

Summary
The fact that a subset of human cancers showed evidence for a spontaneous adaptive immune response as reflected by the T cell-inflamed tumor microenvironment phenotype led to the search for candidate innate immune pathways that might be driving such endogenous responses. Preclinical studies indicated a major role for the host STING pathway, a cytosolic DNA sensing pathway, as a proximal event required for optimal type I IFN production, dendritic cell activation, and priming of CD8+ T cells against tumor-associated antigens. STING agonists are therefore being developed as a novel cancer therapeutic, and a greater understanding of STING pathway regulation is leading to a broadened list of candidate immune regulatory targets. Early phase clinical trials of intratumoral STING agonists are already showing promise, alone and in combination with checkpoint blockade. Further advancement will derive from a deeper understanding of STING pathway biology as well as mechanisms of response versus resistance in individual cancer patients.
Keywords: STimulator of INterferon Genes (STING), cancer immunotherapy, innate immunity, type I interferon, tumor immunity
1. Basic Biology of the STING Pathway
The STING pathway is an innate immune pathway that senses cytosolic DNA and activates several downstream signaling events, the best characterized of which is IRF3 activation and IFN-β gene expression. Components of the STING pathway are regulated by multiple post-translational modifications, are highly dependent on vesicular trafficking for their regulation, and cross-talk with several cellular processes that are relevant for cancer immunotherapy.
1.01. STING Pathway Overview
The STING pathway is triggered by cyclic GMP-AMP (cGAMP) synthase (cGAS) enzyme binding directly to DNA and generates the cyclic dinucleotide 2’−5’ cGAMP (1). Under basal conditions, the STimulator of INterferon Genes (STING) is a transmembrane protein present in the endoplasmic reticulum (ER)(2). Following cGAS activation and cGAMP synthesis, STING is activated by binding directly to cGAMP (3, 4). STING then translocates from the ER to the ER-Golgi intermediate complex, through the Golgi apparatus, and eventually to perinuclear vesicles (5, 6). This vesicular trafficking depends on iRhom2, Sec61b, TRAPb, Sec5, and VPS34 (5–7). siRNA knockdown of any of these factors abrogates STING trafficking and blocks IFN-β gene expression, emphasizing the importance of vesicular trafficking for STING signaling. TANK-binding kinase 1 (TBK1) binds to activated STING and phosphorylates itself as well as STING (8). This appears to happen as STING translocates through the Golgi apparatus because Brefeldin A treatment or knockdown of Sar1 both inhibit ER-to-golgi trafficking and prevent TBK1 binding to STING (9). The vesicles carrying the activated STING-TBK1 complex then translocate to perinuclear regions (5). The transcription factor IRF3 binds the activated STING-TBK1 complex and is phosphorylated by TBK1 (10), whereby phosphorylated IRF3 dissociates from the complex and translocates to the nucleus (5, 11, 12) where it induces expression of target genes including IFN-β.
To prevent chronic signaling, the active STING-TBK1 complex is eventually degraded (13). Immunofluorescent microscopy has shown that, at later time points, STING co-localizes with Rab7-containing vesicles (late endosome/lysosome) but not vesicles containing Rab5 (early endosome) or Rab11 (recycling endosomes). Inhibiting lysosomal function by blocking acidification prevents activation-induced STING degradation (13, 14). To prevent chronic cGAS activation, the STING pathway also induces autophagy and this is thought to help clear cytosolic DNA. Cells deficient in either cGAS or STING fail to induce autophagy in response to cytosolic DNA. In macrophages the autophagosomal marker LC3 co-localizes with transfected DNA as well as cGAS, STING, and TBK1, suggesting a direct role in targeting DNA for autophagosomal degradation (15–18). As another negative regulatory process, cGAMP is degraded by the phosphodiesterase ENPP1 (19).
Thus, cytosolic DNA initiates a cGAS-STING-TBK1-IRF3 signaling cascade that induces a type I IFN response, and signaling becomes resolved by degradation of components of the STING pathway as well as clearance of the stimulatory DNA (14). There are many post-translational modifications of both cGAS and STING that regulate their function and allow cross-talk of the STING pathway with other cellular processes. Understanding these mechanistic details may be important for developing the next generation of STING pathway agonists designed to maximize anti-tumor effects and minimize any undesired effects. A pictorial representation of STING pathway activation is depicted in Figure 1.
Figure 1. STING pathway signaling and regulation.
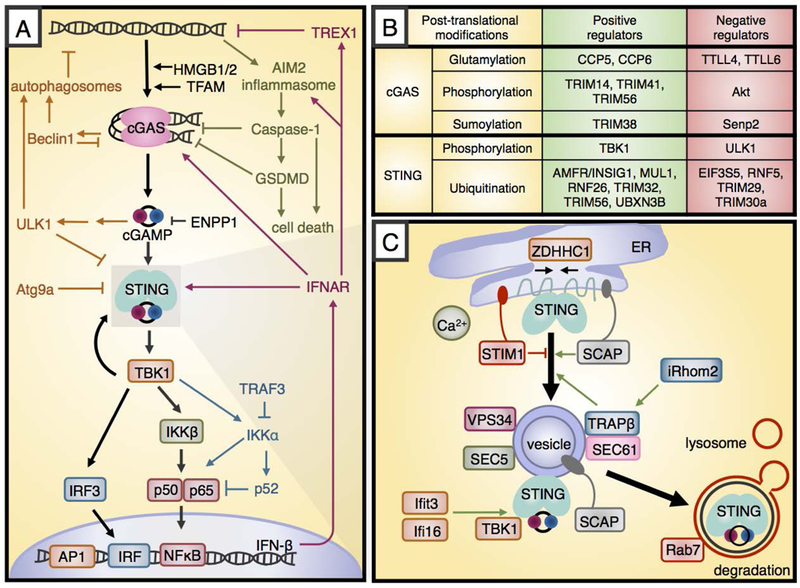
A. Cytosolic DNA activates the enzyme cGAS, which produces the cyclic dinucleotide cGAMP that activates STING. Active STING recruits TBK1 and leads to IFN-β transcription downstream of the transcription factors IRF3 and NF-κB. The STING pathway interacts with various other cellular processes, including autophagy (orange), AIM2 inflammasome signaling (green), non-canonical NFκB signaling (blue) and autocrine signaling downstream of IFN-β (maroon). B. Several post-translational modifications regulate cGAS and STING. The modifying factors that promote pathway activation are shown in green boxes, while those that inhibit are shown in red boxes. C. Upon activation by cGAMP, STING traffics from the ER through the ERGIC to perinuclear vesicles prior to lysosomal fusion and degradation. The black arrows show this motion, and the red and green arrows show positive and negative regulation.
1.02. Mechanistic Insights Into cGAS Function
cGAS is composed of two DNA binding domains and a nucleotidyltransferase domain which converts ATP and GTP into the cyclic dinucleotide cGAMP (20). STING binds the secondary messenger cGAMP with very high (~4 nM) affinity, which means that very small amounts of cGAMP can elicit a type I interferon (IFN) response (21). Because of this, cGAS activity must be tightly controlled in order to accurately respond to cytosolic DNA that arises from a pathogen or cellular damage or stress. Recent molecular studies have shed light on how this regulation occurs at the level of DNA availability, cGAS transcription and posttranslational modification, and the subsequent degradation of cGAS and cGAMP. In the context of cancer therapy, these regulatory mechanisms have the potential to be modulated in order to make the pathway more sensitive to tumor DNA or augment downstream signal transduction.
Defects in DNA repair machinery can lead to aberrant accumulation of self-DNA. SAMHD1 is a dNTPase that acts at stalled replication forks to degrade nascent DNA and allow DNA replication to restart. In its absence, DNA molecules are released from stalled replication forks and can form duplexes in the cytosol that activate cGAS (22). RNAseH2 is another protein involved in DNA repair, and its absence leads to genome instability through a failure to remove incorporated ribonucleotides (23). DNA that fails to segregate properly during mitosis due to loss of RNAseH2 or other sources of damage can lead to the formation of micronuclei. Micronuclear DNA was previously thought to be invisible to immune detection, but it now seems cGAS can access it when micronuclear membranes become unstable during subsequent cell division (24, 25). This implies that cells undergoing cell cycle arrest following DNA damage and micronuclei formation do not activate the STING pathway, whereas cells that continue to divide can undergo STING pathway activation. These tumor cell-intrinsic mechanistic studies provide insight into the ways in which genotoxic agents can sensitize the immune system to tumors and suggest novel ways of regulating DNA repair in tumor cells to render them more immunogenic.
The DNAse TREX1 is another key factor that regulates the amount of DNA accessible to cGAS. TREX1 is a known IFN-stimulated gene, and is required for degrading cytosolic DNA to prevent continuous type I IFN production. Mathematical modeling of the pathway showed that cGAS is not activated by partial inhibition of TREX1 (26), however complete loss can induce Aicardi–Goutières syndrome (27) and other autoimmune disease (28). Oxidized DNA is less susceptible to TREX1 degradation, and can occur as a result of ROS or UV exposure and lead to cGAS activation (29). TREX1 inhibitors are currently being evaluated preclinically and represent a novel approach towards sensitizing the STING pathway to tumor cell-intrinsic cytosolic DNA.
The zinc finger domain within the DNA binding region of cGAS allows it to recognize double stranded DNA as opposed to dsRNA and single stranded nucleic acids (30, 31). DNA binding creates a steric clash with the activation loop of cGAS which is resolved by the induction of a dramatic conformational change exposing the catalytic surfaces that bind ATP and GTP (32). Recent crystallization of cGAS under various conditions revealed that, at physiological ATP concentrations, cGAS adopts a pseudo-active conformation. cGAS becomes fully active when dsDNA stabilizes cGAS residues Gly207-Asn210. A small molecule capable of stabilizing cGAS at these residues could activate cGAS in the absence of DNA and might have therapeutic potential (33).
cGAS binding to DNA does not depend on the DNA sequence but does have a minimum length requirement for optimal activation. This ranges from 45–70 base pairs at 1 mg/mL to 800–2000 bp at lower, more physiological DNA concentrations (34). The binding of cGAS to DNA is cooperative in that when a cGAS dimer binds two strands of DNA, it orients them in such a way that favors binding of subsequent cGAS dimers. The resulting structure resembles a ladder, with cGAS dimers forming rungs between adjacent DNA strands. The structural DNA binding proteins HMGB½ and TFAM may facilitate the initial nucleation step of cGAS2-DNA2 binding by bending DNA into a U shape (35–37). Increasing the expression of these proteins was reported to orient cytosolic DNA in a way that is more favorable for cGAS activation (38).
1.03. cGAS Regulation
The cGAS promoter contains two IFN response elements, and type I IFN sensing increases the expression of cGAS following viral infection or DNA transfection. This is likely because constitutively expressed DNA sensors such as DDX41 produce an initial wave of type I IFNs that, through a positive feedback loop, upregulate cGAS expression and subsequent type I IFN production. DDX41 inhibition and knockout of the type I IFN receptor both were reported to attenuate cGAS mRNA expression in macrophages (39).
Following cGAS expression, several post-translational modifications are capable of regulating its enzymatic activity and degradation. TBK1 downstream of STING phosphorylates and activates Akt (40), which can then phosphorylate cGAS in its catalytic region and block cGAMP production (41). The tubulin tyrosine ligase-like glutamylases TTLL4 and TTLL6 glutamylate cGAS and impair its synthase and DNA-binding abilities, respectively. These glutamates are removed by the carboxypeptidases CCP5 and CCP6, which promote cGAS activation (42). TRIM14, TRIM41, and TRIM56 are members of the tripartite motif E3 ubiquitin ligase family, and all alter cGAS ubiquitination in a way that favors its activation (43–45). TRIM38 sumoylates cGAS and promotes its stability by preventing ubiquitination mediated degradation, and Senp2 desumoylates cGAS to allow its degradation (46). Modulating the proteins that regulate cGAS activity or modulating cGAS itself based on our understanding of its activating and inhibitory posttranslational modifications represents an as yet untapped therapeutic strategy.
Once cGAMP is produced by cGAS, it can be degraded by the phosphatase ENPP1. Inhibiting ENPP1 or generating non-hydrolysable cGAMP variants can lead to more potent downstream STING signaling, and both approaches are being tested in preclinical models (19, 47).
1.04. Mechanistic Insights into STING Function and Regulation
It is important to note that STING is more than just a sensor of cGAMP production, rather it coordinates signals from multiple DNA sensors (48–50). Like cGAS, STING is highly regulated both in terms of expression and activity. At the level of transcription, IFN-sensing components and KDM5 demethylases both regulate STING expression. In keratinocytes, IFN-γ but not IFN-β, TGF-β or TNFα was found to up-regulate STING expression. Phosphorylated STAT1 dimers downstream of IFN-γ sensing can bind and activate the STING promoter (51). In macrophages, on the other hand, type I IFN can induce STING expression, through STAT1/STAT2/p48 binding to a different region of the promoter (52). KDM5 family demethylases carry out additional regulation at the STING promoter by removing H3K4 trimethylation and limiting transcription. STING expression is anticorrelated with KDM5 expression across multiple tumor types in The Cancer Genome Atlas (TCGA), suggesting this is a mechanism by which tumor cells suppress STING signaling. KDM5 inhibitors trigger a robust type I IFN response, and could be evaluated preclinically for immune modulatory effects (53).
The surprising finding that some STING-deficient mice demonstrate worse autoimmune phenotypes than their counterparts (54) led to the investigation of immune-regulatory roles of STING and ultimately to the identification of a novel STING isoform. This isoform, named STING-β, is the product of alternative splicing and dominantly inhibits STING function. It does so by sequestering STING as well as cGAMP, TBK1, and other signaling components. Notably, STING-β expression anticorrelates with type I IFN production, and may rapidly release STING pathway components following stress or infection (55). A third isoform, MITA-related protein (MRP), inhibits IRF3 activation but is capable of activating NF-κB (56). Thus, drugs designed to inhibit the regulatory isoforms may allow the stimulatory isoforms to carry out downstream signaling more successfully.
cGAMP binding to a dimer of STING on the ER membrane induces a STING conformational change that initiates its migration through the ER-Golgi intermediate compartment (ERGIC) to the Golgi. STING recruits TBK1 at the trans-Golgi network where TBK1 phosphorylates multiple substrates before the complex buds off in vesicles that translocate to perinuclear sites, where the complex activates IRF3. Every step of this process is regulated, and has the potential to be tuned therapeutically.
The ER-bound palmitoyl transferase ZDHHC1 promotes STING dimerization, which is a prerequisite for STING activation (57). iRhom2 is essential for STING to traffic from the ER to the Golgi, and iRhom2 deficient cells lose the ability to form perinuclear endosomes containing STING following viral infection or DNA transfection (58). In addition to promoting cGAS activation, the DNA sensor IFI16 actively recruits TBK1 to STING (59, 60). The IFN-inducible gene IFIT3 also has been reported to promote STING-TBK1 binding (61), and SCAP is a scaffold adaptor that travels with STING and recruits IRF3 to the complex with TBK1 (62). Much less is known about the negative regulators of STING translocation, although increases in intracellular calcium concentration have been shown to play a role (63). A new study identified STIM1 as the calcium sensor that anchors STING to the ER membrane (64). In fact, STIM1 deficiency results in spontaneous STING activation and type I IFN production. This makes STIM1 inhibition a potential target for future therapeutic development.
In addition to the binding partners that facilitate STING migration, several proteins promote STING activation by modifying STING ubiquitination, including the AMFR/INSIG1 complex, TRIM32, TRIM56, MUL1, and UBXN3B (65–69). STING palmitoylation is another activating modification required for signal transduction following trafficking. In fact, STING remains palmitoylated even in degradation compartments (70), and small-molecule inhibitors of STING palmitoylation are being tested in autoimmune models (71). The autophagy-associated kinase ULK1 is indirectly activated by STING signaling and phosphorylates STING after it traffics to the Golgi to promote its degradation (72). EIF3S5, TRIM29, TRIM30α, and RNF5 also target STING for degradation by K48-linked ubiquitination (58, 73–75). RNF26 blocks RNF5 ubiquitination by adding a K11-linked ubiquitin (76). STING pathway activation could theoretically be enhanced by inhibiting the negative posttranslational regulators or by activating the positive regulators.
It is worth noting that much of STING pathway regulation remains to be understood. For example, loss of the small ubiquitin-like modifiers SUMO2 and SUMO3 results in spontaneous IFN-β production in the absence of stimuli. However, it appears to do so independently of all known IFN-inducing pathway components, and IRF3 and IRF7 are dispensable for this effect. It is possible the mechanism involves activation of non-essential IFN transcription factors, such as IRF1 and IRF5 or that it somehow alters chromatin accessibility of the Ifnb1 locus (77). Although it is likely that many molecular details of the STING pathway remain to be understood, our existing knowledge of its function and regulation is already providing a rich set of potential nodes for therapeutic intervention.
1.05. STING Pathway Antagonism
The autophagy and inflammasome pathways appear to inhibit STING signaling, and this natural pathway antagonism has the potential to be targeted therapeutically. Beclin1 is an autophagy protein that binds to cGAS following DNA stimulation and blocks cGAMP production. Additionally, this binding interaction releases Rubicon from the Beclin-1 autophagy complex, which is then free to activate PI3K and lead to autophagic degradation of cytosolic DNA (78). Additional inhibition occurs from the autophagy protein Atg9a, which negatively regulates STING translocation from Golgi to endosomes, as well as its association with TBK1 (79).
Caspase-1 activation downstream of the inflammasome can directly bind cGAS and cleave it, abolishing cGAMP synthesis (80). Following non-canonical inflammasome activation induced by LPS, caspases 5,6, and 11 were also capable of cleaving cGAS. Potassium influx following caspase-1 cleavage of gasdermin D directly inhibits cGAS enzyme activity (81). Antiviral and inflammasome programs appear to be mutually inhibitory, as type I IFNs down regulate the NLRP1 and NLRP3 inflammasomes via STAT1 and IL10/STAT3 signaling to reduce pro-IL1 production (82, 83). NLRC3, which is an inflammasome component, also reduces STING translocation and association with TBK1 (84). Several caspase inhibitors have already been developed as drug candidates, and they should be tested for their potential to augment the innate immune response. In general, inhibiting the pathway components that block STING signal transduction might tip the balance in favor of type I IFN production and facilitate an anti-tumor immune response.
1.06. The STING Pathway and the AIM2 Inflammasome
There are several different inflammasome complexes that are activated by a variety of innate immune agonists. The AIM2 inflammasome senses cytosolic DNA and therefore its functional interaction with the STING pathway is important to consider. After binding directly to DNA, AIM2 recruits ASC as well as caspase-1 and this complex catalyzes caspase-1 autoactivation. Typically an initial “signal 1” induces IL-1β and IL-18 expression after which active caspase-1 cleaves IL-1β and IL-18 to convert both into their active forms. Sustained inflammasome signaling ultimately activates gasdermin D which induces necroptosis, a form of inflammatory cell death (85). One study found that cGAMP stimulation could serve as “signal 1” to induce IL-1β gene expression via autocrine type I IFN signaling. A second stimulation with cGAMP after initial priming was then able to induce AIM2 inflammasome activation and IL-1β cleavage independent of cytosolic DNA. Processing IL-1β in this context was dependent on AIM2, NLRP3, ASC, and caspase1 but did not required caspase 11, indicating cGAMP might directly activate the AIM2 inflammasome. In this context, STING−/− cells lost IL-1β processing in response to cGAMP suggesting some crosstalk between signaling molecules downstream of STING with the inflammasome (86).
The AIM2 inflammasome can also regulate cGAS-STING signaling indirectly. Bone marrow-derived DCs and macrophages deficient in AIM2 inflammasome components produced higher levels of IFN-β after cytosolic DNA stimulation. This was associated with increased STING signaling including higher levels of cGAMP as well as greater phosphorylation of TBK1 and IRF3. AIM2 inflammasome-deficient cells also demonstrated reduced cell death as shown by decreased LDH release. These results suggest that cytosolic DNA not only induces cGAS-STING signaling and type I IFN signaling but also activates the AIM2 inflammasome which can silence chronic STING signaling by inducing cell death (87).
Together these results suggest that cGAMP produced by cGAS activation can incite inflammasome activation and this can induce multiple downstream effects including cell death which serve to counter-regulate STING signaling. Finding ways to induce STING activation while avoiding cell death in immune cells represents an opportunity to improve efficacy with STING pathway agonists.
1.07. The STING Pathway and NF-κB
The linear cGAS -> STING -> IRF3 -> IFN-β signaling cascade is a well-established and important consequence of STING signaling for immunity in general and specifically for cancer immunotherapy. However, there are other aspects and outcomes of STING signaling that are less well characterized but are likely important for therapeutic exploitation of the STING pathway.
For example, type I IFNs are induced by STING activation and are an important component of STING pathway biology for antitumor immunity (88). Earlier studies of IFN-β gene expression in response to viral infection revealed a more complex cooperation between IRF3 and additional transcription factors, including NF-κB and AP-1 (89, 90). In certain cellular contexts, STING is capable of activating all 3 of these factors (2, 11, 91), however this is not always the case. DNA stimulation of MEFs has been reported to lead to STING-dependent phosphorylation and nuclear translocation of NF-κBp65 (91). RNAi knockdown of p65 caused a 50% reduction in IFN-β induced by dsDNA, confirming the importance of NF-κB in STING signaling. Over-expression of the signaling adaptors TRAF3 or TRAF6 increased STING-induced NF-κB reporter activity. siRNA knockdown of these adaptors displayed divergent roles with TRAF6 knockdown leading to reduced NF-κBp65 activation while TRAF3 knockdown lead to reduced non-canonical NF-κBp52 signaling, which typically opposes canonical NF-κB signaling (91, 92). Follow up experiments found STING activation in WT MEFs activated non-canonical NF-κBp52, which required IKKα but not TBK1 or IKKβ.
These results are consistent with a study investigating STING activation following radiotherapy. Irradiated tumor cells activate the STING pathway in tumor-infiltrating immune cells which is important for the efficacy of radiation (93). One study demonstrated mice deficient in the non-canonical NF-κB factor RelB respond better to radiotherapy. This response was associated with increased type I IFN production that was explained by removing competition in DNA binding at the IFN-β promoter between canonical and non-canonical NF-κB complexes (94). Together, these results indicate STING can potentially activate both canonical and non-canonical NF-κB, which may regulate IFN-β expression in certain cellular contexts. A greater understanding of the cell-type specific nature of this signaling as well as the specific molecules involved could allow new therapies designed to augment IFN-β expression to maximize therapeutic potential.
1.08. Cell Type-Specific Effects of STING Signaling
The STING pathway has also been linked to cell death in a cell-type specific manner. One of the earliest observations of the mouse STING agonist DMXAA was that it induced endothelial cell apoptosis (95). DMXAA treatment induced endothelial cell death in murine tumors based on TUNEL staining and this led to rapid necrotic tumor cell death. Endothelial cells stimulated in vitro confirmed that DMXAA induced apoptotic cell death and that it was a TNF-independent effect. More recent studies looking at T cells have similarly identified apoptosis as a consequence of STING pathway signaling (96). STING pathway activation in T cells induces IFN-β expression as well as transcriptional up regulation of the pro-apoptotic molecule BAX and down regulation of the anti-apoptotic protein Bcl2. Other reports have also found increased transcription of pro-apoptotic Puma and Noxa (97). STING activation was shown to induce cell death in T cells but not macrophages or DCs (96, 97). T cell death appeared to occur via apoptosis because over-expression of Bcl2 partially rescued viability. STING activation also affected the proliferation and memory formation of T cells. After CD3/CD28 stimulation, T cells co-stimulated with a STING agonist proliferated less. Detailed studies of mitosis found STING activation was associated with lengthened metaphase, which slowed proliferation. Persistent STING signaling reportedly affects T cell function as well. T cells isolated from patients with constitutively activating STING genetic mutations had fewer circulating memory T cells (98). Based on this biology, STING agonists have also been evaluated as a potential therapeutic agent for T cell malignancies. Interestingly, STING agonists delayed tumor growth of T-ALL murine tumors even in STING-deficient hosts, suggesting tumor-intrinsic STING activation could have beneficial anti-tumor effects in T-lineage malignancies (97).
1.09. Tumor Cell-intrinsic STING Signaling
While most studies of STING pathway signaling have investigated effects on host immune cell subsets and demonstrated necessity using STING−/− mice, in the cancer context it is also important to consider a role for STING within tumor cells. Some tumor cells display the presence of cytosolic DNA even in the absence of DNA-damaging therapeutic treatment (99), which might serve as a stimulus for STING pathway activation. Other studies have identified micronuclei that appear adjacent to the nucleus and contain cGAS, suggesting that damaged DNA fragments in these structures might activate the STING pathway (24, 25). These data indicate tumor cells at least have an opportunity for cell-intrinsic STING signaling. Preclinical studies have indicated that tumor cell-intrinsic IFN-β expression driven by a constitutive promoter can leads to tumor regression in vivo. In this case, even mixed tumors that contained only 10% IFN-β expressing cells were rapidly rejected (100). This observation implies there might be selective pressure for tumor cells to lose IFN-β gene expression and/or some components of the STING signaling pathway. Consistent with this idea, colorectal carcinomas and human melanomas reportedly show decreased cGAS and STING expression (101, 102). Interestingly, some tumors might retain cGAS activity but lose STING activity and this residual signaling might explain how STING is activated in tumor-infiltrating host immune cells in certain settings.
1.10. STING Agonist Transfer from Tumor Cells to Immune Cells
Preclinical studies have revealed that STING pathway activation in host immune cells is necessary for production of IFN-β (103). STING signaling and type I IFNs, in turn, were necessary for optimal tumor antigen specific CD8+ T cell cross-priming by Batf3-lineage dendritic cells. Based on these observations, many groups have now developed STING agonists to mimic this activation and facilitate improved anti-tumor immunity. However, one fundamental question that remains poorly understood is how tumor cells lead to activation of STING in host immune cells. A deeper understanding of this mechanism might lead to new therapeutic opportunities to improve anti-tumor immunity.
The two major hypotheses for DC activation by tumor cells are either that tumor-derived cGAMP can become transferred to host antigen-presenting cells (APCs) thereby activating the STING pathway directly via STING, or that tumor-derived DNA can be transferred leading to host STING pathway activation upstream at the level of cGAS. Initial preclinical studies of STING pathway activation in response to tumor cells in vivo revealed the presence of labeled tumor DNA within the cytosol of DCs (103). This observation was consistent with tumor-derived DNA being a stimulus for the STING pathway, a concept further supported by the observation that both host cGAS and STING are necessary for checkpoint blockade immunotherapy in the B16 melanoma model in vivo (104). Similarly, after radiation, tumor cells activate the STING pathway in host immune cells which induces a type I IFN response that is both cGAS- and STING- dependent (93). The mechanism of DNA transfer to host APCs has not yet been elucidated. In principle, tumor-derived DNA would need to avoid extracellular DNAses, bypass lysosomal degradation via the endocytic pathway, and gain access to the cytosol of recipient DCs. One potential mechanism that can achieve all of these is fusion of an extracellular vesicle released by tumor cells with recipient immune cells. Such a process could deliver cargo directly to the cytosol. In vitro studies of irradiated tumor cells showed that tumor-derived exosomes can contain DNA, which can be acquired by DCs. This exosomal DNA was Trex1- sensitive, suggesting that cytosolic DNA after radiation can be packaged in exosomes, transferred to DCs, and ultimately activate the cGAS-STING pathway (105).
Other groups have found changes in the lysosomal compartment are an alternative mechanism to explain how DNA can avoid degradation and stimulate cGAS. Tumor cells transfected with non-degradable DNA, irradiated to induce cell death, and then used to stimulate macrophages in vitro led to cGAS-STING signaling in macrophages. They found that DNAse II was responsible for limiting STING activation suggesting that tumor-derived DNA must avoid degradation by lysosomes in order to stimulate the STING pathway. In that model, tumor cells that expressed cGAS, such as B16-F10, were able to stimulate cGAS−/− macrophages, but at reduced levels compare to WT macrophages. These results argue that, while cGAMP also might be transferred to APCs, DNA is a more potent stimulus. Supporting this notion, when 293T cells lacking cGAS expression were used as a stimulus they failed to activate cGAS −/− macrophages, indicating DNA transfer was required in this system (106).
However, in another model system, STING but not cGAS was required in immune cells for innate immune activation in vivo (107). RMA-S tumor cells and B16-BL6 tumor cells, two NK-cell sensitive tumor models, had faster tumor outgrowth in STING−/− mice but not in cGAS−/− mice. Interestingly, RMA parental cells that are not NK cell sensitive grew out at normal rates in WT or STING−/− mice. RMA-RAE1 cells, that over-express an NK activating ligand to make them NK cell sensitive, grew out more quickly in STING−/− but not in cGAS−/− mice. These results imply there may be something special with tumor cells that are sensitive to NK cell killing that makes them more prone to cGAMP transfer for STING pathway activation. In studies of anti-viral immunity and STING pathway activation, inter-cellular cGAMP transfer also has been implicated via GAP junctions (108) or extracellular vesicles liberated from infected cells (109).
Gaining a greater mechanistic understanding of the process of STING pathway activation within host APCs in the setting of a growing tumor in vivo may enable improved opportunities for therapeutic development.
2. STING PATHWAY INVOLVEMENT IN ANTI-TUMOR IMMUNITY IN PRECLINICAL MODELS
Mouse preclinical models have implicated the host STING pathway is a major innate immune pathway activated by implanted tumors in vivo (103). STING−/− mice displayed defective tumor antigen-specific CD8+ T cell cross-priming and faster tumor outgrowth. This was in contrast to mice lacking other innate immune receptors and adapters including Trif, MyD88, MAVS, TLR4, TLR9, or P2RX7 that had normal anti-tumor CD8+ T cell cross-priming. This defect was paralleled by deficient IFN-β gene expression by tumor-infiltrating immune cells in STING−/− hosts. Previous studies had shown host IFN-β signaling as a critical bridge toward adaptive immune responses against tumors (88, 110). Mixed bone marrow chimera experiments implied that type I IFN signaling had to occur on Batf3-lineage DCs for optimal cross-priming of CD8+ T cells as well as for tumor growth control in vivo (88). Collectively, these data argue for a tumor DNA—cGAS/STING—type I IFN—Batf3DC—CD8+ T cell axis in endogenous anti-tumor immunity against tumors.
Based on these observations of endogenous immune responses, intratumoral injection of exogenous STING agonists has been explored as a therapeutic strategy. Injection of STING agonists led to rapid rejection of murine tumors (111). Maximal tumor control depended on CD8+ T cells, confirming an immune component of this efficacy, although some tumor control was observed in the absence of T cells. STING pathway activation was associated with increased IFN-β expression by tumor-infiltrating immune cells, increased expression of the T cell costimulatory molecule CD86 on APCs, and increased tumor antigen-specific CD8+ T cell cross-priming. These results indicate that additional STING pathway activation through pharmacologic manipulation can lead to augmented type I IFN signaling, APC maturation, CD8+ T cell cross-priming, and recruitment of T cells into the tumor microenvironment where they mediate rejection of tumors. Prior work had revealed that adoptively transferred tumor-specific CD8+ T effector cells failed to accumulate in tumor sites in the absence of Batf3 DCs in the tumor microenvironment (112). Mechanistically this was linked to Batf3 DC production of CXCL9/10, which are induced by STING activation. Therefore, STING pathway activation likely contributes to T cell priming in the tumor-draining lymph node, and also to effector T cell recruitment into the tumor microenvironment. An overview of the STING pathway in anti-tumor immunity is represented in Figure 2.
Figure 2. STING pathway activation initiates the anti-tumor immune response.
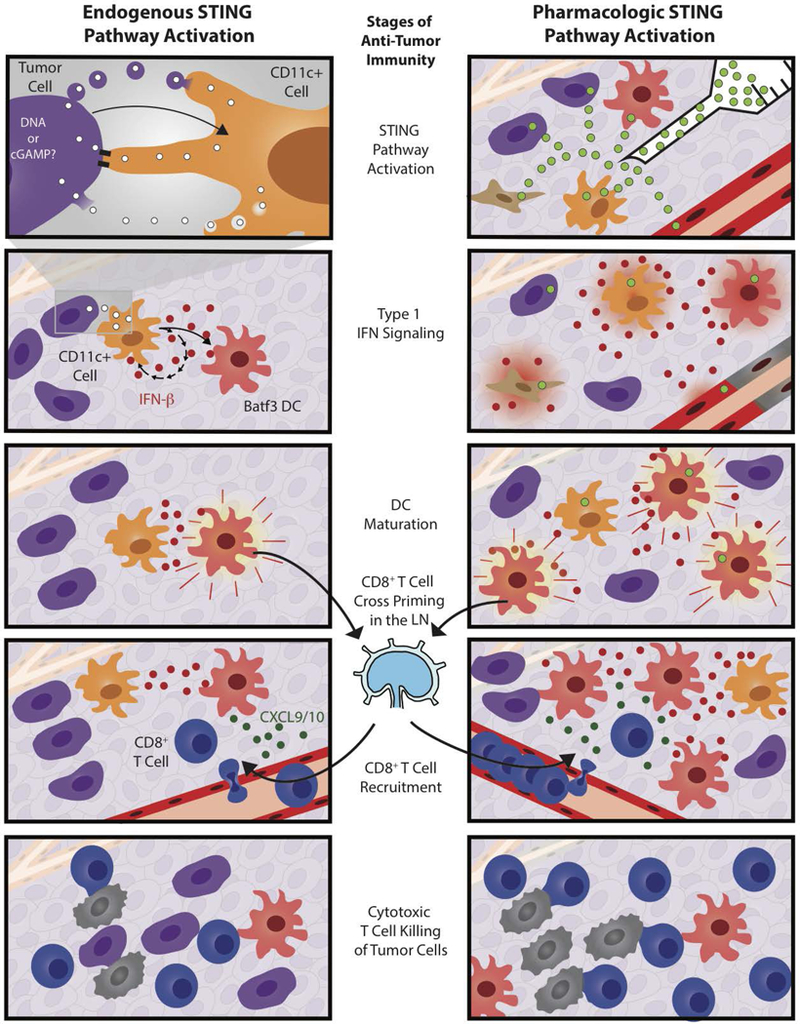
Endogenous (left side) and pharmacologic (right side) STING agonists elicit similar, but slightly different anti-tumor immune responses. The endogenous immune response involves tumor cell (purple cell) activation of CD11c+ cells (orange cells) that leads to type 1 IFN (red dots) signaling and Batf3+ DC (red cell) maturation. This, in turn, leads to CD8+ T cell (blue cell) cross priming and recruitment by CXCL9/10 (green dots). CD8+ T cells can then kill tumor cells (black cells). Treatment with intratumoral STING agonists (green dots) differs because the agonists are membrane permeable and therefore might activate a wider range of cell types including tumor cells, fibroblasts (brown cells), and endothelial cells (red vessels). Pharmacologic agonists also induce endothelial cell death, elicit higher IFN-β expression, and more CD8+ T cell cross-priming, which can lead to tumor clearance in preclinical models.
2.01. Development of STING Agonists as a Cancer Therapeutic
The compound DMXAA was originally characterized as an anti-vascular agent and displayed anti-tumor efficacy in a variety of mouse tumor models in vivo (113, 114). However, the molecular target of DMXAA had not been fully elucidated. Based on potent anti-tumor activity, it nonetheless was advanced to clinical testing, through to phase III studies in lung cancer. However, these efficacy studies in patients were negative, in contrast to the potent effects in mouse models (115, 116). Continued preclinical experimentation revealed that, in addition to its anti-vascular properties, DMXAA induced cytokine production by macrophages (117). Biochemical studies then revealed that DMXAA directly bound to the mouse STING protein (118). Comparison of the structure of mouse and human STING revealed that key amino acid residues in the DMXAA binding pocket were distinct in the human molecule, leading to failed binding of DMXAA to human STING (119), thus potentially explaining lack of efficacy in patients.
In parallel studies, in vivo delivery of DMXAA was found to induce a potent adaptive immune response against tumors, as evidenced by immunologic memory to re-challenge, rejection of distant non-treated tumors, and clearance of metastases. This host response depended on type I IFN signaling and CD8+ T cells, arguing that an adaptive immune response was a major mechanistic component for efficacy (111, 117, 120). The failure of DMXAA efficacy in STING−/− mice, as well as lack of activation of STING−/− APCs in vitro, together argued that DMXAA was indeed a direct STING agonist.
Based on lack of DMXAA binding to human STING, it became desirable to identify new chemical entities with the potential to activate human STING for immune activation in the setting of cancer. Initial drug development has focused on cyclic dinucleotides as a starting point. ADU-S100/MIW815 is a modified cyclic dinucleotide constructed to mimic natural STING agonists but with increased stability and activation of the five major human STING allelic variants. The compound BMS-986301 was also developed to activate human STING variants in addition to mouse STING. BMS-986301 achieved >90% rejection of tested transplantable tumors, while causing low cytotoxicity to CD8+ T cells and less inhibition of their proliferation compared to ADU-S100 (Society for ImmunoTherapy of Cancer Annual Meeting 2018 Poster P525). An additional compound was identified (SB11285) that displayed antitumor activity when delivered via the IV, IP, and IT routes (American Association for Cancer Research 2017 Conference on Tumor Immunology and Immunotherapy Poster A25).
2.02. Novel Strategies for STING Agonist Delivery by the Systemic Route
Several groups have performed chemical compound screens to identify non-nucleotide STING agonists with systemic anti-tumor activity (121–124). Such compounds can be selected for increased cell permeability and also resistance to hydrolysis by ENPP1 (19). One such screen identified several amidobenzimidazole (ABZI)-based compounds. The investigators linked two of these to create a dimeric ligand with even higher STING binding affinity. This compound demonstrated efficacy in the CT-26 colorectal model when administered IV (125).
Packaging cGAMP in liposomal nanoparticles has demonstrated improved cellular uptake and better tumor control in transplantable and genetically engineered triple negative breast cancer models as well as the B16.F10 melanoma model (126). Another study found that liposomal formulated STING agonist can cause loss of APC viability, so the authors chose to load a STING agonist ex vivo into exosomes instead (Exosting). ExoSTING induced superior IFN-β production compared to soluble STING agonists and the responding mice were protected against tumor re-challenge (Society for ImmunoTherapy of Cancer Annual Meeting 2018 Poster P618).
Modifying bacteria is an alternative strategy for developing novel STING agonists that can be delivered systemically. The SYN-STING method introduces a di-nucleotide cyclase gene into E. coli Nissle to generate cyclic-di-AMP in the hypoxic tumor microenvironment. SYN-STING demonstrated robust anti-tumor responses to transplanted tumors and immunologic memory when re-challenged 40+ days after the initial complete response (Society for ImmunoTherapy of Cancer Annual Meeting 2018 Poster P624). Another bacterial approach utilizes a highly attenuated strain of salmonella typhimurium that localizes to tumor due to auxotrophic consumption of immunosuppressive adenosine and delivers TREX1 RNAi to block degradation of cytosolic DNA (STACT-TREX1). Following IV delivery, these bacteria were found to be 1000-fold enriched in the tumor compared to liver and spleen, and demonstrated CD8-dependent tumor growth inhibition and regression in multiple tumor models (Society for ImmunoTherapy of Cancer Annual Meeting 2018 Poster P235).
Other novel preclinical strategies activate the STING pathway by targeting regulatory components that indirectly activate STING. For example, ENPP1 inhibitors reduce cGAMP degradation, thus improving STING pathway activation. They can be delivered systemically to increase sensitivity to endogenous STING agonists (Society for ImmunoTherapy of Cancer Annual Meeting 2018 Poster Poster P410 and (47)). Blocking endolysosome acidification with bafilomycin A1 (BaFA1) can also support STING signaling by preventing STING degradation. Intratumoral injection of cGAMP and BaFA1 in B16 subcutaneous tumors resulted in improved tumor control compared to cGAMP alone (127). As our molecular understanding of the STING pathway and its regulation improves, there will likely be additional opportunities for STING pathway therapeutic targeting beyond administration of cyclic dinucleotides.
2.03. Indirect STING Activation by Other Cancer Therapies
Some cancer therapies originally designed with other intended mechanisms of action can activate the STING pathway. Radiotherapy (RT), certain types of chemotherapy, and some targeted therapies can lead to activation of the STING pathway and this is important for their efficacy in preclinical models. Radiation-mediated tumor regression has historically been linked to cellular damage within tumor cells which promotes cell death and senescence (128). In some settings, the therapeutic effect of RT has been associated with priming of anti-tumor T cell responses in vivo (129, 130). In such instances, innate immune pathways would be imagined to be triggered, and indeed RT has been reported to lead to host STING pathway activation and type I IFN production (93). Irradiated tumor cells demonstrate cytosolic DNA fragments which can serve as agonists for cGAS and might be transferred to host immune cells by tumor-derived exosomes (131). cGAS deficient BM-DCs were not stimulated by irradiated tumor cells in vitro, implying that tumor-derived DNA activated DCs (93). Additional studies have shown that TREX1, which degrades cytosolic dsDNA, is up regulated upon RT treatment and that high doses of radiation induce sufficient TREX1 to clear cytoplasmic dsDNA. However, multiple lower RT doses activated the STING pathway and avoided TREX1 induction (132). These and other similar observations are shaping the design of RT regimens in combination with more targeted immunotherapies.
Certain types of chemotherapies have also been linked to STING signaling. Both cisplatin and etoposide are chemotherapies thought to act by inducing DNA damage and inhibiting DNA repair which leads to DNA adducts (133). Treatment with either compound induced cytosolic DNA that activated cGAS-STING signaling in selected cell models in vitro (134). Such observations suggest that a component of therapeutic efficacy could be linked to STING signaling. A better understanding of which chemotherapies activate STING and the exact mechanism by which it occurs could inform future drug development designed to optimize their ability to activate STING. These observations also raise the possibility of eliciting direct cytotoxicity towards tumor cells along with STING activation, which could provide additional benefits beyond direct STING agonists but will require careful consideration of immune suppressive effects as well.
Targeted therapies like PARP inhibitors (PARPi) have also been linked to STING activation. PARPi are thought to induce DNA damage that leads to double strand breaks in homologous recombination deficient tumor cells that cannot properly repair the DNA damage (135). Homologous repair-deficient tumor cells treated with PARPi in vitro induced cytosolic DNA that lead to STING signaling demonstrated by TBK1 and IRF3 phosphorylation (136–138). Both of these phosphorylation events were reduced after siRNA knockdown of cGAS or STING. When PARP inhibitors were used to treat mouse ovarian tumors, co-treatment with a TBK1 inhibitor abolished efficacy, suggesting that STING signaling was required for efficacy in this model (136). PARPi can be lethal to tumor cells that lack homologous repair but one study reports STING activation in tumor cells regardless of their homologous repair status, implying that STING pathway activation is independent of cell death and may apply to a broader range of molecular subtypes of cancer (138). As predicted from improved immune activation, PARPi combine well with checkpoint blockade antibodies in preclinical models (139).
2.04. Combination Therapies
STING agonists incite inflammatory cytokines that can remodel the tumor microenvironment and promote stronger anti-tumor T cell responses (111). These qualities make STING agonists good candidates for combination with established immunotherapies, such as anti-PD-1/PD-L1 antibodies. In a preclinical model of ovarian cancer, STING agonist treatment induced inflammation including increased expression of several chemokines like CXCL10 and CXCL1. These events were associated with an increase in CD103+ DCs and CD8+ T cells within the tumor (140).
Anti-PD-1/PD-L1 antibody therapies are thought to work best in tumors with evidence on an ongoing immune response that includes infiltration with Batf3-lineage DCs and CD8+ T cells (141–143). Tumors infiltrated with CD8+ T cells show increased expression of key chemokines, including CXCL9 and CXCL10, that likely mediate effector T cell recruitment (88, 112, 143). Preclinical studies have indicated that tumors in mice deficient in Batf3 DCs fail to produce CXCL9/10 and poorly recruit activated CD8+ T cells (112). Tumors that lack Batf3 DCs also poorly respond to adoptive T cell transfer, as the transferred effector T cells fail to accumulate within the tumor site. These observations indicate that T cell activation and expansion in the circulation is not sufficient to reject tumors, because the proper signals for recruitment into tumor sites are still required. STING agonists are capable of inducing both tumor antigen specific CD8+ T cell expansion as well as expression of chemokines known to recruit CD8+ T cells (103, 144). Therefore, STING agonists seem capable of promoting several key processes for important for efficacy of checkpoint blockade immunotherapy. Consistent with this idea, STING agonists combined with PD-1 blockade reduced tumor growth better than either therapy alone (145). In particular, tumor models that poorly responded to checkpoint blockade were sensitized when combined with STING agonists (146). These results are shaping the design of combination clinical trials in cancer patients.
3. STING AGONISTS IN THE CLINC
The initial STING agonist to enter clinical development was ADU-S100/MIW815 with first results reported at the Society for ImmunoTherapy of Cancer meeting in 2018 (147). The study was a phase I, single-arm dose-escalation study evaluating ADU-S100/MIW815 as an intratumoral (IT) injection three weeks out of four per cycle. The objectives for the study included evaluating safety, tolerability, and identifying the recommended phase 2 dose. Secondary objectives included evaluation of preliminary antitumor activity, pharmacokinetics, and pharmacodynamics with exploratory objectives surrounding biomarker analysis. The eligibility population included patients with advanced solid tumors, age of 18 years or older, Eastern Cooperative Oncology Group (ECOG) performance status of 0–1 and at least two cutaneous or sub-cutaneous tumor lesions that could be biopsied, with one accessible for injection. The lesion for injection could measure between 10–100 mm in longest diameter. Pre- and post-treatment biopsies were obtained from both the injected and non-injected lesions. Prior immunotherapy was allowed but not required. Bayesian hierarchical logistic regression model dose escalation was pursued testing does from 50 to 3200 μg.
A total of 41 patients received a dose of ADU-S100/MIW815 with four patients still with on-going treatment at the time of data lock. All but 3 (7.3%) patients had one or more prior lines of treatment with 82.9% having had two or more prior treatments and 53.7% having had prior exposure to checkpoint blockade immunotherapy. Melanoma (excluding uveal) was the most common tumor type making up 19.5% of patients and the most common adverse events included headache, injection site pain, and pyrexia. No maximum tolerated dose was established with elevated lipase being the only grade 3 or higher treatment-related adverse event reported in more than one patient (n=2; 4.9%). Best treatment response by Response Evaluation Criteria in Solid Tumors (RECIST) was partial response observed in two patients (parotid gland adenocarcinoma, PD1 antibody-refractory; Merkel Cell carcinoma, PD1 antibody-naive) with four patients continuing on treatment more than six months. Pharmacokinetic analysis suggested drug exposure to systemic circulation was rapid (within minutes) and generally increased in a dose-dependent manner. Systemic clearance from circulation was rapid with a half-life of 10 to 23 minutes.
Translational analysis was described in the context of two patients who had demonstrated clinical benefit. The first was a patient with collecting duct carcinoma continuing on treatment more than eight months at the 800 μg dose level. In this patient, increases in systemic cytokine levels (i.e. IL-6) were observed consistently following injection, and a more than two-fold increase in gene expression for pre- to post-treatment injected tumor biopsy was observed for IFN-γ, programmed death-ligand 1 (PD-L1), CD8A and a natural killer cell gene set. In the non-injected lesion, an increase in CD8-positive tumor-infiltrating lymphocytes (TIL) and PD-L1 stromal staining also was observed. A second vignette included a patient with esophageal cancer treated with 800 μg for three cycles with best response of stable disease. In this patient increased levels of IFN-β were observed following each injection. Within the injected tumor, biopsy demonstrated more than two-fold increased expression of CD8A and the NK cell gene set as well as an increase in the staining of CD8+ TIL and PD-L1 stromal staining.
Data for a second STING agonist administered IT (MK-1454) were disclosed at the 2018 European Society for Medical Oncology meeting. The study was a phase I, multi-arm dose-escalation study evaluating MK-1454 alone (arm 1; IT injection weekly for 9 weeks then every 3 weeks thereafter) or in combination with pembrolizumab (arm 2; 200 mg IV every 3 weeks). Endpoints for the study were standard, evaluating safety and tolerability, pharmacokinetics and pharmacodynamics, and preliminarily evaluating the overall response rate of the treatment. The eligibility population included subjects with advanced solid tumors, an age of 18 years or older, ECOG 0–1 who had at least two tumor lesions with one accessible for injection. Pre- and post-treatment biopsies were obtained from both of the lesions. Accelerated dose titration was pursued using patient dose cohorts of MK-1454 as monotherapy from 10–90 μg then 3 patients at 270 μg followed by modified toxicity probability interval (MTPI) design including 3–6 patients through 3000 μg. In combination with pembrolizumab, accelerated titration was performed for 90–270 μg then MTPI through 2000 μg. Cross-over of patients from monotherapy to combination was allowed after initial progression.
Results of the study included 26 patients treated with monotherapy and 25 patients treated in combination (9 patients crossed over and not included in the 25). Most patients had one or more lines of treatment though some were treatment naïve including 19.2% and 40% in arm 1 and arm 2, respectively. The most common tumor types were triple-negative breast cancer and head and neck squamous cancer jointly making up 42.2% in arm 1 and 36.0% in arm 2. The most common adverse events included low grade pyrexia, injection site pain, chills and fatigue. No maximum tolerated dose was established, although three dose-limiting toxicities were observed including vomiting at 1500 μg monotherapy as well as erythema multiforme at 540 μg and injection site pain/tumor necrosis at 1500 μg in combination. No RECIST responses were observed in monotherapy with best response of stable disease being observed in a patient with leiomyosarcoma treated with 30 μg for four months. In combination with anti-PD-1, 24% of patients experienced RECIST response (n=6; 3 HNSCC, 1 TNBC, 2 anaplastic thyroid) with most of these ongoing more than six months at the time of data cut-off. Disease control rate was 48% versus 20% in the combination and monotherapy arms. All RECIST responders were treatment-naïve to a PD1/L1 antibody. Pharmacokinetic analysis demonstrated dose-dependent increase in exposure of MK-1454 with a systemic half-life of 1.5 hours. Peripheral blood cytokine analysis demonstrated approximately two-fold induction of IP-10 at 90 μg and apparent plateau of 4–8 fold induction between 270–1500 μg. Similarly, a STING-induced 20-gene signature was induced nearly three-fold at 90 μg and showing a possible plateau between approximately 6–15 fold induction for the 270–1500 μg dose levels.
Multiple next generation STING agonists or inhibitors of STING regulatory proteins are being advanced into clinical development though none yet have reported clinical data. A non-exhaustive list of approaches that highlight the rationale for newer approaches and states of clinical development are highlighted in Table 1. These include approaches that attempt to improve the potency of the molecule while limiting the deleterious impact of STING agonists on activated T cells at high doses, to limit non-canonical NK-κB activation (which lead to elaboration of IL-6 and TNF-α), as well as to facilitate administration approaches that do not require intra-tumoral injection.
Table 1. Novel STING agonism approaches in clinical and pre-clinical development.
IM = intramuscular; IT = intratumoral; IV = intravenous, SITC 2018 = Society for the Immunotherapy of Cancer 2018 Annual Meeting, AACR 2017 = American Association for Cancer Research 2017 Conference on Tumor Immunology and Immunotherapy
| Molecule | Target | Administration route |
Reference / Clinical Trial Number |
|---|---|---|---|
| ADU-S100/MIW815 | STING | IT | NCT02675439 |
| ADU-S100/WIW815 + PDR001 | STING + PD-1 | IT + IV | NCT03172936 |
| MK-1454 | STING | IT | NCT03010176 |
| GSK3745417 | STING | IV | NCT03843359 |
| BMS986301 | STING | IT | SITC 2018 P525 |
| CRD-100 | STING | IT | 123 |
| ExoSTING | STING | IT | SITC 2018 P618 |
| GSK532 | STING | IV | 125 |
| Liposomal cGAMP-NP | STING | IV | 126 |
| MV-626 | ENPP1 | IP | SITC 2018 P410 |
| SB11285 | STING | IP, IT, IV | AACR 2017 P-A25 |
| SRCB-0001 | STING | Oral | 121 |
| STACT-TREX1 | TREX1 | IT, IV | SITC 2018 P235 |
| SYN-STING | STING | IT | SITC 2018 P624 |
4. FUTURE DIRECTIONS
Preclinical studies identified the STING pathway as the innate immune pathway activated by endogenous tumors. Based on these observations, STING agonists were developed and are currently undergoing clinical trials with promising initial results. These preliminary data suggest that not all patients respond to these agonists, raising questions of mechanisms of response versus resistance in individual patients.
A greater mechanistic understanding of the STING pathway may allow for the development of agents to more potently activate it and to selectively induce desired outputs while avoiding immune-suppressive effects. For example, STING pathway activation is linked to multiple inhibitory feedback loops including AIM2 inflammasome activation, autophagy induction, and autocrine IFN signaling which are all thought to help attenuate STING signaling. Agents designed to disrupt one or more of these feedback loops might synergistically interact with STING agonists or act as single agents. These compounds would not directly activate the pathway but rather would serve to increase the sensitivity or magnitude of signaling output of STING signaling activated by endogenous tumors. In this way, these agents may work well as systemic therapies, only increasing sensitivity of the pathway and therefore perhaps only eliciting activation at tumor sites. In addition, with infectious pathogens, activation of a single innate immune pathway rarely occurs in isolation. Rather, multiple innate immune pathways become activated concurrently or sequentially. This idea raises the possibility of co-delivering two innate immune pathway agonists concurrently, such as a STING agonist along with a TLR agonist. Further fundamental studies of how various innate pathways interact to promote transcription of desirable immune target genes will be essential.
It will also be important to better understand the microenvironmental changes induced by type I IFNs elicited by STING pathway activation in both preclinical models and in patients that are responders or non-responders to STING agonists. For example, chronic type I IFN signaling can reportedly have pro-tumor effects including immune suppression and promote metastasis (148). Most STING agonist studies to date haven’t encountered this issue because treatment is performed with 1 or a few doses that lead to a burst of IFN production that favors immune activation. The selection of clinical dose is also critical. Single, low dose intratumoral administration of ADU-S100 appears to be optimal for generating tumor-specific T cell responses in mouse models, and this depends on type I IFN signaling. In contrast, high or repetitive doses of ADU-S100 can mediate tumor destruction, yet impair the anti-tumor T cell response and memory formation. High doses could be promoting tumor regression through direct cytotoxicity, cytokine-mediated toxicity, or antibody-dependent cell-mediated toxicity (ADCC) or cellular phagocytosis. It is possible that administration of a high intratumoral dose in patients could debulk a primary tumor while providing immunogenic doses of STING agonist systemically. Alternatively, lower doses may prove more efficacious due to their advantage in generating an adaptive immune response (149). Loss of STING pathway signaling in tumor cells also needs to be better understood. If selection of STING pathway-deficient tumor cells occurs as a component of carcinogenesis, then understanding those mechanisms and countering them could trigger autonomous tumor cell cell-cycle arrest or death, and perhaps also lead to chemokine and cytokine production for downstream immune priming.
Another caveat of the STING agonists currently being tested in the clinic is that they are delivered via intratumoral injection, which only applies to accessible lesions. New methods of systemic STING agonist delivery are currently being tested in preclinical models and these new agents give the potential to affect a wider range of lesions. This not only presents the opportunity to treat a wider range of patients, but also could elicit STING pathway activation in multiple metastatic sites. As new STING agonists are developed and tested with these emerging concepts in mind, they have the potential to not only increase response rates but perhaps lead to more durable responses as well.
Acknowledgments:
Work involved in this review was supported by R35CA210098. BAF is supported by NIH Medical Scientist Training Program grant T32GM007281 and NIH F30 CA232379–01. EFH is supported by NIH grant T32HD007009.
Footnotes
Conflict of Interest Statement:
BAF, EFH, and SL have no known conflicts of interest to report that might influence our objectivity regarding this work. TFG has received consultancy fees from Merck, Roche-Genentech, Abbvie, Bayer, Jounce, Aduro, Fog Pharma, Adaptimmune, FivePrime, Sanofi; Research support from Roche-Genentech, BMS, Merck, Incyte, Seattle Genetics, Celldex, Ono, Evelo, Bayer, Aduro; Intellectual property/licensing agreements with Aduro, Evelo, and BMS; Co-founder/shareholder with Jounce. JJL: Data and Safety Monitoring Board: TTC Oncology; Scientific Advisory Board: 7 Hills, Actym, Alphamab Oncology, Array, BeneVir, Mavu, Pyxis, Tempest Consultancy: Abbvie, Aduro, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Castle, CheckMate, Compugen, EMD Serono, IDEAYA, Immunocore, Janssen, Jounce, Leap, Merck, Mersana, NewLink, Novartis, RefleXion, Spring Bank, Tempest, Vividio; Research Support: (all to institution for clinical trials unless noted) AbbVie, Array (Scientific Research Agreement; SRA), Boston Biomedical, Bristol-Myers Squibb, Celldex, CheckMate (SRA), Compugen, Corvus, EMD Serono, Evelo (SRA), Delcath, Five Prime, FLX Bio, Genentech, Immunocore, Incyte, Leap, MedImmune, Macrogenics, Novartis, Pharmacyclics, Palleon (SRA), Merck, Tesaro, Xencor; Travel: Array, AstraZeneca, Bayer, BeneVir, Bristol-Myers Squibb, Castle, CheckMate, EMD Serono, IDEAYA, Immunocore, Janssen, Jounce, Merck, Mersana, NewLink, Novartis, RefleXion; Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof).
References
- 1.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013; 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455: 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdette DL, Monroe KM, Sotelo-Troha K, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478: 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339: 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009; 461: 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo W-W, Li S, Li C, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 2016; 17: 1057–1066. [DOI] [PubMed] [Google Scholar]
- 7.Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013; 155: 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015; 347: aaa2630. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa E, Mukai K, Saito K, Arai H, Taguchi T The binding of TBK1 to STING requires exocytic membrane traffic from the ER. Biochem. Biophys. Res. Commun. 2018; 503: 138–145. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 2012; 5: ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abe T, Harashima A, Xia T, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 2013; 50: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stetson DB, Medzhitov R Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006; 24: 93–103. [DOI] [PubMed] [Google Scholar]
- 13.Rueckert C, Rand U, Roy U, Kasmapour B, Strowig T, Guzmán CA. Cyclic dinucleotides modulate induced type I IFN responses in innate immune cells by degradation of STING. FASEB J. 2017; 31: 3107–3115. [DOI] [PubMed] [Google Scholar]
- 14.Gonugunta VK, Sakai T, Pokatayev V, et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be therapeutically targeted to enhance anti-tumor response. Cell Rep 2017; 21: 3234–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen SB, Horan KA, Holm CK, et al. Activation of autophagy by α-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J. Immunol. 2011; 187: 5268–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012; 150: 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Q, Seo GJ, Choi YJ, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014; 15: 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson RO, Bell SL, MacDuff DA, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015; 17: 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Yin Q, Kuss P, et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nature Chemical Biology 2014; 10: 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013; 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Molecular Cell 2013; 51: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coquel F, Silva M-J, Técher H, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018; 557: 57. [DOI] [PubMed] [Google Scholar]
- 23.Williams JS, Gehle DB, Kunkel TA. The role of RNase H2 in processing ribonucleotides incorporated during DNA replication. DNA Repair 2017; 53: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017; 548: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie KJ, Carroll P, Martin C-A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017; 548: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregg RW, Sarkar SN, Shoemaker JE. Mathematical modeling of the cGAS pathway reveals robustness of DNA sensing to TREX1 feedback. Journal of Theoretical Biology 2019; 462: 148–157. [DOI] [PubMed] [Google Scholar]
- 27.Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi–Goutières Syndrome. The Journal of Immunology 2015; 195: 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao D, Li T, Li X-D, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. PNAS 2015; 112: E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gehrke N, Mertens C, Zillinger T, et al. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-Dependent Immune Sensing. Immunity 2013; 39: 482–495. [DOI] [PubMed] [Google Scholar]
- 30.Civril F, Deimling T, Oliveira Mann CC de, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013; 498: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato K, Ishii R, Goto E, Ishitani R, Tokunaga F, Nureki O Structural and Functional Analyses of DNA-Sensing and Immune Activation by Human cGAS. PLOS ONE 2013; 8: e76983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Wu J, Du F, et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Reports 2014; 6: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall J, Ralph EC, Shanker S, et al. The catalytic mechanism of cyclic GMP-AMP synthase (cGAS) and implications for innate immunity and inhibition. Protein Sci. 2017; 26: 2367–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luecke S, Holleufer A, Christensen MH, et al. cGAS is activated by DNA in a length‐dependent manner. EMBO reports 2017; 18: 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009; 462: 99–103. [DOI] [PubMed] [Google Scholar]
- 36.Ngo HB, Kaiser JT, Chan DC. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nature Structural & Molecular Biology 2011; 18: 1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubio-Cosials A, Sidow JF, Jiménez-Menéndez N, et al. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nature Structural & Molecular Biology 2012; 19: 364. [DOI] [PubMed] [Google Scholar]
- 38.Andreeva L, Hiller B, Kostrewa D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature 2017; 549: 394–398. [DOI] [PubMed] [Google Scholar]
- 39.Ma F, Li B, Liu S, et al. Positive Feedback Regulation of Type I IFN Production by the IFN-Inducible DNA Sensor cGAS. The Journal of Immunology 2015; 194: 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ou Y-H, Torres M, Ram R, et al. TBK1 Directly Engages Akt/PKB Survival Signaling to Support Oncogenic Transformation. Molecular Cell 2011; 41: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo GJ, Yang A, Tan B, et al. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Reports 2015; 13: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia P, Ye B, Wang S, et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol 2016; 17: 369–378. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z-S, Zhang Z-Y, Cai H, et al. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci 2018; 8: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo GJ, Kim C, Shin W-J, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nature Communications 2018; 9: 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen M, Meng Q, Qin Y, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Molecular Cell 2016; 64: 105–119. [DOI] [PubMed] [Google Scholar]
- 46.Hu M-M, Yang Q, Xie X-Q, et al. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016; 45: 555–569. [DOI] [PubMed] [Google Scholar]
- 47.Sharma S, Weston A, Thode T, Gomez E, Kaadige M, Vankayalapti H Abstract 1932: Discovery of ENPP1 inhibitors as agonists of STING pathway. Cancer Res 2018; 78: 1932–1932. [Google Scholar]
- 48.Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nature Immunology 2010; 11: 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature Immunology 2011; 12: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012; 1: e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishikawa Y, Matsuzaki Y, Kimura K, Rokunohe A, Nakano H, Sawamura D Modulation of Stimulator of Interferon Genes (STING) Expression by Interferon-γ in Human Keratinocytes. Biochem. Genet. 2018; 56: 93–102. [DOI] [PubMed] [Google Scholar]
- 52.Ma F, Li B, Yu Y, Iyer SS, Sun M, Cheng G Positive feedback regulation of type I interferon by the interferon‐stimulated gene STING. EMBO reports 2015; 16: 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu L, Cao J, Cai WL, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLOS Biology 2018; 16: e2006134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma S, Campbell AM, Chan J, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. PNAS 2015; 112: E710–E717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang P-H, Fung S-Y, Gao W-W, et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res 2018; 46: 4054–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen H, Pei R, Zhu W, et al. An Alternative Splicing Isoform of MITA Antagonizes MITA-Mediated Induction of Type I IFNs. The Journal of Immunology 2014; 192: 1162–1170. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Q, Lin H, Wang S, et al. The ER-Associated Protein ZDHHC1 Is a Positive Regulator of DNA Virus-Triggered, MITA/STING-Dependent Innate Immune Signaling. Cell Host & Microbe 2014; 16: 450–461. [DOI] [PubMed] [Google Scholar]
- 58.Luo W-W, Li S, Li C, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nature Immunology 2016; 17: 1057–1066. [DOI] [PubMed] [Google Scholar]
- 59.Almine JF, O’Hare CAJ, Dunphy G, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 2017; 8: 14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jønsson KL, Laustsen A, Krapp C, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun 2017; 8: 14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Dai M, Cui Y, et al. Association of Abnormal Elevations in IFIT3 With Overactive Cyclic GMP-AMP Synthase/Stimulator of Interferon Genes Signaling in Human Systemic Lupus Erythematosus Monocytes. Arthritis & Rheumatology 2018; 70: 2036–2045. [DOI] [PubMed] [Google Scholar]
- 62.Chen W, Li S, Yu H, et al. ER Adaptor SCAP Translocates and Recruits IRF3 to Perinuclear Microsome Induced by Cytosolic Microbial DNAs. PLoS Pathog. 2016; 12: e1005462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon D, Sesaki H, Kang SJ. Intracellular calcium is a rheostat for the STING signaling pathway. Biochem Biophys Res Commun 2018; 500: 497–503. [DOI] [PubMed] [Google Scholar]
- 64.Srikanth S, Woo JS, Wu B, et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019; 20: 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang L, Wang L, Ketkar H, et al. UBXN3B positively regulates STING-mediated antiviral immune responses. Nature Communications 2018; 9: 2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ni G, Konno H, Barber GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2017; 2: eaah7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsuchida T, Zou J, Saitoh T, et al. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010; 33: 765–776. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Hu M-M, Wang Y-Y, Shu H-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination. J. Biol. Chem. 2012; 287: 28646–28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Q, Liu X, Cui Y, et al. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014; 41: 919–933. [DOI] [PubMed] [Google Scholar]
- 70.Mukai K, Konno H, Akiba T, et al. Activation of STING requires palmitoylation at the Golgi. Nature Communications 2016; 7: 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018; 559: 269. [DOI] [PubMed] [Google Scholar]
- 72.Konno H, Konno K, Barber GN. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013; 155: 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Q, Lin L, Tong Y, et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discovery 2018; 4: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Lian Q, Yang B, et al. TRIM30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLOS Pathogens 2015; 11: e1005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhong B, Zhang L, Lei C, et al. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009; 30: 397–407. [DOI] [PubMed] [Google Scholar]
- 76.Qin Y, Zhou M-T, Hu M-M, et al. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLOS Pathogens 2014; 10: e1004358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crowl JT, Stetson DB. SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. PNAS 2018; 201802114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liang Q, Seo GJ, Choi YJ, et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host & Microbe 2014; 15: 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. PNAS 2009; 106: 20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Y, Ning X, Gao P, et al. Inflammasome Activation Triggers Caspase-1-Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 2017; 46: 393–404. [DOI] [PubMed] [Google Scholar]
- 81.Banerjee I, Behl B, Mendonca M, et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018; 49: 413–426.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guarda G, Braun M, Staehli F, et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011; 34: 213–223. [DOI] [PubMed] [Google Scholar]
- 83.Mayer-Barber KD, Andrade BB, Barber DL, et al. Innate and Adaptive Interferons Suppress IL-1α and IL-1β Production by Distinct Pulmonary Myeloid Subsets during Mycobacterium tuberculosis Infection. Immunity 2011; 35: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang L, Mo J, Swanson KV, et al. NLRC3, a Member of the NLR Family of Proteins, Is a Negative Regulator of Innate Immune Signaling Induced by the DNA Sensor STING. Immunity 2014; 40: 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schroder K, Tschopp J The inflammasomes. Cell 2010; 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 86.Swanson KV, Junkins RD, Kurkjian CJ, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 2017; 214: 3611–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Corrales L, Woo S-R, Williams JB, McWhirter SM, Dubensky TW, Gajewski TF. Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. J Immunol 2016; 196: 3191–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011; 208: 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ford E, Thanos D The transcriptional code of human IFN-β gene expression. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2010; 1799: 328–336. [DOI] [PubMed] [Google Scholar]
- 90.Thanos D, Maniatis T Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995; 83: 1091–1100. [DOI] [PubMed] [Google Scholar]
- 91.Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014; 88: 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He JQ, Zarnegar B, Oganesyan G, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J. Exp. Med. 2006; 203: 2413–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deng L, Liang H, Xu M, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014; 41: 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hou Y, Liang H, Rao E, et al. Non-canonical NF-κB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018; 49: 490–503.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ching L-M, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-Dimethylxanthenone-4-acetic acid. Br. J. Cancer 2002; 86: 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A Activation of STING in T cells induces type I IFN responses and cell death. Journal of immunology (Baltimore, Md. : 1950) 2017; 199: 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gulen MF, Koch U, Haag SM, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun 2017; 8: 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cerboni S, Jeremiah N, Gentili M, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 2017; 214: 1769–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shen YJ, Le Bert N, Chitre AA, et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 2015; 11: 460–473. [DOI] [PubMed] [Google Scholar]
- 100.Spaapen RM, Leung MYK, Fuertes MB, et al. Therapeutic activity of high-dose intratumoral IFN-β requires direct effect on the tumor vasculature. J. Immunol. 2014; 193: 4254–4260. [DOI] [PubMed] [Google Scholar]
- 101.Konno H, Yamauchi S, Berglund A, Putney RM, Mulé JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018; 37: 2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 2016; 14: 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woo S-R, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014; 41: 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H, Hu S, Chen X, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. PNAS 2017; 114: 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diamond JM, Vanpouille-Box C, Spada S, et al. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol Res 2018; 6: 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber GN. Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 2018; 33: 862–873.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018; 49: 754–763.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ablasser A, Schmid-Burgk JL, Hemmerling I, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013; 503: 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gentili M, Kowal J, Tkach M, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 2015; 349: 1232–1236. [DOI] [PubMed] [Google Scholar]
- 110.Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011; 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Corrales L, Glickman LH, McWhirter SM, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Reports 2015; 11: 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017; 31: 711–723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baguley BC, Ching L-M DMXAA: An antivascular agent with multiple host responses. International Journal of Radiation Oncology*Biology*Physics 2002; 54: 1503–1511. [DOI] [PubMed] [Google Scholar]
- 114.Baguley BC. Antivascular therapy of cancer: DMXAA. The Lancet Oncology 2003; 4: 141–148. [DOI] [PubMed] [Google Scholar]
- 115.Früh M, Cathomas R, Siano M, et al. Carboplatin and Paclitaxel Plus ASA404 as First-Line Chemotherapy for Extensive-Stage Small-Cell Lung Cancer: A Multicenter Single Arm Phase II Trial (SAKK 15/08). Clinical Lung Cancer 2013; 14: 34–39. [DOI] [PubMed] [Google Scholar]
- 116.Lara PN, Douillard J-Y, Nakagawa K, et al. Randomized Phase III Placebo-Controlled Trial of Carboplatin and Paclitaxel With or Without the Vascular Disrupting Agent Vadimezan (ASA404) in Advanced Non–Small-Cell Lung Cancer. JCO 2011; 29: 2965–2971. [DOI] [PubMed] [Google Scholar]
- 117.Jassar AS, Suzuki E, Kapoor V, et al. Activation of Tumor-Associated Macrophages by the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid Induces an Effective CD8+ T-Cell–Mediated Antitumor Immune Response in Murine Models of Lung Cancer and Mesothelioma. Cancer Res 2005; 65: 11752–11761. [DOI] [PubMed] [Google Scholar]
- 118.Conlon J, Burdette DL, Sharma S, et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. The Journal of Immunology 2013; 190: 5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shih AY, Damm-Ganamet KL, Mirzadegan T Dynamic Structural Differences between Human and Mouse STING Lead to Differing Sensitivity to DMXAA. Biophysical Journal 2018; 114: 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Prantner D, Perkins DJ, Lai W, et al. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem. 2012; 287: 39776–39788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chin E, Kumar M, Albero AG, Chatterjee A, Lairson L PO-424 Discovery of a novel small molecule sting agonist as a new cancer immunotherapy. ESMO Open 2018; 3: A189–A189. [Google Scholar]
- 122.Berger G, Lawler SE. Novel non-nucleotidic STING agonists for cancer immunotherapy. Future Medicinal Chemistry 2018; 10: 2767–2769. [DOI] [PubMed] [Google Scholar]
- 123.Banerjee M, Middya S, Basu S, et al. Abstract B43: Novel small-molecule human STING agonists generate robust Type I interferon responses in tumors. Cancer Immunol Res 2018; 6: B43–B43. [Google Scholar]
- 124.Khiar S, Lucas-Hourani M, Nisole S, et al. Identification of a small molecule that primes the type I interferon response to cytosolic DNA. Scientific Reports 2017; 7: 2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ramanjulu JM, Pesiridis GS, Yang J, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018; 564: 439. [DOI] [PubMed] [Google Scholar]
- 126.Cheng N, Watkins-Schulz R, Junkins RD, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 2018; 3: e120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gonugunta VK, Sakai T, Pokatayev V, et al. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Reports 2017; 21: 3234–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baumann M, Krause M, Overgaard J, et al. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016; 16: 234–249. [DOI] [PubMed] [Google Scholar]
- 129.Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004; 58: 862–870. [DOI] [PubMed] [Google Scholar]
- 130.Rodriguez-Ruiz ME, Rodriguez I, Garasa S, et al. Abscopal Effects of Radiotherapy Are Enhanced by Combined Immunostimulatory mAbs and Are Dependent on CD8 T Cells and Crosspriming. Cancer Res. 2016; 76: 5994–6005. [DOI] [PubMed] [Google Scholar]
- 131.Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nature Communications 2017; 8: 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vanpouille-Box C, Formenti SC, Demaria S TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology 2017; 6: e1339857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 2014; 0: 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 2014; 5: 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009; 361: 123–134. [DOI] [PubMed] [Google Scholar]
- 136.Ding L, Kim H-J, Wang Q, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 2018; 25: 2972–2980.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chabanon RM, Muirhead G, Krastev DB, et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J Clin Invest 2019; 129: 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 2018; canres.10032018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiao S, Xia W, Yamaguchi H, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res 2017; 23: 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ghaffari A, Peterson N, Khalaj K, et al. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018; 119: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017; 127: 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009; 69: 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Corrales L, Glickman LH, McWhirter SM, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015; 11: 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kinkead HL, Hopkins A, Lutz E, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018; 3: e122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res 2017; 5: 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018). Journal for ImmunoTherapy of Cancer 2018; 6: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018; 553: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sivick KE, Desbien AL, Glickman LH, et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity. Cell Reports 2018; 25: 3074–3085.e5. [DOI] [PubMed] [Google Scholar]