therapy.17
Bile acids have a natural tendency to form self-assembled supramolecular nanomicelles due to their amphiphilic nature.18 In the case of synthetic peptides, availability of peptide linkages for H-bonding allows them to self-assemble into supramolecular aggregates.19 As bile acid and peptide-based self-assembled structures are weak and prone to disintegration, we hypothesize that bioconjugation of peptide and bile acid will provide the required scaffold that can help in forming strong supramolecular self-assembled aggregates. Lithocholic acid (LCA) is the most hydrophobic among all bile acids, can provide much-needed hydrophobic interactions during gelation process, and, therefore, can also help in entrapment of hydrophobic drugs. Presence of free hydroxyl and carboxyl termini in LCA can allow conjugation of dipeptide and aromatic benzyl moieties for providing electrostatic, H-bonding, and π–π interactions required for self-assembly of amphiphiles.
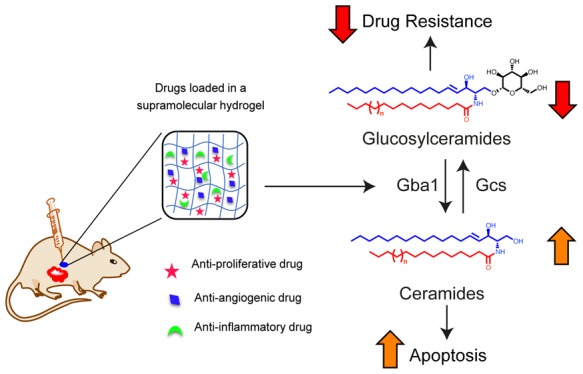
As proliferation, angiogenesis, and inflammation are critical biological processes of TME, we hypothesize that hydrogels maintaining a sustained release of a combination of drugs targeting these processes can avoid multiple chemotherapy cycles, reduce drug-associated toxicity, and minimize the emergence of drug resistance (Figure 1A). Herein, we present the synthesis and screening of 20 LCA–dipeptide conjugates for hydrogelation and characterization of the hydrogels followed by the ability of the most robust hydrogel to deliver a combination of drugs affecting drug resistance (Figure 1A).
Figure 1.

Lithocholic acid-glycine-glycine (A13) forms a robust and biocompatible hydrogel. (A) Schematic representation of the study describing sustained delivery of a combination of antiproliferative, antiangiogenic, and anti-inflammatory drugs from a low molecular weight hydrogel for combating tumor proliferation, angiogenesis, and inflammation. (B) General molecular structure of LCA-dipeptide derived amphiphiles (A1–A20) synthesized and screened for hydrogelation. (C) Rheology characterization of different hydrogels at their minimum gelation concentrations show a comparison of G′ and percentage strain for each gel. (D) Representative images of A13 gel recovered from mice after different days of subcutaneous injection reveal intactness of gel until 21 d. (E, F) H&E (E) and CD45 (F) staining of gel and surrounding tissue retrieved after different days of subcutaneous injection of A13 gel from mice show infiltration of immune cells on day 7 that gets cleared by days 14 and 21, thereby validating the biocompatible nature of gel. Expanded figures with labeling are in Supporting Information as Data Figures ES1 and ES2. (G) Percentage of CD45+ cells quantified by flow cytometry from single-cell suspension of skin tissues with hydrogel isolated on different days after gel injection confirm the biocompatible nature of gel without causing any chronic inflammation. (H, I) Whole-body fluorescence images of BALB/c mice (H) and SD rats (I) after subcutaneous injection of NIR-Gel and NIR-Solution confirm sustained and localized release of dye from NIR-Gel for 21 d in BALB/c mice and for 50 d in SD rats with minimal dye distribution to other organs.
Results
Lithocholic Acid-Glycine-Glycine (A13) Conjugate Forms a Robust Hydrogel
We synthesized 20 LCA–dipeptide conjugates (A1–A20) by tethering of different dipeptides of X-G- motif to 3′-hydroxyl terminal of LCA (Figure 1B, Figure S1). Detailed synthesis and characterization of all the amphiphiles is provided in Supporting Information. We evaluated the ability of each amphiphile to form a hydrogel using inverted vial assay.20 Only glutamic acid (A1), proline (A7), asparagine (A8), threonine (A10), serine (A11), valine (A12), glycine (A13), methionine (A14), and tyrosine (A18)-derived amphiphiles formed gel with minimum gelation concentration of 50–80 mg/mL (Table S1) suggesting that a critical balance of charge and hydrophobicity plays an essential role for amphiphiles to self-assemble for hydrogel formation. Scanning electron micrographs for all hydrogels revealed wax-like texture with highly compact mesh architecture except A8 and A12 hydrogels, which showed fibrous architecture (Figure S2). A11 and A13 hydrogels showed the highest melting temperature (Tm > 80 °C) (Table S1). Rheological characterization of hydrogels at their respective minimum gelation concentration showed highest strengths (G′) of 3 × 105 and 5.3 × 104 Pa (at 1% strain) for A11 and A13 hydrogels with a crossover strain (strain at which gel loses its integrity, G′ = G″) of ∼3 and ∼5%, respectively (Figures 1C, S3, and S4). A11 gel is stronger in terms of having higher G′ than A13 gel but is less elastic (crossover at 3% strain) than A13 gel (crossover at 5% strain). Therefore, we ruled out A11 owing to its inherent nature of disintegrating at lower strain. Minimum gelation concentration of A13 amphiphile is 50 mg/mL (Table S1), and critical micellar concentration of A13 amphiphile revealed by pyrene-based studies is 16 ± 2 μg/mL. Heated solution (sol) of A13 instantly forms hydrogel within a minute on cooling to 25 °C and has Tm of 83 °C (Table S1). Presence of salts affect the hydrogelation process, as A13 spontaneously gets precipitated in phosphate buffer saline (PBS) (pH 7.4).
A13 Gel Is Biocompatible, Biodegradable, and Can Maintain a Sustained Release of Drugs
Incubation of A13 hydrogel with human red blood cells (RBCs) for 6 h showed ∼10% hemolysis (Figure S5A). There was ∼80% cell viability of peripheral blood mononuclear cells (PBMCs) after 6 h of incubation with A13 gel (Figure S5B). A13 gel-coated coverslips did not allow the adsorption of bovine serum albumin (BSA) on its surface after incubation with fluorescein isothiocyanate (FITC)-labeled BSA confirming its nonfouling (inability to adsorb proteins) character (Figure S5C).21 In contrast, positively charged polyethyleneimine (PEI) allows BSA to get adsorbed on its surface (Figure S5C). Esterase- and protease-sensitive biomaterials like A13 amphiphile can get degraded and release chemotherapeutic drugs at tumor site due to high expression of these enzymes at the tumor site (Figure S6A).22 Incubation of A13 hydrogel with PBS at 37 °C in the presence of esterase showed an increase in turbidity of PBS solution with time due to degradation of gel (Figure S6A,B). In contrast, gel incubated with PBS in absence of esterase did not show any change in turbidity (Figure S6B). Liquid chromatography–mass spectrometry (LC-MS/MS) analysis of a turbid solution showed LCA and benzylated LCA (LCA-OBn) as major products of degradation in these simulated conditions (Figure S6C–E). A13 gel can maintain its integrity for 21 d in mice on subcutaneous injection and slowly gets degraded (Figure 1D). Subsequently, we assessed the biocompatibility of A13 gel on subcutaneous injection in BALB/c mice. Hematoxylin and eosin (H&E) staining of gel and surrounding tissue retrieved after different days revealed that there is an influx of immune cells after 7 d that get progressively cleared after 14 and 21 d (Figure 1E). CD45 staining further confirmed the transient infiltration of leukocytes on day 7 that gets cleared on day 21, confirming that A13 gel does not cause any chronic inflammation (Figure 1F). Expanded H&E and CD45 pictures are shown in Supporting Information Figures ES1 and ES2. We also quantified the infiltrated CD45+ cells from skin tissue with gel by flow cytometry after different days of gel injection and observed that there was influx of CD45+ cells on day 7 that gets progressively cleared by day 14 and 21 (Figure 1G). Therefore, these results confirm the biocompatible and biodegradable nature of the A13 hydrogel.
We encapsulated a near-infrared (NIR) fluorescent dye (IR-820) in A13 gel (NIR-Gel) and implanted the NIR-Gel subcutaneously in BALB/c mice (n = 3). Whole-body NIR fluorescence imaging of mice revealed the localized and sustained release of dye at the injection site for 21 d with only minimal amount of dye dissemination to other parts of the body (Figure 1H, upper panel). In contrast, we observed distribution of dye throughout the body on subcutaneous injection of dye solution (Figure 1H, lower panel). Similarly, subcutaneous injection of NIR-Gel in Sprague–Dawley (SD) rats was able to maintain localized and sustained release of dye until 50 d (Figure 1I, upper panel), unlike subcutaneous injection of dye solution without A13 gel (Figure 1I, lower panel). We observed higher fluorescence intensity at day 1 than at 3 h in BALB/c mice and at day 20 in SD rats probably due to gradual release of probe molecules with time and fluorescence quenching during initial time points caused by close vicinity of these molecules on encapsulation.23
TRI-Gel Therapy Induces Tumor Regression with Enhanced Median Survival
We selected three chemotherapeutic drugs, antiproliferative doxorubicin (DOX), antiangiogenic combretastatin A4 (CA4), and anti-inflammatory dexamethasone (DEX) (Figure 2A). Drug loading studies showed that 70 mg of A13 gelator in 1 mL of water can entrap ∼30 mg of each drug while maintaining its integrity and injectability. Heated sols were a little turbid on entrapment of 20 and 30 mg of DEX or CA4, whereas heated sol with DOX was a clear solution even with 30 mg/mL of DOX. Entrapment of DOX enhances gelation rate, while entrapment of CA4 and DEX slows gelation kinetics, but overall heated sol gets converted to gel within a minute.
Figure 2.

TRI-Gel therapy inhibits proliferation, angiogenesis, and inflammation at tumor site. (A) Molecular structures of DOX, CA4, and DEX used in the study. (B) In vitro release profiles (mean ± standard error of mean (SEM), n = 3) of DOX, CA4, and DEX entrapped in A13 gel (TRI-Gel) show sustained release of these drugs over 15 d. (C, D) Tumor growth kinetics (mean ± SEM, n = 7/group) of LLC tumor-bearing mice on different treatments show a significant reduction in the kinetics of tumor growth on TRI-Gel treatment as compared to untreated, TRI-IV, and TRI-TS treated mice. In TRI-TS treated group, mice were given subcutaneous injection of a combination of DOX, CA4, and DEX as suspension near tumor site without hydrogel. In TRI-Gel treated group, mice were treated with subcutaneous injection of a combination of DOX, CA4, and DEX entrapped in A13 gel near the tumor site. In TRI-IV treated group, mice were treated with intravenous injection of DOX and CA4 in saline and oral delivery of DEX on alternate days for 20 d (total 10 doses). (E, F) Final tumor volume (mean ± SEM, n = 7/group) on day 20 of LLC tumor-bearing mice on different treatments show ∼3.5-fold reduction in tumor volume on TRI-Gel therapy as compared to TRI-TS treated mice. (G, H) Kaplan–Meier curve (G) and median survival (H) reveal 13 d increase in median survival of mice on TRI-Gel treatment as compared to TRI-TS treated mice (n = 6/group) and 18 d as compared to untreated mice. (I–L) Flow cytometry analysis of apoptotic (I), Ki67+ (J), CD31+ (K), and CD45+ (L) cells from untreated, TRI-TS, and TRI-Gel-treated tumor tissues confirm significant increase in apoptosis and decrease in proliferation, angiogenesis, and inflammation on TRI-Gel treatment. UT means untreated. Data were analyzed using two-way ANOVA (B–D), unpaired two-tailed Student’s t-test (E,F, I–L) and Log-rank Mantel-Cox test (G).
Lewis lung carcinoma (LLC) in C57BL/6 mice is an ideal syngeneic model to study the effect of chemotherapeutics on TME due to enhanced angiogenesis and inflammation at its tumor site along with proliferating cells.24 We first optimized the dose of each drug entrapped in A13 gel that can cause maximum tumor regression with an increase in median survival in LLC tumor model. We divided LLC tumor-bearing mice into five groups and used a subcutaneous injection of different doses of DOX entrapped in A13 gel (DOX-Gel) for four groups, and no treatment was given to the fifth group of mice. Comparison of tumor regression (Figure S7A), median survival (Figure S7B), and changes in body weight (Figure S7C) of mice led us to decide on 5 mg/kg of DOX-Gel as the most effective dose in regressing tumor growth with a maximum increase in median survival and no significant reduction in body weight. Similarly, LLC tumor-bearing mice treated with subcutaneous injection of different doses of CA4 entrapped in A13 gel (CA4-Gel) revealed a dose of 5 mg/kg as optimized dose (Figure S7D–F). For DEX, we used a dose of 20 mg/kg as reported in an earlier study.25
Rheological characterization of drug-entrapped hydrogels using an optimized dose of drugs showed that entrapment of DOX increased the gel strength and elasticity as percentage strain gets improved from 5 to 9% (Figure S8A–C). Entrapment of CA4 did not induce any significant change in mechanical strength of hydrogel (Figure S8A,D,E). In contrast, entrapment of DEX reduced the gel strength; however, it showed higher elasticity similar to DOX-Gel and CA4-Gel (Figure S8A,F,G). This effect of DEX could be due to its hydrophobicity that might be interfering with supramolecular interactions of hydrogel. A13 gel (TRI-Gel) (70 mg in 1 mL water) was able to entrap the combination of an optimized dose of DOX (0.5 mg), CA4 (0.5 mg), and DEX (2 mg) while maintaining its integrity and injectability. Rheological characterization showed a slight decrease in G′ and G″ of TRI-Gel as compared to neat A13 gel (Figure S8A,H,I). However, TRI-gel showed high elasticity as crossover percentage strain increased from 5 to ∼10%, that can allow it to be used as an injectable hydrogel without compromising gel integrity (Figure S8H,I).
Cumulative drug release profiling from TRI-Gel in PBS at 37 °C revealed the release of DOX, a hydrophilic drug, first, followed by CA4 (Figure 2B). Release of DEX, the most hydrophobic drug, was in a slow and sustained manner (p < 0.0001 with respect to (wrt) DOX and p < 0.001 wrt CA4). We also quantified the actual amount of drug released with time and observed that peak concentration of DOX was achieved on day 1, whereas the highest concentration of DEX was observed on day 15 that is ideal for combating chronic inflammation developed due to chemotherapeutic action of DOX (Figure S9A).26,27 In contrast, CA4 maintained a steady release for 15 d with maximum release on day 1 (Figure S9A). Therefore, TRI-Gel was able to maintain a sustained release of DOX, CA4, and DEX over a period of 15 d (Figures 2B and S9A).
Next, we investigated the effect of TRI-Gel therapy on tumor regression and compared with localized tumor site (represented as TRI-TS) or intravenous/oral (represented as TRI-IV) delivery of the combination of three drugs. In TRI-TS-treated group, mice were treated with subcutaneous injection (200 μL/mouse) of a combination of DOX (5 mg/kg), CA4 (5 mg/kg), and DEX (20 mg/kg) near tumor site without hydrogel. In TRI-Gel treated group, mice were treated with subcutaneous injection of a combination of DOX (5 mg/kg), CA4 (5 mg/kg), and DEX (20 mg/kg) entrapped in A13 gel (200 μL/mouse) near the tumor site. In TRI-IV treated group, mice were treated with intravenous injection of DOX (0.5 mg/kg) and CA4 (0.5 mg/kg) in saline, and with oral delivery of DEX (2 mg/kg) on alternate days for 20 d (total 10 doses). Time growth curves showed a significant reduction in the kinetics of tumor growth on TRI-Gel treatment as compared to untreated, TRI-IV, and TRI-TS treated mice (p < 0.0001 wrt TRI-IV and untreated mice and p < 0.05 wrt TRI-TS) (Figure 2C,D). Nonlinear regression analysis showed that mean doubling time for tumor volume in case of untreated, TRI-IV, and TRI-TS treated groups is 3.83, 3.81, and 3.76 d, respectively. In contrast, mean doubling time for tumor volume is 5.6 d in case of TRI-Gel treatment. On day 20, there was an ∼17-fold decrease in tumor volume of TRI-Gel-treated mice as compared to untreated mice (p < 0.0005) (Figure 2E). In contrast, we observed approximately fourfold reduction on TRI-TS treatment (p < 0.0005) and twofold decrease on TRI-IV treatment (p < 0.005) as compared to untreated mice (Figure 2E). There was an ∼3.5-fold decrease (p < 0.05) in tumor volume on TRI-Gel treatment (126.2 ± 36.8 mm3) as compared to TRI-TS treated mice (447.5 ± 108.3 mm3) (Figure 2F). Therefore, these results confirm that hydrogel-mediated delivery of combination of three drugs (TRI-Gel) induces a significant reduction in rate of tumor growth as compared to TRI-TS treated tumors.
Burst release and diffusion of drugs upon TRI-IV and TRI-TS treatments resulted in high toxicity and no significant increase in mice survivability (Figure 2G). In contrast, TRI-Gel treatment enhanced median survival of mice by 18 d (p = 0.0016) (Figure 2G,H) without any significant change in body weight (Figure S9B). There was a significant reduction in tumor growth kinetics (Figure S9C) and final tumor volume (Figure S9D) in TRI-Gel-treated mice over any of two-drug combination regimens (DOX-CA4-Gel, DOX-DEX-Gel, and DEX-CA4-Gel) entrapped in A13 gel. TRI-Gel treatment induced ∼3.5-fold reduction in tumor volume after 21 d as compared to untreated mice (p < 0.0001) in murine breast cancer (4T1) model (Figure S9E,F), thereby validating the versatility of the system across different tumor models.
Annexin V FITC and propidium iodide staining followed by flow cytometry analysis confirmed significant increase in total apoptotic cells on TRI-Gel treatment as compared to untreated and TRI-TS treated tumors (Figure 2I). TUNEL assay validated the increase in apoptosis on TRI-Gel treatment (Figure S10A). Flow cytometry analysis showed a significant decrease in percentage of Ki67+ (marker for proliferation) (Figure 2J) and CD31+ (used as a marker of endothelial cells gated on CD45– population) cells on TRI-Gel treatment as compared to untreated and TRI-TS treated tumors (Figure 2K). We observed a significant decrease in percentage of CD45+ (marker for leukocytes) cells on TRI-Gel and TRI-TS treatment as compared to untreated samples (Figure 2L). Immunofluorescence staining for Ki67 (Figure S10B), α-smooth muscle actin (α-SMA) (a marker for vascular smooth muscle cells for angiogenesis) (Figure S10C) and CD45 (Figure S10D) further validated the decrease in proliferation, angiogenesis, and inflammation on TRI-Gel treatment. Therefore, these results confirm that TRI-Gel is able to induce significant increase in apoptosis and decrease in proliferation and angiogenesis as compared to TRI-TS treated tumors.
TRI-Gel Therapy Induces Alterations in Sphingolipid Metabolism
TRI-Gel-mediated significant changes in proliferation, apoptosis, angiogenesis, and inflammation inspired us to explore the effect of this treatment on sphingolipid metabolism. We evaluated qualitative and quantitative changes in sphingolipids in TRI-Gel-treated tumor tissues and compared them with untreated ones, using liquid chromatography–mass spectrometry (LC-MS/MS) in multiple reaction monitoring (MRM) mode (Figure 3A).28,29 Lipid profiles showed quantitative alterations in majority of sphingolipids in TRI-Gel-treated tumors relative to untreated controls (Figure 3B and Data Set S1). All ceramide species analyzed showed upregulation, whereas glucosylceramides and ceramide-1-phosphates showed downregulation on TRI-Gel therapy (Data Set S1). There was no significant change in sphingosine-1-phosphate (S1P) and lactosylceramides on TRI-Gel treatment (Data Set S1).
Figure 3.

TRI-Gel therapy alters the levels of key sphingolipids.(A) Simplified scheme of the study showing isolation and analysis of lipids from untreated (n = 4) and TRI-Gel-treated (n = 4) tumors. (B) Heat map representing the altered profile of sphingolipids reveals significant changes in expression of fatty acyl chain-specific ceramides, glucosylceramides, and ceramide-1-phosphates on TRI-Gel treatment as compared to untreated tumors. M1–M4 represent four different mice in a group. (C) Scheme of glucosylceramide-ceramide-ceramide-1-phosphate pathway and key enzymes regulating the pathway. Different enzymes are Gcs: glucosylceramide synthase, Gba1: β-glucocerebrosidase 1, Cerk: ceramide kinase, Lpps: lipid phosphate phosphatase. (D–F) Changes in altered sphingolipids in TRI-Gel-treated tumors (n = 4) in comparison to untreated control (n = 4) (mean ± standard deviation (SD)) confirm significant increase of C18 and C24 ceramides (D) with decrease in C16, C18, and C20 glucosylceramides (E) and C20, C22, and C24:1 ceramide-1-phosphates (F). Data were analyzed by unpaired two-tailed Student’s t-test (D–F).
Ceramides, glucosylceramides, and ceramide-1-phosphates are key sphingolipids that modulate apoptosis, drug sensitivity, angiogenesis, and inflammation of TME (Figure 3C).5−7 Ceramide species like C18 showed more than 1.3-fold increase (p < 0.005), and C24 species increased by more than 1.2-fold (p < 0.05) on TRI-Gel therapy as compared to untreated mice (Figure 3D). There was a significant decrease in levels of glucosylceramides on TRI-Gel treatment with C16 species showing more than 1.25-fold decrease (p = 0.0524), C18 species showing more than 1.6-fold decrease (p < 0.05), and C20 species decreased by ∼1.5-fold (p < 0.005) (Figure 3E). Earlier studies showed that an increase in ceramides is responsible for ceramide-triggered apoptosis and that reduction in glucosylceramides can circumvent glucosylceramide-mediated multidrug resistance.30 TRI-Gel treatment also showed more than 2.4-fold reduction in C20 (p = 0.001), more than fourfold decrease in C22 (p < 0. 05), and more than threefold decrease in C24:1 (p < 0. 05) ceramide-1-phosphates that play a critical role in inducing inflammation (Figure 3F).31 Therefore, an increase in levels of ceramides supports antiproliferative action of TRI-Gel treatment, whereas a decrease of ceramide-1-phosphates helps in combating chronic inflammation at TME site on TRI-Gel therapy. Our data suggest that an overall reduction in levels of glucosylceramides may prevent the tumors to develop drug resistance against TRI-Gel therapy and possibly assists in maintaining tumor regression during the treatment.
TRI-Gel Therapy Stimulates Global Alterations in Alternative Splicing (AS)
Therapeutic efficacy and associated alterations in sphingolipid levels in response to TRI-Gel therapy urged us to investigate the effect of TRI-Gel treatment on tumor transcriptomic profile. Therefore, we performed RNA-sequencing (RNA-seq) and pairwise comparative analysis of untreated, DOX-Gel, DEX-Gel, and CA4-Gel-treated tumors. A clustergram in the form of a heat map analyzed by unsupervised hierarchical clustering shows distinct gene expression profiles in response to each treatment (Figure S11A, Data Set S2). Expanded heat maps are shown in Supporting Information Figures ES3–ES6. In total, there are 169 significantly and differentially expressed genes in TRI-Gel-treated tumors as compared to untreated tumors with 77 upregulated and 92 downregulated genes (≥1.5 with p-value ≤0.05) (Figure S11B,C). We observed 126 genes exclusively expressed in TRI-Gel-treated tumors and an overlap of only five genes among all treated groups (Figure S11B). There were 10 significantly deregulated genes with p-value less than or equal to 0.001 and 44 genes with p-value less than or equal to 0.01 on TRI-Gel treatment (Figure S11C).
Each treatment affects multiple pathways with accentuation of specific processes inherent to the mechanism of action of drug (Figure S11D, Data Sets S3 and S4). TRI-Gel treatment showed enrichment of significantly downregulated genes for functions associated with mRNA processing, alternative splicing, and nuclear export (like Rbm38, Hnrnpa2b1, Mbnl1, Plrg1), vacuolar and endosomal transport (like Arf1, Rab6a, Syntaxin5a), protein catabolic process and localization to organelles (like Rab6a, lft20) (Figures S11D and S11E, Data Sets S3 and S4). In contrast, histone acetylation, chromatin modification, and chromosome organization (like Kat7, Brd4, Chd8), DNA metabolic process, and cell cycle (like Chtf8, Rps3, Rps9, Nde1) were enriched pathways for significantly upregulated genes on TRI-Gel therapy (Figures S11D and S11E, Data Sets S3 and S4). We validated some of the genes that are differentially expressed on TRI-Gel treatment by quantitative real-time PCR (qRT-PCR) (Figure S12A, Table S2). Surprisingly, transcriptomic data did not show any significant alteration in expression of genes of sphingolipid pathway in response to TRI-Gel treatment. Validation of some genes of sphingolipid pathway including Cers1, Cers2, Cers4, Cers5, Cers6 (ceramide synthases) (Figure S12B, Table S2), Gcs (glucosylceramide synthase), Gba1 (β-glucocerebrosidase 1), Glb1 (β-galactosylceramidase 1), and B4galt6/Lcs (β-1,4-galactosyltransferase 6/lactosylceramide synthase) by qRT-PCR did not show any significant change in their expression on TRI-Gel treatment (Figure S12C).
As we observed differential expression in key regulatory genes of alternative splicing (AS) like hnRNPA1/B2, Rbm38, Mbnl1, Gemin8, and Plrg1 on TRI-Gel treatment, we hypothesized that TRI-Gel therapy may play a key role in modulating genome-wide AS in tumor tissues, including genes of sphingolipid pathway. Therefore, we systematically investigated and compared transcriptome-wide AS events in TRI-Gel-treated tumors with DOX-Gel, DEX-Gel, and CA4-Gel-treated tumors to explore mechanisms by which TRI-Gel could alter the levels of bioactive sphingolipids.
Of different AS events known, we analyzed cassette exons (CE) (where an exon is spliced-in or spliced-out) and intron retention (IR) (where retention of an intron occurs in one isoform under a certain condition) events in DOX-Gel, DEX-Gel, CA4-Gel, and TRI-Gel-treated tumors in comparison to untreated tumors as per published bioinformatic pipeline.32 TRI-Gel therapy induced 351 CE events in 285 genes and 1763 IR events in 1411 genes (Figure 4A, Data Sets S5 and S6). Total IR events for TRI-Gel treatment were significantly higher than any other treatment, whereas the number of CE events are similar in all treatment types (Figure 4A). We predicted unique CE events in 84 genes (Figure 4B) and unique IR events in 397 genes (Figure 4C) in TRI-Gel-treated tumor tissues.
Figure 4.

TRI-Gel therapy induces AS of sphingolipid genes. (A) Total number of CE and IR events and corresponding total number of genes altered on different drug treatments reveal a significant increase in the number of IR events on TRI-Gel treatment as compared to other treatments. (B, C) Venn diagrams of genes undergoing AS in response to different drug treatments show 84 unique genes for CE events (B) and 397 unique genes for IR events (C) on TRI-Gel treatment. (D) KEGG pathways and Gene-Ontology-Biological Processes predicted by DAVID Functional Annotation tool for predicted IR events in response to TRI-Gel treatment show enrichment for genes in mRNA processing, RNA splicing, spliceosome, cell cycle, metabolic pathways, protein transport, and DNA repair. The number adjacent to each bar represents number of events identified in that category. (E) Semiquantitative PCR validation of IR events in G3bp1 (two different events), Dennd6a, Topbp1, Rplp0, and Cct3 confirm the effect of TRI-Gel therapy on AS. The two lanes for each representative gene showing an event are biological replicates. For each gene, two alternatively spliced isoforms show differential expression in untreated (UT) and TRI-Gel-treated tumors. A pictorial representation of exon/intron positions, primer sites, and expected product size (bp) are shown alongside. (F) List of all CE and IR events in different genes of sphingolipid biosynthesis pathway in response to different drug treatments show some common events among different treatments and some unique events for a particular treatment.
Data Sets S7 and S8 show an enrichment analysis for biological functions and molecular pathways affected by CE and IR events in response to different treatments. We observed an enrichment of IR events on TRI-Gel treatment for genes in mRNA processing and spliceosome, cell cycle, metabolic, and endocytosis pathways (Figure 4D, Data Set S8). We validated some of the predicted IR events in genes critical for cell proliferation, apoptosis, and metastasis in multiple oncogenic pathways including G3bp1 (GTPase activating protein binding protein 1), Dennd6a (DENN/MADD domain containing 6A, a Rab GEF), Topbp1 (Topoisomerase II binding protein 1), Rplp0 (Ribosomal protein, large, P0), and Cct3 (Chaperonin containing Tcp1, subunit 3) (Figure 4E, Table S3). All of them showed differential isoform expression in TRI-Gel-treated tumors as compared to untreated tumors (Figure 4E).
Interestingly, we observed multiple genes of the sphingolipid pathway that are targets for AS in different drug-treated tumors, indicating post-transcriptional regulatory control of sphingolipid genes in response to chemotherapy (Figure 4F, Data Set S9). We observed some common CE and IR events in response to different drug treatments such as common CE event for hexosaminidase A (HexA) in DOX-Gel and DEX-Gel treated tumors and common IR event for Elovl1 in DOX-Gel and CA4-Gel treated tumors (Figure 4F, Data Set S9). We also observed some unique events specific for a drug treatment like CE event for St6galnac6 on CA4-Gel treatment and IR event for Cers2 on DOX-Gel treatment (Figure 4F, Data Set S9). TRI-Gel treatment induced two unique IR events in glucosidase genes, β-glucocerebrosidase 1 (Gba1), and β-galactosylceramidase (Glb1), that might be playing a role in alteration of key sphingolipids at TME site (Figure 4F).
Differential Isoform Expression in Gba1 Contributes to Tumor Regression
β-Glucocerebrosidase 1 (Gba1) undergoes a unique alternative splicing event on TRI-Gel treatment, where intron 8 is retained in tumor tissues and is removed on TRI-gel therapy (Figure 5A, Figure S13). Therefore, we validated this specific and exclusive IR event using event-specific primer pair (Figure 5B, Table S3). For intron 8-retained transcript, we used the primer pair targeted at intron 8 and at junction of exon 9 and exon 10. For the transcript without intron 8 (full-length native protein-coding transcript (PC)), we used primer pair targeted at exon 8 and at the junction of exon 9 and exon 10 (Figure 5B, Table S3). Isoform-specific semiquantitative PCR showed that expression of Gba1 transcripts with intron 8 retention is more than threefold (p < 0.05) lower in TRI-Gel-treated tumors as compared to untreated tumors (Figure 5B,C). We observed a corresponding approximately twofold (p < 0.05) increase in the full-length native protein-coding transcript by qRT-PCR in TRI-Gel treated tumors as compared to untreated tumors (Figure 5D). Expression of Gba1 tested by qRT-PCR using Gba1 gene-specific primer pair did not show any significant change in overall gene expression (sum total of all transcripts) of Gba1 (Figure S12C, Table S2). Therefore, unique alternative splicing event in Gba1 on TRI-Gel therapy is responsible for significant increase in levels of full-length native protein-coding transcript. Interestingly, we did not find any significant decrease in expression of IR transcript and increase in expression of PC transcripts on TRI-TS treatment (Figure S14A).
Figure 5.

β-Glucocerebrosidase 1 (Gba1) is a potential target of TRI-Gel therapy. (A) Diagrammatic representation of full-length native protein-coding (PC) transcript and IR splicing event in β-glucocerebrosidase 1 (Gba1). (B) Isoform-specific PCR of Gba1 (n = 3) confirms decrease in IR transcript in TRI-Gel-treated tumors as compared to untreated tumors. A pictorial representation of exon/intron position, primer sites, and expected product sizes (bp) is shown alongside. (C) Quantification (mean ± SD, n = 3) of transcripts showing IR event of Gba1 confirm more than threefold decrease in IR transcripts on TRI-Gel treatment. (D) Quantification (mean ± SD, n = 3) of full-length native PC transcripts by qRT-PCR confirm a more than twofold increase in PC transcripts on TRI-Gel treatment. (E) Immunoblot of Gba1 (two replicates) and its quantification (mean ± SD, n = 5) reveal a more than twofold increase in expression of Gba1 on TRI-Gel treatment. Arrow indicates Gba1 (59 kDa). Other bands indicate glycosylated forms. (F) Enzymatic activity (mean ± SD, n = 6) of Gba1 confirm a more than 1.4-fold increase in activity on TRI-Gel treatment. (G) qRT-PCR confirms significant downregulation of Csk, Arrb2, Ctnnb1, Tcf4, A4galt, Mdr1, and Mrp1 on TRI-Gel treatment as compared to untreated tumors. (H, I) Immunofluorescence images (H) and quantification (I) (mean ± SD, n = 3) of P-glycoprotein (P-gp) confirm more than sevenfold downregulation of P-gp on TRI-Gel treatment as compared to untreated samples. (J) Tumor growth kinetics (mean ± SEM) of LLC tumor-bearing mice (n ≥ 5/group) on different treatments show an increase in kinetics of tumor growth on Gba1 siRNA + TRI-Gel therapy as compared to TRI-Gel-treated mice (p < 0.005). (K) Final tumor volume (mean ± SEM) on day 21 of LLC tumor-bearing mice after different treatments show approximately fourfold increase in tumor volume on Gba1 siRNA + TRI-Gel treatment as compared to TRI-Gel therapy. (L, M) Immunofluorescence images (L) and quantification (M) (mean ± SD, n = 3) of P-gp reveal approximately twofold upregulation of P-gp on Gba1 siRNA + TRI-Gel therapy as compared to TRI-Gel therapy. Data were analyzed by unpaired two-tailed Student’s t-test (C–G, I, K, M) and two-way ANOVA (J).
Immunoblotting showed a twofold increase in expression of Gba1 and its glycosylated forms in TRI-Gel treated tumors as compared to untreated tumors (p < 0.005) (Figure 5E). Similarly, a corresponding higher Gba1 enzyme activity was observed in TRI-Gel-treated tumors as compared to untreated tumors (p < 0.05) (Figure 5F). Immunofluorescence staining of tumor sections with anti-Gba1 antibody further confirmed more than threefold higher Gba1 expression in TRI-Gel-treated tumors (p < 0.05) (Figure S14B). As Gba1 catalyzes the hydrolysis of glucosylceramide to ceramide,33 this elevated Gba1 level in response to TRI-Gel therapy is responsible for observed increase in ceramide levels (Figure 3D) and decrease in glucosylceramides (Figure 3E), thereby forcing the cancer cells to undergo apoptosis.
Glucosylceramides can activate cSrc kinase through glycosphingolipid-enriched microdomains. Activated cSrc kinase increases the nuclear localization of β-catenin and upregulates Tcf4 followed by increased expression of Mdr1 and P-gp (P-glycoprotein).34 qRT-PCR showed ∼2.5-fold reduction (p < 0.01) in expression of Csk (C-terminal Src Kinase), more than 50-fold reduction (p < 0.0001) in expression of Arrb2 (β-arrestin-2 that phosphorylates β-catenin), more than twofold decrease (p < 0.005) in Ctnnb1 (for β-catenin), more than 29-fold reduction (p < 0.0001) in Tcf4 (transcription factor 4), and ∼2.2-fold decrease (p < 0.005) in Mdr1 (ABCB1) expression on TRI-Gel treatment as compared to untreated tumors (Figure 5G). Glucosylceramides get converted to lactosylceramides by A4galt, and lactosylceramides are the common precursor of nearly all neutral glycosphingolipids and higher gangliosides that are mediators of drug resistance.35 TRI-Gel treatment also induced more than fivefold decrease (p < 0.0001) in expression of A4galt (Figure 5G). We also checked the expression of multidrug resistance associated protein1 (Mrp1)/ ABCC1 (regulated by glycosphingolipids), as it plays an active role in protecting cancer cells by efflux of a wide array of drugs. We found ∼2.4-fold decrease in expression of Mrp1 (p < 0.05) on TRI-Gel treatment (Figure 5G).36 However, Mdr1 and Mrp1 expressions were not affected significantly in tumors treated with TRI-TS therapy (Figure S14C,D). Immunofluorescence for P-gp confirmed a significant reduction in P-gp levels on TRI-Gel treatment as compared to untreated and TRI-TS treated tumors (Figure 5H,I). Therefore, TRI-Gel therapy-mediated increase in Gba1 activity helps in reducing the expression of P-gp in tumor tissues that is adept to combat the emergence of drug resistance as compared to local injection of all the drugs without hydrogel.
Careful analysis of RNA sequencing data (Data Set 2) showed significant downregulation of a plethora of genes on TRI-Gel treatment that are involved in intrinsic mechanisms of drug resistance like alterations in expression of drug transporters/efflux pumps, changes in vesicular or endosomal trafficking, and activation of antiapoptotic pathways. Genes like Arnt1,37Rab6a,38 and Uhrf1(39) regulate Mdr1 gene expression and sensitize cancer cells to anticancer drugs. Other differentially expressed genes on TRI-Gel treatment like Arf1,40Rbpj,41Trim25,42Usp24,43DNAJB1,44Mia3,45Pex3,46 and Obfc1(47) have been shown to be associated with drug resistance in different cancer models using diverse mechanisms. qRT-PCR confirmed significant downregulation of Arf1 (2.4-fold, p < 0.005), Arnt (3.3-fold, p < 0.05), Uhrf1 (sixfold, p < 0.0001), Trim25 (twofold, p < 0.001), Ndfip2 (1.9-fold, p < 0.05), and Rbpj (42-fold, p < 0.0001) on TRI-Gel treatment as compared to untreated tissues (Figure S14E). In contrast, TRI-TS treatment did not show a significant downregulation of any of these genes, whereas we observed upregulation of Trim25 and Ndfip2 gene expression (Figure S14F). Therefore, these results confirm that TRI-Gel mediated sustained release of drugs, unlike TRI-TS treatment, helps in combating drug resistance through other pathways as well.
To further validate the Gba1-mediated effect of TRI-Gel therapy on tumor regression and drug resistance, we investigated the effect of Gba1 knockdown on TRI-Gel therapy. LLC tumor-bearing mice were randomized into five groups and subjected to different treatments. There was no treatment for group 1 mice, and group 2 mice were subjected to TRI-Gel treatment. Gba1 siRNA along with TRI-Gel therapy was given to group 3 mice, whereas scrambled siRNA with TRI-Gel therapy were used for group 4 mice. Group 5 mice were treated with only Gba1 siRNA. We observed an increase in kinetics of tumor growth on using a combination of Gba1 siRNA and TRI-Gel therapy as compared to only TRI-Gel treatment (p < 0.005) (Figure 5J). However, the use of scrambled siRNA in combination with TRI-Gel therapy did not cause any change in tumor growth kinetics. We observed more than 15-fold decrease in tumor volume on TRI-Gel treatment as compared to untreated tumors (p < 0.0001) (Figure 5K). In contrast, there was only approximately fourfold reduction in tumor volume on using a combination of Gba1 siRNA and TRI-Gel treatment, whereas combination of scrambled siRNA and TRI-Gel induced ∼18-fold reduction in tumor volume after 21 d (Figure 5K). There was a fourfold increase in tumor volume on using a combination of Gba1 siRNA and TRI-Gel treatment (762.7 ± 73.7 mm3) as compared to only TRI-Gel treatment (181.8 ± 32.3 mm3) (Figure 5K). We confirmed Gba1 silencing in tumors by its enzymatic activity assay (Figure S14G). There was a significant increase in P-gp levels in tumors treated with a combination of Gba1 siRNA and TRI-Gel (p < 0.05) suggesting the emergence of drug resistance in response to Gba1 siRNA and the inability of TRI-Gel to control it (Figure 5L,M). Therefore, these results confirm the Gba1-mediated effect of TRI-Gel therapy in tumor regression and combating drug resistance.
Discussion
Angiogenesis is one of the key targets for successful cancer treatment as witnessed by clinical success of different chemical inhibitors and approval of monoclonal antibodies like bevacizumab.48,49 Sengupta et al. have shown that sequential delivery of antiangiogenic CA4 followed by antiproliferating DOX from nanocells help in tumor regression.50 We hypothesized that release of antiproliferative drug followed by antiangiogenic and anti-inflammatory drugs will help in combating the rapid proliferation of cancer cells, temporarily keeping the angiogenesis and acute inflammation intact. Initial blood supply to tumor site can help in delivery of other drugs/imaging agents on intravenous administration in clinical settings. Acute inflammation usually assists in effective action of antiproliferating and antiangiogenic drugs.51 In contrast, chronic inflammation, initiated by enhanced apoptosis, causes increased DNA damage, disruption of DNA repair pathways, inhibition of apoptosis, promotion of angiogenesis, cell proliferation, and tumor recurrence.52 Therefore, sustained release of anti-inflammatory dexamethasone can help in abrogating chronic inflammation subsequent to the action of antiproliferating and antiangiogenenic drugs.53 Recently, Jia et al. have shown that inhibition of NF-κB and Wnt/β-catenin signaling of inflammatory pathway after chemotherapy withdrawal helps in reducing the tumor mass more effectively.53
Mechanical strength of hydrogels is highly dependent on intermolecular interactions between self-assembled molecules, and entrapment of payloads influences these mechanical properties.54 Presence of hydrophilic and hydrophobic moieties in amphiphilic hydrogelators can help these molecules to interact with different drugs efficiently without compromising the mechanical strength.55 Therefore, the presence of LCA and dipeptide in A13 hydrogel allows the entrapment of DOX, CA4, and DEX effectively. Release of hydrophilic drugs is mainly controlled by diffusion, whereas release of hydrophobic drugs is maintained by degradation rate of hydrogels. Therefore, early release of DOX is diffusion driven, and slow degradation of gel maintains sustained release of CA4 and DEX. This hydrogel-mediated sustained release of combination of drugs helps in effective tumor regression and increase in survivability over tumor site (TRI-TS) delivery of combination of drugs without using hydrogel.
In-depth transcriptomic analysis of TRI-Gel-treated tumors showed increased AS, especially for IR events, of many genes responsible for oncogenic transformation including sphingolipid metabolism genes. In most intron-retained transcripts, IR events usually lead to the inclusion of premature termination codons (PTC) that direct the transcripts to Nonsense Mediated Decay (NMD) pathway.56 In some cases, IR transcripts may not be susceptible to NMD, and on demand, these IR transcripts even act as a pool of RNA. This pool of RNAs splice and translate at a much faster rate than de novo transcripts that proceed through transcription and translation.57 This phenomenon provides an additional level of gene regulation by IR transcripts. There was a significant reduction in intron retained Gba1 transcript and a corresponding increase in its full-length native protein-coding transcript on TRI-Gel therapy leading to enhanced Gba1 activity. This is an interesting instance of chemotherapy and pharmacological modulation of AS that triggers a higher expression of functional Gba1 protein in TRI-Gel-treated tumors.
Differential gene expression studies showed upregulation of splicing factors like Hnrnpd and Rbm10 and downregulation of Hnrnpa2b1 and Rbm38 in response to TRI-Gel treatment. TRI-Gel treatment also induces downregulation of Gemin8, Mbnl1, Fmr1, and Plrg1 that are implicated in RNA splicing.58−60 Therefore, TRI-Gel-mediated change in expression of splicing factors can revert the intron retention event in Gba1 mRNA and lead to upregulation of protein-coding transcripts at post-transcriptional level. This upregulation of Gba1 transcends to increased Gba1 protein activity, thereby catalyzing the conversion of glucosylceramides to ceramides. The exact mechanism for alternative splicing of Gba1 in response to TRI-Gel treatment and role of different splicing factors and their regulation by chemotherapeutic drugs need to be investigated.
This intricate and precise AS event in response to TRI-Gel therapy results in a significant increase in cellular ceramides (C18 and C24 chain) with a concomitant decrease in glucosylceramides (C16, C18, and C20).61 It has been reported that accumulation of C18 ceramides by overexpression of mammalian upstream regulator of growth and differentiation factor (mUOG1) causes apoptosis in cancer cells.62 In a similar trend, accumulation of C18 ceramides in TRI-Gel-treated tumors leads to a significant increase in apoptosis. Decrease in glucosylceramides (C16, C18, and C20) lower the P-gp expression in TRI-Gel-treated tumors via downregulating the Tcf4 transcription factor through cSrc kinase pathway. siRNA-mediated knockdown of Gba1 diminished the effect of TRI-Gel therapy by increasing the P-gp expression, thereby validating the Gba1-mediated effect of TRI-Gel therapy. Apart from the decrease in Mdr1/P-gp expression, other drug-resistance genes were found be downregulated on TRI-Gel treatment. Interestingly, we also observed that tumor site injection of a combination of these drugs without hydrogel does not induce alternative splicing in Gba1 and does not downregulate the expression of drug-resistance genes including Mdr1 and Mrp1.63
Taken together, these results suggest that TRI-Gel mediated delivery of combination of three drugs creates a unique milieu that affects attenuation of drug resistance by multiple genes via diverse mechanisms, and alternative splicing of Gba1 is one of them. Therefore, chimeric TRI-Gel therapy presented here opens new directions for exploring hydrogel-mediated delivery of combination of drugs for targeting different biological processes of TME, and in-depth mechanistic understanding of these delivery vehicles is essential for development of future cancer therapeutics.
Methods
Detailed materials and methods have been described in Supporting Information.
Acknowledgments
We are grateful to Dr. A. Mukhopadhyay for critical reading of the manuscript and many insightful discussions. We thank Dr. S. S. Tabrez for discussions and B. Chaterjee for his contribution to the presentation of Figure S11D. We thank Bencos Research Solutions (P) Ltd. for giving commercial support for RNA sequencing and initial data analysis. We are also grateful for the support of DBT e-Library Consortium (DeLCON) for providing access to e-resources, Advanced Technology Platform Centre of NCR Biotech Science Cluster, Amity Lipidomics Research Facility at Amity University, and small-animal facility of National Institute of Immunology.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00551.
Supporting figures (Figures S1−S14), tables (Tables S1−S3), complete list of materials used, and experimental details (PDF)
Expanded H&E (Figure ES1) and CD45 (Figure ES2) staining figures, expanded heat maps (Figures ES3−ES6), and original blots (Figures ES7−8) (PDF)
Data Set 1. Quantification of different sphingolipid species on TRI-Gel treatment as compared to untreated control (XLSX)
Data Set 2. Differentially expressed genes after different treatments as compared to untreated control (XLSX)
Data Set 3. Functions of differentially expressed genes after different treatments as compared to untreated control (XLSX)
Data Set 4. Ontology (GO) analysis of differentially expressed genes after different treatments (XLSX)
Data Set 5. Cassette Exon events predicted after different treatments (XLSX)
Data Set 6. Intron Retention events predicted after different treatments (XLSX)
Data Set 7. BP and KEGG pathways enriched for Cassette Exon (CE) events observed on different treatments (XLSX)
Data Set 8. BP and KEGG pathways enriched for Intron Retention (IR) events observed on different treatments (XLSX)
Data Set 9. Cassette exon and intron retention events in sphingolipid genes on different treatments (XLSX)
Author Present Address
⊖ University of Technology, Sydney, NSW-2007.
Author Present Address
⦶ Department of Chemistry and Biochemistry, Freie University, Berlin, Germany.
Author Contributions
S.K., D.M., and V.K. synthesized the amphiphiles. V.K., S.K., and P.S.Y. performed hydrogel characterization, biocompatibility, and drug-release studies. S.K., P.S.Y., and A.S. performed and analyzed the rheology experiments. A.K. and S.P. performed all immunostaining and confocal microscopy experiments. S.P. and S.K. performed all animal experiments. Z.A.R. and S.P. performed flow cytometry studies, and A.A. supervised and helped in analysis. P.D. helped in H&E and CD45 staining and characterization of skin tissues samples. N.M., D.M., and U.D. performed all lipidomics studies and analyzed the data. S.N. helped in gel degradation experiments. M.P. helped in lipidomics studies. N.M., R.D.S., and U.D. performed transcriptomic data analysis. N.M. and A.K. performed RT-PCR validation experiments. S.S. and A.B. supervised animal studies. U.D. and A.B. wrote the manuscript, conceived the idea, and supervised the whole project.
Author Contributions
⊕ These authors contributed equally.
The support from RCB, Amity University Haryana, NII and IISER-Bhopal core funds and Department of Biotechnology (DBT), India, is greatly acknowledged. Work in A.B. laboratory is supported by the following grants: BT/PR12361/MED/32/361/2014 (DBT), BT/PR21680/NNT/28/1190/2016 (DBT), BT/PR19624/BIC/101/488/2016 (DBT), BT/MED/30/SP11263/2015 (DBT), SR/NM/NS-1108/2015(G) (DST), and INT/Korea/P-49 (DST). U.D. is supported by BT/PR19624/BIC/101/488/2016 (DBT) and ECR/2016/001603 (DST). R.D.S. is supported by BT/PR27108/BID/7/820/2017 (DBT), and A.S. is supported by BT/PR12361/MED/32/361/2014 (DBT). S.S. is supported by BT/MED/30/SP11263/2015 (DBT). Animal work in small animal facility of Regional Centre for Biotechnology is supported by BT/PR5480/INF/22/158/2012 (DBT) and Amity Lipidomics Research Facility at Amity University Haryana is supported by DST-FIST grant, SR/FST/LSI-664/2016. S.P. and P.S.Y. thank UGC, and N.M., S. K., and A.K. thank RCB for research fellowships.
The authors declare no competing financial interest.
Notes
All the raw sequencing data are deposited at https://www.ncbi.nlm.nih.gov with submission ID SUB4518182.
Supplementary Material
References
- Quail D. F.; Joyce J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. 10.1111/j.1349-7006.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Zhuang X.; Lin L.; Yu P.; Wang Y.; Shi Y.; Hu G.; Sun Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015, 13, 45. 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni N. E.; Noel A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. 10.1373/clinchem.2012.185363. [DOI] [PubMed] [Google Scholar]
- Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad S. A. F.; Cabot M. C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- Ogretmen B.; Hannun Y. A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- del Solar V.; Lizardo D. Y.; Li N.; Hurst J. J.; Brais C. J.; Atilla-Gokcumen G. E. Differential regulation of specific sphingolipids in colon cancer cells during staurosporine-induced apoptosis. Chem. Biol. 2015, 22, 1662–1670. 10.1016/j.chembiol.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Rebillard A.; Tekpli X.; Meurette O.; Sergent O.; LeMoigne-Muller G.; Vernhet L.; Gorria M.; Chevanne M.; Christmann M.; Kaina B.; Counillon L.; Gulbins E.; Lagadic-Gossmann D.; Dimanche-Boitrel M. T. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007, 67, 7865–7874. 10.1158/0008-5472.CAN-07-0353. [DOI] [PubMed] [Google Scholar]
- Patwardhan G. A.; Liu Y. Y. Sphingolipids and expression regulation of genes in cancer. Prog. Lipid Res. 2011, 50, 104–114. 10.1016/j.plipres.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S.; Kulkarni A. Design principles for clinical efficacy of cancer nanomedicine: a look into the basics. ACS Nano 2013, 7, 2878–2882. 10.1021/nn4015399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Mooney D. J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde J.; Oliva N.; Zhang Y.; Artzi N. Local triple-combination therapy results in tumour regression and prevents recurrence in a colon cancer model. Nat. Mater. 2016, 15, 1128–1138. 10.1038/nmat4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper E. R.; Adams D. J. Low-molecular-weight gels: The state of the art. Chem. 2017, 3, 390–410. 10.1016/j.chempr.2017.07.012. [DOI] [Google Scholar]
- Zhang S.; Ermann J.; Succi M. D.; Zhou A.; Hamilton M. J.; Cao B.; Korzenik J. R.; Glickman J. N.; Vemula P. K.; Glimcher L. H.; Traverso G.; Langer R.; Karp J. M. An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease. Sci. Transl. Med. 2015, 7, 300ra128. 10.1126/scitranslmed.aaa5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajanayake T.; Olariu R.; Leclère F. M.; Dhayani A.; Yang Z.; Bongoni A. K.; Banz Y.; Constantinescu M. A.; Karp J. M.; Vemula P. K.; Rieben R.; Vögelin E. A single localized dose of enzyme-responsive hydrogel improves long-term survival of a vascularized composite allograft. Sci. Transl. Med. 2014, 6, 249ra110. 10.1126/scitranslmed.3008778. [DOI] [PubMed] [Google Scholar]
- Tian R.; Chen J.; Niu R. The development of low-molecular weight hydrogels for applications in cancer therapy. Nanoscale 2014, 6, 3474–3482. 10.1039/c3nr05414d. [DOI] [PubMed] [Google Scholar]
- Nonappa; Maitra U. Unlocking the potential of bile acids in synthesis, supramolecular/materials chemistry and nanoscience. Org. Biomol. Chem. 2008, 6, 657–669. 10.1039/b714475j. [DOI] [PubMed] [Google Scholar]
- Eskandari S.; Guerin T.; Toth I.; Stephenson R. J. Recent advances in self-assembled peptides: implications for targeted drug delivery and vaccine engineering. Adv. Drug Delivery Rev. 2017, 110–111, 169–187. 10.1016/j.addr.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kundu S.; Reddy A.; Sreekanth V.; Motiani R. K.; Sengupta S.; Srivastava A.; Bajaj A. Injectable small molecule hydrogel for localized and sustained in vivo delivery of doxorubicin. Nanoscale 2014, 6, 12849–12855. 10.1039/C4NR04064C. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Cao Z.; Bai T.; Carr L.; Ella-Menye J. R.; Irvin C.; Ratner B. D.; Jiang S. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. 10.1038/nbt.2580. [DOI] [PubMed] [Google Scholar]
- Dong H.; Pang L.; Cong H.; Shen Y.; Yu B. Application and design of esterase-responsive nanoparticles for cancer therapy. Drug Delivery 2019, 26, 416–432. 10.1080/10717544.2019.1588424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Sun R.; Cheng J.; Liu J.; Gou F.; Xiang H.; Zhou X. Fluorescence aggregation-caused quenching versus aggregation-induced emission: a visual teaching technology for undergraduate chemistry students. J. Chem. Educ. 2016, 93, 345–350. 10.1021/acs.jchemed.5b00483. [DOI] [Google Scholar]
- Eklund L.; Bry M.; Alitalo K. Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol. Oncol. 2013, 7, 259–282. 10.1016/j.molonc.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. J.; Li J.; Hao F.-R.; Yuan Y.; Li J. Y.; Lu W.; Zhou T. Y. Dexamethasone suppresses the growth of human non-small cell lung cancer via inducing estrogen sulfotransferase and inactivating estrogen. Acta Pharmacol. Sin. 2016, 37, 845–856. 10.1038/aps.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiya M.; Niiya K.; Kiguchi T.; Shibakura M.; Asaumi N.; Shinagawa K.; Ishimaru F.; Kiura K.; Ikeda K.; Ueoka H.; Tanimoto M. Induction of TNF-alpha, uPA, IL-8 and MCP-1 by doxorubicin in human lung carcinoma cells. Cancer Chemother. Pharmacol. 2003, 52, 391–398. 10.1007/s00280-003-0665-1. [DOI] [PubMed] [Google Scholar]
- Sauter K. A.; Wood L. J.; Wong J.; Iordanov M.; Magun B. E. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol. Ther. 2011, 11, 1008–1016. 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner R. L.; Allegood J. C.; Park H.; Wang E.; Kelly S.; Haynes C. A.; Sullards M. C.; Merrill A. H. J. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 2009, 50, 1692–1707. 10.1194/jlr.D800051-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta U.; Bamba T.; Chiantia S.; Karim P.; Tayoun A. N. A.; Yonamine I.; Rawat S. S.; Rao R. P.; Nagashima K.; Fukusaki E.; Puri V.; Dolph P. J.; Schwille P.; Acharya J. K.; Acharya U. Ceramide kinase regulates phospholipase C and phosphatidylinositol 4, 5, bisphosphate in phototransduction. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20063–20068. 10.1073/pnas.0911028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senchenkov A.; Litvak D. A.; Cabot M. C. Targeting ceramide metabolism- a strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A.; Presa N.; Gomez-Larrauri A.; Rivera I. G.; Trueba M.; Ordonez M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2016, 61, 51–62. 10.1016/j.plipres.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Tabrez S. S.; Sharma R. D.; Jain V.; Siddiqui A. A.; Mukhopadhyay A. Differential alternative splicing coupled to nonsense-mediated decay of mRNA ensures dietary restriction-induced longevity. Nat. Commun. 2017, 8, 306. 10.1038/s41467-017-00370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitatani K.; Sheldon K.; Rajagopalan V.; Anelli V.; Jenkins R. W.; Sun Y.; Grabowski G. A.; Obeid L. M.; Hannun Y. A. Involvement of acid beta-glucosidase 1 in the salvage pathway of ceramide formation. J. Biol. Chem. 2009, 284, 12972–12978. 10.1074/jbc.M802790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Y.; Gupta V.; Patwardhan G. A.; Bhinge K.; Zhao Y.; Bao J.; Mehendale H.; Cabot M. C.; Li Y. T.; Jazwinski S. M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol. Cancer 2010, 9, 145. 10.1186/1476-4598-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel I.; Klappe K.; Kok J. W. Up-regulation of lactosylceramide synthase in MDR1 overexpressing human liver tumor cells. FEBS Lett. 2005, 579, 3381–3384. 10.1016/j.febslet.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Gouaze-Andersson V.; Cabot M. C. Glycosphingolipids and drug resistance. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 2096–2103. 10.1016/j.bbamem.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Chan Y.; Kalpana S.; Chang W.; Chang W.; Chen B. Expression of aryl hydrocarbon receptor nuclear translocator enhances cisplatin resistance by upregulating MDR1 expression in cancer cells. Mol. Pharmacol. 2013, 84, 591–602. 10.1124/mol.113.087197. [DOI] [PubMed] [Google Scholar]
- Subramani D.; Alahari S. K. Integrin-mediated function of Rab GTPases in cancer progression. Mol. Cancer 2010, 9, 312. 10.1186/1476-4598-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B.; Liu Y.; Yu X.; Yin L.; Peng Y.; Gao Y.; Zhu Q.; Cao T.; Yang Y.; Fan X.; Li X. FOXM1 contributes to taxane resistance by regulating UHRF1-controlled cancer cell stemness. Cell Death Dis. 2018, 9, 562. 10.1038/s41419-018-0631-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Wang Q.; He Y.; Ding L.; Zhong F.; Ou Y.; Shen Y.; Liu H.; He S. ADP-ribosylation factor 1 (ARF1) takes part in cell proliferation and cell adhesion-mediated drug resistance (CAM-DR). Ann. Hematol. 2017, 96, 847–858. 10.1007/s00277-017-2949-2. [DOI] [PubMed] [Google Scholar]
- Xue L.; Wang Z.; Li H.; Li Z.; Chen Q.; Zhang P.; Chen H.; Wang Z.; Chong T. RBPJ contributes to acquired docetaxel resistance in prostate cancer cells. Mol. Cell. Toxicol. 2017, 13, 279–285. 10.1007/s13273-017-0031-8. [DOI] [Google Scholar]
- Qin X.; Qiu F.; Zou Z. TRIM25 is associated with cisplatin resistance in non-small-cell lung carcinoma A549 cell line via downregulation of 14–3-3σ. Biochem. Biophys. Res. Commun. 2017, 493, 568–572. 10.1016/j.bbrc.2017.08.151. [DOI] [PubMed] [Google Scholar]
- Wang Y. C.; Wu Y. S.; Hung C. Y.; Wang S. A.; Young M. J.; Hsu T. I.; Hung J. J. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing P300 and β-TrCP and promotes cancer malignancy. Nat. Commun. 2018, 9, 3996. 10.1038/s41467-018-06178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y.; Choi H. K.; Seo J. S.; Yoo J. Y.; Jeong J. W.; Choi Y.; Choi K. C.; Yoon H. G. DNAJB1 negatively regulates MIG6 to promote epidermal growth factor receptor signaling. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 2722–2730. 10.1016/j.bbamcr.2015.07.024. [DOI] [PubMed] [Google Scholar]
- Gao H.; Cong X.; Zhou J.; Guan M. MicroRNA-222 influences migration and invasion through MIA3 in colorectal cancer. Cancer Cell Int. 2017, 17, 78. 10.1186/s12935-017-0447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahabieh M. S.; Ha Z.; Di Pietro E.; Nichol J. N.; Bolt A. M.; Goncalves C.; Dupéré-Richer D.; Pettersson F.; Mann K. K.; Braverman N. E.; Del Rincón S. V.; Miller W. H. Jr. Peroxisomes protect lymphoma cells from HDAC inhibitor-mediated apoptosis. Cell Death Differ. 2017, 24, 1912–1924. 10.1038/cdd.2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Chai W. Suppression of STN1 enhances the cytotoxicity of chemotherapeutic agents in cancer cells by elevating DNA damage. Oncol. Lett. 2016, 12, 800–808. 10.3892/ol.2016.4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Qin Y.; Wu L.; Yang S.; Li N.; Wang H.; Xu H.; Sun K.; Zhang S.; Han X.; Sun Y.; Shi Y. A Phase I clinical trial assessing the safety and tolerability of combretastatin A4 phosphate injections. Anti-Cancer Drugs 2014, 25, 462–471. 10.1097/CAD.0000000000000070. [DOI] [PubMed] [Google Scholar]
- Weis S. M.; Cheresh D. A. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- Sengupta S.; Eavarone D.; Capila I.; Zhao G.; Watson N.; Kiziltepe T.; Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature 2005, 436, 568–572. 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Coussens L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Roxburgh C. S. D.; McMillan D. C. Cancer and systemic inflammation: treat the tumour and treat the host. Br. J. Cancer 2014, 110, 1409–1412. 10.1038/bjc.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D.; Li L.; Andrew S.; Allan D.; Li X.; Lee J.; Ji G.; Yao Z.; Gadde S.; Figeys D.; Wang L. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. 10.1038/cddis.2017.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr J.; Saldias C.; Diaz D. D. Release of small bioactive molecules from physical gels. Chem. Soc. Rev. 2018, 47, 1484–1515. 10.1039/C7CS00515F. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Maitra U.; Mukhopadhyay S.; Srivastava A.. Advances in molecular hydrogels. In Molecular Gels ;Weiss R. G., Terech P., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp 613–647. [Google Scholar]
- Kurosaki T.; Maquat L. E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. 10.1242/jcs.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley P. T.; Khaladkar M.; Kim J.; Eberwine J. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip. Rev. RNA 2014, 5, 223–230. 10.1002/wrna.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny P.; Stepniak-Konieczna E.; Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. 10.1093/nar/gku767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M.; Mukherjee N.; Bandaru P.; Miller J. B.; Nusbaum J. D.; Corcoran D. L.; Langlois C.; Munschauer M.; Dewell S.; Hafner M.; Williams Z.; Ohler U.; Tuschl T. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinridders A.; Pogoda H. M.; Irlenbusch S.; Smyth N.; Koncz C.; Hammerschmidt M.; Bruning J. C. PLRG1 is an essential regulator of cell proliferation and apoptosis during vertebrate development and tissue homeostasis. Mol. Cell. Biol. 2009, 29, 3173–3185. 10.1128/MCB.01807-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy S.; Meyers-Needham M.; Senkal C. E.; Saddoughi S. A.; Sentelle D.; Selvam S. P.; Salas A.; Ogretmen B. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 6, 1603–1624. 10.2217/fon.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koybasi S.; Senkal C. E.; Sundararaj K.; Spassieva S.; Bielawski J.; Osta W.; Day T. A.; Jiang J. C.; Jazwinski S. M.; Hannun Y. A.; Obeid L. M.; Ogretmen B. Defects in cell growth regulation by C18:0 -ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. 10.1074/jbc.M406920200. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.