

Abstract
Distinct approaches to synthesize bis-azine biaryls are in demand as these compounds have multiple applications in the chemical sciences and are challenging targets for metal-catalyzed cross-coupling reactions. Most approaches focus on developing new reagents as the formal nucleophilic coupling partner that can function in metal-catalyzed processes. We present an alternative approach using pyridine and diazine phosphines as nucleophilic partners and chloroazines where the heterobiaryl bond is formed via a tandem SNAr-phosphorus ligand-coupling sequence. The heteroaryl phosphines are prepared from chloroazines and are bench-stable solids. A range of bis-azine biaryls can be formed from abundant chloroazines using this strategy that would be challenging using traditional approaches. A one-pot cross-electrophile coupling of two chloroazines is feasible, and we also compared the phosphorus-mediated strategy with metal-catalyzed coupling reactions to show advantages and compatibility.
Graphical Abstract
INTRODUCTION
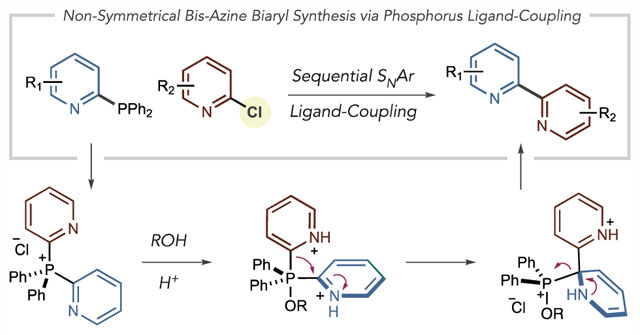
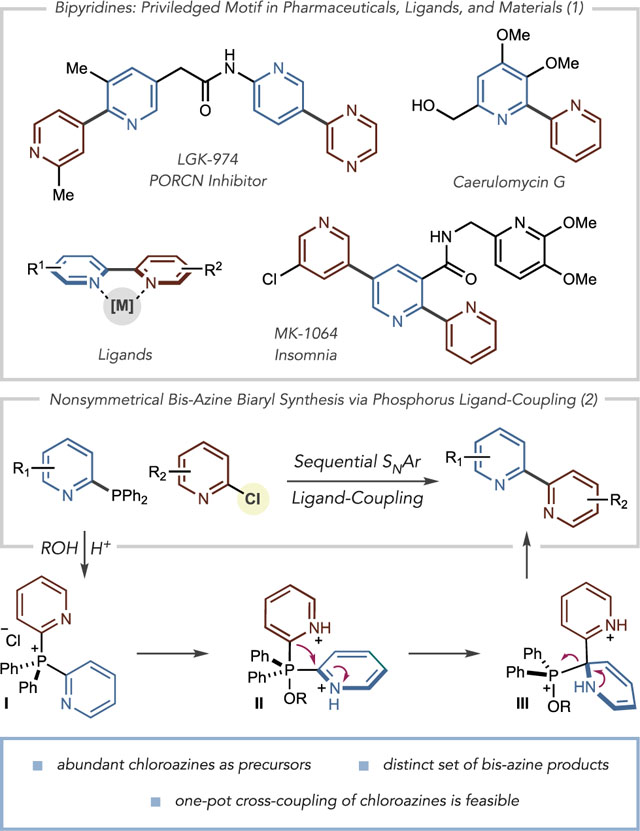
Bisazine biaryls are widely applied in the chemical sciences with applications in drug development, as ligands for metal complexes and as components of materials (eq 1).1 Transition-metal-catalyzed cross-coupling reactions are most commonly used to synthesize nonsymmetrical variants; however, pyridine and diazine couplings represent some of the most challenging cases for this class of reactions.2 Readily available and stable cross-coupling precursors are often limiting factors; haloazines are relatively abundant from commercial sources compared to nucleophilic partners such as pyridyl zincs, stannanes, silanes and Grignards that often have to be prepared and carefully handled.3 Suzuki–Miyaura reactions are the most widely applied in medicinal chemistry, as azine boronic acids are typically bench-stable powders.4 Despite this advantage, halopyridines remain dramatically more commercially available than pyridine boronic acids, and 2-pyridyl boronic acids have a proclivity to decompose during cross-coupling reactions.5,6 Herein we show that heteroarylphosphines can serve as nucleophilic partners and couple with haloazines where the biaryl bond is formed via a phosphorus ligand-coupling reaction.7 These phosphines are straightforward to prepare from haloazines and are bench-stable. Preliminary examples of a net cross-electrophile coupling are shown via a one-pot coupling of two haloazines.
Several groups have developed alternatives to azine boronic acids for metal-catalyzed cross-coupling reactions. Potassium trifluoroborate salts and MIDA boronates display enhanced stability compared to boronic acids, but a general platform for azine-azine coupling has yet to be established using these reagents.8 Willis and co-workers advanced this area by demonstrating that pyridyl sulfinate salts and allyl sulfones are viable alternatives for Suzuki couplings.9 Recently, we reported an alternative strategy to form bis-azine biaryls by constructing bis-azine phosphonium salts from C–H precursors and then exploiting phosphorus ligand-coupling reactions to form the biaryl bond.10 However, the inherent selectivity of the two C–P bond-forming steps restrict access to bis-azine biaryls within some isomeric patterns such as 2,2′-bipyridines (vide infra). Furthermore, certain functional groups and azine substitution patterns are not tolerated. We envisioned a strategy analogous to metal-catalyzed cross-coupling using chloroazines and heteroaryl phosphines as precursors (eq 2). This approach could overcome limitations of current metal-catalyzed processes as well as result in a suite of bis-azine biaryls that are beyond the scope of our original report. Pyridyl and diazinyl phosphines, prepared in one step from chloroazines, can function as nucleophiles and couple with a second chloroazine via a tandem SNAr-ligand-coupling sequence. The mechanism involves forming a bis-heteroarylphosphonium salt (I) that is intercepted in acidic alcohol or aqueous solutions to form a P(V) alkoxyphosphorane intermediate (II). In line with our previous studies, we anticipated that subsequent ligand-coupling would proceed via an asynchronous process, involving a dearomatized species III as a discrete intermediate.
HISTORICAL CONTEXT AND BACKGROUND
Reports of forming bipyridines via phosphorus ligand-coupling reactions date back to the 1940s with seminal works by Mann, Newkome, Uchida, and Oae showing the feasibility of the process using several distinct precursors (Scheme 1).11 In all cases, the ligand-coupling step occurs after forming a P(V) alkoxyphosphorane intermediate, although the reagent-trigger can differ markedly. The earliest report of using halopyridines as precursors for phosphorus ligand-coupling comes from Mann in 1948 (Scheme 1A) where di- and tripyridyl phosphines were treated with methyl iodide in methanol and resulted in the formation of 2,2′-bipyridinium dimers.11a The authors propose a free radical mechanism, but subsequent developments suggest this process occurs via a phosphorus ligand-coupling process. Despite this early report, the next use of this strategy came 30 years later from Newkome (Scheme 1B). By reacting a phosphine oxide containing two 2-pyridyl groups with a sodium alkoxide at 140 °C, a 2,2′-bipyridine product formed.11b Following this report, Uchida and Oae showed that treating a tripyridyl phosphonium salt with acidic water could result in symmetrical bipyridines (Scheme 1C).11c In a different approach, they also demonstrated that organometallic reagents could be added to phosphine oxides to trigger ligand-coupling (Scheme 1D).11d
Scheme 1.

Key Developments in Phosphorus Ligand-Coupling Reactions To Synthesize Bipyridines
Despite these seminal contributions, bipyridine synthesis via phosphorus ligand-coupling strategies received little further attention and, consequently, never found widespread use. Two principal factors can account for the absence of subsequent developments. First, the ligand-coupling precursors lack generality toward a range of pyridine substrates via the methods laid out in the reports described above. Specifically, the methods to form these precursors can require multiple steps and are not practical for cross-coupling reactions of two pyridine coupling partners. Second, and as a result, the reports showed only dimerizations and macrocyclizations that did not highlight the potential of these reactions for pyridine-pyridine cross-coupling. Our laboratory was interested in exploiting these phosphorus ligand-coupling reactions, but, in contrast to the previous reports, we sought to devise a practical system where widely available halopyridines (and other haloazines) partners are coupled to form nonsymmetrical bipyridines. In this way, the disconnection logic would replicate metal-catalyzed cross coupling; the nucleophilic partner is a pyridyl phosphine, derived from the corresponding halide in one step. The electrophilic partner is a halopyridine, and phosphorus ligand-coupling replaces the action of a transition metal catalyst in the biaryl bond-forming step.
Scheme 2 shows specific limitations of our previous approach using azine C–H bonds as precursors and potential solutions using chloroazines.10 In the former case, two C–P bond-forming reactions are required to form bis-heterocyclic phosphonium salt I (Scheme 2A). Fragmentable phosphine IV is used with Tf2O and DBU as a base to form an azinylphosphine (not shown); the process is then repeated with the second azine coupling partner to form salt I. Under these conditions, common functional groups such as alcohols, phenols, and alkyl-substituted amides12,13 are not tolerated due to their propensity to react with Tf2O (Scheme 2B). Pyridines with certain substitution patterns are also not amenable; 2,6-disubstituted pyridines and 2-CF3 pyridines are unsuccessful, as the sp2 nitrogen atom is either too sterically crowded or reduced in nucleophilicity such that reaction with Tf2O is ineffective. For pyridines and quinolines, both C–P bond-forming reactions are inherently 4-selective, whereas 2-position selectivity can only be achieved when the 4-position is blocked.14 Using this sequence to prepare 2,2′-bipyridines, for example, requires 4-position substituents to be present to ensure the correct positional selectivity. Similarly, C–P bond-formation in diazines is also inherently selective; pyrimidines react at the 4-position in this manifold, and this selectivity restricts the possible isomeric patterns in the resulting bis-azine biaryl. The strategy in Scheme 2C can potentially overcome these issues by using SNAr reactions with chloroazines for each C–P bond-forming event. Scheme 2B shows classes of readily available chloroazines where the previous functional group incompatibilities and regiomeric constraints can be addressed.
Scheme 2.

Limitations of Heterobiaryl Synthesis Using C–H Precursors and an Alternative Strategy via Chloroazines
RESULTS AND DISCUSSION
Synthesis of Heteroarylphosphines.
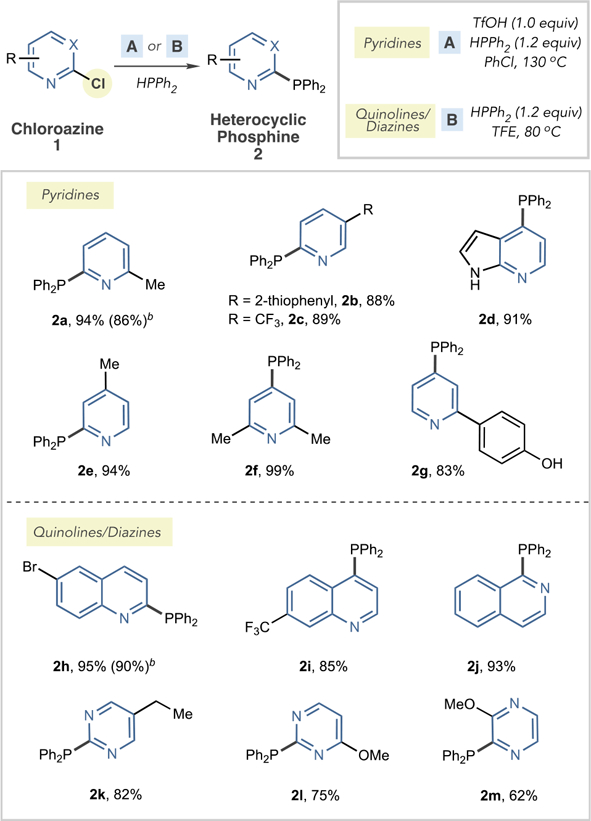
We first developed conditions for SNAr couplings between chloroazines and HPPh2 and intentionally prepared heteroarylphosphines that were precluded from our previous study by steric, electronic, or isomeric constraints (Table 1).10,15 For 2- and 4-chloropyridines, heating in chlorobenzene at 130 °C with one equivalent of TfOH was effective, and pyridylphosphines 2a–2g were formed in high yields (condition set A), addressing the restrictions mentioned above in C–P bond formation (vide supra). For more SNAr active substrates, such as chloroquino-lines, chloroisoquinolines, and chlorodiazines,16 trifluoroethanol (TFE) at 80 °C was sufficient and acid activation was not required (condition set B). Notably, these conditions allow access to phosphines at the 2-position of quinolines and pyrimidines. We found that these heteroarylphosphines are bench-stable, but, as a precaution, they were kept in a−20 °C refrigerator for long-term storage.
Table 1.
SNAr Reactions to Form Heteroarylphosphinesa
|
Isolated yields from 2.0 mmol scale reactions.
Run on 10 mmol scale.
Tandem SNAr-Ligand-Coupling Sequences.
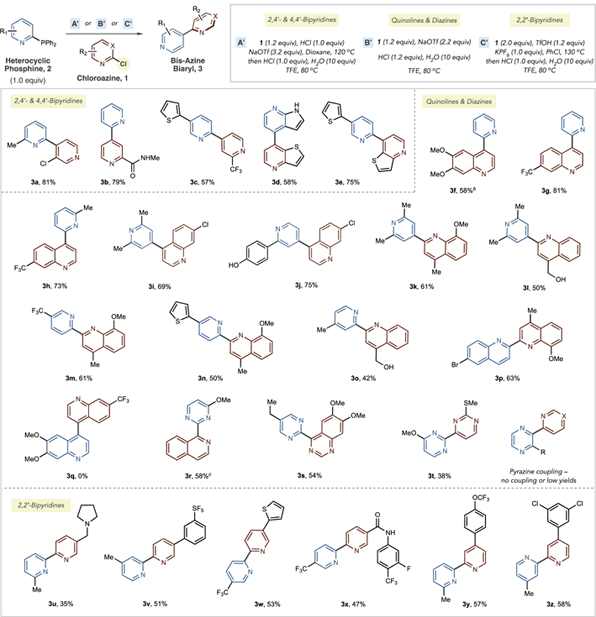
With a set of heteroaryl phosphines in hand, we next developed one-pot procedures to form bis-azine biaryls via the tandem SNAr-ligand-coupling sequence (Table 2). We found that coupling reactions could be separated into three categories depending on the ease of the SNAr process. First, 2,4′-and 4,4′-bipyridines were synthesized in a two-stage process; heating the phosphine and chloropyridine in dioxane at 120 °C with one equivalent each of HCl and NaOTf drives the SNAr reaction to completion and forms the bis-heterocyclic phosphonium salt.17 Then, a further equivalent of HCl, 10 equiv of H2O, and TFE are added, and heating at 80 °C is optimal for the ligand-coupling process (condition set A′). Without NaOTf, the SNAr reaction does not reach full conversion, and we presume that bis-azine phosphonium salt formation is promoted by anion exchange and precipitation of NaCl. By employing these conditions, a set of 2,4- and 4,4′-bipyridines are formed in reasonable yields (3a–3e). Bipyridine 3b is notable as the alkyl amide group was incompatible with our previous protocol. Second, chloroquino-lines and chlorodiazines couple heteroarylphosphines using a single-stage protocol. In line with the observations in Table 1, these heterocycles undergo more facile SNAr processes and the tandem SNAr-ligand-coupling sequence is performed in TFE at 80 °C with H2O, HCl, and NaOTf as additives (condition set B′). By employing these conditions, a set of 2,4- and 4,4′-pyridine-quinolines are formed (3f–3l) and include examples of pyridines with 2,6-disubstitution, 4-position C–H bonds, as well as products containing phenols and alcohols. Furthermore, 2,2-pyridine-quinoline systems 3m–3o can also be synthesized that are challenging to prepare via Suzuki–Miyaura couplings (vide infra). Similarly, 3p is a 2,2′-biquinoline that includes a C–Br bond that would typically be active in transition metal-catalyzed processes. However, an attempt to form 4,4′-biquinoline 3q failed; we presume unfavorable steric interactions occur in the ligand-coupling transition state in this case. Diazines can be used as both formal nucleophilic phosphines and electrophilic chlorides; a 2-pyrimidylphosphine was coupled with a chloroisoquinoline in moderate yield (3r), and diazine–diazine coupling is possible using this approach (3s–3t). At this point, pyrazines perform poorly as coupling partners; a short survey of pyridine, quinoline, and diazine partners resulted in no coupled products or in poor yields (see the Supporting Information). Third, we targeted 2,2′-bipyridines using this strategy due to the challenges in synthesizing these systems using Suzuki–Miyaura cross-coupling reactions (vide supra). Condition set A′ was modified to C′, where changes in solvent, acid, and additive resulted in higher yields of bipyridine products (see the Supporting Information). Examples 3u–3z show a variety of isomeric patterns as well as substituents such as aliphatic amines, SF5 groups, thiophenes, amides, and chlorides. As with all other examples in Table 2, these bipyridine products could not be formed using our previous approach from C–H precursors.10 The moderate yields approximate those in our previous report involving 2,2′-bipyridine couplings, and further investigations into the mechanism of this process are ongoing.
Table 2.
Tandem SNAr-Ligand-Coupling Reactions To Form Nonsymmetrical Bis-Azine Biarylsa
|
Isolated yields are reported.
3f was formed using a two-stage protocol: 1.2 equiv of chloroazine, 2.2 equiv of NaOTf, TFE, 80 °C then 1.2 of equiv HCl and 10 equiv of water were added.
TfOH used instead of HCl.
We developed a one-pot, net cross-electrophile coupling of two chloroazines in a stage-wise protocol involving sequential addition of reagents (Table 3).18 We hypothesized that the heteroarylphosphine could be formed in situ and reacted with the second chloroazine via a tandem SNAr-ligand-coupling sequence from Table 2. The four examples, 3e, 3f, 3i, and 3y, in Table 3 show that pyridine–pyridine and pyridine–quinoline couplings are viable using this process, forming products in reasonable yields, with 2,2′-, 2,4′-, and 4,4′-conectivity between the two heterocycles. Current efforts are focused on increasing the efficiency of these process as well as expanding the scope to other azine-azine couplings.
Table 3.
One-Pot Cross-Coupling of Chloroazinesa
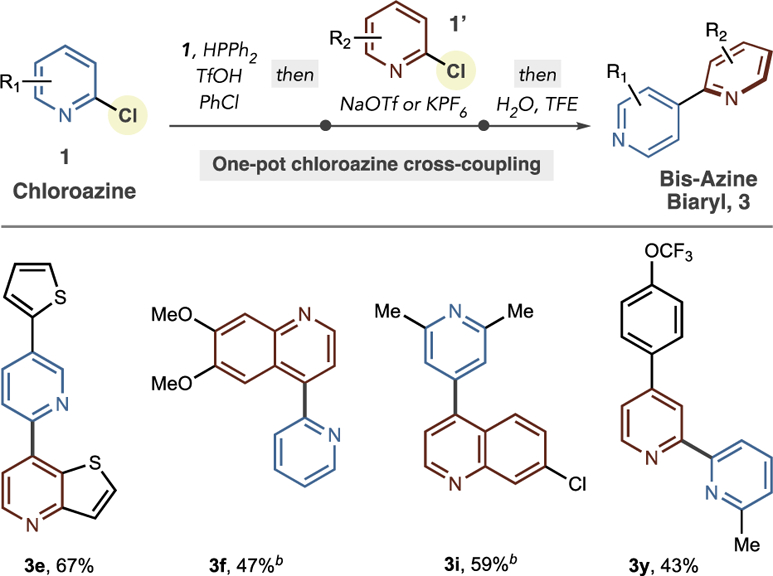
|
Isolated yields are reported.
TFE/toluene 1:1 used in the final stage.
In Scheme 3, we examine other important attributes of the ligand-coupling protocol for bis-azine biary synthesis. First, running the reaction on a gram scale showed minor yield loss when forming 3a (Scheme 3A). Second, there was no change in efficiency when conducting the reaction under an air or nitrogen atmosphere (Scheme 3B). Third, we combined the SNAr method to form heteroarylphosphines with our previously reported protocol to form phosphonium salts from C–H precursors (Scheme 3C). By combining the two approaches, bipyridines can potentially be formed that are outside of the scope of either method alone. As an example, we used phosphine 2f formed from the SNAr protocol in Table 1 (and not amenable to synthesis via the C–H protocol as 2,6-disubstitution prevents reaction with Tf2O), and Loratadine to form bis-pyridyl phosphonium salt 4; subsequent ligand-coupling using acidic ethanol resulted in novel 4,4′-bipyridine 3ab. Interchanging both methods either to make heteroaryl phosphines or for phosphonium salt formation-coupling sequences can, therefore, result in distinct structural and functional group diversity in the bis-azine biaryl products.
Scheme 3. Other Attributes of the Ligand-Coupling Processa.

aIsolated yields are reported. bYields calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
GUIDELINES AND LIMITATIONS
It is important to consider which of the chloroazine partners to convert into the heteroaryl phosphine in the first stage of the coupling sequence. In general, it is advisable to transform the least SNAr active chloroazines into the phosphine (Figure 1A). In that way, lower temperatures are applicable for the salt-formation-ligand-coupling step. For example, to make a 2,2′-pyridine-quinoline, the optimal order is to first form a pyridyl phosphine and then employ this phosphine as a nucleophile with the cholorquinoline (condition set B′ from Table 2).
Figure 1.

Guidelines and limitations for heteroaryl phosphine formation and SNAr-ligand-coupling sequences.
In the opposite order, using a quinolyl phosphine as a nucleophile, condition set C′ is required where phosphonium salt formation requires 130 °C in chlorobenzene. Under these conditions, we have observed side reactions that can reduce yields, including dehalogenation of the chloroazine partner and a chloride-phosphorus metathesis process shown in Figure 1B. We also observed the same metathesis in condition set A′ if we did not store NaOTf in a glovebox or desiccator, presumably because of the influence of water during salt formation. Figure 1C shows problematic substrates for the ligand-coupling process. We have observed that alkoxy substituents are prone to dealkylation during the reaction, although this pathway is mitigated by using TfOH rather than HCl in the ligand coupling step. Bromides and iodides at the 3-position of pyridines undergo dehalogenation during heteroaryl phosphine and phosphonium salt formation. Pyridyl fluorides result in reduced yields when employed in place of chlorides or bromides in the tandem SNAr-ligand-coupling step; we believe that the liberated fluoride anion reacts with the phosphonium salt and C–H products are observed in the LCMS of the crude reaction mixture. Certain functional groups and substitution patterns result in slower ligand-coupling or lower yields of bipyridine products. When one of the pyridine coupling partners contains an electron-donating group, such as hydroxy, alkoxy, and amino groups, reaction rates are reduced. We also found that 2-chloro-6-aryl pyridines and 2-chloro-3-substituted pyridines are sluggish in the coupling step and yields suffer as a consequence. For each additional basic nitrogen atom in the substrate, an extra equivalent of acid is required.19 Finally, the mechanistic principles of this approach dictate that SNAr-inactive substrates, such as 3-halopyridines and 5-halopyrimidines, are not viable.
COMPARISON TO PALLADIUM-CATALYZED CROSS-COUPLING REACTIONS
We chose three cases where the phosphorus ligand-coupling approach for bis-azine biaryl synthesis could have advantages over, or be complementary to, metal-catalyzed cross-coupling reactions. Scheme 4B compares 2,2′-bipyridine synthesis to Suzuki couplings using boronic acid surrogates that are typically more stable under cross-coupling conditions.8 Using 2-pyridyl BF3K salts or MIDA boronates, we obtained low to moderate yields of 3aa, whereas the reaction was more efficient using diphenyl-2-pyridylphosphine.8b,20,21 These results reinforce that 2,2-bipyridine synthesis via Pd-Suzuki reactions are challenging, although we acknowledge that further reaction development could address these problems. Next, we examined whether the PPh2 group could proceed through a Pd cross-coupling reaction in the presence of another nucleophilic partner. Using Fu et al.’s conditions for Suzuki coupling, where PCy3 is a ligand for palladium, biaryl 2n formed in a reasonable yield with the phosphine group available for coupling in a subsequent step (Scheme 4B).22,23 Selectively transforming polyhalogenated azines is an advantage that the phosphorus-mediated approach could hold over metal-catalyzed reactions. We selected quino-line dihalides 5 and 6 to test this hypothesis and used reported Pd-catalyzed methods that specifically target haloazines. A Negishi coupling reaction, using conditions reported by Luzung and Yin, resulted in a mixture of products in favor of bis-bipyridine 3af, even though a 1.0:1.2 ratio of halide to heteroaryl zinc was employed.24 A Stille reaction was selective for 4-isomer 3ac, although products 3ae and 3af were present in the reaction mixture.25 The phosphorus-mediated approach, on the other hand, due to its propensity to select the most SNAr active position on the scaffold resulted in a single regioisomer 3ac.
Scheme 4. Compatibility and Comparison to Metal-Catalyzed Cross-Coupling Reactionsa.

aYields and product ratios calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. bIsolated yield shown.
CONCLUSIONS
In summary, we have developed an alternative strategy to form bis-azine biaryls by coupling azinylphosphines with chloroazines. The reaction proceeds via a tandem SNAr-ligand-coupling sequence, and the heteroaryl phosphines are bench-stable solids that are prepared from chloroazines in a separate step. A diverse set of bis-azine biaryl products can be formed, including substitution patterns such as 2,2′-bipyridines, that are challenging for traditional metal-catalyzed approaches. Abundant chloroazines, simple protocols, and valuable bis-azine biaryl products make this approach useful for medicinal chemists.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by The National Institutes of Health (NIGMS) under Award Number R01 GM124094.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08504.
Experimental procedures and spectral data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Liu J; Pan S; Hsieh MH; Ng N; Sun F; Wang T; Kasibhatla S; Schuller AG; Li AG; Cheng D; Li J; Tompkins C; Pferdekamper AM; Steffy A; Cheng J; Kowal C; Phung V; Guo G; Wang Y; Graham MP; Flynn S; Brenner JC; Li C; Villarroel MC; Schultz PG; Wu X; McNamara P; Sellers WR; Petruzzelli L; Boral AL; Seidel HM; McLaughlin ME; Che J; Carey TE; Vanasse G; Harris JL Targeting Wnt-Driven Cancer through the Inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roecker AJ; Mercer SP; Schreier JD; Cox CD; Fraley ME; Steen JT; Lemaire W; Bruno JG; Harrell CM; Garson SL; Gotter AL; Fox SV; Stevens J; Tannenbaum PL; Prueksaritanont T; Cabalu TD; Cui D; Stellabott J; Hartman GD; Young SD; Winrow CJ; Renger JJ; Coleman PJ Discovery of 5′-Chloro-N-[(5,6-Dimethoxypyridin-2-Yl)Methyl]-2,2′:5′,3′-Terpyridine-3′-Carboxamide (MK-1064): A Selective Orexin 2 Receptor Antagonist (2-SORA) for the Treatment of Insomnia. ChemMedChem 2014, 9, 311–322. [DOI] [PubMed] [Google Scholar]; (c) Fu P; Wang S; Hong K; Li X; Liu P; Wang Y; Zhu W Cytotoxic Bipyridines from the Marine-Derived Actinomycete ActinoalloteichusCyanogriseus WH1–2216–6.J. Nat. Prod 2011, 74, 1751–1756. [DOI] [PubMed] [Google Scholar]; (d) Fletcher NC Chiral 2,2′-Bipyridines: Ligands for Asymmetric Induction. J. Chem. Soc., Perkin Trans. 1 2002, 0, 1831–1842. [Google Scholar]; (e) Kaes C; Katz A; Hosseini MW Bipyridine:The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2’-Bipyridine Units. Chem. Rev 2000, 100, 3553–3590. [DOI] [PubMed] [Google Scholar]; (f) Roberts JM; Fini BM; Sarjeant AA; Farha OK; Hupp JT; Scheidt KA Urea Metal–Organic Frameworks as Effective and Size-Selective Hydrogen-Bond Catalysts. J. Am. Chem. Soc 2012, 134, 3334–3337. [DOI] [PubMed] [Google Scholar]; (g) Suh MP; Cheon YE; Lee EY Syntheses and Functions of Porous Metallosupramolecular Networks. Coord. Chem. Rev 2008, 252, 1007–1026. [Google Scholar]; (h) Corma A; García H; LlabrésiXamena FX Engineering Metal-Organic Frameworks for Heterogeneous Catalysis. Chem. Rev 2010, 110, 4606–4655. [DOI] [PubMed] [Google Scholar]; (i) Newkome GR; Patri AK; Holder E; Schubert US Synthesis of 2,2′-Bipyridines: Versatile Building Blocks for Sexy Architectures and Functional Nanomaterials. Eur. J. Org. Chem 2004, 2004, 235–254. [Google Scholar]
- (2).Campeau L-C; Fagnou K Applications of and Alternatives to π-Electron-Deficient Azine Organometallics in Metal Catalyzed Cross-Coupling Reactions. Chem. Soc. Rev 2007, 36, 1058–1068. [DOI] [PubMed] [Google Scholar]
- (3).(a) Colombe JR; Bernhardt S; Stathakis C; Buchwald SL; Knochel P Synthesis of Solid 2-Pyridylzinc Reagents and Their Application in Negishi Reactions. Org. Lett 2013, 15, 5754–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto Y; Azuma Y; Mitoh H General Method for Synthesis of Bipyridines: Palladium Catalyzed Cross-Coupling Reaction of Trimethylstannyl-Pyridines with Bromopyridines. Synthesis 1986, 1986, 564–565. [Google Scholar]; (c) Blakemore DC; Marples LA Palladium(0)-Catalysed Cross-Coupling of 2-Trimethylsilylpyridine with Aryl Halides. Tetrahedron Lett 2011, 52, 4192–4195. [Google Scholar]
- (4).Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- (5).A Scifinder search of the CAS database for pyridyl boronic acids (all isomers) resulted in 2686 commercially available pyridyl boronic acids versus 747 322 commercially available chloropyridines.
- (6).Cox PA; Reid M; Leach AG; Campbell AD; King EJ; Lloyd-Jones GC Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion. J. Am. Chem. Soc 2017, 139, 13156–13165. [DOI] [PubMed] [Google Scholar]
- (7).(a) Finet J-P Ligand Coupling Reactions with Heteroaromatic Compounds; Pergamon, 1998; Vol. 18, Chapter 4. [Google Scholar]; (b) Oae S; Uchida Y Ligand-Coupling Reactions of Hypervalent Species. Acc. Chem. Res 1991, 24, 202–208. [Google Scholar]
- (8).(a) Molander GA; Canturk B; Kennedy LE Scope of the Suzuki-Miyaura Cross-Coupling Reactions of Potassium Heteroaryltrifluoroborates. J. Org. Chem 2009, 74, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knapp DM; Gillis EP; Burke MD A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lennox AJJ; Lloyd-Jones GC Selection of Boron Reagents for Suzuki–Miyaura Coupling. Chem. Soc. Rev 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (9).(a) Markovic T; Rocke BN; Blakemore DC; Mascitti V; Willis MC Pyridine Sulfinates as General Nucleophilic Coupling Partners in Palladium-Catalyzed Cross-Coupling Reactions with Aryl Halides. Chem. Sci 2017, 8, 4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Markovic T; Murray PRD; Rocke BN; Shavnya A; Blakemore DC; Willis MC Heterocyclic Allylsulfones as Latent Heteroaryl Nucleophiles in Palladium-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2018, 140, 15916–15923. [DOI] [PubMed] [Google Scholar]
- (10).Hilton MC; Zhang X; Boyle BT; Alegre-Requena JV; Paton RS; McNally A Heterobiaryl Synthesis by Contractive C–C Coupling via P(V) Intermediates. Science 2018, 362, 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Mann FG; Watson J Conditions of Salt Formation in Polyamines and Kindred Compounds. Salt Formation in the Tertiary 2-Pyridylamines, Phosphines, and Arsines. J. Org. Chem 1948, 13, 502–531. [Google Scholar]; (b) Newkome GR; Hager DC A New Contractive Coupling Procedure. Convenient Phosphorus Expulsion Reaction. J. Am. Chem. Soc 1978, 100, 5567–5568. [Google Scholar]; (c) Uchida Y; Kozawa H Formation of 2,2′-Bipyridyl by Ligand Coupling on the Phosphorus Atom. Tetrahedron Lett 1989, 30, 6365–6368. [Google Scholar]; (d) Uchida Y; Onoue K; Tada N; Nagao F; Kozawa H; Oae S Reactions of 2-Pyridyl Substituted Phosphine Oxides and Phosphonium Salts with Organo-metallic Reagents and in Aqueous Media. Heteroat. Chem 1990, 1, 295–306. [Google Scholar]
- (12).We have found that N-aryl amides are generally compatible with Tf2O whereas N-alkyl amides can react competitively with azines.
- (13).Barbe G; Charette AB Highly Chemoselective Metal-Free Reduction of Tertiary Amides. J. Am. Chem. Soc 2008, 130, 18–19. [DOI] [PubMed] [Google Scholar]
- (14).(a) Hilton MC; Dolewski RD; McNally A Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc 2016, 138, 13806–13809. [DOI] [PubMed] [Google Scholar]; (b) Dolewski RD; Fricke PJ; McNally A Site-Selective Switching Strategies to Functionalize Polyazines. J. Am. Chem. Soc 2018, 140, 8020–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pyridyl phosphines have been synthesized previously through basic SNAr conditions or metal-catalyzed conditions.; (a) Hintermann L; Dang TT; Labonne A; Kribber T; Xiao L; Naumov P The AZARYPHOS Family of Ligands for Ambifunctional Catalysis: Syntheses and Use in Ruthenium-Catalyzed Anti-Markovnikov Hydration of Terminal Alkynes. Chem. - Eur. J 2009, 15, 7167–7179. [DOI] [PubMed] [Google Scholar]; (b) Newkome GR Pyridylphosphines. Chem. Rev 1993, 93, 2067–2089. [Google Scholar]; (c) Newkome GR; Hager DC Chemistry of Heterocyclic Compounds. 27. An Improved Preparation of Pyridyldiphenylphosphines. J. Org. Chem 1978, 43, 947–949. [Google Scholar]; (d) Yang J; Chen T; Han L-B C–P Bond-Forming Reactions via C–O/PH Cross-Coupling Catalyzed by Nickel. J. Am. Chem. Soc 2015, 137, 1782–1785. [DOI] [PubMed] [Google Scholar]
- (16).Terrier F, Ed. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- (17).Mečiarová M; Toma Š Loupy A; Horváth B Synthesis of Phosphonium Salts—Phosphine Structure and Inorganic Salts Effects. Phosphorus, Sulfur Silicon Relat. Elem 2007, 183, 21–33. [Google Scholar]
- (18).(a) Ackerman LKG; Lovell MM; Weix DJ MultimetallicCatalysed Cross-Coupling of Aryl Bromides with Aryl Triflates. Nature 2015, 524, 454–457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Further details on guidelines and limitations are show in the Supporting Information.
- (20).Ren W; Li J; Zou D; Wu Y; Wu Y Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reaction of Potassium 2-Pyridyl Trifluoroborate with Aryl (Heteroaryl) Halides. Tetrahedron 2012, 68, 1351–1358. [Google Scholar]
- (21).Potassium 2-pyridyl trifluoroborates have been previously noted to perform poorly under Suzuki cross-coupling reaction conditions.; Molander GA; Biolatto B Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions of Potassium Aryl- and Heteroaryltrifluoroborates. J. Org. Chem 2003, 68, 4302–4314. [DOI] [PubMed] [Google Scholar]
- (22).Kudo N; Perseghini M; Fu GC A Versatile Method for Suzuki Cross-Coupling Reactions of Nitrogen Heterocycles. Angew. Chem., Int. Ed 2006, 45, 1282–1284. [DOI] [PubMed] [Google Scholar]
- (23).Other sets of Suzuki cross-coupling conditions gave undesired oxidation of the heteroaryl phosphine or products resulting from insertion into the C–P bond.
- (24).Luzung MR; Patel JS; Yin J A Mild Negishi Cross-Coupling of 2-Heterocyclic Organozinc Reagents and Aryl Chlorides. J. Org. Chem 2010, 75, 8330–8332. [DOI] [PubMed] [Google Scholar]
- (25).Schubert US; Eschbaumer C; Heller M Stille-Type Cross-Coupling An Efficient Way to Various Symmetrically and Unsymmetrically Substituted Methyl-Bipyridines: Toward New ATRP Catalysts. Org. Lett 2000, 2, 3373–63376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.