

Abstract
Chimeric antigen receptor T cell (CART) therapy has dramatically changed the therapeutic prospects for B cell malignancies. Over the last decade CD19-redirected CART have demonstrated the ability to induce deep, long-lasting remissions and possibly cure patients with relapsing B cell neoplasms. Such impressive results with CART19 fostered efforts to expand this technology to other incurable malignancies that naturally do not express CD19, such as acute myeloid leukemia (AML), Hodgkin lymphoma (HL) and multiple myeloma (MM). However, to reach this goal, several hurdles have to be overcome, in particular: i the apparent lack of suitable targets as effective as CD19; ii. the immunosuppressive tumor microenvironment; iii. intra-tumoral heterogeneity and antigen-negative relapses. Therefore, new strategies that allow safer and more potent CART platforms are under development and may provide grounds for new exciting breakthroughs in the field.
The CAR immunotherapy revolution and the CD19 paradigm
Chimeric antigen receptor-based immunotherapy constitutes one of the most significant breakthroughs for the treatment of hematologic malignancies, in particular B cell neoplasms. [1] The revolutionary idea behind the success of this therapy is the development of a synthetic protein, the chimeric antigen receptor (CAR), that is able to redirect otherwise inoffensive T cells against cancer cells. [2,3] A CAR typically includes an antigen binding domain, most commonly a single-chain variable fragment (scFv) obtained from a monoclonal antibody (mAb), a co-stimulatory domain (commonly derived from 4-1BB or CD28) and the intracellular signaling domain of the T cell receptor (CD3 ζ chain) [4]. The introduction of the CAR transgene into patient’s T cells enables them to engage a surface tumor-associated antigen (TAA) triggering T cell activation and cytotoxicity against the malignant cell.
CD19-specific CAR T (CART19) cells have led to unprecedented results in the treatment of B-cell acute lymphoblastic leukemia (B-ALL), with up to 90% complete remissions (CR) and durable molecular responses reported in relapsing/refractory (r/r) patients [5–9]. As a result, the United States of America Food and Drug Administration (FDA) recently approved the University of Pennsylvania/Novartis CART19 product (Kymriah(TM)/tisagenlecleucel, formerly CTL019) for the treatment of children and young adults with r/r ALL. Sustained CR over 4 years after CART19 cell therapy have been described also in a subset of heavily pre-treated chronic lymphocytic leukemia (CLL) patients [10]. Recently, more than 70% responses, including more than 1 year CR, have been also reported in refractory diffuse large B cell lymphoma (DLBCL) [11–13]. Such an impressive success is partly explained by the unique nature of the tumor target CD19. CD19 is a surface antigen, highly and homogeneously expressed on malignant B cells and relatively tumor-restricted. The only non-malignant cells expressing CD19 are normal B lymphocytes, and their depletion (B cell aplasia) is clinically manageable in most patients. Therefore, identification of suitable TAA represents the first step in attaining clinical success in hematologic malignancies that do not express CD19, such as acute myeloid leukemia (AML), Hodgkin lymphoma (HL) and multiple myeloma (MM). However, synthetic biology and gene-editing technologies could increase the therapeutic index of CART for these malignancies using the currently available targets.
1. Extending the CART technology to hematologic malignancies that do not express CD19
Patients with relapsing or refractory hematologic malignancies have usually poor prognosis [14]. The current standard treatments for such patients often have limited clinical impact, thus highlighting an unmet need for more effective therapeutic strategies. Durable remissions and even cures in patients with AML, MM and HL attributed to the immune-mediated graft-versus-tumor effect following allogeneic HSCT (allo-HSCT) [15–25] underscore the notion that the immune system is capable of eradicating these malignancies. This, together with the promising results reported with CD19-specific CAR T cells in B cell neoplasms prompted the pre-clinical development and clinical investigation of CAR-based immunotherapy in other hematologic malignancies that do not express CD19.
2a. Pre-clinical and early clinical experience using conventional CAR-based approaches for AML, HL and MM
Acute myeloid leukemia
Despite significant advances in the understanding the cellular and molecular biology of AML over the past two decades, little progress has been made in the treatment strategies [26]. The development of effective CAR-based immunotherapy for AML is one of the biggest challenges in the field but it is hampered by the lack of suitable targets and the diverse cellular architecture of AML [27,28]. Several targets are being evaluated for CART-based therapy of AML in both the preclinical and clinical setting.
CD33 is a transmembrane receptor of the sialic-acid-binding immunoglobulin-like lectin family involved in inflammatory and immune responses [29]. It is generally expressed on AML blasts [30–32], but also on normal hematopoietic cells, including the hemopoietic stem cell (HSC) and myeloid progenitors (Figure 1), and on Kupffer cells in the liver [33–35]. Gemtuzumab ozogamicin (GO), a calicheamicin-conjugated anti-CD33 antibody, has been associated with potent anti-leukemia activity, but also some clinical toxicity [36,37]. Similarly, CART33 cells featuring a GO-derived scFv exhibited potent activity against AML cell lines and primary AML cells in vitro and in vivo, leading to improved survival in AML xenograft models, although with evidence of hematopoietic toxicity [38]. Two clinical trials (NCT01864902 and NCT02799680) are currently recruiting relapsed/refractory CD33+ AML patients in China. The first patient treated with multiple autologous CART33 cell infusions achieved a transitory partial remission (PR) but rapidly progressed three weeks post CAR T cell transfer. Febrile reactions were reported in association with each cell infusion with high levels of IL-6, TNF-α and INF-γ. Profound pancytopenia and transient, mild hyperbilirubinemia were recorded after escalated CART cell dosing [39].
Figure 1.
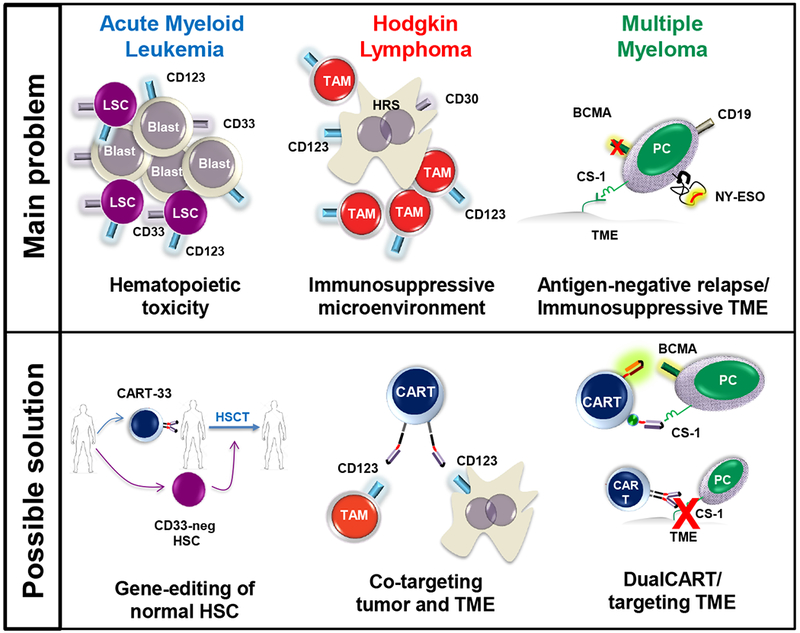
CAR-cell immunotherapy in hematologic malignancies: challenges and novel approaches.
Compared to CD33, the interleukin-3 receptor α chain, CD123, is more frequently expressed on the AML leukemia stem cell (LSC), but it is also present on normal hemopoietic HSC and progenitors (Figure 1) [40], monocytes and some subsets of endothelial cells. Antibody-based targeting of CD123 has shown anti-leukemic activity in vitro and in animal models [41,42] and some initial clinical activity in AML patients [43–48]. However, three fatal events associated with capillary leak syndrome (CLS) were reported following treatment with the SL-401, a fusion molecule composed of the catalytic and translocation domains of the diphtheria toxin fused to IL3 (NTC02113982). This side-effect could potentially be linked to CD123 expression in endothelial cells.Preclinical studies of CART123 cells showed, together with potent anti-AML activity, severe impairment of normal hematopoiesis [40], raising concerns about ‘on-target’, ‘off-tumor toxicity’. Several trials are currently evaluating anti-CD123 CAR immunotherapy. A case report from a Chinese group, showed reduction in BM blasts after infusion of a 4th generation CART123, associated with rigorous chills and fevers, low blood pressure and hypoxemia one day after infusion. [49] A phase I trial (NCT03190278) evaluating 3rd party, CART123 cells (UCART123, Cellectis) for r/r AML, was recently placed on hold by the FDA after the death of the first patient treated with due to CLS. In UCART123 cells, the endogenous TCRα gene is deleted using TALEN-based gene editing to prevent acute graft-versus-host disease [50]. While no GVHD was reported, lethal CLS occurred early post UCART123 infusion, confirming the preliminary clinical evidence with SL-401 suggesting a link between CLS and CD123-targeted immunotherapy. The toxicity of autologous CART123 cells is still under evaluation but safety measures were included to control CART123 toxicity, in particular: transient delivery of CAR constructs through RNA-electroporation (NCT02623582), co-expression of EGFR- (NCT02159495) and CD20- derived (NCT03190278) depletion genes (please also see section 3a) and inducible suicide systems (NCT03125577[49]). Based on the evidence that CAR cell toxicity against normal cells is proportional to potency against malignant cells [28], pursuing CD123 targeting may perhaps require alternative CAR designs employing a different scFv clone associated with lower antileukemic activity in vitro to ensure an acceptable safety profile. [51].
CLEC12A (also known as CLL1) was proposed as a LSC marker [52] and has been recently shown to be expressed on chemorefractory AML blasts. Second generation CLEC12A-specific CAR T cells were highly reactive against CLEC12A+ AML cell lines and primary cells in vitro, leading to eradication of minimal residual disease and prolonged survival in AML-bearing mice that received induction chemotherapy followed by CART-CLEC12A immunotherapy. [53] Anti-CLL-1 CART were proven to be active against a subset of AML samples both in vitro and in vivo, without depleting the HSC but causing toxicity on mature myeloid cells. For this reason the authors suggest that depletion of CART-CLL-1 cells once leukemia is cleared could restore the myeloid compartment thanks to the intact HSC [54].
In keeping with the compelling need for more effective therapeutic targets in AML, the search for new candidates continues and a number of novel potential targets have been proposed [28,55]. The Lewis Y antigen [56–58], the NKG2D ligands [59], CD44v6 [60] and CD133 [61] are expressed by the AML cells and the hematopoietic system and, are also expressed on other types of cancer cells. However, expression of these antigens on healthy tissues calls for careful evaluation of potential toxicity of these CART approaches. Additional targets include the FMS-like tyrosine kinase 3 (FLT3) that is a well characterized antigen with high relevance to AML pathogenesis and strongly associated with poor clinical features. FLT3-targeted CAR T cells have shown promising anti-leukemic activity in vitro and in vivo[62]. As CD7 could be aberrantly expressed in AML blasts, an ongoing trial (NCT02742727) is also evaluating CD7-directed CAR-modified NK-92 cells in AML and T cell-malignancies. Further optimization may require additional manipulation, i.e. CD7-knockout of the desired T/NK effector cells prior to CAR engineering to prevent fratricidal killing and enhance antitumor activity [63].
Hodgkin Lymphoma
Classical Hodgkin lymphoma is a unique entity among mature B cell malignancies. Despite of their B cell origin, Hodgkin Reed-Sternberg (HRS) cells do not typically express B cell antigens like CD19. Therefore, direct tumor killing cannot be achieved with CART19. Instead, nearly all HRS cells express CD30 (Figure 1), a tumor necrosis factor receptor, that delivers pro-survival signals through activation of signaling pathways such as PI3-kinase/Akt/mTOR, ERK/MAPK and NF-κB [64]. CD30 has been the focus for therapeutic targeting by immunoconjugates like brentuximab vedotin with valuable clinical efficacy and acceptable toxicity [65]. CAR30 effectors, including EBV-specific cytotoxic T lymphocytes (CTL), were shown to effectively target CD30+ Hodgkin cells lines in vitro and in vivo [66–69]. Importantly, despite expression of CD30 by a fraction of activated CD30-specific CAR T cells and also by unmodified T cells, no apparent fratricidal killing, impaired cellular immune responses or reduced overall performance of CD30-specific CART cells were observed. On this basis, early clinical trials have been testing the safety and efficacy of CD30-specific CAR T cells in r/r HL patients, using different CAR designs, viral vectors and therapeutic schedules (see Table 1). In one study (NCT01316146), out of nine patients, one experienced CR and another 4 patients had stable disease (SD) resulting in an overall response of 67% [66,70]. In another study involving 18 HL patients with progressive disease, (NCT02259556 [71]) the overall response rate was 72% (13/18, of which 7 PR, 6 stable SD, no CR), including in patients who had undergone previous autoHSCT and treatment with brentuximab [71]. No cases of CRS were observed. Of note, the observed CART30 cell clinical activity was attained without prior chemotherapy preparatory conditioning and required repeated CAR T infusions. Overall, interim analyses from these 3 clinical studies suggest modest clinical efficacy with mild toxicity. Objective responses seemed to correlate with higher CAR T cell doses within all studies.
Table 1.
CAR cell clinical products currently tested in AML, HL and MM patients (as of September 30, 2017)
| Product | Target | Tumor | Start date | Academia | Industry | Vector | Construct (scFv-Hinge-TM-CD-SD) |
Additional features | Active clinical trials |
Reports | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CART-LeY | LeY | Myeloid malignancies | Oct-16 | Southwest Hospital, China | NA | NA | NA | NA | NCT02958384 | NA |
| 2 | CM-CS1 T | NKG2DL | AML, MDS, MM | Mar-15 | DFCI, USA | Ceylad | Retro | NKG2D-CD3ζ | Single infusion of autologous CM-CS1 cells | NCT02203825* | Nikiforow S, 2016[158] |
| 3 | CART33 | CD33 | AML | Apr-13 | Chinese PLA Hospital, China | NA | Retro | AM402974.1-41BB-CD3ζ | Autologous | NCT01864902 | Wang Q-S, 2015[39] |
| 4 | CART33 | CD33 | AML | Oct-15 | Chinese PLA Hospital, China | NA | NA | NA | Allogeneic | NCT02799680 | NA |
| 5 | CD33.CAR T | CD33 | Myeloid malignancies | Oct-16 | Southwest Hospital, China | NA | NA | NA | NA | NCT02958397 | NA |
| 6 | Allo-CARpNK-33 | CD33 | AML | Oct-16 | FPHH, China | PersonGen BioTherapeutics | Viral | NA-CD28-41BB-CD3ζ | NK-92 clinical grade cell line | NCT02944162 | NA |
| 7 | CD33.CAR T | CD33 | AML | Aug-17 | MDACC, USA | NA | Lenti | Autologous, EGFR-derived kill switch | NCT03126864 | Song D, 2016 [99] | |
| 8 | CART-133 | CD133 | Solid tumors1, AML, ALL | Sep-15 | Chinese PLA Hospital, China | NA | Lenti | HW350341.1-CD137-CD3ζ | NA | NCT02541370 | Feng KC, 2017[159] |
| 9 | RNA CART123 | CD123 | AML | Dec-15 | UPenn, USA | Novartis | mRNA | NVS2-41BB-CD3ζ | mRNA | NCT02623582* | NA |
| 10 | CART123 | CD123 | AML | Dec-15 | NCI, USA | NA | Lenti | NA-CD28-CD3ζ-EGFRt | Autologous or donor-derived, EGFRt | NCT02159495 | NA |
| 11 | CD123.CAR T | CD123 | Myeloid malignancies | Oct-16 | Southwest Hospital, China | NA | NA | NA | NA | NCT02937103 | NA |
| 12 | CAR123 T | CD123 | AML | Mar-17 | AHAMMS, China | Shenzhen Geno-Immune Medical Institute | NA | CD123CAR-41BB-CD3zeta-EGFRt | Allogeneic | NCT03114670 | NA |
| 13 | UCART123 | CD123 | AML | Jun-17 | NA | Cellectis | Lenti | NA-41BB-CD3ζ | TALEN-mediated CD52-TRAC ablation | NCT03190278** | NA |
| 14 | Anti-CD7 CAR-pNK | CD7 | AML T cell malignancies | Mar-16 | NA | PersonGen BioTherapeutics (Suzhou) Co., Ltd. | NA-CD28-41BB-CD3ζ | Allogeneic, clinical-grade NK-92 cell line | NCT02742727 | NA | |
| 15 | 4SCAR123 | CD123 | AML | Jul-17 | FAHZU, China | NA | Lenti | NA/CD28/CD137/CD27/CD3ζ-iCasp9 | 4th generation iCaspase9 | NCT03125577 | Luo Y, 2015[49] |
| 16 | CART123 CART33 | CD123 CD33 | AML,MDS ALL | Sep-17 | Zhujiang Hospital, China | NA | NA | NA | Combined with Eps8 peptide specific DC | NCT03291444 | NA |
| 17 | CAR.CD30 | CD30 | HL, NHL | May-11 | BCM, USA | NA | Retro | NA-CD28-CD3ζ | Autologous CAR.CD30 EBV specific-CTLs | NCT01192464* | NA |
| 18 | CAR.CD30 | CD30 | HL | Oct-11 | BCM, USA | NA | Retro | NA-CD28-CD3ζ | NA | NCT01316146* | Ramos, 2015[70] |
| 19 | 4SCAR30273 | CD30 | HL | Mar-14 | Peking University, China | NA | Lenti | NA-CD28-CD137-CD27-CD3ζ | 4th generation iCaspase9 | NCT02274584 | Ying Z-T, 2015[160] |
| 20 | CART30 | CD30 | HL | Oct-14 | Chinese PLA, Hospital, China | CBMG | Lenti | AJ878606.1-CD137ζ -GFP | Repeated infusions | NCT02259556 | Wang C-M, 2017[71] |
| 21 | ATLCAR.CD30 | CD30 | HL, NHL | Jun-16 | BCM, USA | NA | Retro | HRS3-CD28-CD3ζ | 3rd line therapy | NCT02690545, | NA |
| 22 | ATLCAR.CD30 | CD30 | HL, NHL | May-16 | BCM, USA | NA | Retro | HRS3-CD28-CD3ζ | 2nd line therapy | NCT02663297 | NA |
| 23 | CD30.CAR T | CD30 | HL, NHL | Oct-16 | Southwest Hospital, China | NA | NA | NA | NA | NCT02958410 | NA |
| 24 | Anti-CD30 CAR T | CD30 | Hl, NHL | Mar-17 | NCI, USA | NA | Retro | NA | Autologous T cells, fully human anti-CD30 CAR | NCT03049449 | Kochenderfer J, 2017 [161] |
| 25 | CD30.CAR T | CD30 | HL, NHL | May-17 | BCM, USA | NA | Retro | NA-CD28-CD3ζ | No previous autologous tHSCT | NCT02917083 | NA |
| 26 | CART | CD123, CD30, BCMA | Myeloid/lymphoid malignancies | Dec-16 | NA | Hebei Senlang Biotechnology | NA | NA | Autologous | NCT03121625 | NA |
| 27 | Anti-BCMA CAR-T | BCMA | MM | Aug-14 | NCI, USA | NA | Retro | Anti-BCMA-CD28-CD3z | Autologous | NCT02215967 | Ali SA, 2016[80] |
| 28 | CART-BCMA | BCMA | MM | Sep-15 | UPenn, USA | Novartis | NA | Anti-BCMA-41BB-CD3z | Autologous | NCT02546167 | NA |
| 29 | LCAR-B38M | BCMA | MM | Oct-15 | NA | Nanjing Legend Biotech | NA | NA | NA | NCT03090659 | Frank XF, 2017 [162] |
| 30 | bb2121 | BCMA | MM | Jan-16 | NA | Bluebird Bio | Lenti | Anti-BCMA-41BB-CD3z | Autologous | NCT02658929 | Berdeja J, 2017[163] |
| 31 | CART-138/BCMA | CD138/BCMA | MM | Sep-16 | FAHSU, China | NA | NA | NA | Autologous or allogeneic | NCT03196414 | NA |
| 32 | Anti-BCMA CAR-T | BCMA | MM, Leukemia, Lymphoma | Oct-16 | Southwest Hospital, China | NA | NA | NA | NA | NCT02954445 | NA |
| 33 | Anti-BCMA CAR-T | BCMA | MM | Feb-17 | SAHHUTCM, China | NA | Retro | Anti-BCMA-41BB-CD3z | Autologous | NCT03093168 | NA |
| 34 | Anti-BCMA CAR-T | BMCA | MM, PCL | Feb-17 | MSKCC, USA | Juno Therapeutics | NA | EGFRt/Anti-BCMA-41BB-CD3z | NA | NCT03070327 | NA |
| 35 | bb21217 | BCMA | MM | Aug-17 | NA | Bluebird Bio | Lenti | Anti-BCMA-41BB-CD3z | CART cells cultured with a PI3k inhibitor | NCT03274219 | NA |
| 36 | P-BCMA-101 | BCMA | MM | Sep-17 | NA | Poseida Therapeutics | piggyBAC transposon | Anti-BCMA.FN3-CD8a-41BB-CD3z | Selection and safety switch genes | NCT03288493 | Hermanson DL, 2016[164] Barnett BE, 2016[165] |
| 37 | CART-138 | CD138 | MM | Jun-13 | Chinese PLA Hospital, China | NA | Retro | Anti-CD138-41BB-CD3z | Autologous or allogeneic | NCT01886976 | Guo B, 2016[166] |
| 38 | CART-19 | CD19 | MM | May-14 | UPenn, USA | Novartis | Lenti | FMD63scFv-41BB-CD3z | Autologous, r/r MM | NCT02135406 | Garfall A, 2015[167] Garfall A, 2016[168] |
| 39 | CART-19 | CD19 | MM | Jun-16 | UPenn, USA | Novartis | Lenti | FMD63scFv-41BB-CD3z | Autologous, day+60 post first-line autoHSCT | NCT02794246 | NA |
| 40 | CAR-K+ T | κ light chain | MM, CLL, NHL | Jul-09 | BCM | NA | Retro | NA-CD28-CD3z | Autologous | NCT00881920 | Ramos, 2016[169] |
not recruiting;
suspended participant recruitment
Liver, pancreatic, brain, breast, ovarian, colorectal
DFCI, Dana-Farber Cancer Institute; FPHH, The First People’s Hospital of Hefei; MDACC MD Anderson Cancer Center; UPenn University of Pennsylvania; NCI National Cancer Institute; AHAMMS Affiliated Hospital to Academy of Military Medical Sciences; FAHZU, The First Affiliated Hospital of Zhejiang University; BCM Baylor College of Medicine; FAHSU, The First Affiliated Hospital of Soochow University; SAHHUTCM The Second Affiliated Hospital of Henan University of Traditional Chinese Medicine, MSKCC Memorial Sloan Kettering Cancer Center, TM trans-membrane domain, CD costimulatory domain, SD signaling domain, autoHSCT autologous hematopoietic transplantation. NA not available, PCL plasma cell leukemia. Retro: retroviral vector; Lenti: lentiviral vector. r/r: relapsing or refractory; TRAC: TCR-α constant region; DC: dendritic cells; FN3: human tenascin fibronectin type III consensus sequence
Multiple Myeloma
Multiple myeloma is an incurable plasma cell dyscrasia characterized by cellular and genetic heterogeneity. While molecules such as CD138, CD38 and CD56 are expressed on all or the majority of malignant plasma cells, their broader expression on healthy non-hematopoietic tissues makes them potentially problematic as CAR targets. By contrast, B-cell maturation antigen (BCMA), CD19 and CS-1 appear more promising thanks to their reduced off-tumor expression (Figure 1) [72].
BCMA (CD269) is a surface molecule that belongs to tumor necrosis factor receptor family and it is only expressed by late stage B lineage cells [73]. It delivers pro-survival signals to late-stage, mature B cells/plasma cells upon engagement by APRIL or BAFF ligands [74]. BCMA targeting by either therapeutic mAb [75] or CAR T cells [76–79] has generated significant anti-myeloma activity in vitro and in vivo. An ongoing phase I trial at National Cancer Institute (NCI) has demonstrated that BCMA-specific CAR T cells are capable of inducing CR in r/r myeloma patients with high disease burden. Results from 12 patients showed an objective response rate of 33% (1 CR, 2 VGPR and 1 PR), lasting up to at least 26 months, and SD in all remaining cases [80]. Similar to ALL and CLL patients treated with CART19 cells, the depth of remissions correlated with CAR cells dose and persistence as well as occurrence of cytokine-release syndrome (CRS). Importantly, in line with pre-clinical data [79], serum BCMA, which is abundant in myeloma, did not impact clinical outcome. However, tumor escape associated with appearance of BCMA-negative myeloma cells has been observed in 1 out of 12 cases [80]. In an effort to mitigate against immune escape, an APRIL-based CAR which can bind to and target both BCMA and TACI, another APRIL/BAFF receptor heterogeneously expressed on myeloma plasma cells [81,82], is in pre-clinical development [77]. Two recent reports from the American Society of Clinical Oncology (ASCO) annual meeting (see Table 1) showed high rate of responses after BCMA-CART (NCT03090659, NCT02658929). Fan F et al. (#LBA3001) treated 19 MM patients with their LCAR-B38M anti-BCMA CAR-T (Nanjing Legend Biotech Inc.) and obtained 100% ORR (1 PR, 4 VGPR, 14 CR). Berdeja JG et al. (#3010) had 100% ORR in 6 evaluable patients treated with bb2121 anti-BCMA CART (bluebird bio), including 2 sCRs and 2 MRD-negative responses.
The rationale of CAR targeting CD19 in MM is based on the notion that CD19 is expressed on the putative myeloma stem cell, with self-renewal capacity, myeloma-propagating potential and chemo-resistance features [83]. Despite the fact that only a small minority of malignant plasma cells expressed CD19 (as low as 0.05%), a pilot trial (NCT02135406) reported 80% ORR in 10 r/r myeloma patients receiving a single infusion of 5×10e7 CTL019 cells combined with standard auto-HSCT chemotherapy conditioning regimen, including 6 VGPR [84]. Toxicity was mild, with only one case of grade 1 CRS. Ultimately, all patients experienced disease progression, which occurred earlier than after the first HSCT with the exception of 2 patients. Interestingly, the time to progression was found to correlate with CART19 cell peak in the bone marrow rather than their frequency and persistence in PB. On this basis, it has been speculated that CART19 would specifically kill the small CD19+, stem cell like clone while cytotoxic therapies target the bulk tumor. A phase II study is currently assessing the activity of 10-fold higher doses of CART19 cells in high risk patients following autoHSCT (NCT02794246).
CS-1 (SLAM7) is a SLAM receptor involved in the cross-talk between myeloma and surrounding stromal cells which is crucial for tumor initiation and progression [85]. CS1 is highly expressed on both malignant and normal plasma cells although also on activated NK and T lymphocytes [86]. The administration of the anti-CS-1 mAb elotuzumab has been proved to positively impact on clinical responses and outcome in combination with anti-myeloma agents, paving the way for further development of CS-1 targeted immunotherapy in MM [87,88]. Likewise, anti-CS1 CAR T cells have been successfully tested against primary myeloma cells in vitro and exhibited promising anti-tumor activity in vivo [88–91]. Further optimization, including TALEN-induced knock-out of CS-1 in T cells [92], aims to fully exploit the therapeutic potential of CS-1 targeted CAR strategies. This approach is expected to eliminate the risk of CS-1-mediated fratricidal effect of CS-1-specific CAR T cells that could negatively impact on CAR cell manufacturing, immunotherapy outcome as well as homeostasis and function of patients’ immune system.
As mentioned, additional targets with broader off-tumor expression like CD38, CD138 and others [72,93,94] have been developed for MM and are currently being evaluated pre-clinically or in early-phase clinical trials (NCT01886976; NCT02203825).
2. Novel CART cell approaches aimed to increase the therapeutic index of CART for hematologic malignancies
While CD19 has served as an optimal CAR target for B cell malignancies, the search for similar antigens for non-CD19+ hematologic malignancies is still on going. In AML most of the potential targets are also expressed on healthy hemopoietic progenitors and HSC (Figure 1), thus carrying a potential risk of myelotoxicity that could limit their clinical application. For MM, BCMA certainly represents a “CD19-like” antigen, however, BCMA-negative relapses were observed in early BCMA-CART clinical trials [80,95,96](Figure 1) [80]. For HL there is still little experience in the clinic with CART immunotherapy, but CART against CD30 have led to some clinical response and other targets like CD123 are being evaluated. Hence, while the quest for identifying the best antigen target continues, a conceptual shift is already taking place seeking to optimize CAR technology using the currently available targets (Figure 1). These approaches include: a. management of toxicity; b. increasing specificity; c. targeting intracellular targets; d. targeting the tumor microenvironment and e. targeting multiple antigen on tumor cells.
3a. Management of toxicity: controlling CART effector functions
As discussed, most of the currently available antigens to target AML, MM and HL are also expressed in normal tissues. Therefore, the ability to control T cell effector functions over time would be highly beneficial to treat possible toxicities. To this end, three main strategies have been adopted in pre-clinical and clinical settings: i. suicide genes, ii. antibody-mediated depletion and iii. CAR RNA-electroporation.
The inducible caspase9 (iC9) includes a drug-dimerizer binding domain, cloned in frame with human caspase9 [97]. In the presence of the dimerizer drug, (e.g. AP-20187 or the clinical-grade equivalent AP-1903), the caspase9 pro-molecules dimerize and rapidly activate the mitochondrial apoptotic pathway, terminating allo-T cell-mediated GVHD in alloHSCT recipients as early as 30 minutes [98]. Another strategy for CART depletion, is the co-expression of depletion markers, such as truncated, biologically inert CD19, CD20 or EGFR, to allow selection/in vivo ablation by the corresponding mAb [91,92]. Lastly, CAR mRNA electroporation allows transient expression of the CAR as opposed to stable integration and expression following viral delivery. Such strategies have been explored either alone [38,99] or in combination [98] for CART123 and CART33 immunotherapy.
Safety switches may be key to implement clinical development of CAR immunotherapy in AML, MM and HL. However, there are some flaws inherent to each approach. In iC9-based systems, a population of caspase-resistant CAR T cells has been recently described, characterized by overexpression of anti-apoptotic molecules, such as Bcl2, compared to their caspase-sensitive counterparts [98]. By contrast, although not yet tested in the clinic, no ‘escape’ has been observed in association with mAb-based strategies in preclinical models [38,100,101]. Transient expression of the CAR construct after mRNA electroporation may result in either incomplete clearance of the malignant clone or loss of long-term immunosurveillance and consequent relapse. In facts, all depletion strategies can negatively impact CAR immunotherapy outcome by reducing the persistence of CART cells. However, compared to the progressive, stochastic loss of activity following CAR mRNA transfection, depletion genes offer the advantage to exert a temporal control of CART lifespan by triggering active termination exactly at the occurrence of severe adverse events, if any. In addition, it might be possible to identify a ‘window of opportunity’ that could be exploited for short-term interventions [100], allowing enough time to achieve disease eradication before the occurrence of relevant off-target toxicity [100], thus increasing the number of patients with deep and sustained remissions eligible for HSCT [38,101]. More recently, ON/OFF-switch CARs have been developed with the aim to remotely control and manipulate immune cell function. One version is based on constitutively inactive, heterodimeric CARs, consisting of one chain carrying the scFv-binding sequence and a separate chain with the signaling domains and a small molecule-dimerizer domains. Timely regulated administration of appropriate chemical compounds, would allow to control assembly and disassembly of the CARs thus modulating immune cell activation in vivo [102,103]. Similarly, tetracycline (Tet)-inducible systems have been investigated to modulate the expression of CAR constructs inserted into a pRetroX-TetOne third-generation vector. [104]. Another option is represented by downregulatory feedback circuits that dampen immune cell reactivity in response to signals of hyperactivation such as an excess of IL-6, thus limiting the risk of severe toxicity[102]. Although less practical, such technologies would have the advantage of preventing the permanent ablation of CAR T cells and the option of resuming their therapeutic effect and immunosurveillance in the long-term. Lastly, as shown in Figure 1, a novel interesting approach [105] includes genetic engineering of the normal HSC in order to render them invisible to CART33. In a preclinical model, HSC were knocked out for CD33 using the CRISPR-Cas9 technology and engrafted in immunodeficient mice carrying CD33+ AML. Of note, upon CART33 infusion AML cells were cleared but HSC and hemopoiesis in general were spared.
3b. Increasing specificity: modulating CAR affinity and recognition
CAR switches represent the first requirement to improve safety of CAR T immunotherapy. Yet additional strategies need to be considered to improve precision and achieve higher specificity. Modulation of scFv affinity and CAR expression can be used to discriminate between high- (malignant) and low- (normal) antigen-expressing cells in a temporally controlled manner [106–109]. A low affinity CD123-specific CAR was designed to trigger robust lytic response only against targets expressing >1600 CD123 molecules, i.e., typically leukemic cells [110]. Another anti-CD123 CAR characterized by reduced lysis of normal HSC, but full anti-AML activity in vivo was generated by combining different VH and VL chains derived from four CD123-specific mAbs [111]. A similar ‘light chain exchange’ approach has been successfully applied in other contexts, e.g. CD38-specific CAR T cells tested against multiple myeloma cells [112]. Although more functional studies would be required before testing such approach into clinical settings, these preclinical findings suggest that tuning of CAR affinity and antigen expression on malignant cells may represent a valid option to increase the therapeutic index and the range of potential applications of CAR immunotherapy.
Finally, synthetic biology has been recently applied to generate smart CAR platforms and micro-circuits that would provide T cells with the ability of discriminating between healthy and cancer cells in vivo by integrating the information from a defined pattern of antigens. CAR AND-gate circuits consist of two distinct CAR constructs, one endowed with the CD3z activating domain and the other with a costimulatory motif, to allow full activation only against tumor cells expressing the ‘right’ combination of antigens engaging both CARs at the same time [113–115]. Since the presence of a single antigen might still be sufficient to trigger a cytotoxic response [102], in an alternative approach one construct was created to serve as a ‘sensor and primer-CAR’ provided with a Notch-derived regulatory transmembrane region and an intracellular transcriptional activator motif (SynNOTCH) [116,117]. Upon engagement of the first antigen, such CAR undergoes intramembrane cleavage, followed by translocation of the intracellular domain to the nucleus and induction of an ‘effector’ CAR. Further T cell proliferation and killing occur when the opposing cell also presents the ligand for the second receptor. Another strategy to potentially achieve more specific tumor recognition is with CAR NOT-gate circuits combining one conventional activating with one inhibitory CAR (iCAR), i.e., a receptor equipped with an intracellular CTLA-4- or PD-1-derived domain overriding the signal delivered from the first construct, thus preventing reaction against bystander (normal) cells. [118]
3c. Expanding the available antigens: targeting intracellular targets
To circumvent the relative dearth of suitable cell surface targets, T cells could also be redirected against intracellular antigens. Indeed, there is indirect evidence that AML, HL and MM are suitable for such approaches. For instance, adoptive transfer of ex vivo expanded autologous EBV-specific CTLs, recognizing the EBV-derived latent membrane proteins 1 and 2 (LMP1 and LMP2), induce objective responses in patients with r/r EBV+ HL [119,120]. More recently, T cells specific for intracellular tumor-associated antigens, i.e. PRAME, SSX2, MAGEA4, NY-ESO-1 and survivin, were shown to induce complete responses in patients with HL in the absence of conditioning chemotherapy [121,122]. Spontaneous as well as post vaccination humoral and cellular immunity against the NY-ESO157-165/HLAA*02:01 complex have been also observed in myeloma patients [123]. Similarly, antibody and CTL responses have been detected in AML patients against a pool of immunogenic peptides, which are relevant to the tumor biology and correlate with poor prognosis [124,125]. Building upon these encouraging results, considerable efforts have been initially devoted to the clinical development of TCR-like CARs provided with a single scFv recognizing the MHC:peptide complex. Anti-WT1/HLA-A*02:01 CARs have been already successfully tested in AML patients [126–128]. Additional candidates under pre-clinical investigation include cancer testis antigens NY-ESO-1, in MM [129] and AML; LAGE-1, MAGE-A3, PRAME, proteinase 3, RHAMM and Flt3 in AML [28,62,130,131].
3d. Targeting the tumor microenvironment
Targeting the ‘right antigen’ might be not be sufficient to eradicate cancer cells if CAR T cells have to reach and function in highly immunosuppressive contexts. Therefore, it may be beneficial to redirect CAR T cells against components of the TME. In MM, the myeloid-derived suppressive cells (MDSC) may induce with immune paresis, ultimately promoting myelomagenesis and relapse [60,132,133]. Dependence from pro-survival signals within the TME has been equally demonstrated in HL, where the HRS cells typically represent a small minority of the tumor mass, nurtured and sheltered by the surrounding infiltrate. In addition, tumor-associated macrophages (TAM), as well as HRS cells, generally express increased levels of PD-1 ligands in association with a recurrent genetic amplification at chromosome 9p24.1, thus suggesting that the PD-1/PD-L pathway might represent a key determinant of immunosuppressive TME in HL. Accordingly, PD-1 blockade significantly improved the outcome of r/r HL patients, who reported >65% response rate upon treatment with the PD-1 inhibitors nivolumab and pembrolizumab [134–137].
In order to attack tumor cells, T cells must overcome the immunosuppression of the TME to physically reach them. In a preclinical in vivo model of HL co-expression of CD30-specific CAR and exogenous chemokine receptor CCR4 have proved to enhance homing of CAR T cells to the tumor site [138], leading to improved anti-tumor activity with no systemic toxicity [139–141]. Alternatively, CAR-mediated cytotoxicity could be directed against the neighboring immunosuppressive cells. Indeed, CAR T cells recognizing CD123 on TAM within the HL microenvironment as well as in HRS cells effectively killed both TAM and HL cell lines in vitro and displayed potent therapeutic activity against disseminated disease in an in vivo model of HL (Figure 1) [142,143]. Another proposed approach aimed to overcome the TME immunosuppression in HL includes EBV-specific CTL expressing exogenous IL-12 [144]. Given the promising responses to nivolumab and pembrolizumab, strategies combining CAR technology with checkpoint inhibitors might have a higher therapeutic impact.
3e. Avoid antigen-negative escape: targeting multiple antigen on tumor cells
Following the paradigm of multi-agent chemotherapy, in order to mitigate antigen escape and improve discrimination against healthy cells, ongoing efforts focus on developing CAR approaches that target simultaneously more than one TAA [145]. Although yet-to be tested in clinical settings, several co-targeting approaches have been proved superior to mono-specific strategies in solid and hematological malignancies and could be grouped in two categories: i. “pooled CART” - a mixtures of CAR T cells endowed with different specificities [146], or ii. “multi-specific CART” - a single CAR T cell that can engage multiple antigens thanks to the presence of either 2 different CAR molecules (bi- or dual-CART) [147–149] or a single CAR molecule with two recognition regions (tandem CARs) [150–152]. Early preclinical experience suggests that, provided optimal spatial configuration, dual targeting by tandem CAR may prevent immune escape more effectively than the combination of distinct CARs [153]-[154]. Alternatively, switchable CAR like UniCAR, consist of an array of soluble modular, tagged scFv domains, equally fitting a constant CAR stalk and supplied as required, to add extra flexibility and multi-specificity in a timely controlled manner [155]. Such an approach was specifically developed to simultaneously target CD33 and CD123 on AML blasts [156], and potentially allowing the control of myeloablation.
3. Conclusions and future perspectives
Overall, the majority of MM patients, more than half AML patients and 10-15% of patients with HL will develop r/r disease [14,157]. For these patients there is little chance of attaining long-term remission with conventional treatments. The development of CART immunotherapies for these diseases is currently rapidly advancing but it has become clear that significant optimization of the current CART dogma is required in order to reproduce the clinical success of CART19 in the setting of AML, HL and MM. Synthetic biology and gene-editing technologies are now tools available to researchers to generate CART products specifically crafted for a defined disease. Ultimately, as for CART19, early clinical trials will guide the required improvements for the successful development of CART for AML, HL and MM.
Acknowledgments and funding:
A.R. is supported by a Bloodwise Clinical Research Fellowship, A.K. is supported by a Bloodwise Program Grant. AR and AK acknowledge infrastructure support from the Cancer Research UK Imperial Centre, the Imperial Experimental Cancer Medicine Centre and the National Institute for Health Research Imperial Biomedical Research Centre. M.R. is supported by grants from the Univ. of Pennsylvania-Novartis Alliance, the NIH-NCI (K99 CA212302-01A1), the Gabrielle’s Angel Foundation, American Society of Hematology (Scholar Award), the Parker Institute for Cancer Immunotherapy and Tmunity therapeutics.
Footnotes
Competing interests: M.R. works under a research collaboration involving the University of Pennsylvania and the Novartis Institutes of Biomedical Research, Inc. and is inventor of intellectual property licensed by the University of Pennsylvania to Novartis.
References
- 1.Ruella M, June CH. Chimeric Antigen Receptor T cells for B Cell Neoplasms: Choose the Right CAR for You. Curr Hematol Malig Rep 2016. [DOI] [PubMed] [Google Scholar]
- 2.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989;86:10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun 1987;149:960–968. [DOI] [PubMed] [Google Scholar]
- 4.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol 2016;13:370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brentjens RJ, Davila ML, Riviere I, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci Transl Med 2013;5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brudno JN, Somerville RPT, Shi V, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. Journal of Clinical Oncology 2016;34:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet;385:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126:2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015;7:303ra139–303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neelapu SS, Locke FL, Bartlett NL, et al. A Phase 2 Multicenter Trial of KTE-C19 (anti-CD19 CAR T Cells) in Patients With Chemorefractory Primary Mediastinal B-Cell Lymphoma (PMBCL) and Transformed Follicular Lymphoma (TFL): Interim Results From ZUMA-1. Blood 2016;128:998. [Google Scholar]
- 12.Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Molecular Therapy 2017;25:285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. Journal of Clinical Oncology 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SEER Cancer Statistics Review, 1975–2013, National Cancer Institute; Bethesda, MD, [Internet]. Updated September 12, 2016 based on November 2015 SEER data submission, posted to the SEER web site, April 2016 - [cited March 2017]. Available from: http://seer.cancer.gov/csr/1975_2013/ [Google Scholar]
- 15.Brayer JB, Pinilla-Ibarz J. Developing Strategies in the Immunotherapy of Leukemias. Cancer control : journal of the Moffitt Cancer Center 2013;20:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parmar S, Fernandez-Vina M, de Lima M. Novel transplant strategies for generating graft-versus-leukemia effect in acute myeloid leukemia. Curr Opin Hematol 2011;18:98–104. [DOI] [PubMed] [Google Scholar]
- 17.Borchers S, Provasi E, Silvani A, et al. Genetically modified donor leukocyte transfusion and graft-versus-leukemia effect after allogeneic stem cell transplantation. Hum Gene Ther 2011;22:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alyea E, Weller E, Schlossman R, et al. T-cell--depleted allogeneic bone marrow transplantation followed by donor lymphocyte infusion in patients with multiple myeloma: induction of graft-versus-myeloma effect. Blood 2001;98:934–939. [DOI] [PubMed] [Google Scholar]
- 19.Gahrton G, Tura S, Ljungman P, et al. Allogeneic Bone Marrow Transplantation in Multiple Myeloma. New England Journal of Medicine 1991;325:1267–1273. [DOI] [PubMed] [Google Scholar]
- 20.Donato ML, Siegel DS, Vesole DH, et al. The graft-versus-myeloma effect: chronic graft-versus-host disease but not acute graft-versus-host disease prolongs survival in patients with multiple myeloma receiving allogeneic transplantation. Biol Blood Marrow Transplant 2014;20:1211–1216. [DOI] [PubMed] [Google Scholar]
- 21.Tricot G, Vesole DH, Jagannath S, Hilton J, Munshi N, Barlogie B. Graft-versus-myeloma effect: proof of principle. Blood 1996;87:1196–1198. [PubMed] [Google Scholar]
- 22.Anderlini P, Swanston N, Rashid A, Bueso-Ramos C, Macapinlac HA, Champlin RE. Evidence of a Graft-versus-Hodgkin Lymphoma Effect in the Setting of Extensive Bone Marrow Involvement. Biology of Blood and Marrow Transplantation 2008;14:478–480. [DOI] [PubMed] [Google Scholar]
- 23.Porter DL, Stadtmauer EA, Lazarus HM. /`GVHD/’: graft-versus-host disease or graft-versus-Hodgkin’s disease? an old acronym with new meaning. Bone Marrow Transplant 0000;131:739–746. [DOI] [PubMed] [Google Scholar]
- 24.Jones RJ, Ambinder RF, Piantadosi S, Santos GW. Evidence of a graft-versus-lymphoma effect associated with allogeneic bone marrow transplantation. Blood 1991;77:649–653. [PubMed] [Google Scholar]
- 25.Akpek G, Ambinder RF, Piantadosi S, et al. Long-Term Results of Blood and Marrow Transplantation for Hodgkin’s Lymphoma. Journal of Clinical Oncology 2001;19:4314–4321. [DOI] [PubMed] [Google Scholar]
- 26.Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood 2016;127:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program 2004:80–97. [DOI] [PubMed] [Google Scholar]
- 28.Gill S Chimeric antigen receptor T cell therapy in AML: How close are we? Best Pract Res Clin Haematol 2016;29:329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMillan SJ, Crocker PR. CD33-related sialic-acid-binding immunoglobulin-like lectins in health and disease. Carbohydr Res 2008;343:2050–2056. [DOI] [PubMed] [Google Scholar]
- 30.Walter RB, Gooley TA, van der Velden VH, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007;109:4168–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwonzen M, Diehl V, Dellanna M, Staib P. Immunophenotyping of surface antigens in acute myeloid leukemia by flow cytometry after red blood cell lysis. Leuk Res 2007;31:113–116. [DOI] [PubMed] [Google Scholar]
- 32.Hoyer JD, Grogg KL, Hanson CA, Gamez JD, Dogan A. CD33 detection by immunohistochemistry in paraffin-embedded tissues: a new antibody shows excellent specificity and sensitivity for cells of myelomonocytic lineage. Am J Clin Pathol 2008;129:316–323. [DOI] [PubMed] [Google Scholar]
- 33.Pearce DJ, Taussig D, Zibara K, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood 2006;107:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakahata T, Okumura N. Cell surface antigen expression in human erythroid progenitors: erythroid and megakaryocytic markers. Leuk Lymphoma 1994;13:401–409. [DOI] [PubMed] [Google Scholar]
- 35.Maniecki MB, Hasle H, Bendix K, Møller HJ. Is hepatotoxicity in patients treated with gemtuzumabozogamicin due to specific targeting of hepatocytes? Leuk Res 2011;35:e84–e86. [DOI] [PubMed] [Google Scholar]
- 36.Stasi R Gemtuzumab ozogamicin: an anti-CD33 immunoconjugate for the treatment of acute myeloid leukaemia. Expert Opin Biol Ther 2008;8:527–540. [DOI] [PubMed] [Google Scholar]
- 37.Pagano L, Fianchi L, Caira M, Rutella S, Leone G. The role of Gemtuzumab Ozogamicin in the treatment of acute myeloid leukemia patients. Oncogene 2007;26:3679–3690. [DOI] [PubMed] [Google Scholar]
- 38.Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015;29:1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q-s, Wang Y, Lv H-y, et al. Treatment of CD33-directed Chimeric Antigen Receptor-modified T Cells in One Patient With Relapsed and Refractory Acute Myeloid Leukemia. Molecular Therapy 2015;23:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014;123:2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Hussaini M, Rettig MP, Ritchey JK, et al. Targeting CD123 in acute myeloid leukemia using a T-cell–directed dual-affinity retargeting platform. Blood 2016;127:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu SY, Pong E, Chen H, et al. Immunotherapy with Long-Lived Anti-CD123 × Anti-CD3 Bispecific Antibodies Stimulates Potent T Cell-Mediated Killing of Human AML Cell Lines and of CD123+ Cells in Monkeys: A Potential Therapy for Acute Myelogenous Leukemia. Blood 2014;124:2316.25301330 [Google Scholar]
- 43.Smith BD, Roboz GJ, Walter RB, et al. First-in Man, Phase 1 Study of CSL362 (Anti-IL3Rα / Anti-CD123 Monoclonal Antibody) in Patients with CD123+ Acute Myeloid Leukemia (AML) in CR at High Risk for Early Relapse. Blood 2014;124:120. [Google Scholar]
- 44.Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013;122:3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol 2013;161:389–401. [DOI] [PubMed] [Google Scholar]
- 46.Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014;28:1596–1605. [DOI] [PubMed] [Google Scholar]
- 47.Zhou L, Liu X, Wang X, Sun Z, Song XT. CD123 redirected multiple virus-specific T cells for acute myeloid leukemia. Leuk Res 2016;41:76–84. [DOI] [PubMed] [Google Scholar]
- 48.Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014;123:2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo Y, Chang L-J, Hu Y, Dong L, Wei G, Huang H. First-in-Man CD123-Specific Chimeric Antigen Receptor-Modified T Cells for the Treatment of Refractory Acute Myeloid Leukemia. Blood 2015;126:3778–3778. [Google Scholar]
- 50.Release CP. Cellectis submits an application for UCART123, an allogeneic gene edited CAR T-cell product candidate, in AML and BPDCN. https://www.cellectis.com/en/content/cellectis-submits-ind-application-ucart123-allogeneic-gene-edited-car-t-cell-product-0. New York; 2017. [Google Scholar]
- 51.Galetto R Cd123 specific chimeric antigen receptors for cancer immunotherapy. Google Patents; 2015. [Google Scholar]
- 52.van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007;110:2659–2666. [DOI] [PubMed] [Google Scholar]
- 53.Kenderian SS, Ruella M, Shestova O, et al. 766. Leukemia Stem Cells Are Characterised By CLEC12A Expression and Chemotherapy Refractoriness That Can be Overcome By Targeting with Chimeric Antigen Receptor T Cells. ASH annual meeting. San Diego; 2016. [Google Scholar]
- 54.Tashiro H, Sauer T, Shum T, et al. Treatment of Acute Myeloid Leukemia with T Cells Expressing Chimeric Antigen Receptors Directed to C-type Lectin-like Molecule 1. Mol Ther 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rotiroti MC, Arcangeli S, Casucci M, et al. Acute Myeloid Leukemia Targeting by Chimeric Antigen Receptor T Cells: Bridging the Gap from Preclinical Modeling to Human Studies. Hum Gene Ther 2017;28:231–241. [DOI] [PubMed] [Google Scholar]
- 56.Westwood JA, Smyth MJ, Teng MW, et al. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc Natl Acad Sci U S A 2005;102:19051–19056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peinert S, Prince HM, Guru PM, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther 2010;17:678–686. [DOI] [PubMed] [Google Scholar]
- 58.Ritchie DS, Neeson PJ, Khot A, et al. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol Ther 2013;21:2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immun 2013;13:8. [PMC free article] [PubMed] [Google Scholar]
- 60.Casucci M, Falcone L, Camisa B, et al. CD44v6 Is Required For In Vivo Tumorigenesis Of Human AML and MM Cells: Role Of Microenvironmental Signals and Therapeutic Implications. Blood 2013;122:605–605. [Google Scholar]
- 61.Koerner SP, Andre MC, Leibold JS, et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2017;31:459–469. [DOI] [PubMed] [Google Scholar]
- 62.Chien CD, Sauter CT, Ishii K, et al. Preclinical Development of FLT3-Redirected Chimeric Antigen Receptor T Cell Immunotherapy for Acute Myeloid Leukemia. Blood 2016;128:1072. [Google Scholar]
- 63.Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017;130:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clodi K, Younes A, Goodacre A, et al. Analysis of p53 gene deletions in patients with non-Hodgkin’s lymphoma by dual-colour fluorescence in-situ hybridization. Br J Haematol 1997;98:913–921. [DOI] [PubMed] [Google Scholar]
- 65.Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramos CA, Heslop HE, Brenner MK. CAR-T Cell Therapy for Lymphoma. Annu Rev Med 2016;67:165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagle SJ, Garfall AL, Stadtmauer EA. The Promise of Chimeric Antigen Receptor Engineered T Cells in the Treatment of Hematologic Malignancies. Cancer J 2016;22:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hombach A, Heuser C, Sircar R, et al. Characterization of a chimeric T-cell receptor with specificity for the Hodgkin’s lymphoma-associated CD30 antigen. J Immunother 1999;22:473–480. [DOI] [PubMed] [Google Scholar]
- 69.Savoldo B, Rooney CM, Di Stasi A, et al. Epstein Barr virus–specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 2007;110:2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramos CA, Ballard B, Liu E, et al. Chimeric T Cells for Therapy of CD30+ Hodgkin and Non-Hodgkin Lymphomas. Blood 2015;126:185–185.26024876 [Google Scholar]
- 71.Wang C-M, Wu Z-Q, Wang Y, et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clinical Cancer Research 2017. [DOI] [PubMed] [Google Scholar]
- 72.Rotolo A, Caputo V, Karadimitris A. The prospects and promise of chimeric antigen receptor immunotherapy in multiple myeloma. Br J Haematol 2016;173:350–364. [DOI] [PubMed] [Google Scholar]
- 73.Bellucci R, Alyea EP, Chiaretti S, et al. Graft-versus-tumor response in patients with multiple myeloma is associated with antibody response to BCMA, a plasma-cell membrane receptor. Blood 2005;105:3945–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 2004;199:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015;7:1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pulé M, Yong K, Lee L, Draper B. Chimeric antigen receptor. Google Patents; 2015. [Google Scholar]
- 77.Lee SHL, Draper BO, Chaplin N, et al. An APRIL Based Chimeric Antigen Receptor to Simultaneously Target BCMA and TACI in Multiple Myeloma (MM) Has Potent Activity in Vitro and in Vivo. ASH annual meeting. San Diego; 2016. [Google Scholar]
- 78.Kochenderfer JN. Chimeric antigen receptors targeting b-cell maturation antigen. Google Patents; 2013. [Google Scholar]
- 79.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res 2013;19:2048–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ali SA, Shi V, Maric I, et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016;128:1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moreaux J, Cremer FW, Reme T, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005;106:1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Novak AJ, Darce JR, Arendt BK, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood 2004;103:689–694. [DOI] [PubMed] [Google Scholar]
- 83.Hajek R, Okubote SA, Svachova H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol 2013;163:551–564. [DOI] [PubMed] [Google Scholar]
- 84.Garfall AL, Stadtmauer EA, Maus MV, et al. Pilot Study of Anti-CD19 Chimeric Antigen Receptor T Cells (CTL019) in Conjunction with Salvage Autologous Stem Cell Transplantation for Advanced Multiple Myeloma. Blood 2016;128:974. [Google Scholar]
- 85.Tai YT, Soydan E, Song W, Fulciniti M, Kim K, Hong F. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood 2009;113:4309–4318. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res 2008;14:2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Magen H, Muchtar E Elotuzumab: the first approved monoclonal antibody for multiple myeloma treatment. Ther Adv Hematol 2016;7:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. New England Journal of Medicine 2015;373:621–631. [DOI] [PubMed] [Google Scholar]
- 89.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chu J, He S, Deng Y, et al. Genetic Modification of T Cells Redirected towards CS1 Enhances Eradication of Myeloma Cells. Clin Cancer Res 2014;20:3989–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Danhof S, Gogishvili T, Koch S, et al. Preclinical Analysis of Feasibility and Efficacy of CS1 Directed CAR T cell Therapy in Multiple Myeloma in the Autologous Setting. Clinical Lymphoma Myeloma and Leukemia 2015;15:e39. [Google Scholar]
- 92.Juillerat A, Valton J, Gautron A, Duchateau P, Poirot L. Targeted genome modifications for improved adoptive immunotherapy Chimeric Antigen Receptor Therapy in Haematology and Oncology: Current Successes and Challenges, London 2015, P006; 2015. [Google Scholar]
- 93.Atanackovic D, Steinbach M, Radhakrishnan SV, Luetkens T. Immunotherapies targeting CD38 in Multiple Myeloma. Oncoimmunology 2016;5:e1217374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ormhoj M, Bedoya F, Frigault MJ, Maus MV. CARs in the Lead Against Multiple Myeloma. Curr Hematol Malig Rep 2017;12:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghodke K, Bibi A, Rabade N, et al. CD19 negative precursor B acute lymphoblastic leukemia (B-ALL)—Immunophenotypic challenges in diagnosis and monitoring: A study of three cases. Cytometry Part B: Clinical Cytometry 2016:n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 96.Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma 1995;18:385–397. [DOI] [PubMed] [Google Scholar]
- 97.Di Stasi A, Tey S-K, Dotti G, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. New England Journal of Medicine 2011;365:1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Minagawa K, Jamil MO, Al-Obaidi M, et al. In Vitro Pre-Clinical Validation of Suicide Gene Modified Anti-CD33 Redirected Chimeric Antigen Receptor T-Cells for Acute Myeloid Leukemia. PLoS One 2016;11:e0166891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song D, Swartz MH, Biesecker SG, et al. Chimeric Antigen Receptor-Modified T Cells for the Treatment of Acute Myeloid Leukemia Expressing CD33. Blood 2016;128:4058. [Google Scholar]
- 100.Tasian SK, Kenderian SS, Shen F, et al. Optimized Depletion of Chimeric Antigen Receptor T-Cells in Murine Xenograft Models of Human Acute Myeloid Leukemia. Blood 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gill S, Tasian SK, Ruella M. Preclinical targeting of human acute myeloid leukemia and myloablation using chimeric antigen receptor-modified T cells. Blood 2014;123:2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell;168:724–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 2015;350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sakemura R, Terakura S, Watanabe K, et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol Res 2016;4:658–668. [DOI] [PubMed] [Google Scholar]
- 105.Kim MY, Kenderian SS, Schreeder D, et al. 273. Genome Editing Using CRISPR-Cas9 to Increase the Therapeutic Index of Antigen-Specific Immunotherapy in Acute Myeloid Leukemia. Molecular Therapy;24:S108. [Google Scholar]
- 106.Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res 2017;27:38–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Caruso HG, Hurton LV, Najjar A, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res 2015;75:3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X, Jiang S, Fang C, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 2015;75:3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arcangeli S, Bardelli M, Rotiroti MC, et al. Balance of Anti-CD123 Chimeric Antigen Receptor (CAR) Binding Affinity and Density for the Treatment of Acute Myeloid Leukemia. Blood 2016;128:2163–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arcangeli S, Rotiroti MC, Bardelli M, et al. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther 2017;25:1933–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thokala R, Olivares S, Mi T, et al. Redirecting Specificity of T cells Using the Sleeping Beauty System to Express Chimeric Antigen Receptors by Mix-and-Matching of VL and VH Domains Targeting CD123+ Tumors. PLoS One 2016;11:e0159477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Drent E, Themeli M, Poels R, van de Donk NWCJ, Lokhorst HM, Mutis T Reducing on-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors By Affinity Optimization. Blood 2016;128:2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 2013;31:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res 2013;1:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wilkie S, Schalkwyk MC, Hobbs S, Davies DM, Stegen SJ, Pereira AC. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 2012;32. [DOI] [PubMed] [Google Scholar]
- 116.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016;164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016;164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A 2016;113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin’s disease. J Exp Med 2004;200:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lucas KG, Salzman D, Garcia A, Sun Q. Adoptive immunotherapy with allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T-lymphocytes for recurrent, EBV-positive Hodgkin disease. Cancer 2004;100:1892–1901. [DOI] [PubMed] [Google Scholar]
- 121.Leen AM, Tzannou I, Liu H, et al. Immunotherapy for Lymphoma Using T Cells Targeting Multiple Tumor-Associated Antigens. Biology of Blood and Marrow Transplantation 2016;22:S44–S45. [Google Scholar]
- 122.Leen A, Tzannou I, Bilgi M, et al. Immunotherapy for Lymphoma Using T Cells Targeting Multiple Tumor Associated Antigens. Blood 2015;126:186. [Google Scholar]
- 123.van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood 2005;105:3939–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greiner J, Ringhoffer M, Taniguchi M, et al. Characterization of several leukemia-associated antigens inducing humoral immune responses in acute and chronic myeloid leukemia. International Journal of Cancer 2003;106:224–231. [DOI] [PubMed] [Google Scholar]
- 125.Oka Y, Tsuboi A, Taguchi T, et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci U S A 2004;101:13885–13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pegram HJ, Smith EL, Rafiq S, Brentjens RJ. CAR therapy for hematological cancers: can success seen in the treatment of B-cell acute lymphoblastic leukemia be applied to other hematological malignancies? Immunotherapy 2015;7:545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rafiq S, Dao T, Liu C, Scheinberg DA, Brentjens RJ. Engineered T Cell Receptor-Mimic Antibody, (TCRm) Chimeric Antigen Receptor (CAR) T Cells Against the Intracellular Protein Wilms Tumor-1 (WT1) for Treatment of Hematologic and Solid Cancers. Blood 2014;124:2155. [Google Scholar]
- 128.McLaughlin L, Cruz CR, Bollard CM. Adoptive T-cell therapies for refractory/relapsed leukemia and lymphoma: current strategies and recent advances. Ther Adv Hematol 2015;6:295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schuberth PC, Jakka G, Jensen SM, et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther 2013;20:386–395. [DOI] [PubMed] [Google Scholar]
- 130.Rashidi A, Walter RB. Antigen-specific immunotherapy for acute myeloid leukemia: where are we now, and where do we go from here? Expert Review of Hematology 2016;9:335–350. [DOI] [PubMed] [Google Scholar]
- 131.Sasine JP, Schiller GJ. Emerging strategies for high-risk and relapsed/refractory acute myeloid leukemia: Novel agents and approaches currently in clinical trials. Blood Rev 2015;29:1–9. [DOI] [PubMed] [Google Scholar]
- 132.Binsfeld M, Fostier K, Muller J, et al. Cellular immunotherapy in multiple myeloma: Lessons from preclinical models. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2014;1846:392–404. [DOI] [PubMed] [Google Scholar]
- 133.Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013;122:3461–3472. [DOI] [PubMed] [Google Scholar]
- 134.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2015;372:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Younes A, Ansell SM. Novel agents in the treatment of Hodgkin lymphoma: Biological basis and clinical results. Semin Hematol 2016;53:186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Armand P, Shipp MA, Ribrag V, et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. Journal of Clinical Oncology 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moskowitz CH, Ribrag V, Michot J-M, et al. PD-1 Blockade with the Monoclonal Antibody Pembrolizumab (MK-3475) in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure: Preliminary Results from a Phase 1b Study (KEYNOTE-013). Blood 2014;124:290–290. [Google Scholar]
- 138.Enblad G, Karlsson H, Loskog AS. CAR T-Cell Therapy: The Role of Physical Barriers and Immunosuppression in Lymphoma. Hum Gene Ther 2015;26:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009;113:6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 2008;118:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hinrichs CS, Borman ZA, Cassard L, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A 2009;106:17469–17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ruella M, Klichinsky M, Kenderian SS, et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. ASH annual meeting. San Diego; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ruella M, Kenderian SS, Shestova O, et al. Novel Chimeric Antigen Receptor T Cells for the Treatment of Hodgkin Lymphoma. Blood 2014;124:806. [Google Scholar]
- 144.Wagner HJ, Bollard CM, Vigouroux S, et al. A strategy for treatment of Epstein-Barr virus-positive Hodgkin’s disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther 2004;11:81–91. [DOI] [PubMed] [Google Scholar]
- 145.Navai Shoba A, Ahmed N. Targeting the tumour profile using broad spectrum chimaeric antigen receptor T-cells. Biochemical Society Transactions 2016;44:391. [DOI] [PubMed] [Google Scholar]
- 146.Anurathapan U, Chan RC, Hindi HF, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther 2014;22:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest;126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ma Y PINZK, JIANG X, Wada M, Chen K. CHIMERIC ANTIGEN RECEPTORS (CARs), COMPOSITIONS AND METHODS OF USE THEREOF. Google Patents; 2016. [Google Scholar]
- 149.Savoldo B, Rooney CM, Di Stasi A, et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 2007;110:2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grada Z, Hegde M, Byrd T, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2013;2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 2016;4:498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang Z, Wu Z, Liu Y, Han W. New development in CAR-T cell therapy. J Hematol Oncol 2017;10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest 2016;126:3036–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 2016;4:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cartellieri M, Feldmann A, Koristka S, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J 2016;6:e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, von Bonin M. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J 2014;4:e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.San-Miguel JF, Mateos M-V. Can multiple myeloma become a curable disease? Haematologica 2011;96:1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nikiforow S, Werner L, Murad J, et al. Safety Data from a First-in-Human Phase 1 Trial of NKG2D Chimeric Antigen Receptor-T Cells in AML/MDS and Multiple Myeloma. ASH annual meeting. San Diego; 2016. [Google Scholar]
- 159.Feng KC, Guo YL, Liu Y, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol 2017;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ying Z-T, Chang L-J, Kuo H-H, et al. 415. First-In-Patient Proof of Safety and Efficacy of a 4th Generation Chimeric Antigen Receptor-Modified T Cells for the Treatment of Relapsed or Refractory CD30 Positive Lymphomas. Molecular Therapy 2015;23:S164. [Google Scholar]
- 161.KOCHENDERFER JN. Anti-cd30 chimeric antigen receptors. Google Patents; 2017. [Google Scholar]
- 162.Frank XF, Wanhong Z, Jie L, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. 2017 ASCO Annual Meeting. Volume 35: J Clin Oncol; 2017. pLBA3001 [Google Scholar]
- 163.Berdeja JG, Lin Y, Raje NS, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: Updated results. 2017 ASCO Annual Meeting. Volume 35: J Clin Oncol; 2017. [Google Scholar]
- 164.Hermanson DL, Barnett BE, Rengarajan S, et al. A Novel Bcma-Specific, Centyrin-Based CAR-T Product for the Treatment of Multiple Myeloma. Blood 2016;128:2127. [Google Scholar]
- 165.Barnett BE, Wang X, Hermanson DL, Tan Y, Osertag EM, Shedlock DJ. Development of Novel Non-Immunoglobulin Centyrin-Based Cars (CARTyrins) Targeting Human Bcma. Blood 2016;128:4557. [Google Scholar]
- 166.Guo B, Chen M, Han Q, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. Journal of Cellular Immunotherapy 2016;2:28–35. [Google Scholar]
- 167.Garfall AL, Maus MV, Hwang W-T, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. New England Journal of Medicine 2015;373:1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Garfall AL, Stadtmauer EA, Maus MV, et al. Pilot Study of Anti-CD19 Chimeric Antigen Receptor T Cells (CTL019) in Conjunction with Salvage Autologous Stem Cell Transplantation for Advanced Multiple Myeloma. ASH Annual Meeting. Volume Volume 128(22):974–974. San Diego: Blood; 2016. [Google Scholar]
- 169.Ramos CA, Savoldo B, Liu E, et al. Clinical Responses in Patients Infused with T Lymphocytes Redirected to Target Kappa-Light Immunoglobulin Chain. Biology of Blood and Marrow Transplant;20:S26. [Google Scholar]