

Abstract
We have used a novel experimental setup to conduct the first synchrotron-based 61Ni Mössbauer spectroscopy measurements in the energy domain on Ni coordination complexes and metalloproteins. A representative set of samples was chosen to demonstrate the potential of this approach. 61NiCr2O4 was examined as a case with strong Zeeman splittings. Simulations of the spectra yielded an internal magnetic field of 44.6 Tesla, consistent with previous work by the traditional 61Ni Mössbauer approach with a radioactive source. A linear Ni amido complex, 61Ni{N(SiMe3)Dipp}2, was chosen as a sample with an “extreme” geometry and large quadrupole splitting. Finally, to demonstrate the feasibility of metalloprotein studies using synchrotron-based 61Ni Mössbauer spectroscopy, we examined the spectra of 61Ni-substituted rubredoxin in reduced and oxidized forms, along with (Et4N)2[61Ni(SPh)4] as a model compound. For each of the above samples, a reasonable spectrum could be obtained in about 1 day. Given that there is still room for considerable improvement in experimental sensitivity, synchrotron-based 61Ni Mössbauer spectroscopy appears to be a promising alternative to measurements with radioactive sources.
Keywords: Mössbauer, 61Ni, rubredoxin, hydrogenase, synchrotron radiation, nickel
Graphical Abstract
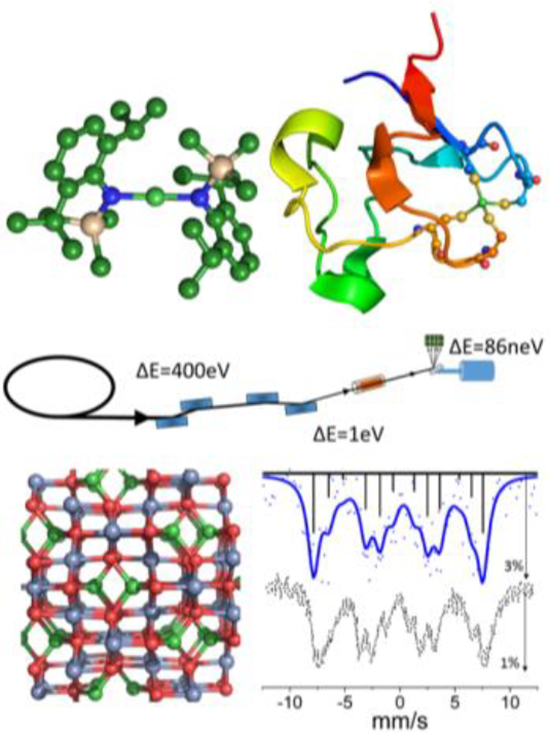
We have performed synchrotron-based 61Ni Mössbauer spectroscopy in the energy domain on a variety of samples. Our results demonstrate sensitivity to internal magnetic fields (via Zeeman splittings) and sensitivity to coordination geometry (via quadrupole splittings). Our spectra on Ni rubredoxin are the first ever 61Ni Mössbauer spectra reported for a metalloprotein.
Introduction
Despite the importance of Ni chemistry to materials1 and battery science,2 catalysis,3 inorganic chemistry,4 and bioinorganic chemistry,5–7 there are relatively few reports on 61Ni Mössbauer spectroscopy. Specifically, there are tens of thousands of papers reporting the application of 57Fe Mössbauer spectroscopy, while the 61Ni Mössbauer literature can be counted in the dozens. The reason for this discrepancy is clear – compared to 57Fe experiments, traditional Mössbauer spectroscopy using the 61Ni 67.4 keV nuclear resonance is logistically difficult.8 The most appropriate parent isotopes, 61Co or 61Cu, have lifetimes of 99m or 3.3h respectively, requiring that experiments be done with frequent source changes and in close proximity to proton cyclotrons or electron accelerators where the sources are produced.8–11
Time-domain approaches using synchrotron radiation12 can in principle yield the same information as energy domain Mössbauer experiments. For example, nuclear forward scattering (NFS) has been used to study Ni metal at low temperatures13 and under high pressure,14 and synchrotron radiation perturbed angular correlation (SRPAC) measurements on Ni metal have also been successful.15 However, the interpretation of such time-domain measurements is not always straightforward, especially for mixtures. The popularity of 61Ni nuclear spectroscopy would certainly benefit from a better energy domain experiment that can be interpreted by standard Mössbauer procedures. Improvements in synchrotron x-ray generation and detection have recently made just such an experiment possible.
Energy-domain synchrotron radiation Mössbauer spectroscopy (SR-MS), first proposed in 1974,16 was first conducted in 198517 using an 57Fe-yttrium iron garnet nuclear Bragg diffraction monochromator.17 Monochromator development continued with work using a single line 57FeBO3 monochromator.18,19 However, this diffraction approach requires near perfect isotopically-enriched single crystals with favorable magnetic properties,17,20–22 and until now has only been achieved with 57Fe. An alternate approach to SR-MS uses a moderately monochromatic synchrotron beam as the source, and a single line Mössbauer isotope as analyzer (Scheme 1). It was first demonstrated in 2009 by Seto and coworkers on 57Fe and 73Ge.23 It has since been applied to 151Eu,24 174Yb,25 125Te,25 and 40K,26 and it has been proposed for nearly 2 dozen lanthanide isotopes.25 Recently, Seto and coworkers have applied 61Ni SR-MS to cathode materials27 and nanoparticles.28 This SR-MS approach is well suited to nuclei with lifetimes shorter than the typical ~10–100 ns electron bunch spacing in a synchrotron i.e. the time between radiation pulses.
Scheme 1.

The synchrotron radiation Mössbauer spectroscopy (SR-MS) experiment.
The 61Ni nucleus, with an excited state t1/2=5.27 ns, is an excellent candidate for SR-MS. Since 61Ni Mössbauer is relatively unfamiliar to chemists, we first compare the spectroscopic properties of this isotope with the more familiar 57Fe. The key nuclear properties are summarized in Table 1, and we briefly discuss some of the spectroscopic consequences below.
Table 1.
Comparison of Mössbauer-relevant nuclear properties for 57Fe and 61Ni.
| abundance (%) | E (keV) | t1/2 (ns) | 2Γ (mm*s−1) | Ig | Ie | αt | μg (n.m.) | μe (n.m.) | Qg (barn) | Qe (barn) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 57Fe | 2.14 | 14.41 | 97.81 | 0.194 | 1/2− | 3/2− | 8.21 | 0.0906 | −0.155 | -- | 0.16 |
| 61Ni | 1.19 | 67.40 | 5.27 | 0.77 | 3/2− | 5/2− | 0.12 | −0.749 | 0.481 | 0.162 | −0.2 |
Lifetime and energy.
The most obvious differences between 61Ni and 57Fe are the energy and lifetimes of the first excited states. The long lifetime (98 ns) and low energy (14.4 keV) of the 57Fe excited state lead to a natural linewidth 2Γ of 9.1 neV, while the shorter lifetime (5.3 ns) and higher energy of the 61Ni resonance (67.4 keV) lead to spectral features with a linewidth of 173 neV, nearly a factor of 20 broader.
Isomer shift (δIS).
For 61Ni, there is a smaller change in nuclear mean square radius (Δ<r2>/<r2>) and higher transition energy, and these lead to a significant reduction in the 61Ni isomer shift range, especially when expressed in terms of mm/s Doppler shift. Whereas common 57Fe δIS values vary from −0.8 to +2.0 mm/s,9 the range for 61Ni δIS is about 30-fold smaller, from −0.06 to +0.04 mm/s.29 This is more than an order of magnitude smaller than the natural linewidth 2Γ = 0.77 mm/s. Moreover, the high energy for this transition (67.4 keV) requires corrections for 2nd order Doppler shifts (SOD) that are of comparable magnitude to the real chemical isomer shifts.29 Although such corrections are possible if the lattice dynamics are well understood, as described by Gütlich,29 that author has noted that so far, “the information concerning chemical bond properties was not very impressive.”29 For these reasons, in this work we have not tried to exploit 61Ni isomer shifts (although others might do so in the future).
Magnetic properties.
The 61Ni nucleus has favorable magnetic properties. It has relatively large magnetic moments in its ground and excited states, μg = −0.75 and μe = 0.48 nuclear magnetons (n.m.),29 compared to μg = +0.09 and μe = −0.15 n.m. for 57Fe (Table 1). Previous work with mixed spinel type oxides, such as Cu0.9Ni0.1Cr2O4, exhibited very large splitting of ±13.6 mm/s.30 Since many of the interesting problems in Ni chemistry can be framed as questions about magnetic properties, this makes observation of Zeeman splittings in 61Ni Mössbauer spectra one of the most promising applications.
Quadrupole splittings.
For 57Fe, the transition is from I=1/2→I=3/2. Since only the excited state is split by an electric field gradient, 57Fe Mössbauer often presents a symmetric doublet spectrum. The 61Ni resonance involves an I=3/2→I=5/2 transition. The quadrupole splitting is more complex, as the ground state (I=3/2) and excited state (I=5/2) yield two and three states respectively (Figure S1) for five non-forbidden transitions. Although the five transitions for 61Ni are not usually resolved, they can in principle provide more robust information than 57Fe through the asymmetry of the quadrupole split spectrum (for axial systems), which is characterized by the sign of the Vzz component of the EFG and the asymmetry parameter η.
In this study we recorded SR-MS on a ferromagnetic nickel spinel – 61NiCr2O4, two coordination complexes with respectively linear and tetrahedral geometries – 61Ni[N(SiMe3)C6H3-2,6-Pr2]2 and (Et4N)2[61Ni(SPh)4], and a Ni protein – Ni-substituted rubredoxin. The data for these samples showcases the sensitivity of 61Ni SR-MS to the Ni internal magnetic hyperfine field, as well as to both the magnitude and the sign of the electric field gradient (EFG). The results also demonstrate that the SR-MS can be used on dilute samples such as Ni proteins – the technique should eventually be useful for catalytic studies of hydrogenase, which contain NiFe in their active site.
Experimental Methods
Synthesis of NiCr2O4.
61NiCr2O4 (1) was produced from 99% enriched 61Ni (Isoflex) and Cr2O3 powders. Following a previously established procedure, the two powders were mixed in an agate mortar and sintered twice in a conventional vertical furnace at atmospheric pressure and 1200° C for 24h in air.31 The sample composition was confirmed by powder x-ray diffraction.
Synthesis of 61NiBr2(OEt2).
In a Schlenk flask, 61Ni metal (0.051 g, 0.84 mmol) in ca. 10 mL Et2O and excess Br2 (1.36 g, 8.51 mmol) were added via a syringe.32 The reaction mixture was stirred for 16 h without light. The insoluble 61NiBr2(OEt2) was allowed to settle and filtered through a filter cannula. The residue was washed twice with Et2O and dried under vacuum to afford 0.206 g (0.699 mmol) of 61NiBr2(OEt2).
Synthesis of 61Ni{N(SiMe3)Dipp}2.
61Ni{N(SiMe3)Dipp}2, (2) (where Dipp= C6H3-2,6-iPr2). 0.375 g (1.47 mmol) LiN(SiMe3)Dipp and 0.206 g (0.699 mmol) 61NiBr2(OEt2) were combined as solids in a Schlenk flask and 30 mL of Et2O was added at ca. 0 °C.33 The reaction mixture quickly turned purple and was allowed to warm to room temperature and stirred for 18 h. All volatile material was removed under vacuum, and the residue was extracted with ca. 30 mL of pentane. The solution was filtered through a filter cannula, and the filtrate was concentrated to incipient crystallization. Storage at – 80 °C for 1 day afforded 0.172 g (0.308 mmol) of crystals.
Synthesis of [Et4N]2[61Ni(SPh)4].
[Et4N]2[61Ni(SPh)4] (3) was prepared as described previously,34 with the following modifications: 61Ni-labeled (Et4N)2(NiCl4) was used as a reagent; [Et4N]2[NiCl4] and [Et4N]SPh were combined in acetonitrile rather than propionitrile and stirred for 4 h at room temperature, isolated by filtration and then washed with cold acetonitrile.
61Ni-substituted rubredoxin purification and preparation.
Pyrococcus furiosus rubredoxin (Pf Rd) was isolated and purified according to Jenney and Adams.35 The Ni form of Pf Rd (Ni Rd) was prepared essentially according to Moura et al.36 except that 61Ni powder was dissolved in aqua regia, then neutralized with NaOH before addition to the denatured protein. Oxidized protein was prepared by treatment with 5 mM K3[Fe(CN)6], which was removed by buffer exchange. The samples were prepared by exchange into Tris buffer (100 mM, pH 8.5) and by repeated concentration/dilution using a Centricon® YM3 concentrator (Amicon). The final sample concentrations were 5–6 mM. Reduced samples contained 10 mM dithionite.
Synchrotron-based 61Ni Mössbauer Spectroscopy.
Samples were prepared in 2.0 mm inner diameter (ID) quartz tubes (for compounds 1, 2 and 3) or 1.45 mm ID (for rubredoxins). The outer diameter was 3 mm. The path lengths were 2 mm for 1, 2 cm for 2 and 3, and 10 cm for the rubredoxin samples. The longer path length for the rubredoxin samples helped compensate for the lower 61Ni concentrations. We note that the small divergence of a synchrotron beam allows the use of long narrow capillaries that would not be practical with radioactive sources (because extreme collimation would reduce the flux throughput from a radioactive source.)
The quartz sample cells were placed inside a 1 cm × 1 cm × 20.2 cm Cu block, which contained a cavity with a diameter of 3.3 mm. A temperature probe was attached to the surface of the cold-finger. The entire setup was shielded by a second Cu case with Al foil windows, which allowed for temperatures as low as 5 K.
All of the spectra were recorded using the approach illustrated in Scheme 1. The SPring-8 storage ring was running in the A bunch mode with 203 bunches spaced at 23.6 ns and a total current of 100 mA. Measurements (except for compound 3) were taken at BL19LXU, where the synchrotron radiation comes from a 27m undulator,37,38 and the experimental setup was similar to that previously described.23 The spectrum for 3 was collected at BL09XU with a 4.5 m long standard undulator and an otherwise identical setup.39,40 The 9th undulator harmonic was first monochromated by a Si(333) high heat load monochromator, followed by a Si(111) low energy filtering monochromator yielding a bandwidth of 2.3 eV with a flux of 2.2×1011 photon s−1 (4.3 eV with a flux of 1.0×1011 photon s−1 for BL09XU) at 67.4 keV. The radiation impinged on the sample in the cold finger, and the transmitted photons then passed to a second cryostat containing a 61Ni0.86V0.14 analyzer foil mounted onto a velocity transducer. The radiative decay of the analyzer including photons and electrons, through nuclear fluorescence and internal conversion, was captured by an 8-element Si avalanche photodiode (APD) array. The APDs were gated to restrict the collection time to between 3 ns and 18 ns after the prompt pulse to discriminate against electronic scattering unrelated to the Mössbauer effect. The resulting spectrum is a function of the analyzer Doppler velocity, and is nearly identical to a conventional Mössbauer spectrum.
The spectra were simulated using a mixed magnetic and quadrupole Hamiltonian as implemented in the MossWinn software.41–43 Fitting parameters can be found in Tables S1, S2, S3, and S4.
Results
61NiCr2O4.
Our synchrotron Mössbauer spectrum for 1 is shown in Figure 1. It exhibits a well-resolved Zeeman splitting indicating a significant internal magnetic field in this ferromagnetic sample, which is a spinel with high-spin Ni(II) in the tetrahedral sites. There are 12 magnetically split transitions for 61Ni and the spectrum can be described as four sets of three partially resolved lines. The fit was fixed to an axial EFG as in previous studies.44,45 The fit yielded an internal hyperfine magnetic field of 44.6 T, in agreement with the value (44.5 T) from previous Mössbauer studies using radioactive 61Co in a 62Ni0.85Cr0.15 source that was activated by bremsstrahlung.46 Vzz was fit to 7.23 V/m2, however the fit was largely invariant of the Vzz value due to the quality of the data and the small effect of the EFG relative to the large internal magnetic field.
Figure 1.

Top: The experimental SR-MS spectrum of NiCr2O4 powder (points) and a simulation (solid line) using an internal hyperfine field of 44.6 T. The pattern of four sets of three partially resolved magnetic hyperfine lines results from Zeeman splitting of I=3/2 ground state and I=5/2 excited state. The sticks represent individual transition contributions, solid line represents the fit and dots indicate actual data. The maximum intensity decrease was about 3%. Bottom: a recreation of the previously reported spectrum obtained with a radioactive source.46
61Ni{N(SiMe3)Dipp}2.
The spectrum for 2 at 4.2K is broad and asymmetric (Figure 2). The spectrum was fit with an axial EFG (Table S2), and from this simulation we found that the asymmetry can be explained by a strong quadrupole splitting with a Vzz EFG component of 11.53×1021 V/m2 at 4.2K (The eQVzz value for the ground state in mm/s is provided in Table S2). We also examined the temperature dependence of the spectral intensity, by taking additional data at 15K, 30K, and 40K (Figure 2 and Figure S4). The rapid decline in intensity with increasing temperature illustrates the importance of the Lamb-Mössbauer factor for this high-energy transition and underlines the importance of conducting experiments at near LHe temperatures. Since linear Ni(II) complexes show pronounced changes in magnetic properties between 4K and 40K,47 the possible relevance of variable Zeeman splittings should be considered for detailed interpretation of the temperature dependent spectra. We decided that better signal-to-noise is required for such an analysis.
Figure 2.

Left: The 61Ni Mössbauer spectrum for the linear nickel complex 2. The solid line represents the fit and the vertical lines represent the energy and relative intensity of the individual transitions. The distance between the furthest transitions is shown above the spectrum in mm/s and neV. Right: A plot of the of the normalized integrated intensity for 4.2K, 15K, 30K, and 40K relative to the 4.2K integrated intensity.
61Ni-Substituted Pf Rubredoxin and 3.
The 61Ni Mössbauer spectra for reduced and oxidized Ni Rd are compared with 3 in Figure 3 and fit parameters can be found in Table S3. The high quality of the spectra allowed them to be fit with a curved baseline to capture some of the well-known time window effect inherent in delayed coincidence Mössbauer and SR-MS.48,49 The simulation for reduced Ni Rd resulted in a negative value for the Vzz component of the EFG, −11.2 × 1021V/m2 (η=0.24). In contrast, the oxidized rubredoxin data were best simulated with a positive Vzz component, however a large asymmetry parameter, (7.6 × 1021V/m2 η=0.83), consistent with higher spherical symmetry of the EFG than the reduced sample. The spectrum for 3 was best simulated by a Vzz component of 10.8 × 1021V/m2 (η=0.000). Our results cannot exclude modest magnetic effects from a slow paramagnetic relaxation and alternative fits are supplied in Table S4.
Figure 3.

Top to bottom: 61Ni SR-MS spectra for 3, reduced, and oxidized 61Ni Pf Rd. The sticks represent individual transition contributions to the simulation using parameters from Table S3.
Discussion
In this work, we have reported the first energy domain synchrotron Mössbauer spectra for 61Ni coordination complexes and metalloproteins, with samples chosen for extremes of Zeeman splitting, quadrupole splitting, or dilution. Our first sample was nickel chromite, NiCr2O4, 1. This is a normal spinel with high-spin Ni2+ in the tetrahedral A sites and Cr3+ in the octahedral B sites. Apart from applications as an NOx sensor, a high emissivity coating, and as a catalyst, it has a rich variety of structural and magnetic phase transitions.31 For our current purposes, it served as a test for the ability of SR-MS to span a wide range of resonances caused by a large Zeeman splitting.
As was documented in Figure 1, our instrument was capable of recording Zeeman splittings out to ±10 mm/s, which arise from the internal magnetic field of ~44.6 T. The signal-to-noise could certainly have been improved with more beam time. However, an unexpected benefit of the wide range of the data was that the magnetic field derived from a least squares refinement was tightly constrained by the intensity in the wings of the spectrum and in good agreement with the previous result of 44.5 T.46 In the future, SR-MS will be a useful probe of magnetic properties.
Our second sample was 61Ni{N(SiMe3)Dipp}2, 2, a high-spin linear Ni(II) complex. It belongs to a class of 2-coordinate open-shell transition metal complexes with interesting magnetic and catalytic properties (see50 for references). For our current purposes this served as a system likely to have a strong quadrupole splitting. Although we could not resolve discrete transitions, we observed a strong asymmetry in the 61Ni Mössbauer spectrum for 2, especially at ~4.2K (Figure 2). The magnitude and sign of Vzz derived from the simulation (+11.5×1021 V/m2) is consistent with our predicted value. For 2, using the d-electron orbital energy order described previously,33 the expectation value prediction of Vzz=+(4/7)e<r−3>3d. However, it is important to note, for the interpretation of real data the sign of Vzz loses meaning as η approaches unity. Our attempts to record and interpret the temperature dependence of the spectra for 2 were less convincing. The signal dropped more than 3-fold between 4 and 40 K, illustrating the dramatic temperature dependence of the Lamb-Mössbauer factor at 67.4 keV .
Our final set of samples consisted of Ni(II) and Ni(III) Pf rubredoxin (Ni Pf Rd) and the model compound 3. In the small (6 kDa) Pf Rd protein, the natural Fe center has been replaced with Ni, yielding a high-spin Ni(II) center with four cysteine thiolate ligands in an elongated tetrahedral conformation.51 The Ni site bears some resemblance to the active site Ni in NiFe H2ases, where the Ni also has 4 cysteine ligands, two of them bridged to Fe.52 Ni(II) Pf Rd can be oxidized to Ni(III) with ferricyanide,53 and based on similar EPR signals, oxidized Ni Rd has been proposed as a model for the electron paramagnetic resonance (EPR) active Ni-C form of [NiFe] hydrogenases.51 It was recently shown that Ni Pf Rd shows high hydrogen turnover frequencies (20−100 s−1), admittedly with a large overpotential of 540mV.54 Our final sample, 3 has been proposed as a model for Ni(II) Rd, as it also has a high-spin Ni(II) with distorted tetrahedral thiolate ligation.55 For 3, the distortion is an axial compression as opposed to the elongation in Ni(II) Pf Rd. For our current purposes, although interesting in their own right, these three samples are also obvious stepping stones to 61Ni Mössbauer of NiFe H2ases and other Ni enzymes.
As mentioned in the Introduction, 61Ni has quadrupole moments of 0.16 and −0.20 barns respectively for the I=3/2 ground and I=5/2 nuclear excited states. This leads to five allowed transitions in a purely quadrupole split spectrum (Supporting Information Figures S1 and S2). Using a single electron approximation for d-electrons and a simple crystal field model, as discussed previously,29 we can predict the sign of the expectation value for Vzz based on geometry. Using such a model, the prediction for a tetragonally elongated geometry becomes Vzz=-(8/7)e<r−3>)3d. A tetragonally compressed Ni(II) complex is predicted by the same procedure to have Vzz=+(4/7)e<r−3>3d. The net result for a tetrahedral system with an axial elongation is a stronger Mössbauer signal at high mm/s, while the converse results for a tetrahedral system with an axial elongation.
For the approximately tetrahedral Ni(II) site in Pf Rd, we observed an asymmetry in the spectral envelope (a greater dip in transmission at lower mm/s) (Figure 3). In contrast, for 3 there is also an asymmetric envelope, but this time with a greater dip in transmission at lower high mm/s. These results indicate an elongated tetrahedral distortion in Ni(II) Rd and an axial compression in 3. Such findings are consistent with the results from optical MCD spectroscopy,55 Raman,56 and the crystal structures57. The structural predictions are less clear for the Ni(III) sample, where we saw a loss in the asymmetry of the Mössbauer transmission. This argues for a more spherically symmetric EFG, perhaps due to a combination of geometrical effects and a different electronic configuration.51 A high-resolution crystal structure could test this hypothesis.
Overall, we find that despite the limited resolution for 61Ni, quadrupole effects can produce a useful broadening and asymmetry of the overall spectral envelope. The asymmetry can in turn be used to determine the sign of Vzz for axial systems and thus make predictions about the type of asymmetry at the Ni site.
Summary
Overall, we have demonstrated that 61Ni SR-MS can be an effective tool in studying the magnetic properties and local geometries of 61Ni sites in solid-state materials, coordination complexes and in Ni metalloproteins. For such dilute samples, the small spot size and low divergence of a synchrotron source are an important factor. One can use cylindrical samples that are several cm long and yet only one mm in diameter, allowing for modest volumes of precious enzymes. Such a sample configuration is not possible with radioactive sources because of the large source size and divergence. As is usual with synchrotron-based experiments, there is still room for significant improvement in this technique, through brighter sources as well as faster and more efficient detectors. For energy domain experiments, 61Ni SR-MS should thus be a strong competitor to the conventional approach with radioactive sources. At synchrotron sources around the world, both energy- and time-domain nuclear techniques promise exciting opportunities for learning more about Ni chemistry through the special properties of 61Ni.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (grant GM-65440 to SPC), the National Science Foundation (grant CHE-1112035 to CGR and grant CHE-1263760 to PPP), and the Deutsche Forschungsgemeinschaft (DFG, Cluster of Excellence UniCat to LL). Preparation of Ni-rubredoxin samples was supported by the U.S. Department of Energy, Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences (DE-FG05–95ER20175 to M.W.W.A.). Additional support came from PCOM (to FEJ), and JSPS KAKENHI (grant 24221005 to M. Seto). The 61Ni Mössbauer experiments were performed at BL09XU and BL19LXU of SPring-8 with the approval JASRI and RIKEN (Proposal No. 2014A1384, 2014B1047, 2015B1175 and 2015B0103). We thank Elizabeth Wille and Susan Kauzlarich for assistance in the preparation of 61NiCr2O4. We thank Zoltán Klencsár for assistance with Mosswinn.
Footnotes
Supporting Information. Nuclear energy levels and lifetimes for 61Ni and typical radioactive sources. Energy levels for pure Zeeman and quadrupole splittings and intermediate case. Predicted spectra for pure Zeeman splittings as a function of magnetic field. Predicted spectra for pure quadrupole splittings. PXRD spectrum for NiCr2O4. The spectral temperature dependence of of (2). Fit parameters for (1) and (2). Fit parameters for (3) and the rubredoxins. Fit parameters for (2) using an unshared Vzz value. Fit with non-negligible magnetic field fit parameters for (2). Non-negligible magnetic field fit parameters for the rubredoxins and (3).
References
- (1).Nickel Alloys; Heubner U, Ed.; CRC Press: New York, 2000. [Google Scholar]
- (2).Ralph J,B In Batteries for Sustainability; Ralph J,B, Ed.; Springer: New York, 2013, p 423–443. [Google Scholar]
- (3).Tasker SZ; Standley EA; Jamison TF Nature. 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Collinson SR; Schröder M In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd: 2011. [Google Scholar]
- (5).Ragsdale SW J. Biol. Chem 2009, 284, 18571–18575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Can M; Armstrong FA; Ragsdale SW Chemical Reviews. 2014, 114, 4149–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Boer JL; Mulrooney SB; Hausinger RP Arch. Biochem. Biophys 2014, 544, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Obenshain FE; Wegener HHF Phys. Rev 1961, 121, 1344–1349. [Google Scholar]
- (9).Gütlich P; Link R; Trautwein AX Mössbauer Spectroscopy and Transition Metal Chemistry; Springer-Verlag: Berlin, 1978; Vol. 3. [Google Scholar]
- (10).Audi G; Bersillon O; Blachot J; Wapstra AH Nuclear Physics A. 1997, 624, 1–124. [Google Scholar]
- (11).Audi G; Wapstra AH; Thibault C Nuclear Physics A. 2003, 729, 337–676. [Google Scholar]
- (12).Gerdau E Hyperfine Interact. 1994, 90, 301–312. [Google Scholar]
- (13).Sergueev I; Chumakov AI; Beaume-Dang THD; Ruffer R; Strohm C; van Burck U Phys. Rev. Lett 2007, 99. [DOI] [PubMed] [Google Scholar]
- (14).Sergueev I; Dubrovinsky L; Ekholm M; Vekilova OY; Chumakov AI; Zajac M; Potapkin V; Kantor I; Bornemann S; Ebert H; Simak SI; Abrikosov IA; Ruffer R Phys. Rev. Lett 2013, 111, 157601. [DOI] [PubMed] [Google Scholar]
- (15).Sergueev I; Leupold O; Wille H-C; Roth T; Chumakov AI; R. R Physical Review B. 2008, 78. [Google Scholar]
- (16).Ruby SL J. Phy. Colloq 1974, 35, 209–211. [Google Scholar]
- (17).Gerdau E; Rüffer R; Winkler H; Tolksdorf W; Klages CP; Hannon JP Phys. Rev. Lett 1985, 54, 835–. [DOI] [PubMed] [Google Scholar]
- (18).Chumakov AI; Zelepukhin MV; Smirnov GV; van Bürck U; Rüffer R; Hollatz R; Rüter HD; Gerdau E Physical Review B. 1990, 41, 9545–9547. [DOI] [PubMed] [Google Scholar]
- (19).Smirnov GV; van Bürck U; Chumakov AI; Baron AQR; Rüffer R Physical Review B. 1997, 55, 5811–5815. [Google Scholar]
- (20).Smirnov GV; Zelepukhin MV; Vanburk W JETP Lett. 1986, 43, 352–355. [Google Scholar]
- (21).Mitsui T; Seto M; Masuda R; Kiriyama K; Kobayashi Y Jap. J. App. Phys. Lett 2007, 46, L703–L705. [Google Scholar]
- (22).Mitsui T; Seto M; Masuda R Jap. J. App. Phys. Lett 2007, 46, L930–L932. [Google Scholar]
- (23).Seto M; Masuda R; Higashitaniguchi S; Kitao S; Kobayashi Y; Inaba C; Mitsui T; Yoda Y Phys. Rev. Lett 2009, 102, 217602. [DOI] [PubMed] [Google Scholar]
- (24).Matsuoka T; Fujihisa H; Hirao N; Ohishi Y; Mitsui T; Masuda R; Seto M; Yoda Y; Shimizu K; Machida A; Aoki K Phys. Rev. Lett 2011, 107. [DOI] [PubMed] [Google Scholar]
- (25).Masuda R; Kobayashi Y; Kitao S; Kurokuzu M; Saito M; Yoda Y; Mitsui T; Iga F; Seto M Applied Physics Letters. 2014, 104. [Google Scholar]
- (26).Nakano T; Fukuda N; Seto M; Kobayashi Y; Masuda R; Yoda Y; Mihara M; Nozue Y Physical Review B. 2015, 91. [Google Scholar]
- (27).Segi T; Masuda R; Kobayashi Y; Tsubota T; Yoda Y; Seto M Hyperfine Interact. 2016, 237, 1–5. [Google Scholar]
- (28).Masuda R; Kobayashi Y; Kitao S; Kurokuzu M; Saito M; Yoda Y; Mitsui T; Hosoi K; Kobayashi H; Kitagawa H; Seto M Scientific Reports. 2016, 6, 20861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gütlich P; Bill E; Trautwein AX In Mossbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer-Verlag: Berlin, 2011, p 235–390. [Google Scholar]
- (30).Okada T; Noro Y; Kobayashi Y; Kitazawa H; Ambe F Phys. Lett. A 1995, 209, 241–245. [Google Scholar]
- (31).Klemme S; van Miltenburg JC Phys. Chem. Min 2002, 29, 663–667. [Google Scholar]
- (32).Ducelliez F; Raynaud A Compt. Rend. Chim 1914, 158, 2002–2003. [Google Scholar]
- (33).Lin CY; Guo JD; Fettinger JC; Nagase S; Grandjean F; Long GJ; Chilton NF; Power PP Inorg. Chem 2013, 52, 13584–13593. [DOI] [PubMed] [Google Scholar]
- (34).Rosenfield SG; Armstrong WH; Mascharak PK Inorg. Chem 1986, 25, 3014–3018. [Google Scholar]
- (35).Jenney FE Jr.; Adams MWW Methods Enzymol. 2001, 334, 45–55. [DOI] [PubMed] [Google Scholar]
- (36).Moura I; Teixeira M; Moura JJG; LeGall J Journal of Inorganic Biochemistry. 1991, 44, 127–139. [DOI] [PubMed] [Google Scholar]
- (37).Yabashi M; Mochizuki T; Yamazaki H; Goto S; Ohashi H; Takeshita K; Ohata T; Matsushita T; Tamasaku K; Tanaka Y; Ishikawa T Nucl. Inst. Meth. A 2001, 467, 678–681. [Google Scholar]
- (38).Hara T; Yabashi M; Tanaka T; Bizen T; Goto S; Marechal XM; Seike T; Tamasaku K; Ishikawa T; Kitamura H Rev. Sci. Instrum 2002, 73, 1125–1128. [Google Scholar]
- (39).Hara T; Tanaka T; Tanabe T; Marechal X-M; Okada S; Kitamura H Journal of Synchrotron Radiation. 1998, 5, 403–405. [DOI] [PubMed] [Google Scholar]
- (40).Yoda Y; Yabashi M; Izumi K; Zhang XW; Kishimoto S; Kitao S; Seto M; Mitsui T; Harami T; Imai Y; Kikuta S Nuclear Instruments & Methods in Physics Research Section A-Accelerators Spectrometers Detectors & Associated Equipment. 2001, 467, 715–718. [Google Scholar]
- (41).Matthias E; Schneider W; Steffen RM Phys. Rev 1962, 125, 261–268. [Google Scholar]
- (42).Kündig W Nucl. Inst. Meth 1967, 48, 219–228. [Google Scholar]
- (43).Klencsár Z Hyperfine Interact. 2013, 217, 117–126. [Google Scholar]
- (44).Göring J Zeit. Nat. A 1971, 26, 1931. [Google Scholar]
- (45).Göring J; Wurtinger W; Link R J. App. Phys 1978, 49, 269–272. [Google Scholar]
- (46).Gütlich P; Hasselbach KM; Rummel H; Spiering H J. Chem. Phys 1984, 81, 1396–1405. [Google Scholar]
- (47).Bryan AM; Merrill WA; Reiff WM; Fettinger JC; Power PP Inorg. Chem 2012, 51, 3366–3373. [DOI] [PubMed] [Google Scholar]
- (48).Kobayashi T; Shimizu S Phys. Lett. A 1975, 54, 311–312. [Google Scholar]
- (49).Seto M; Masuda R; Higashitaniguchi S; Kitao S; Kobayashi Y; Inaba C; Mitsui T; Yoda YJ Phys. Conf. Ser 2010, 217, 012002. [DOI] [PubMed] [Google Scholar]
- (50).Lin CY; Fettinger JC; Grandjean F; Long GJ; Power PP Inorg. Chem 2014, 53, 9400–9406. [DOI] [PubMed] [Google Scholar]
- (51).Huang YH; Park JB; Adams MWW; Johnson MK Inorg. Chem 1993, 32, 375–376. [Google Scholar]
- (52).Lubitz W; Ogata H; Rudiger O; Reijerse E Chemical Reviews. 2014, 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- (53).Saint-Martin P; Lespinat PA; Fauque G; Berlier Y; LeGall J; Moura I; Teixeira M; Xavier AV; Moura JJG Proc. Nat. Acad. Sci 1988, 85, 9378–9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Slater JW; Shafaat HS J. Phys. Chem. Lett 2015, 6, 3731–3736. [DOI] [PubMed] [Google Scholar]
- (55).Kowal AT; Zambrano IC; Moura I; Moura JJG; LeGall J; Johnson MK Inorg. Chem 1988, 27, 1162–1166. [Google Scholar]
- (56).Huang YH; Moura I; Moura JJG; LeGall J; Park JB; Adams MWW; Johnson MK Inorg. Chem 1993, 32, 406–412. [Google Scholar]
- (57).Maher M; Cross M; Wilce MC; Guss JM; Wedd AG Acta Crys. D 2004, 60, 298–303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.