

Abstract
The A51V variant of human cytochrome c is linked to thrombocytopenia 4 (THC4), a condition that causes decreased blood platelet counts. A 1.82 Å structure of the A51V variant shows only minor changes in tertiary structure relative to the wild type (WT) protein. Guanidine hydrochloride denaturation demonstrates that the global stability of the A51V variant is 1.3 kcal/mol less than that of the WT protein. The midpoint pH, pH1/2, of the alkaline transition of the A51V variant is 1 unit less than that of the WT protein. Stopped-flow pH jump experiments show that the A51V substitution affects the triggering ionization for one of two kinetically distinguishable alkaline conformers and enhances the accessibility of a high spin heme transient. The pH1/2 for acid unfolding of the A51V variant is 0.7 units higher than for that of the WT protein. Consistent with the greater accessibility of non-native conformers for the A51V variant, the kcat values for its peroxidase activity increase by 6 to 15 fold in the pH range 5 to 8 versus those of the WT protein. These data along with previously reported data for the other THC4–linked variants, G41S and Y48H, underscore the role of Ω-loop C (residues 40 – 57) in modulating the peroxidase activity of cytochrome c early in apoptosis.
Graphical Abstract
INTRODUCTION
The role of mitochondrial cytochrome c (Cytc) as an electron carrier in the electron transport chain is well-established.1 More recently Cytc has been shown to have diverse cellular functions,2 and in particular plays an important role in the intrinsic apoptotic pathway.3–4 Under apoptotic stimuli, Cytc can oxygenate polyunsaturated acyl chains of the mitochondrial lipid cardiolipin (CL)5–6 leading to Cytc dissociation from the membrane. Upon passage from the mitochondria into the cytoplasm, Cytc interacts with apoptotic protease activating factor 1, Apaf-1, to form a complex known as the apoptosome. The apoptosome cleaves procaspase-9 to its active form, caspase-9, ultimately leading to cell death.7
Cytc contains a single covalently attached heme group. Cys14 and Cys17 bind the heme via two thioether bonds, while His18 and Met80 are the axial ligands of the heme iron.8 Thus, the heme iron is six-coordinate and low spin. Heme ligation remains the same in both ferricytochrome c and ferrocytochrome c, making Cytc an efficient electron transporter. When Cytc interacts with CL, Cytc can become a peroxidase leading to oxygenation of CL.5 It is known that heme peroxidases need a pentacoordinate heme or an available heme coordination site to allow the Fe(III) to react with H2O2.9 Thus, to convert Cytc from an electron carrier to a classical heme peroxidase, the heme ligation of Cytc needs to be changed. Previously, we have demonstrated that modest structural rearrangement of Ω-loop D permits loss of Met80-heme ligation allowing Cytc to gain peroxidase function.10 We have also shown that the hydrocarbon chains of detergents can act as structural mimics for CL. These detergents facilitate dimerization of iso-1-Cytc to create an available heme coordination site,11 leading to enhancement of the peroxidase activity of Cytc.12
Given the essential biochemical functions of Cytc, few naturally occurring variants have been documented in humans. Thrombocytopenia 4 (THC4; OMIM 612004) is an inherited autosomal-dominant disease whose symptom is a decrease in the number of platelets in circulating blood.13–14 THC4 is caused by mutation in the Cytc gene (CYCS). Three Cytc variants (G41S, Y48H and A51V) have been identified so far that cause THC4.13–16 G41S and Y48H human (Hu) Cytc have been extensively studied.17–24 Both the G41S and Y48H variants of Hu Cytc show increased flexibility of Ω-loop C (residues 40 – 57) and Ω-loop D (residues 71 – 85)19–20 as well as more accessible alkaline conformers, consistent with the enhanced peroxidase activity of both variants. Interestingly, all three naturally occurring mutations are located in Ω-loop C, which is the least stable substructure of Cytc.25 Recent in vivo studies indicate that the enhanced peroxidase activity of the THC4-linked Cytc variants is unlikely the cause of THC4.14 However, these variants remain excellent model systems for probing the structural factors that control the peroxidase activity that mediates the early stages of apoptosis.5
In the present study, we probe the effect of the naturally occurring A51V variant on the properties of Hu Cytc. We present an X-ray structure of A51V Hu Cytc and characterize the global stability, the local stability and the dynamics of Ω-loop D as measured by the alkaline conformational transition, the acid unfolding and the peroxidase activity of A51V Hu Cytc.
EXPERIMENTAL METHODS
Mutagenesis and Protein Purification.
A51V and A51V-r mutagenesis primers (Invitrogen; see Table S1) were used to add the A51V mutation via PCR-based mutagenesis (QuikChange Lightening, Agilent) to the WT Hu Cytc gene in the pBTR(HumanCc) plasmid.26 The pBTR(HumanCc) vector is a derivative of the pBTR1 expression vector27–28 with the yeast iso-1-Cytc gene replaced with a synthetic Hu Cytc gene. It co-expresses yeast heme lyase allowing covalent attachment of heme to the CSQCH heme recognition sequence of Hu Cytc in the cytoplasm of Escherichia coli. Sequencing to confirm the mutation required for the A51V variant was performed by Eurofins Genomics (Louisville, KY).
The A51V variant of Hu Cytc was expressed in BL21(DE3) E. coli cells (BioRad, phage T1 resistant strain) transformed with the A51V pBTR(HumanCc) plasmid. Purification was carried out as previously reported.29–32 Briefly, cells were broken using a Q700 sonicator (Qsonica, LLC), and the lysate was cleared via centrifugation. Following 50% ammonium sulfate saturation, precipitates were removed via centrifugation, and the supernatant was dialyzed against 12.5 mM sodium phosphate, pH 7.2, 1 mM EDTA, 2 mM β-mercaptoethanol (β-ME). Protein was then batch absorbed to CM-Sepharose Fast Flow resin pre-equilibrated to 50 mM sodium phosphate buffer, pH 7.2, 1 mM EDTA, 2 mM β-ME, and then eluted with a linear gradient of 0 – 0.8 M NaCl in 50 mM sodium phosphate buffer, pH 7.2, 1 mM EDTA, 2 mM β-ME. After concentration and exchange into 50 mM sodium phosphate, pH 7, by ultrafiltration, 1.5 mL aliquots of ~3 mg/mL protein were flash frozen in liquid nitrogen and stored at −80 °C. Prior to experiments, proteins were purified using a HiTrap SP HP 5.0 mL column coupled to an ÄKTAprime plus chromatography system (GE Healthcare Life Sciences). Protein samples were concentrated by ultrafiltration and oxidized with K3[Fe(CN)6]. Separation of oxidized Cytc from the oxidizing agent was accomplished by Sephadex G25 chromatography using a running buffer appropriate to the planned experiment.
Global Stability Measurements by Guanidine Hydrochloride Denaturation.
Global stability measurements were performed using guanidine hydrochloride (GdnHCl) as a denaturant. Measurements were performed with an Applied Photophysics Chirascan circular dichroism (CD) spectrometer interfaced with a Hamilton Microlab 500 Titrator at 25 °C, as previously discussed.29–33 Briefly, the A51V variant at 4 μM in 20 mM Tris, pH 7.5, 40 mM NaCl and ~6 M GdnHCl was titrated into protein at the same concentration in 20 mM Tris, pH 7.5, 40 mM NaCl in a 4 mm pathlength cuvette (Hellma Analytics, Part No. 119004F-10–40) containing a stir bar. After each addition, the sample was stirred to mix, followed by data collection at 222 and 250 nm. Baseline correction was accomplished by subtracting the ellipticity at 250 nm from the ellipticity at 222 nm (θ222corr = θ222 − θ250). Plots of θ222corr versus GdnHCl concentration for A51V Hu Cytc were fit to a two-state model using nonlinear least-squares methods (SigmaPlot v. 13; Systat Software, Inc.), assuming a linear free energy relationship and with native and denatured state baselines that depend linearly on GdnHCl concentration, eq 1.34
| (1) |
In eq 1, θN and mN are the intercept and the slope of the native state baseline, θD and mD are the intercept and the slope of the denatured state baseline, m is the rate of change (slope) of ΔGu with respect to GdnHCl concentration and ΔGuo’(H2O) is the free energy of unfolding extrapolated to 0 M GdnHCl. ΔGuo’(H2O) and m are used to calculate the concentration of GdnHCl at the unfolding midpoint, Cm (= ΔGuo’(H2O)/m). Reported parameters are the average and standard deviation of three independent trials.
Measurement of the Alkaline Conformational Transition.
A Beckman Coulter DU 800 spectrophotometer was used for pH titrations monitored at 695 nm and 22 ± 2 °C to measure the alkaline conformational transition, as previously described.35 Briefly, a 600 μL protein stock solution of ~200 μM oxidized A51V Hu Cytc in 200 mM NaCl was prepared. The protein stock solution and Milli-Q water were mixed 1:1 to produce a working solution of ~100 μM oxidized A51V in 100 mM NaCl. pH titrations were carried out by adding equal volumes of either NaOH or HCl solutions of appropriate concentration and the protein stock solution so that protein concentration in the working solution did not change during the titration. pH was measured with a Denver Instrument UB-10 pH/mV meter using an Accumet double junction semi-micro pH probe (Fisher Scientific, Cat. No. 13-620-852). Absorbance at 750 nm was subtracted from absorbance at 695 nm to correct for baseline drift (A695corr = A695 − A750) during the titration. A695corr was converted to an extinction coefficient, ε695corr, by dividing by protein concentration determined using the published extinction coefficients for the isosbestic points at 526.5 and 541.75 nm for spectra near neutral pH.36
Plots of ε695corr versus pH for the A51V variant were fit to a modified form of the Henderson−Hasselbalch equation, eq 2.
| (2) |
In eq 2, εN is the corrected extinction coefficient at 695 nm for the native state with Met80 bound to the heme, εalk is the corrected extinction coefficient at 695 nm for the alkaline state with lysine as the alkaline state heme ligand,28, 37 pH1/2 is the midpoint pH of the alkaline transition, and n is the number of protons linked to the alkaline transition.
Acid unfolding of A51V Hu Cytc.
Acid unfolding of the A51V variant of Hu Cytc was monitored in the 500 – 750 nm spectral region with a Beckman Coulter DU 800 spectrophotometer. The initial sample was prepared as for the alkaline conformational transition. A protein stock solution of ~200 μM oxidized protein in 200 mM NaCl was prepared and mixed with an equal volume of MilliQ water producing a ~100 μM working solution of the A51V variant in 100 mM NaCl. The working solution was titrated from pH 7.0 to pH 2.0 by adding equal volumes of HCl solutions of appropriate concentration and the protein stock solution so that the protein concentration remained unchanged in the working solution during the titration. After each addition, a spectrum was acquired and pH was measured with a Denver Instrument UB-10 pH/mV meter using an Accumet double junction semi-micro pH probe. The titration was carried out at room temperature (20 ± 2 °C).
Data were fit as for the alkaline transition using an equation analogous to eq 2 to extract the midpoint pH, pH1/2, for acid unfolding. Plots of extinction coefficient versus pH were also fit to a pH dependent observed equilibrium constant, Kobs (pKobs = pKC(A/N)/[(1+10n(pKa-pH)]), between the acid unfolded state, A, and the native state, N, using eq 3.
| (3) |
In eq 3, ελcorr is the corrected extinction coefficient at the wavelength being monitored (ελ – ε750), εA is the corrected extinction coefficient of the acid state and εN is the corrected extinction coefficient of the native state at that wavelength, pKa is the acid dissociation constant of the group which protonates upon acid unfolding and pKC(A/N) (= −logKC(A/N)) is the conformational equilibrium constant at high pH for formation of the native state where the ionizable group is fully deprotonated. Given the pH range of acid unfolding, we assume pKa is 4, corresponding to ionization of a carboxylate group such as a heme propionate.38
pH Jump Stopped-flow Kinetics of the Alkaline Transition.
As previously reported,39–40 pH jump stopped-flow experiments were executed at 25 °C using an Applied Photophysics SX20 stopped-flow spectrometer. A total of 5000 data points were collected on a logarithmic time scale monitoring at 398 nm. Data were collected on a 100 ms to a 100 s time scale depending on final pH. Both upward and downward pH jump data were collected in increments of 0.25 pH units. Initial sample conditions for upward pH jumps were 20 μM A51V Hu Cytc in 0.1 M NaCl (pH 6), which was mixed in a 1:1 ratio with 50 mM buffer of the desired pH (pH 7.5 – 11) in 0.1 M NaCl producing a 10 μM protein solution in 25 mM buffer in 0.1 M NaCl. Downward pH jumps were carried out in a similar manner beginning at pH 10.5 and jumping to the pH regime 7 – 8. Effluent was collected, and the final pH was measured with a Denver Instrument UB-10 pH/mV meter using an Accumet double junction semi-micro pH probe. The pH of the effluent was within error of the pH of the buffer used for mixing in all cases. Buffers were as follows: MES (pH 6.0−6.5), NaH2PO4 (pH 6.75−7.5), Tris (pH 7.75−8.75), H3BO3 (pH 9−10), and CAPS (pH 10–11). A minimum of 5 trials were collected at each pH. Data were fit to the appropriate exponential function using SigmaPlot v. 13.
Crystallization and Structure Determination of Human A51V Cytc.
A51V Hu Cytc was oxidized with K3[Fe(CN)6], followed by separation from the oxidizing agent and exchange into 50 mM sodium phosphate pH 7 using Sephadex G-25 chromatography. It was then concentrated to ~13.5 mg/mL using centrifuge ultrafiltration (Amicon Ultra-4 10,000 MWCO). Screening for crystallization conditions was carried out using the JCSG screening kits. The ratio of protein to reservoir solution in the drops in the 96 well sitting drop screening plates was 1:1. We obtained crystals from JCSG core II, well D6 (0.1 M sodium citrate pH 5.5, 40% w/v PEG 600). Additional vapor diffusion crystallization experiments were set up in a 24-well plate by expanding upon the pH and precipitant concentration of this initial condition. After 3 days to 2 weeks of equilibration at 20 °C, crystals diffracting to 1.82 Å in the C2221 space group were obtained from a drop containing 1 μL of protein and 1 μL of 40% w/v PEG 600, 0.1 M sodium citrate pH 5.7. Crystals were cryoprotected with 20% glycerol and flash frozen in liquid N2 for data collection.
The X-ray diffraction dataset was collected under cryogenic conditions at 100 K using the SMB beamline 9–2 of the Stanford Synchrotron Radiation Lightsource (SSRL) and a Pilatus 6 M detector. Diffraction data were processed using iMOSFLM41 and scaled and merged using Aimless.42 The initial electron density map was determined using phases obtained with the molecular replacement method using MOLREP,43 incorporated in the CCP4i2 software suite,44 and the coordinates of the K72A human Cytc structure (PDB ID: 5TY3)45 as a search model. The initial model was built into a continuous electron density map and subsequently refined using REFMAC46 and PHENIX.47 The structure model was further refined to 1.82 Å resolution by multiple rounds of manual model rebuilding with COOT48 and restrained refinement with REFMAC and PHENIX using 5 % of reflections for calculation of Rfree. Data collection and refinement statistics of the final model are summarized in Table 1.
Table 1.
X-ray Crystallography and Data Collection and Refinement Statistics
| PDB code | 6DUJ |
| Beamline | SSRL SMB 9–2 |
| Wavelength (Å) | 1.07 |
| Resolution range (Å) | 33.32–1.82 (1.89–1.82)* |
| Space group | C 2 2 21 |
| Unit cell dimensions | |
| a, b, c (Å) | 62.4, 184.4, 35.7 |
| α, β, γ (°) | 90, 90, 90 |
| Unique reflections | 18770 (1878)* |
| Multiplicity | 6.5 (5.3)* |
| Completeness (%) | 98.7 (99)* |
| Mean I/σ(I) | 24.5 (6.1)* |
| Wilson B-factor | 21.98 |
| Rsym† | 0.07 (0.27)* |
| Refinement | |
| Rwork§ | 0.184 (0.292)* |
| Rfree§ | 0.235 (0.317)* |
| Number of total atoms | 1787 |
| protein | 1610 |
| heme | 86 |
| solvent | 91 |
| Total protein residues | 210 |
| RMS (bonds, Å) | 0.02 |
| RMS (angles, °) | 1.62 |
| Ramachandran favored (%)†† | 98 |
| Rotamer outliers (%)†† | 0 |
| Average B-factor (Å2) | 32.34 |
| macromolecules (Å2) | 33.00 |
| heme (Å2) | 13.90 |
| solvent (Å2) | 38.80 |
Data for highest resolution shell are given in brackets.
Rsym =∑hkl ∑i | Ii (hkl)- 〈I (hkl)〉|/ ∑hkl ∑i Ii (hkl) where Ii (hkl) is the ith observation of the intensity of the reflection hkl.
Rwork =∑hkl || Fobs|-|Fcalc||/ ∑hkl |Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes for each reflection hkl. Rfree was calculated with 5% of the diffraction data that were selected randomly and excluded from refinement.
Calculated using MolProbity.52
Guaiacol Assay of Peroxidase Activity.
Peroxidase activity was measured with the colorimetric reagent, guaiacol, using previously reported conditions and procedures.40, 45 The reaction was monitored at 25 °C using an Applied Photophysics SX20 stopped-flow apparatus. The formation of tetraguaiacol from guaiacol and H2O2 in the presence of Cytc was monitored at 470 nm. Cytc (4 μM) in 50 mM buffer, 0.1 M NaCl was mixed in a 1:1 ratio with guaiacol at four-fold the desired final guaiacol concentration in 50 mM buffer, 0.1 M NaCl to produce Cytc/guaiacol stock solutions in 50 mM buffer, 0.1 M NaCl. Each Cytc/guaiacol stock solution was mixed 1:1 with 100 mM H2O2 in 50 mM buffer, 0.1 M NaCl by the stopped-flow instrument yielding a final solution containing 1 μM Cytc, 50 mM H2O2 and guaiacol at the desired concentration in 50 mM buffer, 0.1 M NaCl. Concentration was determined using the extinction coefficients of H2O2 (ε240 = 41.5 M−1 cm−1; average of published values49–50) and guaiacol (ε274 = 2,150 M−1 cm−1).51 Buffers used for peroxidase experiments were the same as those used in pH-Jump experiments. Final concentrations of guaiacol after mixing were 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 80, and 100 μM.
The segment of the A470 versus time data with the greatest slope following the initial lag phase was used to obtain initial velocity, v, at each guaiacol concentration. The data were fit to a linear equation and the slope from five repeats was averaged. The slope (dA470/dt) was divided by the extinction coefficient of tetraguaiacol at 470 nm (ε470 = 26.6 mM−1cm−1)53 and multiplied by 4 (4 guaiacol consumed per tetraguaiacol produced) to give the initial rate of guaiacol consumption, v. The initial rate, v, was divided by Cytc concentration, plotted against guaiacol concentration and fit (SigmaPlot v. 13) to the Michaelis-Menten equation, eq 4, to obtain Km and kcat values.
| (4) |
RESULTS
Structure of A51V Hu Cytc.
A51V Hu Cytc crystallized in acetate buffer at pH 5.8. The crystals diffracted to 1.82 Å in the C2221 space group with two Cytc molecules in the asymmetric unit. The overall fold of A51V Cytc (PDB entry 6DUJ: Rwork and Rfree values of 0.18 and 0.24, respectively) is similar to that of WT Hu Cytc (PDB entry 3ZCF) with an all-atom root-mean-square-deviation (RMSD) of 0.835 Å (using molecule A of each structure) between the two X-ray structures (Figure 1). The all-atom RMSD between the two molecules in the asymmetric unit of the A51V Hu Cytc structure is slightly higher (1.104 Å). The X-ray structures of the other THC4 variants of Hu Cytc, G41S22 and Y48H,20 are also very similar to the WT X-ray structure.
Figure 1.

Overlay of the A51V (PDB ID: 6DUJ, chain A, dark gray) and WT (PBD ID: 3ZCF, chain A, light gray) Hu Cytc structures. Ω-loop C (residues 40 – 57) is shown in wheat (WT) and light purple (A51V). Ω-loop D (residues 70 – 85) is shown in light pink (A51V) and salmon (WT). Stick models are used for the heme and selected side chains. Buried water molecules are shown as red spheres. Hydrogen bonds are shown as yellow dashed lines.
There are small, localized structural changes. In particular, the larger steric size of valine versus alanine pushes the main chain of Ω-loop D away from Ω-loop C. The largest main chain shift is at Gly77 (Figure 1). This shift propagates across the loop to Ile81. There is also a change in the ring pucker of Pro76. However, the hydrogen bond (H-bond) network that couples Met80 to two buried waters and heme propionates 6 and 7 (HP6 and HP7) through the side chains of Tyr67, Thr78, Trp59, Asn52, Thr49, Tyr48 and Arg38 is largely unchanged in the A51V variant relative to that of the WT protein (Figure 1). The slight shift of the main chain of Ω-loop D around Gly77 does allow invasion of water between Ω-loops C and D (Figure 2). For the A51V structure, the carbonyl oxygen of Val51 is linked with the carbonyl oxygen of Pro76 through a network of three water molecules that are not observed in the WT structure (Figure 2). The same is observed for the other molecule in the asymmetric unit of the A51V variant (Figure S1).
Figure 2.

Hydrogen bond network (yellow dashed lines) across Ω-loop D in (a) A51V (chain A) and (b) WT (chain A) Hu Cytc. Coloring of Ω-loops C and D is as in Fig. 1. Ω-loop D is shown with stick models. Waters are shown as red spheres.
A closer examination of Ω-loop D shows that the cross-loop network of direct and water mediated H-bonds is much more sparse for the A51V variant versus that observed for WT Hu Cytc (Figure 2). Both WT and A51V Hu Cytc share main chain to main chain L68 to I85 and I75 to T78 H-bonds. Both also have a water mediated cross-loop H-bond network involving Asn 70, Lys72 and the carbonyl of Phe82. Water-mediated H-bonds are also largely retained on the periphery of the loop for the A51V variant as observed for the WT protein. However, the extensive network of water mediated H-bonds connecting the side chain of Lys72, and the main chain atoms of Phe82, Met80, Thr78 and Gly77 observed with the WT protein is missing in the A51V variant. The cross-loop H-bond network is even more sparse for the other molecule of A51V Hu Cytc in the asymmetric unit (Figure S1, chain C).
This observation suggests that Ω-loop D may be more dynamic for the A51V variant than for WT Hu Cytc. Crystallographic B-factors can provide a qualitative estimate of differential dynamics for variants of the same protein. Based on the difference in resolution alone (WT, 1.65 Å; A51V, 1.82 Å) a 20% increase in the crystallographic B-factors would be expected for the A51V variant.54 For WT Hu Cytc, the average backbone B-factor is 18.4 ± 3.1 Å2 compared to 27.6 ± 5.5 Å2 for that of the A51V variant, a 50% increase. The largest increase in B-factors occurs in the C-terminal half of the protein following the site of the A51V substitution, with consistently higher B-factors in Ω-loop D (Figure S2). The large standard deviation of the B-factors in this region may reflect differences in intermolecular contacts between the two molecules in the asymmetric unit. Thus, the upper limit of the error bars in Figure S2 may be more reflective of the differences in dynamics in Ω-loop D for WT versus A51V Hu Cytc.
Our previous structural study of a K72A variant of yeast iso-1-Cytc (PDB entry 4MU8) shows that Arg38 can act as a gate that mediates access to a buried water channel.10 A similar effect has been also observed in the structure of G41S Hu Cytc (PDB entry 3NWV).22 The movement of Arg38 away from the heme allows access of water (and possibly H2O2) to the heme. Both proteins exhibit enhanced peroxidase activity. However, the Arg38 side chain in the A51V structure is in the same position as in the WT structure (i.e., no heme access) and that of the Y48H variant of Hu Cytc.20
Global Stability of A51V Hu Cytc.
Global unfolding thermodynamics of A51V Hu Cytc was monitored by CD at 25 °C and pH 7.5 with the use of GdnHCl as denaturant. Figure 3 shows a plot of θ222corr versus GdnHCl concentration for the A51V variant compared to WT Hu Cytc. Thermodynamic parameters from fits to a two-state model are given in Table 2. ΔGu°’(H2O) of the A51V variant is lower than that of the WT protein by ~1.3 kcal mol−1. The data for the G41S and Y48H variants were acquired at 15 °C20 versus 25 °C for the A51V variant (Table 2). ΔGu°’(H2O) for WT Hu Cytc is ~1.2 kcal/mol larger at 15 °C than at 25 °C (Table 2). If the temperature dependence of ΔGu°’(H2O) is assumed to be similar for the variants, then ΔGu°’(H2O) of A51V is larger than those for the other two naturally occurring variants linked to THC4, G41S and Y48H, by ~2.6 kcal mol−1 and ~4.3 kcal mol−1, respectively. Within error, the midpoint GdnHCl concentration for unfolding, Cm, is unchanged for the A51V variant relative to that of WT Hu Cytc, whereas Cm of the G41S and Y48H variants are decreased by ~0.4 M and ~0.6 M, respectively, relative to that of WT Hu Cytc.
Figure 3.

GdnHCl denaturation of A51V and WT Hu Cytc at 25 °C and pH 7.5. Corrected ellipticity at 222 nm, θ222corr, is plotted versus GdnHCl concentration. Solid curves are fits to eq 1 in Experimental Procedures. Data shown with open circles were not used in the fits to the two-state model. Parameters obtained from the fits are given in Table 1. The data for WT Hu Cytc are from ref.29 as reanalyzed in ref45.
Table 2.
Thermodynamic Parameters for Global Unfolding of Hu Cytc Variants
In Figure 3, the unfolding transition appears to be broader for the A51V variant than for that of WT Hu Cytc. Consistent with this observation, fits of the A51V data to a two-state model yield a decrease of about 0.7 kcal mol−1 M−1 for the GdnHCl m-value compared to that of WT Hu Cytc (Table 2). The m-values of the A51V and G41S variants are the same and about 0.4 kcal mol−1 M−1 larger than that of the Y48H variant. Thus, a common feature of the variants linked to THC4 is a loss in the cooperativity of the folding of Hu Cytc.
Local Stability Assessed by the Alkaline Conformational Transition.
Cytc undergoes a conformational change at moderately alkaline pH.55–56 In the terminology of Theorell and Åkesson,57 this conformational change is the conversion from state III (native state) to state IV (alkaline state) and involves replacement of the Met80 heme ligand with either Lys73 or 79.37, 58 For mammalian cytochromes c, the apparent pKa of this transition is typically between 9 and 9.5.59 Above pH 12, state V forms with replacement of the lysine ligand with hydroxide in mammalian cytochromes c.57, 60 However, the less stable yeast iso-1-cytochrome c forms states IV and V at lower pH.37, 61 The alkaline transition correlates well with the local stability of Ω-loop D.33, 58 The stability of Cytc with respect to the alkaline conformational transition and the accessibility of states which promote peroxidase activity often correlate well.18, 40 Thus, the local unfolding thermodynamics for the alkaline conformational transition of A51V Hu Cytc was determined by pH titration, monitored at 695 nm to follow the loss of Met80-heme iron ligation8 when a lysine (Lys72, Lys73 or Lys79) from Ω-loop D binds to the heme. Comparison of data for the A51V variant to previously published data29 for WT Hu Cytc (Figure 4) show that the alkaline transition is shifted to significantly lower pH. The fits of the data for A51V to eq 2 (Experimental Methods) show that the number of protons, n, linked to the conformational change is approximately equal to 1 (Table 3), consistent with a one proton process as normally observed for the alkaline transition of Cytc.55–56 The A51V substitution causes a decrease in the midpoint pH, pH1/2, of the alkaline transition of about 1 unit relative to WT Hu Cytc. Within error, the pH1/2 of the A51V variant is similar to those of the G41S and Y48H variants, obtained by Worrall and coworkers also using data at 695 nm (Table 3).20 However, the pH1/2 reported by Bren, Ledgerwood and coworkers for the G41S variant using data at 695 nm is somewhat lower (Table 3).21 It should be noted that the alkaline transition is sensitive to buffer conditions,59 which vary for these different measurements.
Figure 4.

Plots of ε695corr versus pH for the alkaline transition of the A51V variant versus WT Hu Cytc.29 Data were collected at room temperature (22 ± 2 °C) in 0.1 M NaCl solution. Solid lines are fits to eq 2 in Experimental Methods.
Table 3.
Thermodynamic Parameters for the Alkaline Transition of WT and THC4-linked Variants of Hu Cytc
| Variant | pH1/2 | n |
|---|---|---|
| A51Va | 8.56 ± 0.04 | 0.9 ± 0.1 |
| WT | 9.54 ± 0.03b | 1.03 ± 0.02b |
| 9.3 ± 0.2c | -d | |
| G41S | 8.5 ± 0.2c | -d |
| 7.8 ± 0.3e | -d | |
| Y48H | 8.4 ± 0.1c | -d |
Data were collected at 22 ± 2 °C in 0.1 M NaCl.
Parameters are from ref.29 and were collected at 22 ± 2 °C in 0.1 M NaCl.
Parameters are from ref20 for data collected at 695 nm and room temperature in 20 mM sodium phosphate.
Data were fit assuming a one proton process.
Parameters are from ref21 for data collected at 695 nm and 25 °C in the 50 mM sodium phosphate.
Kinetics of the Alkaline Conformational Transition of A51V Hu Cytc.
In order to determine the actual rates of interconversion between the native and alkaline conformations, stopped-flow pH jump methods were employed. Both upward and downward pH jumps were monitored at 398 nm to follow heme-ligand changes linked to rearrangement of Ω-loop D. Because there are three lysines, Lys72, Lys73, and Lys79, present in Ω-loop D, as many as three kinetic phases are possible. In upward pH jump experiments, the A51V variant shows two slow phases from pH 7.5 to 10 (kobs,1, A1u) or 10.5 (kobs,2, A2u) and a fast phase (kobs,3, A3u) from pH 8.5 to 11 (Figures S3, 5 and 6). Previous studies on the G41S and Y48H variants linked to THC4 also observed deviation from single-exponential behavior for the slow phase.62 However, the deviation was considered small so the slow phase was fit as a single first-order kinetic process. The study of the G41S and Y48H variants did not acquire downward pH jump data. We observe two slow phases in downward pH jump data, as well. Therefore, we have elected to fit the entire data set assuming the presence of two slow phases.
Figure 5.

Rates constants for the alkaline transition of the A51V variant of Hu Cytc at 25 °C in 0.1 NaCl. Plots of (a) kobs,1, (b) kobs,2, and (c) kobs,3 versus pH from upward (filled symbols) and downward (open symbols) pH jump experiments. Solid lines are fits of the combined upward and downward kobs versus pH data to eq 5 in the Discussion.
Figure 6.

Plots of amplitude versus pH for the three kinetic phases observed for the alkaline conformational transition of A51V Hu Cytc. Color scheme is the same as for Figure 5. Filled symbols are used for upward pH jumps (A1u, A2u and A3u). Open symbols are used for downward pH jumps from pH 10.5 (A1d, A2d and A3d). Data points are the average and standard deviation of a minimum of five trials. A1u versus pH and A2u versus pH data were fit to eq 6 in the Discussion (solid lines). In these fits, kb and kf in eq 6 were set equal to the values determined from the fits of kobs versus pH to eq 5 in Figure 5. Data points connected with dashed lines were not included in these fits. A3u versus pH data were fit to Henderson-Hasselbalch equation (solid line).
The rate constant for one of the two slow phases, kobs,1 remains relatively constant below pH 8.25, with values near 0.04 s−1 (Table S2). Above pH 8.25, kobs,1 increases to ~0.35 s−1 as pH approaches 10 (Table S2 and Figure 5). The amplitude for this phase A1u increases from around 0.007 at pH 7.5 to 0.057 at pH 8.75, and then begins to decrease above pH 8.75 (Table S2 and Figure 6) as the other slow phase (kobs,2, A2u) becomes the dominant phase.
The rate constant for the other slow phase, kobs,2, also is relatively constant below pH 8.25, with values near 0.09 s−1 (Table S2 and Figure 5). Above pH 8.25, kobs,2 increases to 3.6 s−1 as pH approaches 10.5. The amplitude for this phase, A2u, increases from around 0.005 at pH 7.5 to 0.09 at pH 9.5, and then decreases above pH 9.5 (Table S2 and Figure 6).
At pH 8.5 and above a fast phase (kobs,3, A3, Table S2) grows in. The rate constant for this fast phase, kobs,3, cannot be obtained with high precision between pH 8.5 and 9.5. Values near 23 s−1 begin increasing above pH 8.5, approach 50 s−1 at pH 9.5, and remain around 30 s−1 above pH 10. The amplitude for the fast phase, A3u, is small initially, but grows rapidly above pH 9.25, becoming the dominant phase as the two slow phases disappear above pH 10 (kobs,1, A1u) and pH 10.5 (kobs,2, A2u).
To obtain accurate rate constants in the lower pH regime, downward pH jump experiments were carried out beginning at pH 10.5 and jumping to pH 7–8 (Figure 5, Table S3). Downward pH jump data were fit to a triple exponential equation (Figure S4). The observed rate constants for the two slow phases in the downward pH jumps correspond well to kobs,1 and kobs,2, observed in upward pH jumps (Figures 5A and 5B). The observed rate constants for the fast phase in the downward pH jumps corresponds well to kobs,3 (Figure 5C).
Acid Unfolding of A51V and WT Hu Cytc.
The pH of intermembrane space of mitochondria is more acidic (pH = 6.88 ± 0.08) than the matrix (7.8 ± 0.2) or the cytosol (7.59 ± 0.01).63 Thus, it is important to determine the stability of A51V Hu Cytc with respect to pH to evaluate the role of Ω-loop C with regard to the accessibility of alternate conformers with the high spin heme believed to be necessary for peroxidase activity. The unfolding thermodynamics for the acid unfolding of A51V Hu Cytc was determined by pH titration. Figure 7 shows plots of the corrected extinction coefficients at 695 nm (Met80 ligation), ε695corr, and 622 nm (high spin heme), ε622corr, versus pH for the acid unfolding of A51V compared to previously reported data for WT Hu Cytc.64 The plots show that the A51V variant is significantly less stable to acid unfolding than WT Hu Cytc. For both proteins, the data monitored at 622 nm yield a broader transition than the data at 695 nm. The somewhat different behaviors of the 695 nm data and the 622 nm data are a result of the complexity of acid unfolding. Acid unfolding leads to sequential formation of states II and I from the native state (state III). While state II is compact, state I is disordered.65 High ionic strength stabilizes state II with respect to state I, but under the low ionic strength conditions of our experiments, the two transitions are not well-separated.38, 65 State II is a mix of low spin and high spin (Met 80 replaced by H2O) states.38, 66 Originally, the low spin state was thought to involve Met80 ligation,38 however, more recent resonance Raman data suggest that ligation by a histidine (likely His 33) is involved in the low spin state II conformer.66 The loss of the 695 nm band can be due to both formation of another low spin species and a high spin state and so prefect overlap of acid unfolding monitored at 695 nm and 622 nm (high spin) is not expected.
Figure 7.

Plots of ε695corr and ε622corr versus pH for acid unfolding of WT and A51V Cytc. Data were acquired at 22 ± 2 °C in the presence of 100 mM NaCl. The solid curves are fits to Henderson-Hasselbalch equation. Data for the WT protein are from ref.64.
Fits of the data to the Henderson-Hasselbalch equation show that the midpoint pH, pH1/2, for acid unfolding shifts ~0.7 unit to higher pH for the A51V variant relative to that of WT Hu Cytc (Table 4). A modest decrease in pH1/2 (although within error for the A51V variant) is also observed for data monitored at 622 nm relative to 695 nm (Table 4). The number of protons, n, linked to acid unfolding for data monitored at 622 nm versus 695 nm also decreases (Table 4). Consistent with these observations, an isosbestic point near 647 nm is lost at pH values at or just below pH1/2 for acid unfolding for both proteins (Figure S5). Loss of this isosbestic point may signal the onset of the overlapping state II to state I transition of acid unfolding.38, 65 Data were also fit to eq 3 in Experimental Methods to provide an equilibrium constant for the acid to native state transition (pKC(A/N), Table 4).
Table 4.
Thermodynamic Parameters for the Acidic Transition of WT and A51V Hu Cytc at 22 ± 2 °C
| 695 nm | 622 nm | |||||
|---|---|---|---|---|---|---|
| Variant | pH1/2 | pKC(A/N)a | n | pH1/2 | pKC(A/N)a | n |
| A51V | 3.19 ± 0.09 | −1.4 ± 0.3 | 1.8 ± 0.4 | 3.13 ± 0.06 | −1.1 ± 0.2 | 1.3 ± 0.1 |
| WTb | 2.50 ± 0.03 | −2.50 ± 0.07 | 1.64 ± 0.07 | 2.41 ± 0.03 | −1.73 ± 0.06 | 1.13 ± 0.08 |
Assumes the pKa of the ionizable group(s) is 4.
Parameters are taken from ref64.
Peroxidase Activity of A51V Hu Cytc.
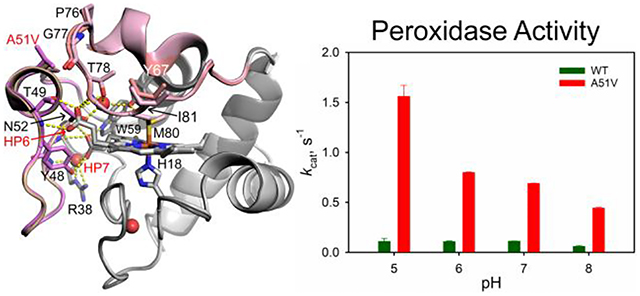
The peroxidase activity of A51V Hu Cytc was measured by monitoring the formation of tetraguaiacol from guaiacol in the presence of H2O2. Michaelis-Menten plots with respect to guaiacol concentration were generated to permit extraction of kcat and Km across the pH range 5 to 8 (Figure 8a). The A51V variant shows a significant increase in kcat values relative to those of WT Hu Cytc (Figure 8b). The increase in kcat values for the A51V variant relative to those of WT Hu Cytc is more pronounced as pH decreases. kcat increases ~7-fold at pH 6 versus ~14-fold at pH 5 for kcat of the A51V variant versus kcat of WT Hu Cytc (Table 5). Relative to the Km values of the WT protein, those of A51V Cytc are not strongly affected (Table 5).
Figure. 8.

Peroxidase activity of WT and A51V Hu Cytc. (a) Michaelis-Menten plots for the A51V variant of Hu Cytc at pH values from 5 to 8. The solid curves are fits to eq 4 (Experimental Methods). Data were acquired at 25 °C in 50 mM buffer. (b) kcat versus pH for WT (previously published,45, 64 dark green bars) and A51V (red bars) Hu Cytc. Error bars are the standard deviation from three independent experiments.
Table 5.
Michaelis-Menten Parameters as a Function of pH at 25 °C for the Peroxidase Activity of WT and A51V Hu Cytc
DISCUSSION
Effect of the A51V Substitution on the Global and Local Stability and Structure of Hu Cytc.
Consistent with previous work performed on two other naturally occurring variants, G41S and Y48H,20 the A51V variant of Hu Cytc shows both decreased global stability and decreased stability of the native state relative to subglobal unfolding of Ω-loop D to form the alkaline state (Tables 2 and 3). The thermodynamic properties of the foldons of Cytc are consistent with sequential unfolding.67 In particular, mutations that stabilize or destabilize a foldon affect the stability of more stable foldons that unfold after the mutated foldon by the same amount as the mutated foldon, whereas the stabilities of less stable foldons that unfold before the mutated foldon are unaffected.68–70 A good correlation between mutation-induced changes in the stability of Ω-loop D as monitored by the alkaline transition and the effect of these mutations on global stability has been observed.33, 58 The A51V substitution leads to a decrease in the pH1/2 of the alkaline transition of about 1 unit, similar to the decreases in pH1/2 observed for the other THC4-linked variants of Hu Cytc (Table 3). A decrease in pH1/2 by 1 unit corresponds to ~1.3 kcal/mol (ΔΔGalk = 2.3RTΔpH1/2) very close to the change in global stability for the A51V variant versus WT Hu Cytc (Table 2) and consistent with sequential unfolding of the foldons of Cytc. We previously obtained similar correspondence between changes in global stability and changes in the stability of Ω-loop D as determined via the alkaline transition for I52A and I52T variants of yeast iso-1-Cytc.33 Comparing the data in Tables 2 and 3, there is obviously a poor correlation between local and global stability for the other two THC-linked variants of Hu Cytc.20 This observation likely results from the incomplete coupling between the stabilities of the two least stable foldons, the Ω-loop C foldon and the Ω-loop D foldon, that has been noted in studies on horse Cytc.68 For example, it was shown that destabilization of Ω-loop C of horse Cytc by a T47G mutation within the loop or by decreasing pH destabilizes all more stable foldons by the same amount, except for the Ω-loop D foldon, which is destabilized by about half the amount of the other foldons. These results indicate that unfolding of the Ω-loop D foldon sometimes occurs independently of the unfolding of the Ω-loop C foldon and sometimes occurs after unfolding of the Ω-loop C foldon such that stability changes in Ω-loop C may only be partially communicated to Ω-loop D, while fully affecting the global stability of the protein. Therefore, the relative effect of an Ω-loop C mutation on global stability versus the stability of the alkaline state relative to the native state will depend on how the particular mutation affects the proportion of independent versus sequential unfolding of the Ω-loop C and Ω-loop D foldons.
Previous studies showed a significant loss in the cooperativity of unfolding for THC4-linked Hu Cytc variants as measured by the magnitude of the denaturant m-value. Both the A51V and G41S substitutions cause a 20% decrease in the GdnHCl m-value whereas the Y48H variant causes a 30% decrease in the GdnHCl m-value relative to that of WT Hu Cytc (Table 2). Because GdnHCl unfolding monitored by circular dichroism at 222 nm (α-helix) is not strongly sensitive to unfolding of Ω-loops, the smaller m-values could reflect partial unfolding of Ω-loop C (and possibly Ω-loop D) prior to global unfolding (loss of helical structure). This type of behavior has been observed for histidine variants (K73H and K79H) in Ω-loop D of yeast iso-1-Cytc.30, 39, 71–74 Loss of structure in Ω-loop C prior to the major unfolding transition monitored by CD would indicate destabilization of this substructure by the A51V, G41S and Y48H substitutions. We note that decreased m-values can also reflect a more compact denatured state.75
The loss of ordered water structure around Ω-loop D of the A51V variant (Figure 2) and the overall increase in the magnitude of crystallographic B-factors for the A51V variant are (Figure S2) consistent with destabilization of Ω-loops C and D. H/D exchange experiments on the G41S and Y48H variants also support this interpretation.19–20
The nature of out-of-plane distortion of the heme cofactor appears to be conserved within different classes of heme proteins.76 Ruffling is predominant in c-type cytochromes,76–77 whereas peroxidases exhibit strong saddling and moderate ruffling.76 Ruffling affects the electronic structure of c-type cytochromes and has been shown to be correlated to redox potential.77–78 X-ray structures of WT,18 G41S22 and Y48H20 human Cytc along with the A51V structure presented here allow the effect of the substitutions in all three THC-linked variants on heme out-of-plane distortion, Doop, to be compared. Using the NSD program79–80 to analyze Doop of the heme and its normal mode components, we find that the magnitude of Doop of the heme ranges from 0.6 – 1 Å and follows the order: G41S > WT ≈ Y48H > A51V (Table S4). In all cases, ruffling (B1u component) is the predominate form of distortion as expected for c-type cytochromes,77 following the order: G41S > WT > Y48H > A51V (Table S4). Saddling (B2u component), while a much smaller component of Doop, does contributes significantly to heme distortion. Relative to WT human Cytc, saddling increases for the Y48H and G41S variants and decreases for the A51V variant (Table S4). Thus, the A51V substitution causes a significant decrease in Dopp, primarily due to a decrease in ruffling, but with a notable contribution from a decrease in saddling. However, overall there no clear trend for the effect of the THC-linked mutations on heme distortion, suggesting that it may not be the predominate factor in the relationship to disease or the enhanced peroxidase activity observed for these variants.
Effect of the A51V Substitution on the Slow Phases of the Alkaline Transition.
The kinetic data for the alkaline transition of A51V Cytc follows the standard mechanism, a rapid deprotonation equilibrium (KH or pKH), which triggers a conformational rearrangement in which a lysine replaces the Met80 heme ligand. For this mechanism,81 the observed rate constant, kobs, increases with pH and then levels off (Figure 5, eq 5).
| (5) |
The forward rate constant, kf, corresponds to formation of the alkaline state from the native state following the triggering deprotonation, and the backward rate constant, kb, is for the return to the native conformer from the alkaline state. Eq 6 describes the pH dependence of the amplitude for this mechanism,81 where ΔAt is the total amplitude when the alkaline transition goes to
| (6) |
completion. The other parameters are as defined for eq 5. In good agreement with this kinetic model, both kobs,1 and kobs,2 increase as pH increases. Parameters for the fits of the kobs,1 and kobs,2 versus pH to eq 5 (Figure 5) are given in Table 6. pKH can also be evaluated with eq 6 from the plots of A1u and A2u versus pH in Figure 6 using the kf and kb values obtained from the fits to eq 5 (Table 6). The pKH values obtained from rate constant and amplitude (values in brackets in Table 6) data are within error the same for kobs,1, indicating that kf and kb obtained from rate constant data are reliable. The values of kf, kb and pKH from kinetic data can also be used to calculate the pH1/2, pH1/2calc. For kobs,1, pH1/2calc is within error the same as the pH1/2 of 8.56 ± 0.04 obtained from equilibrium measurements.
Table 6.
Kinetic Parameters for the Alkaline Transition of A51V Hu Cytc at 25 °C
| Variant | Rate constant | kf, s−1 a | kb, s−1 a | pKH a | pKC b | pH1/2calc c |
|---|---|---|---|---|---|---|
| A51V | kobs,1 | 0.32 ± 0.03 | 0.0368 ± 0.008 | 9.3 ± 0.1 (9.5 ± 0.3)d |
−0.9 ± 0.1 | 8.4 ± 0.2 (8.6 ± 0.3)e |
| kobs,2 | 6.2 ± 0.3 | 0.11 ± 0.02 | 10.37 ± 0.04 (10.9 ± 0.1)d |
−1.75 ± 0.08 | 8.61 ± 0.09 (9.1 ± 0.1)e |
|
| I81Af | kobs,1 | 1.6 ± 0.2 | 0.011 ± 0.003 | 10.7 ± 0.2 (10.8 ± 0.1)d |
−2.2 ± 0.1 | 8.6 ± 0.2 (8.7 ± 0.2)e |
| kobs,2 | 3.8 ± 0.2 | 0.07 ± 0.01 | 10.8 ± 0.1 | −1.74 ± 0.08 | 9.1 ± 0.1 | |
| V83Gf | kobs,1 | 0.7 ± 0.2 | 0.039 ± 0.003 | 11.2 ± 0.1 | −1.2 ± 0.1 | 9.9 ± 0.2 |
| kobs,2 | 6 ± 1 | 0.13 ± 0.01 | 11.2 ± 0.1 | −1.68 ± 0.08 | 9.6 ± 0.2 |
From fit of data in Fig. 5 to eq 5. Errors are the standard errors in the parameters from the fit to eq 5.
pKC = −LogKC, where KC = kf/kb, and KC is the conformational equilibrium constant after the triggering deprotonation. Error is from standard propagation of the errors in kf and kb.
pH1/2calc = pKH – pKC.
Calculated using pKH from amplitude data.
Parameters are from ref.64.
For kobs,2, kf, kb and pKH obtained from rate constant versus pH data yield pH1/2calc that is also similar to the pH1/2 from equilibrium data (Table 6). However, the pKH obtained from amplitude versus pH data is considerably higher (as is the pH1/2calc, Table 6), suggesting that the parameters obtained from either the rate constant or amplitude data may be less reliable for the kobs,2, A2 slow phase. Constraining kf in the fits of kobs,2 versus pH to the range of 10 – 12 s−1 returns values of kb (0.16 – 0.17 s−1) that are consistent with kb obtained in downward pH jump experiments (Table S3) and yields values of pKH from 10.7 – 10.8. Values of pKH obtained from the amplitude data with these values of kf and kb remain near 10.9. While there may be some uncertainty in the value of pKH for kobs,2, its value is 1 – 1.5 units higher than pKH for kobs,1 allowing the alkaline conformer corresponding to the kobs,1 phase to populate at lower pH despite a less favorable conformational equilibrium (by a factor of ~7, see pKC in Table 6).
Recent work compared the alkaline transition kinetics of WT Hu Cytc and V83G and I81A variants in Ω-loop D to the naturally occurring Y48H and G41S variants in Ω-loop C.62 The value of pKH was observed to decrease by about 1 unit for the Ω-loop C variants relative to WT Hu Cytc and the Ω-loop D variants. For WT and all variants, pKC was unaffected with a value near −2.62 The absolute values of the kinetic parameters reported here cannot be compared directly because our data were fit to two slow phases rather than a single slow phase. However, we have recently reported kinetic data for the V83G and I81A variants of Hu Cytc, which we fit assuming two slow phases.64 Values of pKH for both the kobs,1 and kobs,2 phases were within the range of 10.7 – 11.2 for kinetic data fit with two slow phases for these variants (Table 6).64 As for the G41S and Y48H variants, the increased access to alternate conformers for the A51V variant appears to result from a decrease in the pKH of the triggering deprotonation. For the A51V variant, the decrease in pKH appears to operate only on the alkaline state corresponding to kobs,1 (Table 6) suggesting that the triggering deprotonation controlling the dynamics of different parts of Ω-loop D is not the same.
The I81A variant of Hu Cytc also shows enhanced kinetic accessibility of only one of the two kinetically distinguishable alkaline conformers in 0.1 M NaCl.64 In this case, pKH for kobs,1 is unchanged whereas pKC is more favorable because kf is larger and kb is smaller (Table 6). Notably, kf and kb derived from kobs,1 for the V83G variant are similar to those of the A51V variant yielding a similar less favorable pKC for these two variants (Table 6). For all three variants, the parameters (kf, kb, pKH and pKC) obtained for kobs,2 are similar. These data indicate that the dynamics of Ω-loop D is bipartite with the conformational dynamics corresponding to kobs,1 being more sensitive to mutation. Our studies of an A81I variant of yeast iso-1-cytochrome c were consistent with assignment of the alkaline conformer affected by this mutation to the one with Lys79 bound to the heme.40 Thus, it is tempting to assign kobs,1 to the Lys79-mediated alkaline transition. However, as noted in our report on the I81A and V83G variants,64 additional mutagenesis studies would be required to confirm this assignment. In the case of the Ω-loop D variants, the stability of the part of Ω-loop D that mediates the dynamics corresponding to kobs,1 is affected, whereas for Ω-loop C variants, it appears that the pKH of the triggering ionization corresponding to kobs,1 is affected.
Effect of the A51V Substitution on the Fast Phase of the Alkaline Transition.
Of equal interest, is the observation that the fast phase reported for mammalian Cytc near pH 10 and above45, 62, 64, 82–84 appears near pH 8.5 for A51V Hu Cytc. This phase is often attributed to a transient process involving weakening of the heme-Met80 bond or displacement of Met80 by hydroxide (which could be viewed as a transient form of state V).83–84 EPR data for the transient formed by the Y48H variant at pH 11 show that it is a high spin species and thus relevant for peroxidase activity.62 The A51V variant appears to affect the fast phase differently than the G41S and Y48H variants. For the A51V variant, kobs,3 decreases from pH 9 to 10.5 (Figure 5), consistent with deprotonation occurring after formation of the fast phase species as given by eq 7,62, 81–82 where kobs decreases to the magnitude of kf for pH > pKH. This behavior also is
| (7) |
observed for the WT protein.62, 81–82 For both the G41S and Y48H variants, kobs,3 increases with pH consistent with deprotonation preceding formation of the fast phase species (eq 5).62 In this regard, the A51V variant is less perturbative than the G41S and Y48H variants relative to WT Hu Cytc. For A51V Hu Cytc, kobs,3 increases below pH 10 (pH 11 for WT) consistent with the pKH for the ionization of the group controlling formation of the transient species being less than 10.
In the study on the G41S and Y48H variants,62 it was noted that the pKa of His18 is expected to be near 11.85 These authors suggested that redistribution of electron density into the His18 heme ligand when the Met80 heme ligand dissociates during the conformational change associated with formation of the transient could occur for WT Hu Cytc. This electron density redistribution could decrease the pKa for deprotonation of the pyrrole-NH of His18 to near 9 causing release of a proton after the conformational change. This proposal also is consistent with our data for the A51V variant in Figure 5C. In the case of the Y48H and G41S variants,62 it was proposed that the greater dynamics of Ω-loop C provides direct access of bulk water to the pyrrole-NH of His18 and thus direct deprotonation of this group, with a pKa near 11,62, 85 occurs before Met80 dissociation leading to kinetics consistent with eq 5. The higher global stability of the A51V variant, compared to the G41S and Y48H variants, would likely decrease the dynamics of Ω-loop C for the A51V variant relative to the G41S and Y48H variants preserving the mechanism for the fast phase observed for WT Hu Cytc.
For the A51V variant, the transient phase is also observed in downward pH jump experiments indicating that there is a measureable equilibrium population of a species that is normally a kinetic transient. For yeast iso-1-Cytc, the midpoint pH for formation of state IV is ~8.637, 61 similar to that of A51V human Cytc. State V, which involves replacement of lysine-heme ligation with hydroxide, forms with a midpoint pH between 10.5 and 11 for the yeast protein.61 Given that our downward pH jump experiments start at pH 10.5, the presence of the fast phase in downward pH jumps may indicate equilibrium population of state V for the A51V variant at pH 10.5 and above.
Our data from downward pH jump experiments indicate that kb is 15 – 20 s−1 (Figure 5, Table S3). Based on eq 7, kf is near 25 s−1 (kobs,3 near pH 10.5, Figure 5C, Table S2), yielding KC ~ 1.5 for formation of the transient species. For the Y48H and G41S variants, kf for formation of the transient high spin species is ~150 s−1 and kb is ~20 s−1, yielding KC ~ 7.5 for these variants.62 For WT, kf ~ 20 s−1 and kb > 100 s−1 indicating that KC is < 0.2.62 Thus, the stability of the high spin transient follows the order WT < A51V < G41S ≈ Y48H. The amplitude, A3u, of the fast phase as a function of pH for A51V Hu Cytc (Figure 6) yields a midpoint pH, pH1/2, of 10.23 ± 0.03 when fit to the Henderson-Hasselbalch equation. For WT Hu Cytc, pH1/2 is ~11 for population of the transient.62 Thus, the transient species populates more effectively and is available at lower pH for the Ω-loop C variants. These data suggest that this high spin species will be nearer in energy to the native state structure at pH values above 7 than it is for the WT protein. For enzymes, often alternate conformations that are nearby in energy to the native state drive catalysis.86–87 Given that the fast phase corresponds to heme with an available coordination site,83 the greater availability of this species might also enhance the peroxidase activity of the THC4-linked variants, as proposed previously.62
Effect of the A51V Substitution on Acid Unfolding of Hu Cytc.
The acid unfolding of Cytc is a complex, multiproton process38, 65, 88–89 typically involving 2 – 3 protons.8, 73 However, cooperativity of acid unfolding is readily altered by amino acid substitutions.35, 39–40, 90 We have shown that a G83V substitution in iso-1-Cytc leads to biphasic acid unfolding which appears to be linked to increased peroxidase activity at mildly acidic pH relative to WT iso-1-Cytc.40 For G83V iso-1-Cytc, the loss in cooperativity of acid unfolding appears to weaken the heme-Met80 bond at mildly acidic pH. Acid unfolding of WT and A51V Hu Cytc exhibit monophasic behavior (Figure 7). The pH1/2 of the A51V variant is ~0.7 units higher than that of WT Hu Cytc. The decrease in stability of the native state toward acid unfolding to a high spin state (622 nm data) for the A51V variant relative to WT Hu Cytc is 1.8 ± 0.3 kcal/ mol (ΔΔG°’A51V/WT = 2.3RT[(pKC(A/N)(WT) - pKC(A/N)(A51V)] = −1.8 ± 0.3 kcal/mol).
The pH of intermembrane space of mitochondria is more acidic than the matrix or the cytosol,63 and cytosolic pH decreases further to 5.8 during apoptosis.91 The greater accessibility of the high spin conformer for the A51V variant versus the WT protein, as indicated by the acid unfolding data in Table 4, demonstrates the importance of Ω-loop C for controlling access to conformers competent for peroxidase activity at biologically relevant pH values. The increase in kcat for peroxidase activity for A51V versus that of WT Hu Cytc at pH 5 supports this contention (Figure 8). Even though the high spin conformer may not be populated at equilibrium at pH 5 (Figure 7), the A51V substitution brings the high spin state closer to the energy of the native state at pH 5, making it a more accessible alternate conformer that can drive catalysis.86–87 Recent work shows that horse Cytc bound to CL vesicles populates conformers with the high spin heme necessary for peroxidase activity as pH decreases below 7.92 Thus, the effect of the A51V substitution might be more pronounced at acidic pH in the presence of CL-containing membranes, further indicating the potential importance of Ω-loop C for controlling access to peroxidase-competent conformers. We note that the broader transition observed at 622 versus 695 nm may indicate the presence of a low spin His-heme intermediate during acid unfolding,66 which may somewhat lower access to high spin conformers below pH 7.
Effect of the A51V Substitution to Hu Cytc on Apoptotic Peroxidase Activity.
Previous studies on the peroxidase activity of the THC4-linked variants of Hu Cytc, G41S and Y48H, show that they induce 3- and 7-fold increases in peroxidase activity at pH 6.5 with 2,2-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) as substrate.20, 23, 62 These results are consistent with the magnitude of enhancement of peroxidase activity that we observe at pH 6 and 7 for the A51V variant with guaiacol as substrate. For the G41S variant,23 peroxidase activity was measured over a broad pH range. Consistent with our results for the A51V variant (Figure 8, Table 5), a significant increase in peroxidase activity relative to WT Hu Cytc was observed as pH decreased from 7.5 to 5. Given the lower pH in the intermembrane space and in the cytosol during apoptosis noted above, these studies on the naturally occurring Ω-loop C variants highlight the importance of this loop in modulating the peroxidase activity of Cytc early in apoptosis.
The increase in peroxidase activity of A51V Hu Cytc relative to that of WT Hu Cytc is lowest at pH 7 (~6-fold) and somewhat higher at both pH 6 and 8 (~7-fold, Table 5), suggesting that different mechanisms might provide access to peroxidase competent conformers above and below pH 7. Increased peroxidase activity is often linked to greater accessibility of alkaline conformers,18–21, 40 although the correlation is not perfect.23, 62 All three THC4-linked variants show a similar increase in the accessibility of the alkaline state (pH1/2 decreases by 1 unit, Table 3), which appears to be linked to a decrease in the pKH for the triggering ionization but ~1 unit (Table 6).62 Based on the X-ray structures of the G41S22–23 and Y48H62 variants, which both show disruptions to the hydrogen bond network around heme propionate 7 (HP7), a role for HP7 in this ionization was proposed.62 For the A51V variant, this network is less affected (Figure 1), but because of the increase in the backbone B-factors (Figure S2), the environment of HP7 may be affected for the A51V variant, too. A number of studies indicate that one of the heme propionates of mitochondrial Cytc has a pKa >9, while the other has a pKa < 4.5.8, 93–94 Initially, the pKa < 4.5 was assigned to heme propionate 7 (HP7) because of the nearby positively-charged Arg38 (see Figure 1).8, 93 However, mutagenesis studies on Arg38 of yeast iso-1-Cytc95 and the recent work on the G41S and Y48H variants suggest that HP7 may be the propionate with the pKa > 9. Given the interconnectedness of the buried hydrogen-bond network, it has been suggested that loss of a proton occurs from the network rather than from a specific group.62 Regardless of the exact source of the proton, the ionization corresponding to pKH appears to be more facile in THC4-linked Hu Cytc variants enhancing access to the alkaline state. This decrease in the pKH of the group that triggers the alkaline transition also likely makes alternate conformers of Cytc that are nearby in energy to the native state more accessible above pH 7 so they can promote peroxidase activity.86–87 The strong increase in peroxidase activity of the A51V variant relative to that of WT Hu Cytc at pH 8 is consistent with this contention.
Native-state H/D-exchange studies on horse Cytc,68 show that the stability of Ω-loop C decreases from a stability of ~4 kcal/mol at pH 7 to a stability of 2.5 – 3 kcal/mol at pH 5. The decrease in the stability of Ω-loop C across this pH range has been linked to protonation of either His26 or a heme propionate,68 presumably HP6. H/D exchange studies on the G41S and Y48H variants,19–20 indicate that this loop is destabilized. The greater susceptibility of the A51V variant to acid unfolding (Fig. 7, Table 4) may also be linked to destabilization of Ω-loop C. The lower local stability of Ω-loop C may lead to functionally-significant population of peroxidase-competent conformers for the THC4-linked variants of Hu Cytc as pH drops from 7 to 5 and His26 or a heme propionate is protonated.
CONCLUSION
Studies on A51V Hu Cytc, like the other THC4-linked variants of Hu Cytc, G41S and Y48H, provide considerable insight into the mechanism by which Cytc accesses peroxidase-competent conformers important in the early stages of apoptosis. All have lower global and local stability than that of WT Hu Cytc. The A51V variant also is less stable to acid unfolding. The kinetics of the alkaline conformational transition indicate that enhanced access to the heme crevice needed for peroxidase activity results from a decrease in the pKH of the trigger group for the alkaline conformational transition. The decrease in pKH caused by these substitutions to Ω-loop C may be due changes in the environment of HP7 or more broadly to their effects on release of a proton from the buried H-bond network of Hu Cytc. By contrast, substitutions in Ω-loop D appear not to affect the pKH of the triggering ionization and thus must destabilize Ω-loop D if enhanced peroxidase activity is to result. For A51V, the pKH decreases for only one alkaline conformer, suggesting that the dynamics of the loop are bipartite and have different triggering ionizations. A similar bipartite effect on the stability of Ω-loop D is observed for Ω-loop D variants.64 The enhancement of peroxidase activity below pH 7 appears to result from destabilization of Ω-loop C and may be linked to protonation of HP6 or His26. The latter effect may be more important for understanding the role of Ω-loop C in biologically relevant peroxidase activity given the pH of the intermembrane space.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NSF grant CHE-1609720 (B.E.B). The Macromolecular X-ray Diffraction Core Facility at the University of Montana was supported by a CoBRE grant from the National Institute of General Medical Sciences (P20GM103546). We thank the staff at the Stanford Synchrotron Radiation Lightsouce (SSRL) SMB for assistance with data collection. Use of the SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract DEAC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
Footnotes
Supporting Information
Figure S1 shows the hydrogen bond networks in Ω-loop D for chain C of the A51V Hu Cytc structure (PDB ID: 6DUJ). Figure S2 provides a plot of backbone B-factors versus sequence position for the A51V (PDB ID: 6DUJ) and the WT (PDB ID: 3ZCF) Hu Cytc structures. Figures S3 and S4 show typical pH jump stopped-flow data with fits for A51V Hu Cytc. Figure S5 shows acid unfolding data as monitored by absorbance spectroscopy for A51V and WT Hu Cytc. Table S1 provides the sequences of oligonucleotides used for mutagenesis. Tables S2 and S3 provide rate constants and amplitudes obtained from pH jump stopped-flow data for A51V Hu Cytc. Table S4 tabulates the heme distortion and its normal mode components for WT human Cytc and the three THC-linked variants extracted from X-ray structural data using the NSD program.
The authors declare no competing financial interest.
REFERENCES
- 1.Dickerson RE; Timkovich R, Cytochromes c In The Enzymes, 3rd ed.; Boyer PD, Ed. Academic Press: New York, 1975; Vol. 11, pp 397–547. [Google Scholar]
- 2.Hütteman M; Doan JW; Goustin A-S; Sinkler C; Mahapatra G; Shay J; Liu J; Elbaz H; Aras S; Grossman LI, et al. , Regulation of cytochrome c in respiration, apoptosis, neurodegeneration and cancer: the good, the bad and the ugly In Cytochromes b and c: biochemical properties, biological functions and electrochemical analysis, Thom R, Ed. Nova Science Publishers: New York, NY, 2014; pp 1–38. [Google Scholar]
- 3.Liu X; Kim CN; Yang J; Jemmerson R; Wang X Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X; Wang X Cytochrome c-mediated apoptosis. Annu. Rev. Biochem 2004, 73, 87–106. [DOI] [PubMed] [Google Scholar]
- 5.Kagan VE; Tyurin VA; Jiang J; Tyurina YY; Ritov VB; Amoscato AA; Osipov AN; Belikova NA; Kapralov AA; Kini V, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol 2005, 1, 223–232. [DOI] [PubMed] [Google Scholar]
- 6.Kapralov AA; Kurnikov IV; Vlasova II; Belikova NA; Tyurin VA; Basova LV; Zhao Q; Tyurina YY; Jiang J; Bayir H, et al. The hierarchy of structural transitions induced in cytochrome c by anionic phospholipids determines its peroxidase activation and selective peroxidation during apoptosis in cells. Biochemistry 2007, 46, 14232–14244. [DOI] [PubMed] [Google Scholar]
- 7.Zhou M; Li Y; Hu Q; Bai X.-c.; Huang W; Yan C; Scheres SHW; Shi Y Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015, 29, 2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore GR; Pettigrew GW, Cytochromes c: Evolutionary, Structural and Physicochemical Aspects. Springer-Verlag, New York: 1990. [Google Scholar]
- 9.Poulos TL Heme enzyme structure and function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McClelland LJ; Mou T-C; Jeakins-Cooley ME; Sprang SR; Bowler BE Structure of a mitochondrial cytochrome c conformer competent for peroxidase activity. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 6648–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClelland LJ; Steele HBB; Whitby FG; Mou T-C; Holley D; Ross JBA; Sprang SR; Bowler BE Cytochrome c can form a well-defined binding pocket for hydrocarbons. J. Am. Chem. Soc 2016, 138, 16770–16778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z; Matsuo T; Nagao S; Hirota S Peroxidase activity enhancement of horse cytochrome c by dimerization. Org. Biomol. Chem 2011, 9, 4766–4769. [DOI] [PubMed] [Google Scholar]
- 13.Morison IM; Cramer Bordé EM; Cheesman EJ; Cheong PL; Holyoake AJ; Fichelson S; Weeks RJ; Lo A; Davies SMK; Wilbanks SM, et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet 2008, 40, 387–389. [DOI] [PubMed] [Google Scholar]
- 14.Ledgerwood EC; Dunstan-Harrison C; Ong L; Morison IM CYCS gene variants associated with thrombocytopenia. Platelets 2019, 30, 672–674. [DOI] [PubMed] [Google Scholar]
- 15.De Rocco D; Cerqua C; Goffrini P; Russo G; Pastore A; Meloni F; Nicchia E; Moraes CT; Pecci A; Salviati L, et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [DOI] [PubMed] [Google Scholar]
- 16.Johnson B; Lowe GC; Futterer J; Lordkipanidzé M; MacDonald D; Simpson MA; Sanchez-Guiú I; Drake S; Bem D; Leo V, et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica 2016, 101, 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez-Paggi D; Hannibal L; Castro MA; Oviedo-Rouco S; Demicheli V; Tórtora V; Tomasina F; Radi R; Murgida DH Multifunctional cytochrome c: learning new tricks from an old dog. Chem. Rev 2017, 117, 13382–13460. [DOI] [PubMed] [Google Scholar]
- 18.Rajagopal BS; Edzuma AN; Hough MA; Blundell KLIM; Kagan VE; Kapralov AA; Fraser LA; Butt JN; Silkstone GG; Wilson MT, et al. The hydrogen-peroxide-induced radical behaviour in human cytochrome c–phospholipid complexes: implications for the enhanced pro-apoptotic activity of the G41S mutant. Biochem. J 2013, 456, 441–452. [DOI] [PubMed] [Google Scholar]
- 19.Karsisiotis AI; Deacon OM; Wilson MT; Macdonald C; Blumenschein TMA; Moore GR; Worrall JAR Increased dynamics in the 40–57 Ω-loop of the G41S variant of human cytochrome c promote its pro-apoptotic conformation. Sci. Rep 2016, 6, 30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deacon OM; Karsisiotis AI; Moreno-Chicano T; Hough MA; Macdonald C; Blumenschein TMA; Wilson MT; Moore GR; Worrall JAR Heightened dynamics of the oxidized Y48H variant of human cytochrome c increases its peroxidatic activity. Biochemistry 2017, 56, 6111–6124. [DOI] [PubMed] [Google Scholar]
- 21.Josephs TM; Liptak MD; Hughes G; Lo A; Smith RM; Wilbanks SM; Bren KL; Ledgerwood EC Conformational change and human cytochrome c function: mutation of residue 41 modulates caspase activation and destabilizes Met-80 coordination. J. Biol. Inorg. Chem 2013, 18, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liptak MD; Fagerlund RD; Ledgerwood EC; Wilbanks SM; Bren KL The proapoptotic G41S mutation to human cytochrome c alters the heme electronic structure and increases the electron self-exchange rate. J. Am. Chem. Soc 2011, 133, 1153–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Josephs TM; Morison IM; Day CL; Wilbanks SM; Ledgerwood EC Enhancing the peroxidase activity of cytochrome c by mutation of residue 41: implications for the peroxidase mechanism and cytochrome c release. Biochem. J 2014, 458, 259–265. [DOI] [PubMed] [Google Scholar]
- 24.Ong L; Morison IM; Ledgerwood EC Megakaryocytes from CYCS mutation-associated thrombocytopenia release platelets by both proplatelet-dependent and -independent processes. Br. J. Haematol 2017, 176, 268–279. [DOI] [PubMed] [Google Scholar]
- 25.Krishna MM; Lin Y; Rumbley JN; Englander SW Cooperative omega loops in cytochrome c: role in folding and function. J. Mol. Biol 2003, 331, 29–36. [DOI] [PubMed] [Google Scholar]
- 26.Olteanu A; Patel CN; Dedmon MM; Kennedy S; Linhoff MW; Minder CM; Potts PR; Deshmukh M; Pielak GJ Stability and apoptotic activity of recombinant human cytochrome c. Biochem. Biophys. Res. Commun 2003, 312, 733–740. [DOI] [PubMed] [Google Scholar]
- 27.Rosell FI; Mauk AG Spectroscopic properties of a mitochondrial cytochrome c with a single thioether bond to the heme prosthetic group. Biochemistry 2002, 41, 7811–7818. [DOI] [PubMed] [Google Scholar]
- 28.Pollock WB; Rosell FI; Twitchett MB; Dumont ME; Mauk AG Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry 1998, 37, 6124–6131. [DOI] [PubMed] [Google Scholar]
- 29.Goldes ME; Jeakins-Cooley ME; McClelland LJ; Mou T-C; Bowler BE Disruption of a hydrogen bond network in human versus spider monkey cytochrome c affects heme crevice stability. J. Inorg. Biochem 2016, 158, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cherney MM; Junior C; Bowler BE Mutation of trimethyllysine-72 to alanine enhances His79-heme mediated dynamics of iso-1-cytochrome c. Biochemistry 2013, 52, 837–846. [DOI] [PubMed] [Google Scholar]
- 31.Redzic JS; Bowler BE Role of hydrogen bond networks and dynamics in positive and negative cooperative stabilization of a protein. Biochemistry 2005, 44, 2900–2908. [DOI] [PubMed] [Google Scholar]
- 32.Wandschneider E; Hammack BN; Bowler BE Evaluation of cooperative interactions between substructures of iso-1-cytochrome c using double mutant cycles. Biochemistry 2003, 42, 10659–10666. [DOI] [PubMed] [Google Scholar]
- 33.Kristinsson R; Bowler BE Communication of stabilizing energy between substructures of a protein. Biochemistry 2005, 44, 2349–2359. [DOI] [PubMed] [Google Scholar]
- 34.Santoro MM; Bolen DW Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrysin using different denaturants. Biochemistry 1988, 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- 35.Baddam S; Bowler BE Thermodynamics and kinetics of formation of the alkaline state of a Lys 79->Ala/Lys 73->His variant of iso-1-cytochrome c. Biochemistry 2005, 44, 14956–14968. [DOI] [PubMed] [Google Scholar]
- 36.Margoliash E; Frohwirt N Spectrum of horse-heart cytochrome c. Biochem. J 1959, 71, 570–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosell FI; Ferrer JC; Mauk AG Proton-linked protein conformational switching: definition of the alkaline conformational transition of yeast iso-1-ferricytochrome c. J. Am. Chem. Soc 1998, 120, 11234–11245. [Google Scholar]
- 38.Dyson HJ; Beattie JK Spin state and unfolding equilibria of ferricytochrome c in acidic solutions. J. Biol. Chem 1982, 257, 2267–2273. [PubMed] [Google Scholar]
- 39.Bandi S; Baddam S; Bowler BE Alkaline conformational transition and gated electron transfer with a Lys 79 -> His variant of iso-1-cytochrome c. Biochemistry 2007, 46, 10643–10654. [DOI] [PubMed] [Google Scholar]
- 40.Lei H; Bowler BE Humanlike substitutions to Ω-loop D of yeast iso-1-cytochrome c only modestly affect dynamics and peroxidase activity. J. Inorg. Biochem 2018, 183, 146–156. [DOI] [PubMed] [Google Scholar]
- 41.Powell HR; Johnson O; Leslie AGW Autoindexing diffraction images with iMosflm. Acta Crystallogr., Sect. D: Biol. Crystallogr 2013, 69, 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans PR; Murshudov GN How good are my data and what is the resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vagin A; Teplyakov A Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- 44.Potterton L; Agirre J; Ballard C; Cowtan K; Dodson E; Evans PR; Jenkins HT; Keegan R; Krissinel E; Stevenson K, et al. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 2018, 74, 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nold SM; Lei H; Mou T-C; Bowler BE Effect of a K72A mutation on the structure, stability, dynamics and peroxidase activity of human cytochrome c. Biochemistry 2017, 56, 3358–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murshudov GN; Skubák P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson DP; Kiesow LA Enthalpy of decomposition of hydrogen peroxide by catalase at 25 °C (with molar extinction coefficients of H2O2 solutions in the UV). Anal. Biochem 1972, 49, 474–478. [DOI] [PubMed] [Google Scholar]
- 50.Noble RW; Gibson QH The reaction of ferrous horseradish peroxidase with hydrogen peroxide. J. Biol. Chem 1970, 245, 2409–2413. [PubMed] [Google Scholar]
- 51.Goldschmid O The effect of alkali and strong acid on the ultraviolet absorption spectrum of lignin and related compounds. J. Am. Chem. Soc 1953, 75, 3780–3783. [Google Scholar]
- 52.Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diederix REM; Ubbink M; Canters GW The peroxidase activity of cytochrome c-550 from Paracoccus versutus. Eur. J. Biochem 2001, 268, 4207–4216. [DOI] [PubMed] [Google Scholar]
- 54.Carugo O Atomic displacement parameters in structural biology. Amino Acids 2018, 50, 775–786. [DOI] [PubMed] [Google Scholar]
- 55.Cherney MM; Bowler BE Protein dynamics and function: making new strides with an old warhorse, the alkaline conformational transition of cytochrome c. Coord. Chem. Rev 2011, 255, 664–677. [Google Scholar]
- 56.Wilson MT; Greenwood C, The alkaline transition in ferricytochrome c In Cytochrome c: A Multidisciplinary Approach, Scott RA; Mauk AG, Eds. University Science Books: Sausalito, CA, 1996; pp 611–634. [Google Scholar]
- 57.Theorell H; Åkesson Å Studies on cytochrome c. II. The optical properties of pure cytochrome c and some of Its derivatives. J. Am. Chem. Soc 1941, 63, 1812–1818. [Google Scholar]
- 58.Maity H; Rumbley JN; Englander SW Functional role of a protein foldon - an Ω-loop foldon controls the alkaline transition in ferricytochrome c. Proteins: Struct. Funct. Bioinform 2006, 63, 349–355. [DOI] [PubMed] [Google Scholar]
- 59.Osheroff N; Borden D; Koppenol WH; Margoliash E Electrostatic interactions in cytochrome c. J. Biol. Chem 1980, 255, 1689–1697. [PubMed] [Google Scholar]
- 60.Soffer J; Schweitzer-Stenner R Near-exact enthalpy–entropy compensation governs the thermal unfolding of protonation states of oxidized cytochrome c. J. Biol. Inorg. Chem 2014, 19, 1181–1194. [DOI] [PubMed] [Google Scholar]
- 61.Döpner S; Hildebrandt P; Rosell FI; Mauk AG Alkaline conformational transitions of ferricytochrome c studied by resonance Raman spectroscopy. J. Am. Chem. Soc 1998, 120, 11246–11255. [Google Scholar]
- 62.Deacon OM; Svistunenko DA; Moore GR; Wilson MT; Worrall JAR Naturally occurring disease-related mutations in the 40−57 Ω-loop of human cytochrome c control triggering of the alkaline isomerization. Biochemistry 2018, 57, 4276–4288. [DOI] [PubMed] [Google Scholar]
- 63.Porcellia AM; Ghelli A; Zanna C; Pinton P; Rizzuto R; Rugolo M pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun 2005, 326, 799–804. [DOI] [PubMed] [Google Scholar]
- 64.Lei H; Nold SM; Jung Motta L; Bowler BE Effect of V83G and I81A substitutions to human cytochrome c on acid unfolding and peroxidase activity below neutral pH. Biochemistry 2019, 58, 2921–2933. [DOI] [PubMed] [Google Scholar]
- 65.Stellwagen E; Babul J Stabilization of the globular structure of ferricytochrome c by chloride in acidic solvents. Biochemistry 1975, 14, 5135–5140. [DOI] [PubMed] [Google Scholar]
- 66.Indiani C; de Sanctis G; Neri F; Santos H; Smulevich G; Coletta M Effect of pH on axial ligand coordination of cytochrome c from Methylophilus methylotrophus and horse heart cytochrome c. Biochemistry 2000, 39, 8234–8242. [DOI] [PubMed] [Google Scholar]
- 67.Bai Y; Sosnick TR; Mayne L; Englander SW Protein folding intermediates: native-state hydrogen exchange. Science 1995, 269, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishna MM; Maity H; Rumbley JN; Lin Y; Englander SW Order of steps in the cytochrome c folding pathway: evidence for a sequential stabilization mechanism. J. Mol. Biol 2006, 359, 1410–1419. [DOI] [PubMed] [Google Scholar]
- 69.Xu Y; Mayne L; Englander SW Evidence for an unfolding and refolding pathway in cytochrome c. Nat. Struct. Biol 1998, 5, 774–8. [DOI] [PubMed] [Google Scholar]
- 70.Maity H; Maity M; Englander SW How cytochrome c folds, and why: submolecular foldon units and their stepwise sequential stabilization. J. Mol. Biol 2004, 343, 223–33. [DOI] [PubMed] [Google Scholar]
- 71.Cherney MM; Junior CC; Bergquist BB; Bowler BE Dynamics of the His79-heme alkaline transition of yeast iso-1-cytochrome c probed by conformationally gated electron transfer with Co(II)bis(terpyridine). J. Am. Chem. Soc 2013, 135, 12772–12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bandi S; Bowler BE A cytochrome c electron transfer switch modulated by heme ligation and isomerization of a peptidyl-prolyl bond. Biopolymers (Pept Sci) 2013, 100, 114–124. [DOI] [PubMed] [Google Scholar]
- 73.Godbole S; Bowler BE Effect of pH on formation of a nativelike intermediate on the unfolding pathway of a Lys 73 -> His variant of yeast iso-1-cytochrome c. Biochemistry 1999, 38, 487–95. [DOI] [PubMed] [Google Scholar]
- 74.Godbole S; Dong A; Garbin K; Bowler BE A lysine 73->histidine variant of yeast iso-1-cytochrome c: evidence for a native-like intermediate in the unfolding pathway and implications for m value effects. Biochemistry 1997, 36, 119–126. [DOI] [PubMed] [Google Scholar]
- 75.Shortle D Staphylococcal nuclease: a showcase of m-value effects. Adv. Protein Chem. 1995, 46, 217–245. [DOI] [PubMed] [Google Scholar]
- 76.Jentzen W; Ma J-G; Shelnutt JA Conservation of the conformation of the porphyrin macrocycle in hemoproteins. Biophys. J 1998, 74, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kleingardner JG; Bren KL Biological significance and applications of heme c proteins and peptides. Acc. Chem. Res 2015, 48, 1845–1852. [DOI] [PubMed] [Google Scholar]
- 78.Kleingardner JG; Levin BD; Zoppellaro G; Andersson KK; Elliott SJ; Bren KL Influence of heme c attachment on heme conformation and potential. J. Biol. Inorg. Chem 2018, 23, 1073–1083. [DOI] [PubMed] [Google Scholar]
- 79.Amanda B Graves MTG, and Matthew D Liptak Measurement of heme ruffling changes in MhuD using UV−vis spectroscopy. J. Phys. Chem. B 2016, 120, 3844–3853. [DOI] [PubMed] [Google Scholar]
- 80.Jentzen W; Song X-Z; Shelnutt JA Structural characterization of synthetic and protein-bound porphyrins in terms of the lowest-frequency normal coordinates of the macrocycle. J. Phys. Chem. B 1997, 101, 1684–1699. [Google Scholar]
- 81.Davis LA; Schejter A; Hess GP Alkaline isomerization of oxidized cytochrome c. Equilibrium and kinetic measurements. J. Biol. Chem 1974, 249, 2624–2632. [PubMed] [Google Scholar]
- 82.Hoang L; Maity H; Krishna MM; Lin Y; Englander SW Folding units govern the cytochrome c alkaline transition. J. Mol. Biol 2003, 331, 37–43. [DOI] [PubMed] [Google Scholar]
- 83.Saigo S Kinetic and equilibrium studies of alkaline isomerization of vertebrate cytochromes c. Biochim. Biophys. Acta 1981, 669, 13–20. [DOI] [PubMed] [Google Scholar]
- 84.Kihara H; Saigo S; Nakatani H; Hiromi K; Ikeda-Saito M; Iizuka T Kinetic study of isomerization of ferricytochrome c at alkaline pH. Biochim. Biophys. Acta 1976, 430, 225–243. [DOI] [PubMed] [Google Scholar]
- 85.Gadsby PMA; Peterson J; Foote N; Greenwood C; Thomson AJ Identification of the ligand-exchange process in the alkaline transition of horse heart cytochrome c. Biochem. J 1987, 246, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Henzler-Wildman KA; Lei M; Thai V; Kerns SJ; Karplus M; Kern D A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [DOI] [PubMed] [Google Scholar]
- 87.Henzler-Wildman K; Kern D Dynamic personalities of proteins. Nature 2007, 450, 964–972. [DOI] [PubMed] [Google Scholar]
- 88.Robinson JB Jr.; Strottmann JM; Stellwagen E A globular high spin form of ferricytochrome c. J. Biol. Chem 1983, 258, 6772–6776. [PubMed] [Google Scholar]
- 89.Drew HR; Dickerson RE The unfolding of the cytochromes c in methanol and acid. J. Biol. Chem 1978, 253, 8420–8427. [PubMed] [Google Scholar]
- 90.Baddam S; Bowler BE Mutation of asparagine 52 to glycine promotes the alkaline form of iso-1-cytochrome c and causes loss of cooperativity in acid unfolding. Biochemistry 2006, 45, 4611–4619. [DOI] [PubMed] [Google Scholar]
- 91.Nilsson C; Kågedal K; Johansson U; Öllinger K Analysis of cytosolic and lysosomal pH in apoptotic cells by flow cytometry. Methods Cell Sci. 2003, 25, 185–194. [DOI] [PubMed] [Google Scholar]
- 92.Milorey B; Malyshka D; Schweitzer-Stenner R pH dependence of ferricytochrome c conformational transitions during binding to cardiolipin membranes: evidence for histidine as the distal ligand at neutral pH. J. Phys. Chem. Lett 2017, 8, 1993–1998. [DOI] [PubMed] [Google Scholar]
- 93.Hartshorn RT; Moore GR A denaturation-induced proton-uptake study of horse ferricytochrome c. Biochem. J 1989, 258, 595–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tonge P; Moore GR; Wharton CW Fourier-transform infra-red studies of the alkaline isomerization of mitochondrial cytochrome c and the ionization of carboxylic acids. Biochem. J 1989, 258, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cutler RL; Davies AM; Creighton S; Warshel A; Moore GR; Smith M; Mauk AG Role of arginine-38 in regulation of the cytochrome c oxidation-reduction equilibrium. Biochemistry 1989, 28, 3188–3197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.