

Abstract
Cardiovascular disease (CVD), with atherosclerosis as the major underlying factor, remains the leading cause of death worldwide. It is well established that cholesterol ester-enriched foam cells are the hallmark of atherosclerotic plaques. Multiple lines of evidence support that enhancing foam cell cholesterol efflux by high density lipoprotein (HDL) particles, the first step of reverse cholesterol transport (RCT), is a promising antiatherogenic strategy. Yet, excitement towards the therapeutic potential of manipulating RCT for the treatment of CVD has faded because of the lack of the association between CVD risk and what was typically measured in intervention trials, namely HDL cholesterol (HDL-C), which has an inconsistent relationship to HDL function and RCT. In this review, we will summarize some of the reasons for this inconsistency, update the mechanisms of RCT, and highlight conditions in which impaired HDL function or RCT contributes to vascular disease. On balance, the evidence still argues for further research to better understand how HDL functionality contributes to RCT in order to develop prevention and treatment strategies to reduce the risk of CVD.
Keywords: Reverse cholesterol transport, Cardiovascular disease, Foam cells, Cholesterol efflux, Atherosclerosis, Diabetes
Introduction
The Framingham Heart Study in the 1960s was the first study to report inverse associations between cardiovascular (CV) risk and plasma high density lipoprotein cholesterol (HDL-C)1. This landmark discovery inspired investigations into the mechanisms by which HDL confers atheroprotection, leading to the identification of the reverse cholesterol transport (RCT) pathway2. RCT is defined as the process by which cholesterol moves out of cells in peripheral tissues (including foam cells in atherosclerotic plaques), enters the circulation, and is excreted in the feces. HDL’s CV protective effect has conventionally been attributed to its ability to act as both the acceptor of cholesterol from cells and as the cholesterol carrier in the RCT pathway, including delivery to the liver. It has been estimated from observational studies that CV risk decreases by approximately 2–3% per 1‐mg/dL increase in HDL‐C3. Implicit in this view is that the level of HDL-C in the plasma is a faithful biomarker of the ability of the HDL particles to mediate RCT. In 2019, this is now recognized to be an oversimplification as HDL-C measurements do not necessarily reflect either the overall abundance of HDL particles, the distribution of HDL subspecies4, or RCT capacity5. Additionally, data from human genetic studies6 and a host of negative HDL-raising clinical trials have led to much controversy over the “HDL hypothesis.” This controversy, however, should not negate the strong experimental evidence that a major function of HDL particles is to mediate RCT, and that an increased understanding of the mechanisms by which this is accomplished represents a chance to revise and refine the HDL hypothesis. The framework of this review is illustrated in the Figure, with the points made in the legend discussed in detail below.
Cholesterol Efflux and Peripheral Cells
All mammalian cells require cholesterol, with the highest concentration in the plasma membrane and the lowest in the endoplasmic reticulum (ER) membrane. The amount of free cholesterol is maintained in a relatively narrow range, making cellular cholesterol homeostasis essential for normal cell function. Based on pioneering data from many laboratories2,7, RCT came to be described as the process by which HDL acts as the specific cholesterol acceptor that transports excess cholesterol stores within peripheral tissues to the plasma, and then delivers it to the liver, where it can be directly excreted into the bile or be metabolized into bile acids/salts before excretion. These studies naturally stimulated much research in a variety of in vitro systems, to investigate at the cellular level the initial step of RCT. Herein we will focus on the roles of macrophages and vascular smooth muscle cells in the early steps of the RCT process, given their crucial role in the development of CV diseases, especially atherosclerosis. We will also discuss other aspects of the RCT pathway, including its quantitative assessment in vitro and in vivo.
Macrophages
Macrophage extracellular cholesterol is primarily derived from the internalization of plasma lipoproteins or from the efferocytosis of apoptotic cells, which enter the cellular pool together with newly synthesized cholesterol. To prevent toxicity, surplus cholesterol is effluxed from the cells to extracellular acceptors or converted to cholesteryl ester (CE) and stored in cytosolic lipid droplets (LDs). There are many mechanisms by which cells are defended against cholesterol toxicity. For example, the liver X receptors (LXR), key sterol-sensitive transcription factors in macrophages that regulate intracellular cholesterol (reviewed in8), are induced by excess cholesterol. LXRs, in turn, drive the expression of numerous genes within the efflux pathway, including ATP-binding cassette (ABC) proteins A1 and G1 (ABCA1/G1), which are key cellular cholesterol transporters. Adding to the effort to reduce cellular sterol content, LXR also controls the expression of the inducible degrader of the low-density lipoprotein receptor (IDOL), an E3 ubiquitin ligase that promotes the degradation of the low-density lipoprotein receptor (LDLR). This limits further uptake of exogenous cholesterol via LDLR9. Another defensive response to elevated cell cholesterol is the inhibition of the processing of the sterol regulatory element-binding protein (SREBP), leading to decreased expression of genes that regulate cholesterol syhthesis (HMGCR) and uptake (LDLR). Yet another consequence of cellular cholesterol excess is the reduced expression of a small microRNA encoded in SREBP’s intronic region, miR-33, which among its targets of translational repression are the mRNAs encoding numerous factors in the RCT pathway (ABCA1, NPC1, ABC11, ATP8B1)10–12. miR-33a is encoded in the intron of the SREBP-2 gene in mice and humans, while its isoform miR-33b is encoded within the SREBP-1 gene in higher mammals12. Notably, inhibition of miR-33 in mice and non-human primates holds therapeutic promise as it has been shown to enhance RCT12–14, protect against atherosclerosis15–17 and promote atherosclerosis regression14,18, though some controversy surrounds its role in regulating hepatic triglyceride and fatty acid metabolism19–22. A recent report by the Fernandez-Hernando group demonstrates that repression of ABCA1 is the primary mechanism by which miR-33 regulates macrophage cholesterol efflux and atherogenesis23. Although miR-33 is the most characterized of the miRNAs that regulate RCT, there are at least 10 others that also have targets in this pathway (reviewed in24).
ABCA1 and ABCG1 are critical receptors for the initial step of RCT in atherosclerotic plaques, i.e., cholesterol efflux out of foam cells25–28. Foam cells are traditionally thought to be cholesterol-laden macrophages originating from monocytes, but as reviewed below, they can also be macrophage-like cells originating from cholesterol-laden vascular smooth muscle cells29–33. Prior to efflux, cholesterol must be in its ‘free’ (unesterified) form to be pumped out of cells. This is the case in vitro and in vivo, including in atherosclerosis, where the rate-limiting step in RCT is hydrolysis of lipid droplets (LDs) in vascular foam cells to generate free cholesterol for efflux34–36. Consistent with this are several studies reporting that stimulation or inhibition of macrophage foam cell CE hydrolysis regulates RCT and atherosclerosis37–41. LD cholesterol undergoes constitutive cycles of hydrolysis and re-esterification. Free cholesterol released from LDs via CE hydrolysis can either traffic to the plasma membrane and be effluxed to a cholesterol acceptor, or, in a “futile cycle” be re-esterified by the ER-resident protein acyl-CoA:cholesterol acyl transferase (ACAT)42,43. Original studies by Brown and Goldstein characterizing this ‘futile cycle’ indicated that cytoplasmic CE hydrolysis in macrophage foam cells was mediated by extra-lysosomal, cytoplasmic neutral CE hydrolases42,44. Nevertheless, knocking down or out potential CE hydrolases in macrophages never entirely abolishes cellular CE hydrolysis36. The importance of addressing the missing regulators of CE hydrolysis is underscored by the many studies to date showing that increasing the hydrolysis of LD CE increases cholesterol efflux and is antiatherogenic36.
A key insight into CE hydrolysis came from the observation that the loading of macrophages with pro-atherogenic lipoproteins can activate autophagy, promote sequestration and delivery of LDs to lysosomes for degradation, and enhance RCT from macrophage foam cells45. Autophagy is a ubiquitous cellular process by which cytoplasmic components are degraded within lysosomes. There are three types of autophagy in mammalian cells: 1) macroautophagy, where cargo is sequestered in de novo formed autophagosomes that subsequently fuse with lysosomes, 2) microautophagy, where cargo is taken into lysosomes by invagination and pinching of the lysosomal membrane into the lysosome lumen, and 3) chaperone-mediated autophagy, where single proteins are recognized by chaperones and delivered to lysosomes via a membrane translocation complex46. Macroautophagy - referred to as autophagy hereafter – is the subtype most relevant to this review and can sequester cytosol ‘in bulk’ or selectively. This pathway depends on numerous autophagy (Atg) proteins that organize into functional complexes that orchestrate the autophagic process, first generating the limiting membranes or phagophores that elongate in cup-shaped structures that engulf cytoplasmic cargo, fusing to become autophagosomes47. Subsequent autophagosome fusion with lysosomes releases the ‘autophagic body’, or in the case of RCT – LDs, into the lysosome lumen where they are degraded. Lysosomal acid lipase (LAL), encoded by the LIPA gene in humans, is responsible for the hydrolysis of LD-associated CE to generate free cholesterol for efflux45. Genome-wide association studies have identified several loss of function mutations in LIPA as causative of Wolman disease, cholesterol ester storage disease (CESD) and coronary artery disease (CAD)48.
Targeted LD degradation by autophagy, or ‘lipophagy’, represents a novel pathway to regulate RCT, and it is thought that enhancing autophagy holds promise to promote lipid clearance from the atherosclerotic vascular wall. In particular, atherosclerosis development is associated with a progressive defect in autophagy in cells positive for macrophage markers MOMA-2 and CD11b in the plaque49, and defective clearance of cargo tagged by the autophagy marker p62/SQSTM1 is readily observed by detection of its accumulation in whole aortic protein lysates49,50. Further, inhibition of autophagy pathways in mice promotes atherosclerosis development by reduced lipophagy and lysosome-mediated cholesterol cellular efflux which contributes to inflammasome hyperactivation, elevated cell death and defective efferocytosis within plaques36,49,51. A critical role for autophagy and lysosomal biogenesis to suppress atherosclerosis development is supported by studies showing that systemic miR-33 inhibition or macrophage overexpression of the master transcriptional regulator of autophagy and lysosomal genes, TFEB, restores plaque macrophage autophagy, improves efferocytosis and inflammation, and ultimately reduces atherosclerosis burden50,52.
The routes by which free cholesterol generated at the site of lipid lipolysis (within lysosomes or at the LD surface) reaches the ABCA1 and ABCG1 cholesterol transporters on the plasma membrane depends on both vesicular and non-vesicular trafficking pathways, although the precise mechanisms are poorly characterized53,54. The general working hypothesis is that cholesterol transporters sit at the plasma membrane and await delivery of cholesterol to be effluxed; but, this is an oversimplication as these can be motile, as exemplified by ABCA1 that continuously shuttles between the plasma membrane and endolysosomal compartments55. This shuttling is a regulated process that is impeded by hypoxia56,57. ABCG1 re-localizes from the Golgi and ER to the plasma membrane following LXR activation to stimulate efflux to HDL58. This involves ABCG1 concentrating on intracellular endocytic vesicles (e.g. recycling endosomes) to apparently redistribute sterols to the plasma membrane outer leaflet upon fusion, so that cholesterol desorbs to exogenous lipid acceptors such as HDL59. Transporters that possess distinct subcellular localizations likely preferentially efflux cholesterol from specific intracellular pools; for instance, apoA-I/ABCA1 retroendocytosis is required for efficient cholesterol efflux under lipid-loaded conditions60 and conversely, ABCA1-mediated cholesterol efflux is primarily dependent on autophagy for its cholesterol source45.
Another cholesterol trafficking pathway is mediated by oxysterol-binding (OSBP)-related proteins (ORPs). They constitute a family of lipid binding/transfer proteins that can facilitate non-vesicular transfer of cholesterol between lipid bilayers, increasing the efficiency of cholesterol transport between sub-cellular membranrous organelles. Roles for many of the ORPs within this family (12 members in total) as sterol sensors or transporters at distinct subcellular sites have been recently reviewed61. Recently, ORP6 was found to regulate cholesterol efflux and HDL homeostasis, suggesting that it may represent a novel regulator of the RCT pathway62, yet mechanisms by which ORP6 and other ORP members may regulate this pathway are poorly understood. Other key mediators of inter-organelle lipid trafficking that may represent potential therapeutic enhancers of RCT include the soluble lipid transfer proteins steroidogenic acute regulatory protein (StAR) D4, MLN64, and Niemann-Pick Type C (NPC) proteins63. More recently, Aster proteins have emerged as novel mediators of non-vesicular cholesterol transport at contact sites between the plasma membrane and ER, providing a new mechanism by which HDL-derived cholesterol can be mobilized through the selective HDL cholesterol uptake pathway64.
Vascular Smooth Muscle Cells
While much of the focus on the early steps of RCT has been on defining mechanisms of efflux from macrophages, there have also been investigations on vascular smooth muscle cells (VSMC). VSMC plasticity in atherosclerosis is well recognized; e.g., in the media of atherosclerotic arteries, they are considered to be contractile and can become proliferative, migrate to the intima, where they are synthetic, and exhibit the loss of a number of markers of the VSMC in the contractile state, such as smooth muscle cell actin and myosin heavy chain30. Though it has been long appreciated that VSMC in the intima can also take up cholesterol through a variety of pathways29,65,66, phenotypic changes in these VSMC-foam cells at the molecular level had not been systematically studied. In 2003, it was shown that loading mouse primary VSMC with cholesterol in vitro resulted in the concurrent loss of VSMC marker expression and the gain of macrophage-associated gene expression67.
One implication of these findings is that conventional histological markers used to identify macrophages in plaques would include cells of VSMC lineage. Three studies using lineage-marking approaches in mice31,32,68 and a variety of assays for human plaques69 were published in quick succession to confirm that, indeed, there are macrophage-appearing cells of VSMC origin in human and mouse plaques. These results have also been replicated in studies examining the clonality of VSMC in atherosclerotic plaques in mice70,71. The percent of the macrophage-marker positive cells of VSMC origin varied among the studies, but it was substantial in all, ranging from 30–70% (increasing with the stage of disease). The molecular regulation of this “transition” process has been studied mainly in vitro, where it was shown that cholesterol loading suppresses miR-143/145, resulting in reduced expression of the canonical VSMC regulatory transcription factors, serum response factor (SRF) and myocardin. Suppression of miR-143/145 also increased levels of KLF4, a regulator of macrophage gene expression72. Consistent with the in vitro implication of KLF4 in the acquisition of macrophage features is the work from the Owens lab, which showed that in VSMC-specific KLF4-deficient mice, the percentage of macrophage-like cells derived from VSMC in atherosclerotic plaques was approximately 50% versus those in control mice32. A recent study has also implicated integrin β3 in the transition of VSMC to macrophage-like cells in mouse atherosclerotic plaques71.
The relevance of cholesterol efflux to the macrophage-like transition of VSMC is suggested by the results in vitro that by providing cholesterol acceptors (HDL or apoA-I) to cholesterol-loaded cells. This completely reversed their macrophage-like phenotypes to the pre-loaded VSMC state31. Evidence that impaired efflux may be operating in vivo, and thereby sustaining the effects of cholesterol-loading, comes from two strands of evidence. First, as Francis and colleagues have shown, ABCA1 expression is reduced in intimal-like VSMC (derived from arteries of Wistar-Kyoto rats), and in vitro these cells exhibit less binding of apoA-I compared to those isolated from the medial layer73. Similarly, ABCA1 expression was found to be low in human intimal VSMC, more so in advanced relative to early atherosclerosis69. Very recently, CD45- cells (presumably VSMC-derived) from ApoE−/− mice were also found to exhibit reduced ABCA1 expression relative to CD45+(presumably monocyte-derived) foam cells33.
Such changes would be likely to make cholesterol taken up by intimal VSMC linger and not readily enter the RCT pathway. It should be noted, however, that in other studies, cholesterol loading of murine VSMC increased their ABCA1 mRNA expression31,32,67. Reconciling the apparent discrepancies between the results, which may have been caused by species differences, experimental conditions, etc, will require more investigation, but whatever the level of ABCA1 expression there is, it does not appear to be sufficient to prevent accumulation in vitro or in vivo when VSMC are exposed to elevated levels of cholesterol.
Second, VSMC-derived foam cells exhibit impaired phagocytosis relative to macrophages, compared to contractile VSMC, they are considerably more active (>5-fold )31. The cholesterol content of the phagocytosed cells would, therefore, be expected to contribute to VSMC-foam cell formation beyond the effects of hypercholesterolemia. In contrast to the data for phagocytosis, efferocytosis was not different in vitro between VSMC and cholesterol-loaded VSMC, suggesting that autophagic capacity may be submaximal in VSMC compared to macrophages31. As autophagy is an important factor providing intracellular cholesterol to the efflux pathway45, its potential limitation might be a contributor to impaired VSMC-foam cell cholesterol efflux. Further aggravating the cellular cholesterol imbalance in VSMC-foam cells is that apparently another homeostatic mechanism to promote efflux when cells are cholesterol-loaded, namely, the induction of LXR-regulated pathways, fails to become activated in vitro73.
If the current speculation that transitioned VSMC have negative contributions to atherosclerosis are true, then the value of restoring cholesterol efflux to intimal VSMC becomes clear. An interesting question arises: how do the efflux capacities of distinct foam cell populations differ from one another? The answer would have implications in designing therapeutic strategies to target all or a subset of foam cells in the plaque to maximally promote RCT.
Reverse Cholesterol Transport
Though HDL is thought to have many functions74–76, overwhelmingly its ability to promote RCT is considered key to its atheroprotection. This has stimulated much research in enhancing RCT. Although this pathway has been actively studied for several years, mechanistic understanding of ABC family-mediated lipid export and nascent HDL biogenesis remains incomplete, with basic pieces of the puzzle such as structural information of the “ABC subfamily” only just emerging77. While ABCA1 preferentially lipidates small HDL particles, specifically apoA-I to form nascent HDL78,79, ABCG1 stimulates net cholesterol efflux to larger HDL, but not to lipid-poor apoA-I80. Moreover, as alluded to above, ABCA1 trafficking between the cell surface and late endocytic vesicles is functionally important to stimulate cholesterol efflux out of endosomal/lysosomal compartments to lipid-free apoA-I55,81,82, while ABCG1 is an intracellular sterol transporter that promotes cholesterol frafficking from the ER to the plasma membrane59. In turn, efflux to HDL involves passive diffusion of cholesterol as well as active cholesterol transfer, and ABCA1, ABCG1 as well as unrelated scavenger receptor class B type 1 (SR-B1) mediate lipid transfer to HDL83–86. Following cholesterol transfer to HDL particles, the next step in HDL biology is esterification of the acquired cholesterol by lecithin:cholesterol acyltransferase (LCAT) to form CE, giving rise to mature HDL. Remodeling of HDL particles can occur through the hydrolysis of HDL triglycerides and phospholipids, mediated by hepatic lipase and endothelial lipase, respectively87. In humans (but not mice) CE in the HDL core can be transferred to triglyceride-rich lipoproteins by cholesteryl ester transfer protein (CETP) for elimination via hepatic clearance in the liver through the LDLR, or selectively taken up via SR-B1 acting as a hepatic receptor for CE on HDL. Therefore, RCT to the liver of cholesterol derived from peripheral cells in humans involes two routes; 1) direct (HDL-SR-B1) and 2) indirect (HDL-LDL/VLDL-liver LDLR). In the liver, the CE is hydrolyzed and the free cholesterol is either converted to bile acids or transported by ABCG5 and ABCG8 into the bile for excretion into the feces.
Three conceptual approaches to enhancing RCT have been proposed: 1) improve macrophage cholesterol efflux, 2) improve HDL functionality (i.e., its capacity to accept or transport cholesterol), and, 3) improve hepatic cholesterol uptake and biliary/intestinal excretion88. As research has continued, this third possibility has been informed by mounting evidence that several HDL-independent routes can promote RCT, and that cholesterol removal from the body may not require hepatobiliary cholesterol excretion89. Thus, the term RCT currently encompasses all potential routes of net cholesterol flux from peripheral tissues into the feces90, including artificial ones that have therapeutic potential. For example, non-HDL particles of 2-hydroxypropyl-β-cyclodextrin (CD) are artificial cholesterol acceptors and have been shown in vivo to mediate RCT and atheroprotection91,92.
Other modalities to increase RCT include liposomes93–95, the red blood cell compartment, which can act as a cholesterol sink to increase RCT96, microparticle-mediated cholesterol efflux97, and synthetic nanoparticles and HDL mimetics that not only serve to package and deliver therapeutic drugs such as LXR agonists or statins to the arterial wall to stimulate cholesterol efflux, but can also extract plaque cholesterol98–100. There are also efforts to increase LCAT activity so that more free cholesterol can be esterified, increasing the amount loaded into HDL101. There is renewed interest in hepatic SR-B1 as a target, based on the work from Rader and colleagues showing loss of function SR-B1 mutations in people are associated with increased CV risk, despite elevated HDL-C102. This is consistent with mouse models in which deficiency of SR-B1 increased HDL-C but paradoxically increased atherosclerosis103. In these studies, SR-B1 deletion or loss of function impaired RCT, consistent with the growing body of evidence highlighting that HDL function and cholesterol flux are ulitimately better determinants of atheroprotection than absolute HDL-C concentrations. However, it should be noted that another study found that rare mutations that disrupt SR-B1 function associate with HDL-C but not CAD risk104.
In addition to the hepatobiliary route of cholesterol elimination, there is also transintestinal cholesterol efflux (TICE)90. While hepatobiliary cholesterol secretion involves transfer of cholesterol from hepatocytes into the bile canaliculus105, in TICE cholesterol is transported directly from blood, through the enterocytes, into the lumen of the intestine106. These fecal cholesterol routes- hepatobiliary and TICE- are estimated to account for 65% and 35% of cholesterol elimination in humans106, respectively. The nuclear hormone receptors LXR and farnesoid X receptor (FXR) are important regulators of cholesterol excretion, by controlling the transcription and activity of numerous cholesterol transporters and bile synthesis enzymes105–107.
Although cholesterol itself can be secreted into the bile for excretion from the body, synthesis and excretion of bile acids comprise the major cholesterol catabolism pathway in mammals108. Thus, LXR and FXR both represent potential therapeutic targets to stimulate TICE and biliary cholesterol secretion and promote RCT107,109. Because hepatic LXR activation also stimulates lipogenesis, leading to steatohepatitis110, devising a strategy to selectively activate nuclear receptors in the intestinal lumen to promote TICE without inducing hepatic lipogenesis may represent a targeted approach to circumvent this issue. In addition, miRNAs add an extra level of regulation to cholesterol metabolism by exerting post-transcriptional negative control of certain genes, including ABCB11 and ATP8B1111, suggesting anti-miRNA therapies.
Quantification of RCT
The controversy surrounding HDL-C as a reliable biomarker of HDL function, including the promotion of RCT, does not contradict the view held by many that increasing RCT will contribute to reducing atherosclerosis and the risk of CV events75,112,113. Indeed, an independent inverse association between HDL cholesterol efflux capacity (CEC) and incident cardiovascular events has been shown both in the Dallas Heart Study and in the European Prospective Investigation of Cancer‐Norfolk study114,115. In addition, quantification of cholesterol mass efflux capacity (CMEC) in CAD and stroke cohorts derived from the Multi-Ethnic Study of Atherosclerosis (MESA) indicate a protective role for HDL-mediated efflux in patients with CAD albeit not those with stroke116. Thus, considerable efforts have been made to develop measurements of RCT in vitro and in vivo, especially with an eye to test approaches to increase it, such as those suggested above.
In vitro, commonly employed are assays of the first step in RCT, the efflux of cellular cholesterol. In this type of assay, cells are first incubated with radioactive [3H or 14C]-cholesterol or, alternatively, fluorescent BODIPY-cholesterol to label intracellular cholesterol pools, after which transfer of the labeled cholesterol from the cells to an extracellular cholesterol acceptor, such as apoA-I or HDL, is measured over time117–119. One must consider several factors when designing a cholesterol efflux experiment120, for example, the exogenous cholesterol acceptor and label to be used, keeping in mind how this might affect net cholesterol flux given that efflux to α-HDL may be bi-directional, so that the correlation of BODIPY-cholesterol efflux and that of 3H-cholesterol to pre-β-HDL and α-HDL may differ118. A variation of these assays, originally developed by Rothblat, Rader and colleagues5, has been used to assess the CEC of HDL isolated from human subjects to determine its correlation between HDL CEC and CV risk114,115,121,122. A number of such studies (but not all123) have found an independent inverse association between HDL CEC and incident cardiovascular events, supporting HDL CEC as a metric of CV risk superior to HDL-C. Nevertheless, side-by-side comparisons of the radiolabeled and the fluorescently labelled cholesterol method is necessary to determine if this accounts for differences among studies and the correlation of HDL CEC with HDL-C.
Turning to assays in vivo, a simple assay developed by Rader and colleagues has been used to quantify RCT in experimental mouse models. This consists of injecting macrophages loaded with radiolabeled cholesterol into the peritoneal cavity of mice, and measuring the appearance of the radiolabel into the plasma, liver and feces over time124. The major limitation of this assay is that it does not consider the bidirectional movement of cholesterol in and out of macrophages, and thus one cannot draw conclusions about the net outward flux of cholesterol mass. To circumvent this, macrophage-specific RCT might be better quantified using techniques in which macrophages are trapped into the site of injection using semipermeable hollow fibers or Matrigel plugs, and these implants are removed so that cholesterol mass content may be determined at the end of the assay125,126. More recently, Cuchel et al. adapted the conventional RCT method to allow for quantification of RCT in humans. This method involves intravenous delivery of 3H-cholesterol nanoparticles, followed by blood and sample collection to quantify tracer counts in plasma, non-HDL and HDL fractions, as well as fecal fractions127. This is an exciting advance in the field, providing a feasible approach to quantify RCT in vivo in humans. A combination of this methodology along with HDL CEC quantification and advanced modalities in imaging, such as intravascular ultrasonography (IVUS), optical coherence tomography (OCT) and near-infrared spectroscopy (NIRS) to facilitate in situ plaque imaging may together provide a better assessment of whole-body RCT capacities in humans and allow for clinical testing of new drugs for the treatment of CAD128.
Selected Topics in Cholesterol Efflux, HDL Biology, and RCT
Atherosclerosis prevention and regression
A chronic inflammatory disease, atherosclerosis begins with the accumulation of apoB-containing lipoproteins and their cholesterol in the artery wall. In response to arterial lipoprotein/lipid buildup and retention, macrophages are recruited to the intima and take up the modified lipoproteins and their lipids by multiple processes129, leading to the formation of foam cells that secrete inflammatory mediators and promote the development of early atherosclerotic lesions. These lesions develop into disease-causing advanced plaques in the process commonly referred to as atherosclerosis progression. Once advanced atherosclerotic plaques are established, the process by which they undergo a reduction in one or more standard parameters (size, lipid content, foam cell content, macrophage inflammation) is termed atherosclerosis regression. Macrophage RCT is the mechanism by which atherosclerotic plaques may rid themselves of cholesterol, and, as noted earlier, is still considered as an essential target to inhibit atherosclerosis progression and promote atherosclerosis regression.
Besides cholesterol efflux, macrophage RCT may also involve cholesterol removal from plaques by another mechanism, namely by the emigration of the macrophages themselves. Plaque foam cell population numbers are determined by cell recruitment, proliferation in situ, emigration and cell death130. Historically, atherosclerosis progression studies have placed a major emphasis on understanding mechanisms of monocyte recruitment into the vascular wall and devising strategies to block their influx into plaques130. Recent studies, however, show that there are also factors that determine macrophage retention within and egress from plaques130, and if these are manipulated appropriately, can lead to reductions in macrophage numbers and the cholesterol they contain, resulting in regression in murine models. One of the emigration factors is the C-C chemokine receptor type 7 (CCR7)131, whose transcription is regulated in part by a sterol response element (SRE) in its promoter132. When HDL levels were raised in ApoE−/− mice, the SREBP pathway in plaque macrophages was activated and macrophage emigration was stimulated132.
Another study reported that raising HDL in ApoE−/− mice as a consequence of using an apoE-encoding adenovirus to reduce non-HDL hyperlipidemia decreased plaque macrophage content by 74% after 4 weeks of apoE complementation. This was attributed to a marked reduction in monocyte recruitment to plaques, but not to CCR7-dependent egress of macrophages from plaques133. The role of CCR7 in some models of murine atherosclerosis regression was confirmed in a recent study, in which it was shown that deficiency of LRP1 increased RCT and CCR7 expression in plaque macrophages, and promoted atherosclerosis regression, which was associated by the appearance of plaque macrophages in lymph nodes134. Thus, egress of macrophages and perhaps other leukocytes from plaques is likely a significant contributor to net RCT in certain, but not all, contexts (see below on Lymphatics and RCT). Whether foam cells of VSMC origin can also emigrate from plaques and the extent to which they may do so relative to ‘classical’ macrophage foam cells remains to be determined.
Lymphatics and RCT
There is a growing interest in the role of lymphatics in RCT. Lymphatic capillaries have been localized in the adventitia of atherosclerotic plaques, where they play an important role in the drainage of local inflammatory cells and cytokines and protect against atherosclerosis development135. The lymphatic vasculature is also critical for the removal of cholesterol from macrophages in RCT, accounting for 50% of cholesterol delivery from cholesterol-loaded macrophages into the plasma compartment136. Moreover, lymphatic insufficiency in mice disrupts proper lipoprotein metabolism (e.g., elevated cholesterol and triglyceride levels in VLDL and LDL fractions) and vascular homeostasis, leading to accelerated atherosclerosis137. These findings are in agreement with previous studies showing that interstitial fluid supports RCT; whereas plasma mainly contains α-HDL particles that are the predominant carriers of CE to hepatocytes, interstitial fluid provides a metabolic environment that drives the conversion of α-HDL to pre-β-HDL, the main acceptor of free cholesterol from peripheral tissues138.
HDL particles can be partitioned into several subclasses according to the specific isolation or separation technique applied139. By ultracentrifugation, two HDL subclasses can be obtained: HDL2 (1.063–1.125g/mL) and HDL3 (1.125–1.21g/mL). In turn, agarose gel electrophoresis separates HDL based on surface charge and shape into α- or preβ-migrating particles (α-HDL or pre-β-HDL). Pre-β-HDL primarily consists of poorly lipidated apoA-I and is the substrate for the ATP-binding cassette (ABC) transporters A1 (ABCA1) that transfers phospholipids and cholesterol to apoA-I to generate nascent discoidal HDL140. In turn, α-HDL represents mature HDL that arises from the esterification of free cholesterol into cholesterol ester (CE) by LCAT, and α-HDL can subsequently be further lipidated through the action of ABCG1 and SR-B1 (Figure 1).
Figure 1. Key Steps of Reverse Cholesterol Transport.
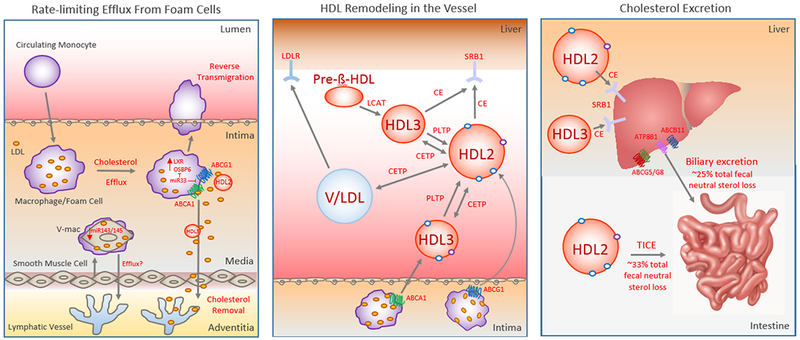
Reverse Cholesterol Transport (RCT) begins with the removal of cholesterol from arterial foam cells that are of vascular smooth muscle cell (V-mac) or macrophage origin (left panel). This is the rate-limiting step of the RCT pathway, and requires the efflux of free cholesterol to cholesterol acceptors such as nascent or mature HDL along with macrophage egress from the plaques. While RCT from macrophage foam cells requires the cholesterol pumps ABCA1 and ABCG1, mechanisms regulating RCT from intimal vascular smooth muscle cells that have transdifferentiated to macrophage-like foam cells (V-mac) are not well understood, though cholesterol efflux from V-macs appears defective relative to macrophage foam cells. Intimal-derived HDL cholesterol can reach the liver 1. directly through binding the hepatic HDL receptor SR-B1 that selectively removes CE from HDL2 and HDL3 or 2. indirectly via apoB-containing lipoproteins (VLDL or LDL [V/LDL])-- to which cholesterol is transferred by the action of cholesterol ester transfer protein (CETP)– that are cleared by hepatic LDLR (middle panel). Phospholipid Transfer Protein (PLTP) also plays an important role in regulating HDL metabolism through HDL remodeling. Finally, the last step of the RCT pathway is cholesterol excretion into the feces (right panel). This can occur through biliary cholesterol excretion or trans-intestinal cholesterol efflux (TICE) that mediate approximately 25% and 33% of total fecal neutral sterol loss, respectively.
Whether apoB-containing lipoproteins, which can also serve as cholesterol acceptors to facilitate RCT depending on the gradient, also enter peripheral tissues and drain into the lymph to regulate RCT remains to be investigated. In addition, more research is needed to understand how artery tertiary lymphoid organs (ATLOs) form in the adventitia during atherosclerosis and to determine their role in regulating the immune response during atherosclerosis and how they may modulate RCT flux. For example, these lymphoid aggregates secrete chemokines that may promote foam cell retention, which may in turn increase plaque lipid burden141.
Diabetes
As an example in which impairment in one or more components of RCT may underlie increased CVD risk, we will discuss diabetes. Diabetes, both type 1 (T1DM) and type 2 (T2DM), represent significant global health issues, with CVD accounting for 65% of mortality in this population142. Additionally, the metabolic syndrome (MetS), a disorder associated with increased risk of developing T2D, is unequivocally linked to increased risk for premature CVD and death143. T2DM and the MetS have a number of associated pathologies, including insulin resistance, obesity and high plasma triglycerides, and, relevant to RCT, low levels of HDL-C and apoA-I, reduced HDL particle (HDL-P) number and dysfunctional HDL-Ps144–150. Thus, there are a number of facets of RCT which likely contribute to heightened CVD risk in diabetic and MetS patient populations.
One mechanism for the impaired HDL-P function may be related to the formation of advanced glycation endproducts (AGEs), which are non-enzymatic modifications of proteins that occur in vivo in diabetic patients. Glycation of HDL and apoA-I is proposed to impair their functionality by reducing both their CEC and antioxidant capacity18,151–156. Additionally, in vitro, high glucose and AGE-modified proteins impair macrophage CEC by downregulation of the transporters ABCA1 and ABCG1, attributable to increased local production of reactive oxygen species 79,157–160. Consistent with this, numerous murine and human studies report decreased expression of ABCA1 and ABCG1 in monocytes and macrophages isolated from diabetic mice and people, translating to decreased myeloid CEC99,161–164.
Mechanistically, reduced CEC transporter levels under diabetic conditions in vivo are mediated, in part, by the receptor for advanced glycation end products (RAGE)161,163, which would be expected to be stimulated by the AGE-production noted above. This was recently highlighted by Daffu et al. who reported that incubation of murine macrophages or human THP-1 cells with the model glycated protein CML-AGE reduced Abca1 and Abcg1 mRNA and protein expression via its interaction with RAGE. Reductions in the expression levels of these receptors resulted in decreased cholesterol efflux to apoA-I and HDL163. Further, consistent with other studies165–169, it was found that diabetes enhanced both atherosclerosis progression and impaired regression, and that global deletion of RAGE overcame these defects by restoration of ABCA1 and ABCG1, promoting macrophage CEC despite ongoing hyperglycemia163,170.
Restoration of global and myeloid ABCA1/ABCG1 expression and improvements to CVD outcomes under diabetic conditions is likely to be multifaceted. In addition to being essential for the removal of cholesterol from plaque macrophages28, ABCA1 and ABCG1 regulate the proliferation of hematopoietic stem and progenitor cells to control the abundance of blood monocytes27. Given the link between myelopoiesis and CVD risk130,171, suppression of this process is likely to directly inhibit the progression of atherosclerotic lesions, and promote lesion regression166. Diabetes can suppress hematopoietic precursor cell ABCA1 and ABCG1 levels, promoting myelopoiesis and atherosclerosis165. Furthermore, inhibition of miR-33, a negative regulator of cellular ABCA1 and ABCG1, suppresses leukocytosis and reduces plaque macrophage inflammation in diabetic mice165. Despite persisant hyperglycemia, suppression of miR-33 not only restored essential cholesterol transporters and reduced myelopoiesis, but it also promoted inflammation resolution in established plaques. Additionally, unpublished work from the Fisher lab has found that raising apoA-I/HDL levels in diabetic mice, in the absence of glucose control, can restore atherosclerosis regression, in part, by overcoming defective CEC in hematopoietic stem cells (Barrett et al, In Revision). These complementary studies highlight the importance of effective CEC at both the level of the bone marrow and plaque under diabetic settings to reduce CVD morbidity and mortality risk.
As with non-diabetic populations, the relationship of HDL-C to effective RCT in diabetic patients with CVD risk remains to be conclusively determined. However, given that macrophage CEC to plasma from diabetic subjects is overwhelmingly reported to be reduced compared to healthy controls139,172–175, and the inverse relationship between glucose tolerance and plasma CEC115,176, it is tempting to speculate that either restoring or enhancing in vivo RCT capacity within this population would reduce the incidence of CVD-linked disorders.
Functional properties of HDL in cholesterol efflux, RCT, and beyond
Factors to consider about the functionality of HDL include its pleiotropic actions besides cholesterol efflux. The bases for these actions likely involve the heterogeneity of HDL particles. For example, independent of its ability to mediate RCT by serving as a cholesterol acceptor, HDL is also known to exert potent antioxidant and anti-inflammatory effects that can improve RCT, retard plaque progression, and promote plaque regression75,177. Indeed, overexpression of an HDL-associated protein that confers antioxidant properties to HDL, paraoxonase 1, improves the efflux capacity of HDL and drives RCT in mice178. HDL exists as subpopulations, classified based on their physicochemical properties: density (HDL2, HDL3), shape (discoidal, spherical), protein (apoA-I, A-II or both), surface charge, and size139. The bulk of RCT is linked to apoA-I, which cycles between lipid-poor (pre-β-HDL) and –rich (α-HDL) forms of HDL, a remodeling event that, as noted earlier, can occur in the interstitial fluid of tissues to generate pre-β-HDL. This process is essential to RCT given that just 5% of plasma apoA-I exists as pre-β-HDL, the principal acceptors of cholesterol from peripheral cells138,179,180. Proteomic analyses reveal that the composition of HDL is more complex than anticipated, containing ~200 diverse proteins distributed among various HDL subclasses. In addition to protein and lipid cargo, HDL can transport functional non-coding RNAs, such as miRNAs, and this pool of lipoprotein-associated RNA can be altered in disease181,182. It is now appreciated that how specific HDL functions (in CEC/RCT, thrombosis, inflammation, etc.) are related to HDL compositional heterogeneity in humans and how HDL subspecies may be altered during CAD could lead to the identification of new diagnostic tools and therapies139,183–185.
Concluding Remarks
The quest for HDL raising therapies has been long-standing in the fields of lipoprotein metabolism and CVD, as reflected in the past by physicians routinely prescribing drugs to boost HDL-C in patients. These therapies are now thought to be ineffective in reducing CVD risk186. In addition, several clinical studies failed to show that raising HDL-C levels (e.g., by niacin187,188 or CETP inhibition189) improves CVD outcomes, and Mendelian randomization studies also find that HDL-C levels are not predictive of CVD events183. These and other studies highlight that while we have observed numerous successes in the development of multiple LDL-cholesterol lowering therapies that have translated into beneficial clinical outcomes, comparable advances in RCT-enhancing strategies through raising HDL-C are lacking. An example of the need for such enhancement independent of HDL-C may be found in the data from the CETP inhibitor trials. In particular, it may be more than a coincidence that the failure of torcetrapib to lower CVD events despite raising HDL-C by ~72%190 was also associated with its failure to promote whole body RCT in a fecal sterol excretion assay191.
It should be noted that in none of the studies mentioned above and in many similar ones has HDL function been ascertained, leaving open the possibility that HDL function is the key attribute for CVD risk reduction113. HDL function as a clinically important factor has found traction not only in the aforementioned CEC studies, but also finds some support from some, but not all (e.g.192) infusion studies of recombinant HDL and HDL-like particles. Notably, however, all of the studies to date are of limited significance, as they have been either too short to assess effects on CVD outcomes, very small in subject number, or both. For example, in one small study, the intravenous infusion of a single dose of reconstituted HDL led to acute changes in plaques in the superficial femoral artery, with a reduction in lipid content, macrophage size, and measures of inflammation, but there were only 20 subjects193. In a larger study of subjects post acute coronary syndromes (ACS; 47 subjects completed the protocol), 5 weekly injections of a recombinant HDL-like particle (designated ETC-216) containing ApoA-Imilano194 resulted in a 4.2% decrease from baseline in coronary atheroma volume as measured by intravascular ultrasound.
A similar study was conducted with a formulation of wild-type ApoA-I (designated CSL111)195. The results were similar to the ETC-216 trial, in that plaque volume was decreased, but to a lesser extent (3.4%), perhaps because of a shorter course of treatment (4 weeks) or other differences between the studies. Again, there are no CVD outcome data in either trial. There is great interest, therefore, in the AEGIS II trial, in which apoA-I in a proprietary formulation of lipids to simulate HDL particles (CSL112), is being administered to subjects with acute coronary syndrome (ACS). With an estimated enrollment of 17400, participants will be randomized to receive either CSL112 or a placebo, administered through IV infusion once a week for four consecutive weeks. The primary endpoint is the first occurrence of a major adverse cardiovascular event (MACE), CV death, MI or stroke within 90 days, and the expected completion date is 2022187.
In spite of the controversies, on-balance we believe that raising levels of functional HDL in those at risk for CVD events may yet represent a viable therapy to suppress atherosclerosis progression and promote atherosclerosis regression. This belief is based on the established biological effects of functional HDL that we have summarized, as well as the encouragement from the clinical studies (114,193–195), although at present they fall short as definitive trials, especially with regard to the relationship between raising levels of functional HDL and MACE. This raises the parallel need for more trials of the type that AEGIS II represents, as well as mechanistic studies to further understand the factors that regulate HDL’s impact on CVD independent of the plasma concentration of HDL-C.
Acknowledgments
Sources of Funding: This work was supported by funding from the Canadian Institutes for Health Research (PJT-391187 and Canada Research Chair to MO), the Heart and Stroke Foundation of Canada (MO), the American Heart Association (18CDA34110203AHA; TJB), the National Institutes of Health (DK095684, HL084312, HL129433, HL092969, HL122728, HL117226, and HL131481; EAF), and the Department of Defense (W81XWH-15–1-0374, W81XWH-16–1-0255; EAF).
Footnotes
Disclosures: None
References
- 1.Wilson PW et al. Prevalence of coronary heart disease in the Framingham Offspring Study: role of lipoprotein cholesterols. Am J Cardiol 46, 649–654 (1980). [DOI] [PubMed] [Google Scholar]
- 2.Glomset JA, Janssen ET, Kennedy R & Dobbins J Role of plasma lecithin:cholesterol acyltransferase in the metabolism of high density lipoproteins. J Lipid Res 7, 638–648 (1966). [PubMed] [Google Scholar]
- 3.Gordon DJ et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79, 8–15 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Hutchins PM et al. Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin Chem 60, 1393–1401, doi: 10.1373/clinchem.2014.228114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de la Llera-Moya M et al. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol 30, 796–801, doi: 10.1161/ATVBAHA.109.199158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voight BF et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380, 572–580, doi: 10.1016/S0140-6736(12)60312-2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pittman RC & Steinberg D Sites and mechanisms of uptake and degradation of high density and low density lipoproteins. J Lipid Res 25, 1577–1585 (1984). [PubMed] [Google Scholar]
- 8.Hong C & Tontonoz P Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 13, 433–444, doi: 10.1038/nrd4280 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Reue K, Fong LG, Young SG & Tontonoz P Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol 32, 2541–2546, doi: 10.1161/ATVBAHA.112.250571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquart TJ, Allen RM, Ory DS & Baldan A miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A 107, 12228–12232, doi: 10.1073/pnas.1005191107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Najafi-Shoushtari SH et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328, 1566–1569, doi: 10.1126/science.1189123 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rayner KJ et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573, doi: 10.1126/science.1189862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rayner KJ et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407, doi: 10.1038/nature10486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rayner KJ et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 121, 2921–2931, doi: 10.1172/JCI57275 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horie T et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. J Am Heart Assoc 1, e003376, doi: 10.1161/JAHA.112.003376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouimet M et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest 125, 4334–4348, doi: 10.1172/JCI81676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rotllan N, Ramirez CM, Aryal B, Esau CC & Fernandez-Hernando C Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice--brief report. Arterioscler Thromb Vasc Biol 33, 1973–1977, doi: 10.1161/ATVBAHA.113.301732 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duell PB, Oram JF & Bierman EL Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes 40, 377–384 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Goedeke L et al. Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol Med 6, 1133–1141, doi: 10.15252/emmm.201404046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horie T et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun 4, 2883, doi: 10.1038/ncomms3883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karunakaran D et al. Therapeutic Inhibition of miR-33 Promotes Fatty Acid Oxidation but Does Not Ameliorate Metabolic Dysfunction in Diet-Induced Obesity. Arterioscler Thromb Vasc Biol 35, 2536–2543, doi: 10.1161/ATVBAHA.115.306404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price NL et al. Genetic Dissection of the Impact of miR-33a and miR-33b during the Progression of Atherosclerosis. Cell Rep 21, 1317–1330, doi: 10.1016/j.celrep.2017.10.023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price NL et al. Specific Disruption of Abca1 Targeting Largely Mimics the Effects of miR-33 Knockout on Macrophage Cholesterol Efflux and Atherosclerotic Plaque Development. Circ Res 124, 874–880, doi: 10.1161/CIRCRESAHA.118.314415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feinberg MW & Moore KJ MicroRNA Regulation of Atherosclerosis. Circ Res 118, 703–720, doi: 10.1161/CIRCRESAHA.115.306300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuchel M & Rader DJ Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation 113, 2548–2555, doi: 10.1161/circulationaha.104.475715 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Westerterp M et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res 112, 1456–1465, doi: 10.1161/CIRCRESAHA.113.301086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yvan-Charvet L et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693, doi: 10.1126/science.1189731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yvan-Charvet L et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest 117, 3900–3908, doi: 10.1172/JCI33372 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allahverdian S, Chaabane C, Boukais K, Francis GA & Bochaton-Piallat ML Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res 114, 540–550, doi: 10.1093/cvr/cvy022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez D & Owens GK Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95, 156–164, doi: 10.1093/cvr/cvs115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vengrenyuk Y et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 35, 535–546, doi: 10.1161/ATVBAHA.114.304029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shankman LS et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637, doi: 10.1038/nm.3866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y et al. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol, ATVBAHA119312434, doi: 10.1161/ATVBAHA.119.312434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldberg IJ et al. Deciphering the Role of Lipid Droplets in Cardiovascular Disease: A Report From the 2017 National Heart, Lung, and Blood Institute Workshop. Circulation 138, 305–315, doi: 10.1161/CIRCULATIONAHA.118.033704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh S, Zhao B, Bie J & Song J Macrophage cholesteryl ester mobilization and atherosclerosis. Vascul Pharmacol 52, 1–10, doi: 10.1016/j.vph.2009.10.002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouimet M & Marcel YL Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol 32, 575–581, doi: 10.1161/ATVBAHA.111.240705 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Igarashi M et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res 107, 1387–1395, doi: 10.1161/CIRCRESAHA.110.226613 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Son SH et al. Enhanced atheroprotection and lesion remodelling by targeting the foam cell and increasing plasma cholesterol acceptors. Cardiovasc Res 109, 294–304, doi: 10.1093/cvr/cvv241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao B et al. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest 117, 2983–2992, doi: 10.1172/JCI30485 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh S Early steps in reverse cholesterol transport: cholesteryl ester hydrolase and other hydrolases. Curr Opin Endocrinol Diabetes Obes 19, 136–141, doi: 10.1097/MED.0b013e3283507836 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Sekiya M et al. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab 10, 219–228, doi: 10.1016/j.cmet.2009.08.004 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Brown MS, Goldstein JL, Krieger M, Ho YK & Anderson RG Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J Cell Biol 82, 597–613 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGookey DJ & Anderson RG Morphological characterization of the cholesteryl ester cycle in cultured mouse macrophage foam cells. J Cell Biol 97, 1156–1168 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown MS, Ho YK & Goldstein JL The cholesteryl ester cycle in macrophage foam cells. Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J.Biol.Chem. 255, 9344–9352 (1980). [PubMed] [Google Scholar]
- 45.Ouimet M et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13, 655–667, doi: 10.1016/j.cmet.2011.03.023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh R & Cuervo AM Autophagy in the cellular energetic balance. Cell Metab 13, 495–504, doi: 10.1016/j.cmet.2011.04.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamasaki M & Yoshimori T Where do they come from? Insights into autophagosome formation. FEBS Lett 584, 1296–1301, doi: 10.1016/j.febslet.2010.02.061 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Zhang H & Reilly MP LIPA Variants in Genome-Wide Association Studies of Coronary Artery Diseases: Loss-of-Function or Gain-of-Function? Arterioscler Thromb Vasc Biol 37, 1015–1017, doi: 10.1161/ATVBAHA.117.309344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razani B et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15, 534–544, doi: 10.1016/j.cmet.2012.02.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ouimet M et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler Thromb Vasc Biol 37, 1058–1067, doi: 10.1161/ATVBAHA.116.308916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao X et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15, 545–553, doi: 10.1016/j.cmet.2012.01.022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sergin I et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun 8, 15750, doi: 10.1038/ncomms15750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iaea DB & Maxfield FR Cholesterol trafficking and distribution. Essays Biochem 57, 43–55, doi: 10.1042/bse0570043 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Holtta-Vuori M & Ikonen E Endosomal cholesterol traffic: vesicular and non-vesicular mechanisms meet. Biochem Soc Trans 34, 392–394, doi: 10.1042/BST0340392 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Chen W, Wang N & Tall AR A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem 280, 29277–29281, doi: 10.1074/jbc.M505566200 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Parathath S et al. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res 109, 1141–1152, doi: 10.1161/CIRCRESAHA.111.246363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parathath S, Yang Y, Mick S & Fisher EA Hypoxia in murine atherosclerotic plaques and its adverse effects on macrophages. Trends Cardiovasc Med 23, 80–84, doi: 10.1016/j.tcm.2012.09.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang N, Ranalletta M, Matsuura F, Peng F & Tall AR LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler Thromb Vasc Biol 26, 1310–1316, doi: 10.1161/01.ATV.0000218998.75963.02 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Tarling EJ & Edwards PA ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci U S A 108, 19719–19724, doi: 10.1073/pnas.1113021108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Azuma Y et al. Retroendocytosis pathway of ABCA1/apoA-I contributes to HDL formation. Genes Cells 14, 191–204, doi: 10.1111/j.1365-2443.2008.01261.x (2009). [DOI] [PubMed] [Google Scholar]
- 61.Kentala H, Weber-Boyvat M & Olkkonen VM OSBP-Related Protein Family: Mediators of Lipid Transport and Signaling at Membrane Contact Sites. Int Rev Cell Mol Biol 321, 299–340, doi: 10.1016/bs.ircmb.2015.09.006 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Ouimet M et al. miRNA Targeting of Oxysterol-Binding Protein-Like 6 Regulates Cholesterol Trafficking and Efflux. Arterioscler Thromb Vasc Biol 36, 942–951, doi: 10.1161/ATVBAHA.116.307282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mesmin B & Maxfield FR Intracellular sterol dynamics. Biochim Biophys Acta 1791, 636–645, doi: 10.1016/j.bbalip.2009.03.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandhu J et al. Aster Proteins Facilitate Nonvesicular Plasma Membrane to ER Cholesterol Transport in Mammalian Cells. Cell 175, 514–529 e520, doi: 10.1016/j.cell.2018.08.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolfbauer G, Glick JM, Minor LK & Rothblat GH Development of the smooth muscle foam cell: uptake of macrophage lipid inclusions. Proc Natl Acad Sci U S A 83, 7760–7764 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frontini MJ et al. Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res 104, 832–841, doi: 10.1161/CIRCRESAHA.108.187302 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Rong JX, Shapiro M, Trogan E & Fisher EA Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 100, 13531–13536, doi: 10.1073/pnas.1735526100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feil S et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 115, 662–667, doi: 10.1161/CIRCRESAHA.115.304634 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Allahverdian S, Chehroudi AC, McManus BM, Abraham T & Francis GA Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559, doi: 10.1161/CIRCULATIONAHA.113.005015 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Chappell J et al. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ Res 119, 1313–1323, doi: 10.1161/CIRCRESAHA.116.309799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Misra A et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun 9, 2073, doi: 10.1038/s41467-018-04447-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao X et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest 121, 2736–2749, doi: 10.1172/JCI45444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi HY et al. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation 119, 3223–3231, doi: 10.1161/CIRCULATIONAHA.108.841130 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Choi HY, Hafiane A, Schwertani A & Genest J High-Density Lipoproteins: Biology, Epidemiology, and Clinical Management. Can J Cardiol 33, 325–333, doi: 10.1016/j.cjca.2016.09.012 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Fisher EA, Feig JE, Hewing B, Hazen SL & Smith JD High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 32, 2813–2820, doi: 10.1161/ATVBAHA.112.300133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rye KA & Barter PJ Cardioprotective functions of HDLs. J Lipid Res 55, 168–179, doi: 10.1194/jlr.R039297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qian H et al. Structure of the Human Lipid Exporter ABCA1. Cell 169, 1228–1239 e1210, doi: 10.1016/j.cell.2017.05.020 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Assmann G & Gotto AM Jr. HDL cholesterol and protective factors in atherosclerosis. Circulation 109, III8–14, doi: 10.1161/01.cir.0000131512.50667.46 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Tang C & Oram JF The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta 1791, 563–572, doi: 10.1016/j.bbalip.2009.03.011 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Wang N, Lan D, Chen W, Matsuura F & Tall AR ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A 101, 9774–9779, doi: 10.1073/pnas.0403506101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neufeld EB et al. Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem 276, 27584–27590, doi: 10.1074/jbc.M103264200 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Chen W et al. Preferential ATP-binding cassette transporter A1-mediated cholesterol efflux from late endosomes/lysosomes. J Biol Chem 276, 43564–43569, doi: 10.1074/jbc.M107938200 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Phillips MC, Johnson WJ & Rothblat GH Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta 906, 223–276 (1987). [DOI] [PubMed] [Google Scholar]
- 84.Rothblat GH & Phillips MC High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol 21, 229–238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kennedy MA et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 1, 121–131, doi: 10.1016/j.cmet.2005.01.002 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Vaughan AM & Oram JF ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem 280, 30150–30157, doi: 10.1074/jbc.M505368200 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Annema W & Tietge UJ Role of hepatic lipase and endothelial lipase in high-density lipoprotein-mediated reverse cholesterol transport. Curr Atheroscler Rep 13, 257–265, doi: 10.1007/s11883-011-0175-2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khera AV & Rader DJ Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep 12, 73–81, doi: 10.1007/s11883-009-0080-0 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Temel RE & Brown JM A new framework for reverse cholesterol transport: non-biliary contributions to reverse cholesterol transport. World J Gastroenterol 16, 5946–5952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brufau G, Groen AK & Kuipers F Reverse cholesterol transport revisited: contribution of biliary versus intestinal cholesterol excretion. Arterioscler Thromb Vasc Biol 31, 1726–1733, doi: 10.1161/ATVBAHA.108.181206 (2011). [DOI] [PubMed] [Google Scholar]
- 91.Mendelsohn AR & Larrick JW Preclinical Reversal of Atherosclerosis by FDA-Approved Compound that Transforms Cholesterol into an Anti-Inflammatory “Prodrug”. Rejuvenation Res 19, 252–255, doi: 10.1089/rej.2016.1849 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Zimmer S et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med 8, 333ra350, doi: 10.1126/scitranslmed.aad6100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pownall HJ & Ehnholm C Enhancing reverse cholesterol transport: the case for phosphatidylcholine therapy. Curr Opin Lipidol 16, 265–268 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Rodrigueza WV et al. Large versus small unilamellar vesicles mediate reverse cholesterol transport in vivo into two distinct hepatic metabolic pools. Implications for the treatment of atherosclerosis. Arterioscler Thromb Vasc Biol 17, 2132–2139 (1997). [DOI] [PubMed] [Google Scholar]
- 95.Stein O, Oette K, Haratz D, Halperin G & Stein Y Sphingomyelin liposomes with defined fatty acids: metabolism and effects on reverse cholesterol transport. Biochim Biophys Acta 960, 322–333 (1988). [DOI] [PubMed] [Google Scholar]
- 96.Hung KT, Berisha SZ, Ritchey BM, Santore J & Smith JD Red blood cells play a role in reverse cholesterol transport. Arterioscler Thromb Vasc Biol 32, 1460–1465, doi: 10.1161/ATVBAHA.112.248971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hafiane A & Genest J ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis 257, 90–99, doi: 10.1016/j.atherosclerosis.2017.01.013 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Sanchez-Gaytan BL et al. HDL-mimetic PLGA nanoparticle to target atherosclerosis plaque macrophages. Bioconjug Chem 26, 443–451, doi: 10.1021/bc500517k (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tang J et al. Immune cell screening of a nanoparticle library improves atherosclerosis therapy. Proc Natl Acad Sci U S A 113, E6731–E6740, doi: 10.1073/pnas.1609629113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duivenvoorden R et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun 5, 3065, doi: 10.1038/ncomms4065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hafiane A & Genest J HDL, Atherosclerosis, and Emerging Therapies. Cholesterol 2013, 891403, doi: 10.1155/2013/891403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zanoni P et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171, doi: 10.1126/science.aad3517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoekstra M SR-BI as target in atherosclerosis and cardiovascular disease - A comprehensive appraisal of the cellular functions of SR-BI in physiology and disease. Atherosclerosis 258, 153–161, doi: 10.1016/j.atherosclerosis.2017.01.034 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Helgadottir A et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur Heart J 39, 2172–2178, doi: 10.1093/eurheartj/ehy169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dikkers A & Tietge UJ Biliary cholesterol secretion: more than a simple ABC. World J Gastroenterol 16, 5936–5945 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paalvast Y, de Boer JF & Groen AK Developments in intestinal cholesterol transport and triglyceride absorption. Curr Opin Lipidol 28, 248–254, doi: 10.1097/MOL.0000000000000415 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Xu Y et al. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology 64, 1072–1085, doi: 10.1002/hep.28712 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Russell DW The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72, 137–174, doi: 10.1146/annurev.biochem.72.121801.161712 (2003). [DOI] [PubMed] [Google Scholar]
- 109.Pelton PD, Patel M & Demarest KT Nuclear receptors as potential targets for modulating reverse cholesterol transport. Curr Top Med Chem 5, 265–282 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Jakobsson T, Treuter E, Gustafsson JA & Steffensen KR Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci 33, 394–404, doi: 10.1016/j.tips.2012.03.013 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Ouimet M & Moore KJ A big role for small RNAs in HDL homeostasis. J Lipid Res 54, 1161–1167, doi: 10.1194/jlr.R036327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rader DJ & Tall AR The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med 18, 1344–1346, doi: 10.1038/nm.2937 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Hewing B, Moore KJ & Fisher EA HDL and cardiovascular risk: time to call the plumber? Circ Res 111, 1117–1120, doi: 10.1161/CIRCRESAHA.112.280958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rohatgi A et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med 371, 2383–2393, doi: 10.1056/NEJMoa1409065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saleheen D et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol 3, 507–513, doi: 10.1016/S2213-8587(15)00126-6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shea S et al. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler Thromb Vasc Biol 39, 89–96, doi: 10.1161/ATVBAHA.118.311366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rothblat GH, de la Llera-Moya M, Favari E, Yancey PG & Kellner-Weibel G Cellular cholesterol flux studies: methodological considerations. Atherosclerosis 163, 1–8 (2002). [DOI] [PubMed] [Google Scholar]
- 118.Sankaranarayanan S et al. A sensitive assay for ABCA1-mediated cholesterol efflux using BODIPY-cholesterol. J Lipid Res 52, 2332–2340, doi: 10.1194/jlr.D018051 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Waddington EI, Boadu E & Francis GA Cholesterol and phospholipid efflux from cultured cells. Methods 36, 196–206, doi: 10.1016/j.ymeth.2004.12.002 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Robichaud S & Ouimet M Quantifying Cellular Cholesterol Efflux. Methods Mol Biol 1951, 111–133, doi: 10.1007/978-1-4939-9130-3_9 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Khera AV et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127–135, doi: 10.1056/NEJMoa1001689 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Khera AV et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 135, 2494–2504, doi: 10.1161/CIRCULATIONAHA.116.025678 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li XM et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 33, 1696–1705, doi: 10.1161/ATVBAHA.113.301373 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y et al. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 108, 661–663, doi: 10.1161/01.CIR.0000086981.09834.E0 (2003). [DOI] [PubMed] [Google Scholar]
- 125.Annema W & Tietge UJ Regulation of reverse cholesterol transport - a comprehensive appraisal of available animal studies. Nutr Metab (Lond) 9, 25, doi: 10.1186/1743-7075-9-25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weibel GL et al. Novel in vivo method for measuring cholesterol mass flux in peripheral macrophages. Arterioscler Thromb Vasc Biol 31, 2865–2871, doi: 10.1161/ATVBAHA.111.236406 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cuchel M et al. A novel approach to measuring macrophage-specific reverse cholesterol transport in vivo in humans. J Lipid Res 58, 752–762, doi: 10.1194/jlr.M075226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chhatriwalla AK & Rader DJ Intracoronary Imaging, Reverse Cholesterol Transport, and Transcriptomics: Precision Medicine in CAD? J Am Coll Cardiol 69, 641–643, doi: 10.1016/j.jacc.2016.12.003 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Tabas I, Williams KJ & Boren J Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116, 1832–1844, doi: 10.1161/CIRCULATIONAHA.106.676890 (2007). [DOI] [PubMed] [Google Scholar]
- 130.Moore KJ, Sheedy FJ & Fisher EA Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13, 709–721, doi: 10.1038/nri3520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Trogan E et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A 103, 3781–3786, doi: 10.1073/pnas.0511043103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Feig JE et al. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One 6, e28534, doi: 10.1371/journal.pone.0028534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Potteaux S et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest 121, 2025–2036, doi: 10.1172/JCI43802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mueller PA et al. Deletion of Macrophage Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Accelerates Atherosclerosis Regression and Increases C-C Chemokine Receptor Type 7 (CCR7) Expression in Plaque Macrophages. Circulation 138, 1850–1863, doi: 10.1161/CIRCULATIONAHA.117.031702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Milasan A, Ledoux J & Martel C Lymphatic network in atherosclerosis: the underestimated path. Future Sci OA 1, FSO61, doi: 10.4155/fso.15.61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martel C et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest 123, 1571–1579, doi: 10.1172/JCI63685 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vuorio T et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol 34, 1162–1170, doi: 10.1161/ATVBAHA.114.302528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miller NE et al. Lipoprotein remodeling generates lipid-poor apolipoprotein A-I particles in human interstitial fluid. Am J Physiol Endocrinol Metab 304, E321–328, doi: 10.1152/ajpendo.00324.2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kontush A et al. Structure of HDL: particle subclasses and molecular components. Handb Exp Pharmacol 224, 3–51, doi: 10.1007/978-3-319-09665-0_1 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Wang S & Smith JD ABCA1 and nascent HDL biogenesis. Biofactors 40, 547–554, doi: 10.1002/biof.1187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yin C, Mohanta SK, Srikakulapu P, Weber C & Habenicht AJ Artery Tertiary Lymphoid Organs: Powerhouses of Atherosclerosis Immunity. Front Immunol 7, 387, doi: 10.3389/fimmu.2016.00387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haffner SM, Lehto S, Ronnemaa T, Pyorala K & Laakso M Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 339, 229–234, doi: 10.1056/NEJM199807233390404 (1998). [DOI] [PubMed] [Google Scholar]
- 143.Okada K et al. Association between blood glucose variability and coronary plaque instability in patients with acute coronary syndromes. Cardiovascular diabetology 14, 111, doi: 10.1186/s12933-015-0275-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Taskinen MR Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46, 733–749, doi: 10.1007/s00125-003-1111-y (2003). [DOI] [PubMed] [Google Scholar]
- 145.Garg A Dyslipoproteinemia and diabetes. Endocrinol Metab Clin North Am 27, 613–625, ix-x (1998). [DOI] [PubMed] [Google Scholar]
- 146.Wilson PW et al. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med 167, 1068–1074, doi: 10.1001/archinte.167.10.1068 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Hwang YC, Ahn HY, Park SW & Park CY Association of HDL-C and apolipoprotein A-I with the risk of type 2 diabetes in subjects with impaired fasting glucose. Eur J Endocrinol 171, 137–142, doi: 10.1530/EJE-14-0195 (2014). [DOI] [PubMed] [Google Scholar]
- 148.Lachin JM, Orchard TJ, Nathan DM & Group, D. E. R. Update on cardiovascular outcomes at 30 years of the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 37, 39–43, doi: 10.2337/dc13-2116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Asleh R & Levy AP Divergent effects of alpha-tocopherol and vitamin C on the generation of dysfunctional HDL associated with diabetes and the Hp 2–2 genotype. Antioxidants & redox signaling 12, 209–217, doi: 10.1089/ars.2009.2829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Patel DC et al. Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS One 6, e22142, doi: 10.1371/journal.pone.0022142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Curtiss LK & Witztum JL Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 34, 452–461 (1985). [DOI] [PubMed] [Google Scholar]
- 152.Hedrick CC et al. Glycation impairs high-density lipoprotein function. Diabetologia 43, 312–320, doi: 10.1007/s001250050049 (2000). [DOI] [PubMed] [Google Scholar]
- 153.Ferretti G, Bacchetti T, Marchionni C, Caldarelli L & Curatola G Effect of glycation of high density lipoproteins on their physicochemical properties and on paraoxonase activity. Acta Diabetol 38, 163–169 (2001). [DOI] [PubMed] [Google Scholar]
- 154.Low H et al. Advanced glycation end-products (AGEs) and functionality of reverse cholesterol transport in patients with type 2 diabetes and in mouse models. Diabetologia 55, 2513–2521, doi: 10.1007/s00125-012-2570-9 (2012). [DOI] [PubMed] [Google Scholar]
- 155.Nobecourt E et al. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol 30, 766–772, doi: 10.1161/ATVBAHA.109.201715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hoang A et al. Advanced glycation of apolipoprotein A-I impairs its anti-atherogenic properties. Diabetologia 50, 1770–1779, doi: 10.1007/s00125-007-0718-9 (2007). [DOI] [PubMed] [Google Scholar]
- 157.Passarelli M et al. Diminished rate of mouse peritoneal macrophage cholesterol efflux is not related to the degree of HDL glycation in diabetes mellitus. Clin Chim Acta 301, 119–134 (2000). [DOI] [PubMed] [Google Scholar]
- 158.Ohgami N et al. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. Journal of Biological Chemistry 276, 13348–13355, doi:DOI 10.1074/jbc.M011613200 (2001). [DOI] [PubMed] [Google Scholar]
- 159.Isoda K, Folco EJ, Shimizu K & Libby P AGE-BSA decreases ABCG1 expression and reduces macrophage cholesterol efflux to HDL. Atherosclerosis 192, 298–304, doi: 10.1016/j.atherosclerosis.2006.07.023 (2007). [DOI] [PubMed] [Google Scholar]
- 160.Hussein MA et al. LXR-Mediated ABCA1 Expression and Function Are Modulated by High Glucose and PRMT2. PLoS One 10, e0135218, doi: 10.1371/journal.pone.0135218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mauldin JP et al. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation 117, 2785–2792, doi: 10.1161/CIRCULATIONAHA.107.741314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Passarelli M et al. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 54, 2198–2205 (2005). [DOI] [PubMed] [Google Scholar]
- 163.Daffu G et al. RAGE Suppresses ABCG1-Mediated Macrophage Cholesterol Efflux in Diabetes. Diabetes 64, 4046–4060, doi: 10.2337/db15-0575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Machado-Lima A et al. Advanced glycated albumin isolated from poorly controlled type 1 diabetes mellitus patients alters macrophage gene expression impairing ABCA-1-mediated reverse cholesterol transport. Diabetes Metab Res Rev 29, 66–76, doi: 10.1002/dmrr.2362 (2013). [DOI] [PubMed] [Google Scholar]
- 165.Distel E et al. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res 115, 759–769, doi: 10.1161/CIRCRESAHA.115.304164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nagareddy PR et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 17, 695–708, doi: 10.1016/j.cmet.2013.04.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Parathath S et al. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes 60, 1759–1769, doi: 10.2337/db10-0778 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kanter JE et al. A Novel Strategy to Prevent Advanced Atherosclerosis and Lower Blood Glucose in a Mouse Model of Metabolic Syndrome. Diabetes 67, 946–959, doi: 10.2337/db17-0744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Willecke F et al. Effects of High Fat Feeding and Diabetes on Regression of Atherosclerosis Induced by Low-Density Lipoprotein Receptor Gene Therapy in LDL Receptor-Deficient Mice. PLoS One 10, e0128996, doi: 10.1371/journal.pone.0128996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Senatus LM et al. Role of Receptor for Advanced Glycation End Products (RAGE) in Regression of Diabetic Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 37:A48 (2017). [Google Scholar]
- 171.Barrett TJ, Murphy AJ, Goldberg IJ & Fisher EA Diabetes-mediated myelopoiesis and the relationship to cardiovascular risk. Ann N Y Acad Sci 1402, 31–42, doi: 10.1111/nyas.13462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Syvanne M et al. Cholesterol efflux from Fu5AH hepatoma cells induced by plasma of subjects with or without coronary artery disease and non-insulin-dependent diabetes: importance of LpA-I:A-II particles and phospholipid transfer protein. Atherosclerosis 127, 245–253 (1996). [DOI] [PubMed] [Google Scholar]
- 173.Autran D, Attia N, Dedecjus M, Durlach V & Girard-Globa A Postprandial reverse cholesterol transport in type 2 diabetic patients: effect of a lipid lowering treatment. Atherosclerosis 153, 453–460 (2000). [DOI] [PubMed] [Google Scholar]
- 174.Attia N et al. Increased phospholipid transfer protein activity associated with the impaired cellular cholesterol efflux in type 2 diabetic subjects with coronary artery disease. Tohoku J Exp Med 213, 129–137 (2007). [DOI] [PubMed] [Google Scholar]
- 175.Zhou H, Shiu SW, Wong Y & Tan KC Impaired serum capacity to induce cholesterol efflux is associated with endothelial dysfunction in type 2 diabetes mellitus. Diab Vasc Dis Res 6, 238–243, doi: 10.1177/1479164109344934 (2009). [DOI] [PubMed] [Google Scholar]
- 176.Annema W et al. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: the CODAM study. Sci Rep 6, 27367, doi: 10.1038/srep27367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Feig JE, Hewing B, Smith JD, Hazen SL & Fisher EA High-density lipoprotein and atherosclerosis regression: evidence from preclinical and clinical studies. Circ Res 114, 205–213, doi: 10.1161/CIRCRESAHA.114.300760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ikhlef S, Berrougui H, Kamtchueng Simo O, Zerif E & Khalil A Human paraoxonase 1 overexpression in mice stimulates HDL cholesterol efflux and reverse cholesterol transport. PLoS One 12, e0173385, doi: 10.1371/journal.pone.0173385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Liang HQ, Rye KA & Barter PJ Cycling of apolipoprotein A-I between lipid-associated and lipid-free pools. Biochim Biophys Acta 1257, 31–37 (1995). [DOI] [PubMed] [Google Scholar]
- 180.Wroblewska M, Kortas-Stempak B, Szutowicz A & Badzio T Phospholipids mediated conversion of HDLs generates specific apoA-II pre-beta mobility particles. J Lipid Res 50, 667–675, doi: 10.1194/jlr.M800399-JLR200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Michell DL et al. Isolation of High-density Lipoproteins for Non-coding Small RNA Quantification. J Vis Exp, doi: 10.3791/54488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD & Remaley AT MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13, 423–433, doi: 10.1038/ncb2210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Heinecke J HDL and cardiovascular-disease risk--time for a new approach? N Engl J Med 364, 170–171, doi: 10.1056/NEJMe1012520 (2011). [DOI] [PubMed] [Google Scholar]
- 184.Heinecke JW The HDL proteome: a marker--and perhaps mediator--of coronary artery disease. J Lipid Res 50 Suppl, S167–171, doi: 10.1194/jlr.R800097-JLR200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shah AS, Tan L, Long JL & Davidson WS Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res 54, 2575–2585, doi: 10.1194/jlr.R035725 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Keene D, Price C, Shun-Shin MJ & Francis DP Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ 349, g4379, doi: 10.1136/bmj.g4379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Behring C Study to Investigate CSL112 in Subjects With Acute Coronary Syndrome (AEGIS-II). https://clinicaltrials.gov/ct2/show/NCT03473223.
- 188.Investigators, A.-H. et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 365, 2255–2267, doi: 10.1056/NEJMoa1107579 (2011). [DOI] [PubMed] [Google Scholar]
- 189.Armitage J, Holmes MV & Preiss D Cholesteryl Ester Transfer Protein Inhibition for Preventing Cardiovascular Events: JACC Review Topic of the Week. J Am Coll Cardiol 73, 477–487, doi: 10.1016/j.jacc.2018.10.072 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Barter PJ et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357, 2109–2122, doi: 10.1056/NEJMoa0706628 (2007). [DOI] [PubMed] [Google Scholar]
- 191.Brousseau ME et al. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol 25, 1057–1064, doi: 10.1161/01.ATV.0000161928.16334.dd (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nicholls SJ et al. Effect of Serial Infusions of CER-001, a Pre-beta High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: A Randomized Clinical Trial. JAMA Cardiol 3, 815–822, doi: 10.1001/jamacardio.2018.2121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shaw JA et al. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res 103, 1084–1091, doi: 10.1161/CIRCRESAHA.108.182063 (2008). [DOI] [PubMed] [Google Scholar]
- 194.Nissen SE et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290, 2292–2300, doi: 10.1001/jama.290.17.2292 (2003). [DOI] [PubMed] [Google Scholar]
- 195.Tardif JC et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297, 1675–1682, doi: 10.1001/jama.297.15.jpc70004 (2007). [DOI] [PubMed] [Google Scholar]