

Abstract
We report a family of hybrid [2]rotaxanes based on inorganic [Cr7NiF8(O2CtBu)16]− (“{Cr7Ni}”) rings templated about organic threads that are terminated at one end with pyridyl groups. These rotaxanes can be coordinated to [Cu(hfac)2] (where Hhfac = 1,1,1,5,5,5-hexafluoroacetylacetone), to give 1:1 or 1:2 Cu:{Cr7Ni} adducts: {[Cu(hfac)2](py-CH2NH2CH2CH2Ph)[Cr7NiF8(O2CtBu)16]}, {[Cu(hfac)2][py-CH2NH2CH2CH3][Cr7NiF8(O2CtBu)16]}, {[Cu(hfac)2]([py-CH2CH2NH2CH2C6H4SCH3][Cr7NiF8(O2CtBu)16])2}, {[Cu(hfac)2]([py-C6H4-CH2NH2(CH2)4Ph][Cr7NiF8(O2CtBu)16])2}, and {[Cu(hfac)2]([3-py-CH2CH2NH2(CH2)3SCH3][Cr7NiF8(O2CtBu)16])2}, the structures of which have been determined by X-ray diffraction. The {Cr7Ni} rings and CuII ions both have electronic spin S = 1/2, but with very different g-values. Continuous-wave EPR spectroscopy reveals the exchange interactions between these dissimilar spins, and hence the communication between the different molecular components that comprise these supramolecular systems. The interactions are weak such that we observe AX or AX2 type spectra. The connectivity between the {Cr7Ni} ring and thread terminus is varied such that the magnitude of the exchange interaction J can be tuned. The coupling is shown to be dominated by through-bond rather than through-space mechanisms.
Introduction
There is a growing literature on the design, synthesis, and study of molecular systems as potential qubits for quantum information science (QIS) using electron spins. The first such proposal was for a Mn12 cluster,1 a single-molecule magnet, but most subsequent studies have focused on S = 1/2 systems that provide a simple two-level quantum system. Such molecules require sufficiently long phase memory (or decoherence) times (Tm) to allow spin manipulation without loss of quantum phase information to the environment, and many studies have focused on maximizing Tm. Systems studied include nitroxyls,2 fullerenes,3 lanthanide ions,4,5 VIV and CuII complexes.6−8 Such work has recently been extended to examine a molecular qudit which allows the implementation of the Grover algorithm in a TbIII sandwich complex via manipulation of the metal hyperfine states.9
The next challenge for molecular systems is to move beyond single qubit gates, requiring the introduction of controllable coupling between two or more qubits. In principle, such coupling between molecular spin qubits can be controlled very precisely via the chemistry. Attempts have been made toward this in systems including organic radicals,2,10 lanthanide dimers,11 vanadyl dimers,12 and fullerene dimers.13 For some QIS proposals it is a requirement to be able to address the individual spin qubits selectively.2,10 One approach is to couple two different spins such that the interaction (J) is much weaker than the difference in Zeeman energies (|J| ≪ ΔgμBB), defining a weakly coupled “AX” spin system. Takui has referred to this as “g-engineering”.10,14 We recently demonstrated that such an approach can be built into coordination cages exploiting supramolecular chemistry.15 This is based on systems of the general formula (cation)[Cr7NiF8(O2CR′)16],16,17 which consist of a ring of seven CrIII and one NiII ion, with each edge bridged by one fluoride and two carboxylates; the cation is typically a protonated secondary amine. {Cr7Ni} rings have a well-isolated S = 1/2 ground state, arising from strong antiferromagnetic nearest neighbor exchange,18 with coherence times at low temperatures that can be optimized by choice of carboxylate (R′) and cation.19,20 The chemical robustness resulting from the CrIII (d3) oxidation state enables a remarkable range of chemistry to be performed on these rings,17 for example, site-specific ligand substitutions, introducing functional groups that allow building into more complex structures. The largest we have characterized to date contains 24 {Cr7Ni} rings centered on a Pd12 cage.21
In the (cation)[Cr7NiF8(O2CR′)16] structures, the cation sits in the center of the {Cr7Ni} ring. If suitable extended and sterically demanding R groups are chosen then [2]rotaxane structures are formed. If instead an extended diammonium thread is used, for example [RNH2(CH2)nNH2R]2+, then a {Cr7Ni} ring can form on each ammonium group to form [3]rotaxanes which are {Cr7Ni}2 homodimers.22 We have previously shown that we can detect the very weak {Cr7Ni}···{Cr7Ni} interactions in a range of these [3]rotaxanes by pulsed EPR methods.23,24 More recently, we used a related approach to couple {Cr7Ni} to a heterospin center.15 This involved introducing a pyridyl (py) headgroup to an extended amine py-CH2NHCH2CH2Ph (thread A, Table 1) which can then form the [2]rotaxane (HA)[Cr7NiF8(O2CtBu)16] (1) where the head of the thread is now functionalized (Figure S1a). This can be reacted with Lewis acid metal species, for example, [Cu(hfac)2] (Hhfac = 1,1,1,5,5,5-hexafluoroacetylacetone), to form {[Cu(hfac)2](HA)[Cr7NiF8(O2CtBu)16]} (2). We showed by continuous wave EPR that the S = 1/2 CuII ion and the S = 1/2 {Cr7Ni} ring (which have g-values of ca. 2.1 and 1.8, respectively), are weakly coupled (at Q-band magnetic fields) giving distinct resonances for the two components, each split by a J-coupling in a manner more commonly associated with NMR spectroscopy. Hence, we demonstrated that we had an AX electron spin system based on supramolecular chemistry principles. However, the mechanism for this interaction was unclear, being too strong and isotropic for through-space dipolar interactions, yet with no obvious exchange pathway other than through H-bonding between the ring F atoms and the ammonium groups.
Table 1. List of Threads: A–E.

In this work we systematically vary the thread that links the {Cr7Ni} ring and CuII site in order to probe the mechanism(s) of the spin–spin interaction. This introduces a number of changes including the through-space and through-bond distances, and the types of bonds (saturated or unsaturated) between the S = 1/2 centers.
Results
Synthesis and Structural Characterization
Four of the five threads used were prepared by Schiff-base condensations followed by reduction, adapting literature methods.25 The fifth thread (B) was commercially available. Thread A (Table 1) forms the [2]rotaxane {[HA][Cr7NiF8(O2CtBu)16]} 1 (Figure S1a) and subsequently its adduct {[Cu(hfac)2][HA][Cr7NiF8(O2CtBu)16]} 2 (Figure 1) via a 1:1 reaction with [Cu(hfac)2]. Crystals of 2 can be grown from warm acetone, and crystallize in the orthorhombic Pnna space group. The pyridyl group of the thread binds to [Cu(hfac)2] at the apical site of a square-pyramidal geometry at the CuII ion. The ammonium of HA lies at the {Cr7Ni} centroid with a Nam···Cu through-space distance of 7.16(3) Å [8.99(2) Å through-bond distance]. The average Cu···{Cr7Ni} distance (average over individual metal ions) is 8.46(3) Å with an average Nam–Cu–M angle (θ) of 31.38(15)° (Table 2).
Figure 1.
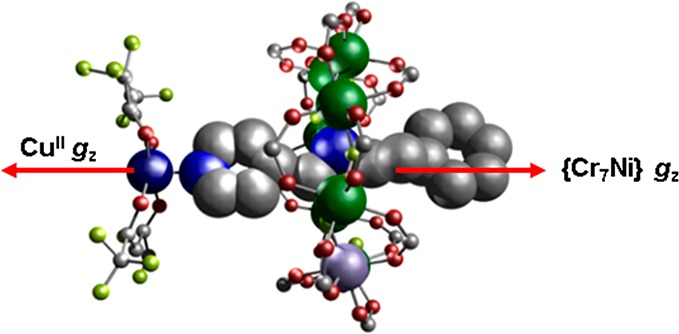
Crystal structure of 2, the red arrows show the unique z axis for the CuII and {Cr7Ni}. For 2 they are parallel with one another. Atom colors: blue (N), red (O), gray (C), green (Cr), lilac (Ni), navy (Cu), yellow (F). tBu(pivalate) groups and hydrogens omitted for clarity
Table 2. Structural Data for Compounds 2, 4, 6, 8, and 10.
| 2 | 4 | 6 | 8 | 10 | |
|---|---|---|---|---|---|
| Cu–Nam dist. via space (Å) | 7.16(3) | 7.14(3) | 8.59(8) | 11.44(10) | 7.79(4) |
| Cu–Nam dist. via bonds (Å) | 8.99(2) | 9.09(2) | 10.60(8) | 14.76(4) | 9.21(4) |
| avg. Cu–M dist. via space (Å) | 8.46(3) | 8.31(3) | 9.69(3) | 12.30(4) | 9.49(4) |
| avg. θ Nam–Cu–M (deg) | 31.38(15) | 32.06(18) | 27.35(14) | 20.95(15) | 27.64(14) |
In order to vary the Cu···{Cr7Ni} separations, four more [2]rotaxanes were synthesized with varying threads (Figure S1b–e). Similar chemistry with thread B (Table 1) gives {[HB][Cr7NiF8(O2CtBu)16]} 3 (Figure S1b) then {[Cu(hfac)2][HB][Cr7NiF8(O2CtBu)16] 4 (Figure 2). Threads A and B only differ by the end group at the far end of the chain from the pyridyl, with A terminated with a phenyl group, providing significant steric bulk to prevent slippage of the {Cr7Ni} ring off the thread (as shown by EPR, see later). In HB the terminus is a methyl group: despite the smaller size presenting no obvious steric barrier to dissociation (hence making 3 and 4 “pseudorotaxanes”), again solution EPR proves the integrity of 4 in solution (see later). Presumably this is a result of the H-bonding between the fluorides of the {Cr7Ni} ring and the ammonium protons. 4 crystallizes in the tetragonal P4/ncc space group, resulting in disorder of the pyridyl···[Cu(hfac)2] over four equivalent sites.
Figure 2.
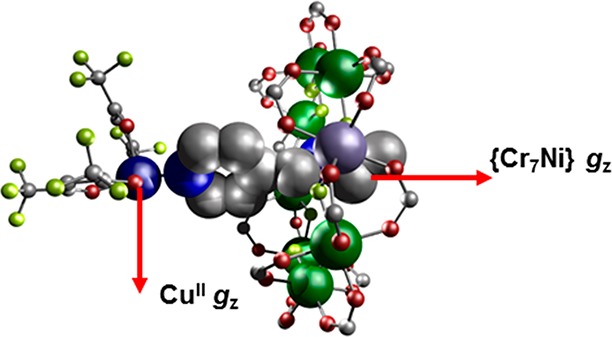
Crystal structure of 4, the red arrows show the unique z axis for the CuII and {Cr7Ni}. For 4 they are perpendicular to one another. Colors as for Figure 1.
X-ray crystallography shows that, in contrast to 2, the thread pyridyl group does not bind at the apical site of the square pyramidal geometry at Cu. Instead, it binds in an equatorial site, with one of the Cu···O bonds now defining the apical site. Hence, the Cu coordination geometry is rotated by 90° with respect to the {Cr7Ni} ring cf. 2 (at least in the solid state). Given the otherwise similar linkages there are minimal structural differences to 2, with through-bond and through-space Nam···Cu distances of 9.09(2) and 7.14(3) Å, respectively. However, the average Cu···{Cr7Ni} distance is 8.31(3) Å [average θ of 32.06(18)°], slightly shorter than in 2.
To extend the Cu···{Cr7Ni} distance an extra CH2 group was introduced between the pyridyl and amine groups in C (Table 1), which forms the [2]rotaxane {[HC][Cr7NiF8(O2CtBu)16]} 5 (Figure S1c). In contrast to the systems based on A and B, rotaxane 5 reacts with [Cu(hfac)2] to produce the 2:1 complex {[Cu(hfac)2]([HC][Cr7NiF8(O2CtBu)16])2} 6 (Figure 3). 6 crystallizes in the monoclinic I2/a space group. The increased py···amine separation allows a second molecule of 5 to bind at the CuII site, giving trans-{(py)2(hfac)2} octahedral coordination at the CuII ion. The through-bond and through-space Nam···Cu distances are 10.57(8) Å and 8.59(8) Å respectively, with an average Cu···{Cr7Ni} distance of 9.69(3) Å (average θ of 27.35(14)°).
Figure 3.
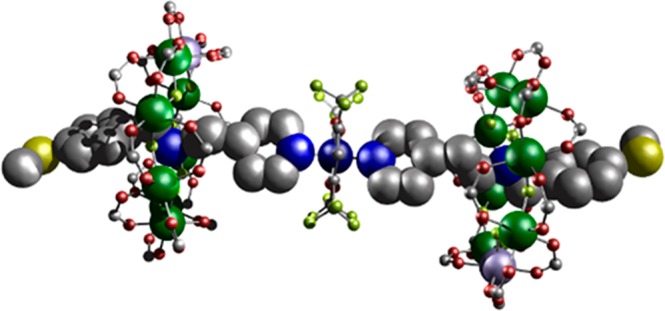
Crystal structure of 6. Colors as for Figure 1, with dull yellow (S).
To further increase the Cu···{Cr7Ni} distance, thread D was prepared, incorporating an p-C6H4 arene spacer between the pyridyl and amine (Table 1). Thread D gives the [2]rotaxane {[HD][Cr7NiF8(O2CtBu)16]} 7 (Figure S1d). Reaction of 7 with [Cu(hfac)2] gives {[Cu(hfac)2]([HD][Cr7NiF8(O2CtBu)16])2} 8 (Figure 4). 8 crystallizes in the orthorhombic P21/c space group. As with 6, the greater separation of the [Cu(hfac)2] and {Cr7Ni} fragments enables formation of the 2:1 product. However, in contrast to 6, in 8 the two molecules of 7 bind in a cis fashion at the six-coordinate CuII ion. The through-bond and through-space Nam···Cu distances are 14.76(4) and 11.44(10) Å, respectively, with an average Cu···{Cr7Ni} distance of 12.30(4) Å (average θ of 20.95(15)°).
Figure 4.
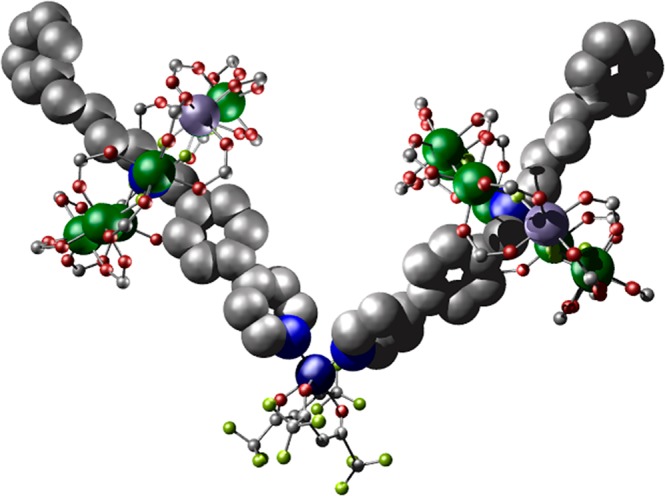
Finally, thread E was synthesized (Table 1), which has the same amine···py linkage as in C except the pyridyl group is now substituted in the 3-position. Thread E produces {[HE][Cr7NiF8(O2CtBu)16]} 9 (Figure S1e), which then reacts with [Cu(hfac)2] to produce the 2:1 adduct {[Cu(hfac)2]([HE][Cr7NiF8(O2CtBu)16])2} 10 (Figure 5). 10 crystallizes in the monoclinic space group C2/c. Again, in contrast to 6, the two molecules of 9 bind cis to one another at the six-coordinate CuII ion. This is accommodated by buckling of the two threads. The through-bond and though-space Nam···{Cr7Ni} distances are 9.47(4) and 7.74(4) Å, respectively, with an average Cu···{Cr7Ni} distance of 9.49(4) Å (average θ = 27.64(14)°).
Figure 5.
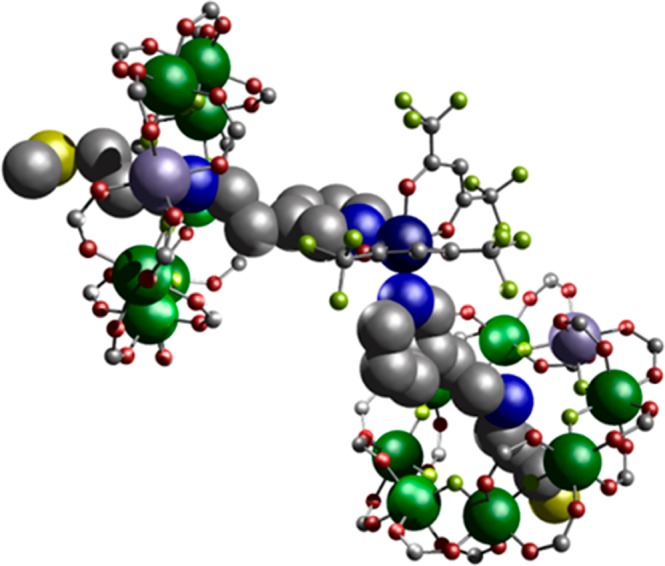
The synthetic work produces five new heterospin systems, which vary in the coordination number and geometry at the copper site, and in the relative orientation of the z-axis at the Cu site, and the unique axis of the ring, which is the direction normal to the plane defined by the eight metal sites. In 2 the CuII ion has a 5-coordinate square-based pyramidal environment with the local z axis pointing to the apex where the [2]rotaxane pyridine is bound. Hence the unique axes of the ring and Cu are almost colinear. In 4, the [2]rotaxane is bound in the equatorial plane of the Cu square-based pyramid; hence, the unique axes of the Cu and {Cr7Ni} are almost perpendicular. In 6, which has a trans-{(py)2(hfac)2} six coordination at Cu, there is a marked elongation along one of the O–Cu–O axes (Cu–O 2.20 Å), which defines the Cu z-axis, and this is orthogonal to the Cu–N direction and to the unique axes of the {Cr7Ni} rings. In both 8 and 10, which have cis-{(py)2(hfac)2} arrangements, there is a marked elongation along the sole trans O–Cu–O direction (2.215–2.280 Å; other Cu–O and Cu–N distances 1.980–2.069 Å); therefore, here the Cu z-axis is again orthogonal to the unique {Cr7Ni} axes.
EPR Spectroscopy
Continuous wave (CW) Q-Band EPR (ca. 34 GHz) spectroscopy measurements were performed on the supramolecular complexes 2, 4, 6, 8 and 10 as powders and as 3 mM solutions in dry (1:1) CH2Cl2:toluene at 5 K. The solution spectra have significantly narrower line widths than those from powders (Figure S5). At 5 K the EPR spectra of the isolated (cation){Cr7Ni} species are dominated by the S = 1/2 ground state of the {Cr7Ni} ring.18
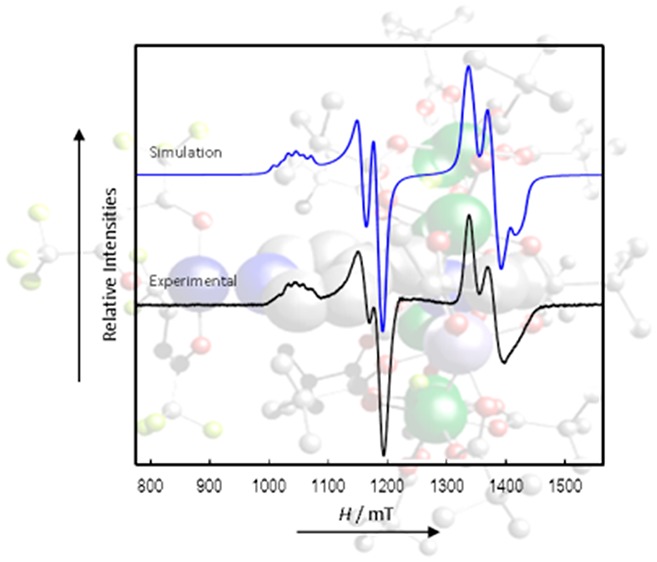
We start by describing the simplest spectrum, which is observed for 8 (Figure 6). Complex 8 has the longest separation of the Cu and {Cr7Ni} components, and the CW EPR spectrum appears as a simple superposition of the independent spectra of the components. Simulation26 (with a 1:2 weighting of the Cu:{Cr7Ni} components) gives g-values that are typical for these species: gx,y,z(Cu) = 2.041, 2.041, 2.287, and gx,y,z(Cr7Ni) = 1.778, 1.755, 1.714, where x, y, z refer to the localg-frames of the two components. The g(Cu) values are consistent with tetragonal coordination with gz corresponding to the elongated O–Cu–O direction (see above). For isolated {Cr7Ni} rings, single crystal studies show that gz (the unique axis, approximating to axial symmetry) corresponds to the normal to the {Cr7Ni} plane.18 The spectra are very sharp, and there is good resolution of the 63,65Cu hyperfine (I = 3/2) interaction on the gz(Cu) component, giving a well-defined quartet with Az = 450 MHz. Since there is no effect of any Cu···{Cr7Ni} coupling in these CW spectra, simulations are independent of the relative orientation of g(Cu) and g(Cr7Ni).
Figure 6.
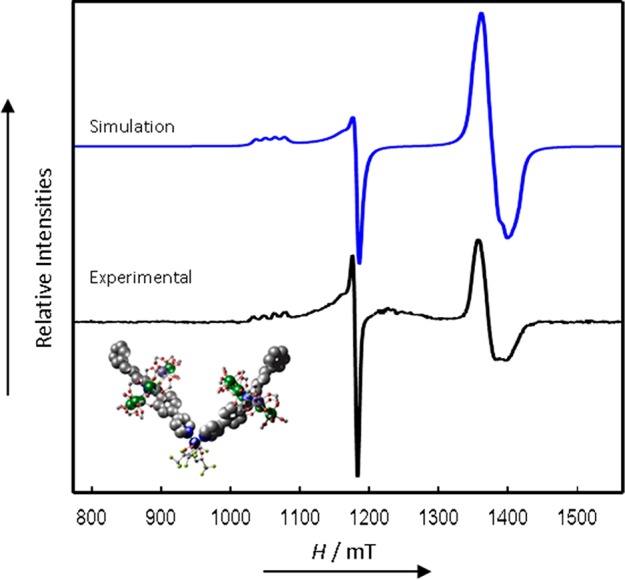
CW Q-Band (ca. 34 GHz) EPR spectrum of 8 in solution at 5 K (black) and simulation (blue).
Complexes 2 and 4, based on a very similar thread, have the shortest Cu···{Cr7Ni} separations. The crystal structure of 4 gives the apical position of the Cu coordination sphere as perpendicular to the Cu···py direction and hence, surprisingly, different to 2. However, this is complicated by disorder of the hfac groups in the structure. We have performed single crystal EPR measurements on 4 to test the orientation of gz(Cu) with respect to the gz(Cr7Ni). An indexed crystal was aligned such that a plane of data could be measured where the applied magnetic field (B) could be rotated from perpendicular to the {Cr7Ni} ring to being in the {Cr7Ni} plane. The resulting angular variation (Figure S12) shows that the maxima in g(Cu) (i.e., lowest resonance fields) correspond the maxima in g(Cr7Ni). Because gz(Cu) > gx,y(Cu) and gz(Cr7Ni) < gx,y(Cr7Ni) this is consistent with the crystal structure model.
However, there is no guarantee that this structure is retained in solution and EPR spectra of 2 and 4 in frozen solution are very similar (Figures 7 and 8, respectively). Each component of both g(Cu) and g(Cr7Ni) is now further split into spectroscopic doublets. This arises from the J-coupling between the dissimilar spins (Figure S6). The J-coupling, being well resolved, does not serve to broaden the line widths (which, therefore, are very similar to those of 8) and the Cu hyperfine interaction on gz(Cu) is still well resolved. Analysis of the multiline pattern shows that it now consists of two overlapping hyperfine quartets; hence, the J-coupling is of similar magnitude to this component of the Cu hyperfine interaction. The spectra can be simulated using a simple spin-Hamiltonian incorporating only the individual g-matrices, the Cu hyperfine interaction, and an isotropic exchange interaction:
Figure 7.
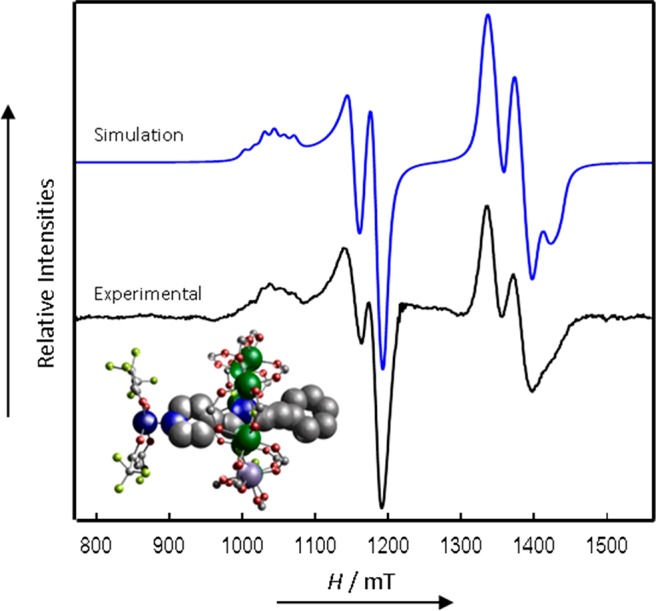
CW Q-Band (ca. 34 GHz) EPR spectra of 2 in solution at 5 K (black) and simulation (blue).
Figure 8.
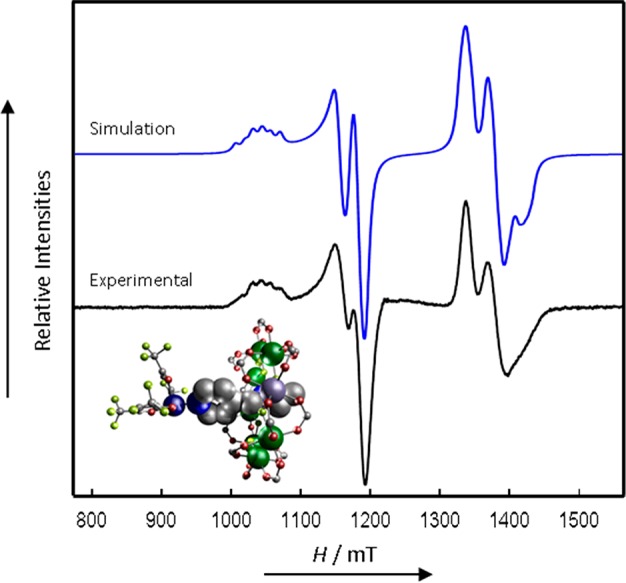
CW Q-Band (ca. 34 GHz) EPR spectrum of 4 in solution at 5 K (black) and simulation (blue).
All g-values (and hyperfines) were initially fixed using values from equivalent isolated Cu and {Cr7Ni} species, with J adjusted as the only free variable; all parameters were then refined within narrow limits to produce the best agreement. For 2, the apical Cu···py direction is parallel to the {Cr7Ni} normal, hence we have set the gz components of the two spins to be coparallel. However, we find that the calculated spectra are insensitive to this relative orientation for the small (with respect to the difference in Zeeman energy) and isotropic |J| values that we observe here. Introducing a slight g-rhombicity gives a better fit to the relative intensities. The final parameters are gx,y,z(Cr7Ni) = 1.782, 1.767, 1.712; gx,y,z(Cu) = 2.065, 2.045, 2.327; and Az(Cu) = 450 MHz. Excellent agreements with the experimental spectra of 2 and 4 are found with isotropic exchange interactions of J = −0.015 and −0.013 cm–1 (450 and 390 MHz), respectively (Table 3). The calculated relative intensities within the exchange split doublets are sensitive to the sign of J, and we find better agreement with an antiferromagnetic interaction.
Table 3. EPR CW Q-Band Spectroscopy Dataa for Compounds 2, 4, 6, 8 and 10.
| 2 | 4 | 6 | 8 | 10 | |
|---|---|---|---|---|---|
| gx,y,z{Cr7Ni} | 1.780, 1.765, 1.710 | 1.782, 1.767, 1.712 | 1.772, 1.750, 1.717 | 1.778, 1.755, 1.714 | 1.772, 1.750, 1.698 |
| gx,y,z(Cu) | 2.065, 2.045, 2.325 | 2.065, 2.045, 2.327 | 2.041, 2.030, 2.278 | 2.041, 2.041, 2.287 | 2.041, 2.030, 2.278 |
| ACuz (MHz) | 450 | 450 | 450 | 450 | 450 |
| J (MHz) | –450 | –390 | ±150 | <±30 | ±90 |
3 mM solutions in CH2Cl2:toluene at 5 K.
The observation of an exchange-split spectrum for the pseudorotaxane 4 is definitive proof that the thread and ring do not dissociate in solution (in this solvent system), despite the lack of a bulky stopper on the thread. This is an indication of the importance of the H-bonding interactions between the secondary ammonium protons of the thread with the bridging fluorides of the {Cr7Ni} ring. The very similar J-coupling for 2 and 4 also demonstrates that the relationship between the Cu and {Cr7Ni} are near identical in the two complexes. This either implies that they relax to the same geometry at Cu in solution, or that the exchange coupling is not very sensitive to the position of the pyridyl group binding site at Cu. The latter seems unlikely, given that the Cu magnetic orbital (dx2–y2 in the local axis system) would either be orthogonal to or in the plane of the Cu···{Cr7Ni} interaction. Hence, the former explanation seems more likely, we can further speculate that the very slightly smaller |J| in 4 than in 2 might reflect a minimal partial slippage, or greater flexibility, in 4 due to the lack of a bulky stopper. However, even if true, this is a very minor effect.
The simple form of the spectra for 2 and 4 arises because the difference in Zeeman energies of the two spins (ΔgμBB) is much greater than the exchange interaction between them, and the latter acts as a perturbation on the former. Approximating to g = 2.1 and 1.8 for Cu and {Cr7Ni}, respectively, for an applied field of B = 1.2 T (roughly g = 2.0 for 34 GHz) ΔgμBB = 0.17 cm–1 (or 5 GHz), which is five to six times larger than |2J|. Hence, 2 and 4 are well described as AX spin systems under these conditions.
Spectra of 6 (Figure 9) do not show the clear resolution of J that is evident in 2 and 4. Hence the Cu···{Cr7Ni} interactions are significantly weaker than in 2 and 4, due to the additional CH2 group between the ammonium and pyridyl groups in thread C. However, the spectrum is noticeably broadened compared to that of 2 and 4, and also to that of 8 (which is also a 1:2 Cu:{Cr7Ni} species). Given the latter (the strongest and weakest coupled systems studied here) it is safe to assume that the broadening is a manifestation of the intramolecular Cu···{Cr7Ni} interactions (all species are studied at the same concentrations). The broadening arises because |J| is of a similar magnitude to the intrinsic line widths. [For example, the Cu hyperfine interaction is more poorly resolved for 6 in solution than in, for example, 2 and 8 in the solid state; Figure S5.] Hence, we have estimated |J| for 6 by simulations, with fixed line widths, where |J| is increased systematically from zero. The Hamiltonian is now:
Figure 9.
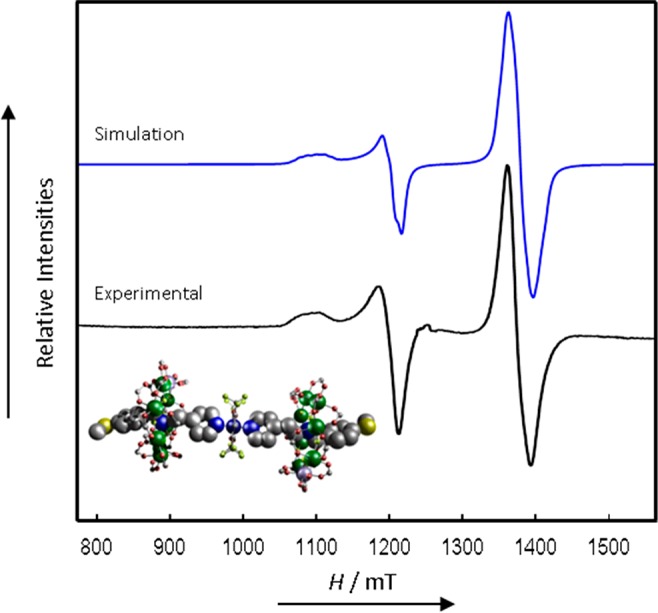
CW Q-Band (ca. 34 GHz) EPR spectrum of 6 in solution at 5 K (black) and simulation (blue).
The gz(Cu) and gz(Cr7Ni) orientations are assumed to be perpendicular, given that the elongated O–Cu–O axis is perpendicular to the unique {Cr7Ni} axis. However, as above, the calculated spectra are only sensitive to this for much larger |J| (>0.05 cm–1, or 1.5 GHz, with our line widths). We find reasonable agreement with |J| = 0.005 cm–1 (150 MHz), although this is obviously less well-defined than in 2 and 4 (and is insensitive to the sign of J, Figure S7). Note that 6 is behaving as an AX2 spin system, and in principle the Cu spectrum is now being split into 1:2:1 triplets by the weak J-coupling with the two equivalent {Cr7Ni} rings. We have previously reported spectra of {[Cu(NO3)2(Me2CO)]([pyCH2NH2CH2CH2Et][Cr7NiF8(O2CtBu)16])2},15 where |J| is bigger at −0.010 cm–1 (300 MHz; note the shorter link), and the triplet structure is resolved more clearly.
Finally, spectra of 10 (Figure 10), where the position of substitution at the pyridyl group is changed, have resolution that lies between those observed for 6 and 8: again, this is seen most clearly in the Cu hyperfine structure. Simulations, following the same model and method as above, give |J| = 0.003 cm–1 (90 MHz). The results and modeling show that complexes 6 and 10 are close to the detection limit of |J| by CW EPR, with our experimental line widths of ca. 12 mT. This intrinsic line width can then be used to define an upper limit for |J| in 8, and calculated spectra show that |J| must be < ≈0.001 cm–1 (30 MHz) for this complex.
Figure 10.
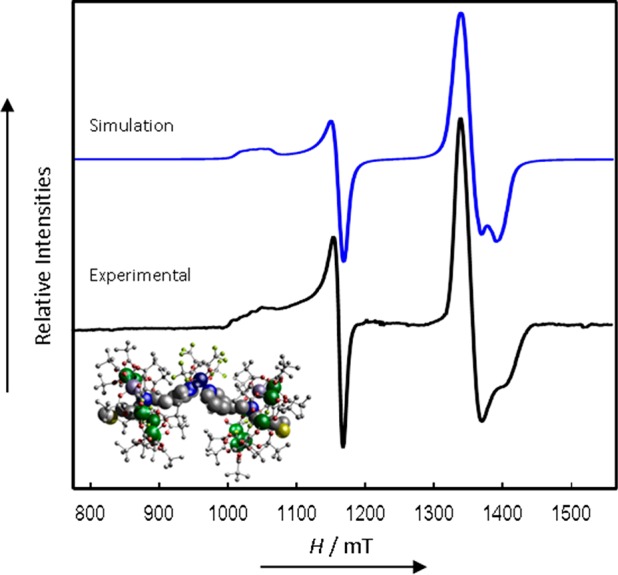
CW Q-Band (ca. 34 GHz) EPR spectrum of 10 in solution at 5 K (black) and simulation (blue).
Relaxation Measurements by Pulsed EPR
We have previously reported electron spin relaxation studies on compound 2, which has the shortest linker between the Cu and {Cr7Ni} groups and the largest |J| of the compounds studied here. In order to test if there is significant variation in relaxation behavior with |J| and/or linker we now report such measurements on compound 8 which has the longest linker and the weakest |J|.
The phase memory time (Tm) was determined by a standard Hahn echo decay sequence [π/2−τ–π–τ–echo] at Q-band for 8 in dry toluene:DCM (1:1) at 5 K for 3 mM solutions, and at 3 and 5 K for 0.2 mM solutions (Table 4). Measurements were taken at different static magnetic fields (B0; Table 4). Here, we discuss those taken at the maxima in the echo detected field swept (EDFS) spectra (Figure S8) for both the Cu (B0 = 1162 mT) and {Cr7Ni} components (B0 = 1353 mT), corresponding to orientations in the local gx,y plane for either component. We find Tm = 250 and 254 ns for the Cu and {Cr7Ni} components, respectively, for the 3 mM solution at 5 K: these extend to 514 and 327 ns, respectively, for the 0.2 mM solution at the same temperature, showing that Tm is still limited by intermolecular effects at the higher concentration. On cooling the 0.2 mM solution to 3 K, Tm increases to 912 and 469 ns for Cu and {Cr7Ni}, respectively (Figures S9, S10). Spin–lattice relaxation (T1) times were determined by inversion recovery measurements [π–T–π/2−τ–π–τ–echo] under the same conditions (Table 4, Figure S11), giving T1 = 281 and 126 μs, for the Cu and {Cr7Ni} components, respectively.
Table 4. Relaxation Times for 8 from Q-Band Pulsed EPR.
| features | gz(Cu) | Mid gz(Cu)/gx,y(Cu) | gx,y(Cu) | gx,y{Cr7Ni} |
|---|---|---|---|---|
| field position (mT) | 1067 | 1115 | 1162 | 1353 |
| concentration, temperature | Tm (ns) | |||
| 3 mM, 5 K | – | 273(15) | 254(7) | 254(4) |
| 0.2 mM, 5 K | 493(5) | 503(5) | 514(3) | 327(1) |
| 0.2 mM, 3 K | 739(1) | 822(1) | 912(1) | 469(0.4) |
| T1 (μs) | ||||
| 0.2 mM, 3 K | 345(7) | 400(8) | 281(4) | 126(1) |
The {Cr7Ni} relaxation time constants (T1 and Tm) are similar to those for isolated (cation)[Cr7NiF8(O2CtBu)16] rings (Tm 400–700 ns, depending on the cation;20T1 of ca. 100 μs for (Me2NH2)[Cr7NiF8(O2CtBu)16]).19 Data for isolated square-pyramidal [Cu(hfac)2(py)] or trans-[Cu(hfac)2(py)2] complexes, equivalent to our Cu moieties in 2 and 8, have not been reported but a related monomeric six-coordinate complex [Cu(hfac)2(4,4′-Me2-2,2′-bipy)] has Tm ≈ 3 μs and T1 ≈ 1000 μs.27 These are substantially longer than {Cr7Ni}, as expected given than the latter are strongly exchange coupled clusters of high-spin ions (CrIII and NiII). They are also longer than the Cu ions in 2 and 8, although we cannot compare directly given that structural factors can have significant effects on relaxation in CuII monomers.28 (We are unable to measure the Cu spin over a wide enough temperature range in order to fit T1 to a mechanistic model; see below.) The important observation is that the slower relaxation of the monometallic Cu than the {Cr7Ni} ring is preserved in the supramolecular adducts.
The effect of a faster relaxing spin on a slower relaxing spin depends critically on the relative magnitude of the coupling and the difference in resonance frequency of the two spins.27 If the coupling is substantial in this regard then the 1/T1 relaxation rate of the slow relaxing system will be enhanced to match that of the fast relaxing spin. We do not observe this here, hence the relaxation behavior of these supramolecular adducts is also consistent with description as weakly coupled AX(2) spin systems. However, even when the interaction is weak it can still significantly enhance the relaxation of the slow spin (see the equations given in ref (27)) and it is possible that this is the reason that the Cu components in 2 and 8 relax faster than in monomeric [Cu(hfac)2(4,4′-Me2-2,2′-bipy)].
Given this, we performed complementary X-band relaxation measurements on 8 (Figures S13, S14): the lower resonance fields give a smaller difference in resonance frequency which should enhance the effect of the interaction on the slow relaxing Cu spin. At 3 K, we find T1 for the Cu component (measured at the equivalent B0) does indeed decrease, from 281 to 175 μs (Q- and X-band, respectively). However, surprisingly, we find that T1 for the {Cr7Ni} component also decreases: there is clearly more than one contributing factor to the relaxation behavior, and this will be studied in depth in a future paper.
The transverse relaxation times of compound 8 are comparable to those of 2 which has Tm ca. 1 μs and 600 ns for Cu and {Cr7Ni}, respectively, under similar conditions.15 (Note care needs to be taken in comparing the 1:1 adduct 2 with the 2:1 adduct 8 as this changes the local concentrations of spins which can effect relaxation.29) This implies that both compounds are in the “slow exchange” regime30 where the 1/T1 relaxation rate of the fast relaxing spins is slow compared to the interaction frequency. This is consistent with 1/T1 {Cr7Ni} ≈ 0.01 MHz (T1 ≈ 100 μs at base temperature) and interactions in the MHz regime. In this case Tm of the slow relaxing spin is determined by other factors, as for the isolated spin. On increasing the temperature, 1/T1 for {Cr7Ni} increases rapidly, enhancing 1/Tm of the Cu. Hence, we can only measure the Cu spin over a limited temperature range.
Discussion
A series of new hybrid [2]rotaxanes (1, 3, 5, 7, and 9) has been made, each comprising a {Cr7Ni} ring about an asymmetric, long-chain secondary ammonium thread functionalized with a pyridyl group at one end. Each of these [2]rotaxanes binds to [Cu(hfac)2] to form the extended [2]rotaxane adducts 2, 4, 6, 8, and 10, respectively. In these structures, the distance between the CuII ion and {Cr7Ni} ring(s) is dictated by the spacing between the secondary ammonium site and the pyridyl group in the thread (Table 1). These separations also control the stoichiometry of the adducts. In 2 and 4, the spacer is a single CH2 group, and the resulting proximity of the bulky {Cr7Ni} pivalates and the hfac-groups only allows a single [2]rotaxane to coordinate at Cu, forming 1:1 adducts. The resulting square pyramidal geometry at Cu has the hfac groups folded away from the {Cr7Ni}, preventing coordination of a further [2]rotaxane at Cu. [For 2 and 4, the square pyramidal geometries at Cu have different orientations (and apical ligands) in the solid state, but may relax to the same structure in solution.] In contrast, in 6, 8, and 10 the spacers in the threads are longer [(CH2CH2) or (C6H4CH2)]. This relieves the strain between {Cr7Ni} and Cu such that a second [2]rotaxane can coordinate at Cu to form 2:1 adducts, with pseudo-octahedral coordination at Cu. There is no obvious reason why, of the 2:1 adducts, 6 should favor trans and 8 and 10 should favor cis geometries at Cu.
CW EPR gives the magnitude, or upper limit, of the exchange coupling between the heterospin (S = 1/2) CuII ion and {Cr7Ni} ring(s). These order as 2 ≈ 4 > 6 > 10 > 8, with the range spanning |2J| = 0.030 to <0.002 cm–1 (900 to <60 MHz). Given that these interactions are much weaker than the difference in Zeeman energy (ca. 5 GHz at Q-band magnetic fields), these are well described as AX or AX2 spin systems. Resolution of such spectra are actually rather rare in CW EPR because, in general, when these conditions are met the J-splitting tends to lie within the experimental line width.31 In the materials studied here the resolution is aided by the very different intrinsic g-values of the {Cr7Ni} ring and CuII ion. The weak coupling in all these systems is consistent with the electron spin relaxation behavior, being very similar between the strongest (2) and weakest (8) coupled materials, and the retention of the slower relaxation times of Cu in the presence of the faster relaxing {Cr7Ni} rings.
The magnitude of the Cu···{Cr7Ni} interaction |J| varies inversely with the distance between the two components (Table 3). The exception to this trend is compound 10 where |J| is smaller than for 6 despite the shorter Cu···{Cr7Ni} distances (see later). This trend would be expected of both through-space (dipolar) or through-bond (exchange) interactions. We have calculated the dipolar interactions in 2, 4, 6, 8, and 10 based on the crystal structures, and using projection factors for the S = 1/2 ground state onto the individual metal ions of the {Cr7Ni} ring calculated32 by ITO (irreducible tensor operator) techniques, with spin Hamiltonian parameters previously defined for the parent (Me2NH2)[Cr7NiF8(O2CtBu)16] ring,18 and that match experimentally determined values from 53Cr NMR.33
Even for 4, with the shortest Cu···{Cr7Ni} distances, the largest magnitude component of the dipolar matrix is −0.0034 cm–1 (100 MHz) (Table S2), almost an order of magnitude smaller than the experimentally observed |2J|. Calculated CW EPR spectra including only the dipolar interaction matrix are indistinguishable from the sum of the isolated spins for all compounds studied here (using a fixed line width of 12 mT). Hence, dipolar coupling is not dominating in these compounds. Moreover, for 2 and 4, where there is clear resolution of the exchange-splitting in both gz and gx,y components of the spectra, we can experimentally observe the isotropic nature of J: an isotropic interaction is not consistent with dipolar coupling. The separation of Cu and {Cr7Ni} is too far for direct orbital overlap, hence the exchange must be through-bond. These pathways involve hydrogen bonds (of two fluorides of the ring to the two ammonium protons on the thread), which must limit the magnitude of the exchange. In contrast, we have previously reported {Cr7Ni} rings bound to CuII via a functionalized −O2C-py carboxylate on the ring,34 providing a through-bond pathway entirely via covalent (or coordination) bonds. In that case, we observed strong coupling in the EPR spectra (i.e., |2J| ≫ ΔgμBB; with |2J| = −0.44 cm–1 or 13 GHz).
The anomalous coupling in compound 10 is consistent with a through-bond mechanism. These two complexes have similar py-CH2CH2NH2R connectivity between Cu and {Cr7Ni} other than the position of substitution of the pyridyl: this is para for 6 and meta for 10. It is well understood from studies of electronic coupling via conjugated pathways that stronger coupling is observed for para than meta linked centers.35,36 We have noted related effects in a family of covalently bound {Cr7Ni} dimers linked via di-immines coordinated directly to the Ni ions.37 For 8, which has the longest linker, we observe no evidence of coupling in the CW EPR spectra and this defines an upper limit to any possible interaction (|2J| < 0.002 cm–1 or 60 MHz). The largest magnitude component of the calculated dipolar matrix for 8 is −0.0012 cm–1 (40 MHz; Table S2), hence for this length of linker the dipolar interactions are likely to be dominant or at least significant with respect to J.
Very weak interactions, exchange and/or dipolar, are potentially measurable by pulsed dipolar EPR spectroscopy. We have previously measured the interactions between two {Cr7Ni} rings in different dimeric structures using double electron–electron resonance (DEER) spectroscopy,23 with interaction frequencies in the range ca. 6–1 MHz ({Cr7Ni}···{Cr7Ni} separations 16–31 Å). In the materials studied here, the spectral separation of the two components is too large for DEER methods. An alternative technique is relaxation induced dipolar modulation (RIDME) which measures the oscillations in the echo decay of a slow relaxing spin due to T1 flipping of a faster relaxing spin. We have previously used RIDME to measure the interaction in a [2]rotaxane containing a {Cr7Ni} ring about a tempo-terminated thread ([Ph(CH2)2NH2CH2(C6H4)2-tempo][Cr7NiF8(O2CtBu)16]).38 This gave a dipole-dominated frequency of 9 MHz (0.0003 cm–1; tempo···{Cr7Ni} distance of 16.8 Å) with a vanishingly small exchange interaction.
We attempted Q-band RIDME measurements on 8, observing on the slower relaxing Cu spectrum (five-pulse π/2−τ1–π–τ1–t–π/2–T–π/2−τ2–t–π–τ2–refocued echo sequence; 0.2 mM in 1:1 CH2Cl2:toluene at 3 K). However, we did not observe any oscillations. The strength of interaction that can be detected by such methods is limited by the excitation bandwidth of the pulses,39 but we observed no oscillations for pulses as short as 12 ns. Hence, we believe the Cu···{Cr7Ni} interaction for 8 lies in a “blind-spot” between being detectable by CW EPR line broadening and RIDME experiments with conventional microwave pulses. This blind-spot is more significant for these sorts of materials than in organic diradicals40 because of the smaller intrinsic line widths of the latter. It may be possible to overcome this by exploiting broad-band excitation methods enabled by pulse shaping technologies.41 We are also currently designing Cu···{Cr7Ni} rotaxanes with longer and more rigid linkers that should put the interaction energy in the range appropriate for RIDME experiments, and diamagnetic analogues42 that will allow such experiments on aligned single crystals.43
Conclusion
In conclusion, we have reported a family of hybrid inorganic–organic rotaxanes based on paramagnetic inorganic rings templated about organic threads. The rotaxanes can be coordinated to CuII complexes, via a pyridyl terminal group on the threads, to provide dissimilar electron spin S = 1/2 species on the ring and the thread components. CW EPR studies show these electron spins—localized on the different molecular components of the supramolecular structures—to behave as weakly coupled AX(2) systems, at the magnetic fields associated with Q-band EPR spectroscopy. The interaction is shown to be through-bond and this must mean it is mediated by the H-bonding between the secondary ammonium site of the thread and the internal bridging fluorides of the ring. While there are many examples of H-bonds contributing to superexchange, these examples tend to involve H-bonds between molecules, typically water, bound in the first coordination sphere of the metal spin centers.44 Here the superexchange interaction is over a much greater distance and significantly weaker. This shows the advantage of EPR methods in measuring weak exchange interactions.
We have shown that by design of the thread we can control the magnitude of this supramolecular interaction. In the future we will extend the series into the regime of weaker couplings that can be measured by pulsed EPR techniques, where the effects of dipolar and exchange effects will be competitive. This work has wider relevance to the study of the weak molecule···molecule interactions in supramolecular systems in general.
Acknowledgments
This work was supported by the EPSRC through the award of a doctoral training grant to SJL and funding of the EPSRC National EPR Facility at Manchester (NS/A000055/1). This work was supported by the EPSRC(UK) (grant number EP/L018470/1) and a Established Career Fellowship (EP/R011079/1) to REPW. We also thank EPSRC (UK) for funding an X-ray diffractometer (EP/K039547/1). We also thank the European Research Council for an Advanced Grant (ERC-2017-ADG-786734). We thank Diamond Light Source for access to synchrotron X-ray facilities. We also thank Dr N.F. Chilton (Manchester) for help with calculations using PHI.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05590.
Experimental details, X-ray crystallography, further structural figures, powder CW EPR, EPR simulations, dipolar calculations, relaxation data, single crystal EPR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Leuenberger M.; Loss D. Quantum computing in molecular magnets. Nature 2001, 410, 789–793. 10.1038/35071024. [DOI] [PubMed] [Google Scholar]
- Sato K.; Nakazawa S.; Rahimi R.; Ise T.; Nishida S.; Yoshino T.; Mori N.; Toyota K.; Shiomi D.; Yakiyama Y.; Morita Y.; Kitagawa M.; Nakasuji K.; Nakahara M.; Hara H.; Carl P.; Hofer P.; Takui T. Molecular electron-spin quantum computers and quantum information processing: pulse-based electron magnetic resonance spin technology applied to matter spin-qubits. J. Mater. Chem. 2009, 19, 3739–3754. 10.1039/b819556k. [DOI] [Google Scholar]
- Morton J. J. L.; Tyryshkin A. M.; Ardavan A.; Benjamin S. C.; Porfyrakis K.; Lyon S. A.; Briggs G. A. D. Bang-bang control of fullerene qubits using ultra-fast phase gates. Nat. Phys. 2006, 2, 40–43. 10.1038/nphys192. [DOI] [Google Scholar]
- Shiddiq M.; Komijani D.; Duan Y.; Gaita-Ariño A.; Coronado E.; Hill S. Enhancing coherence in molecular spin qubits via atomic clock transitions. Nature 2016, 531, 348–351. 10.1038/nature16984. [DOI] [PubMed] [Google Scholar]
- Pedersen K. S.; Ariciu A.-M.; McAdams S.; Weihe H.; Bendix J.; Tuna F.; Piligkos S. Toward molecular 4f single-ion magnet qubits. J. Am. Chem. Soc. 2016, 138, 5801–5804. 10.1021/jacs.6b02702. [DOI] [PubMed] [Google Scholar]
- Zadrozny J. M.; Niklas J.; Poluektov O. G.; Freedman D. E. Millisecond coherence time in a tunable molecular electronic spin qubit. ACS Cent. Sci. 2015, 1, 488–492. 10.1021/acscentsci.5b00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzori M.; Tesi L.; Morra E.; Chiesa M.; Sorace L.; Sessoli R. Room-temperature quantum coherence and Rabi oscillations in vanadyl phthalocyanine: Toward multifunctional molecular spin qubits. J. Am. Chem. Soc. 2016, 138, 2154–2157. 10.1021/jacs.5b13408. [DOI] [PubMed] [Google Scholar]
- Bader K.; Dengler D.; Lenz S.; Endeward B.; Jiang S.; Neugebauer P.; van Slageren P. Room temperature quantum coherence in a potential molecular qubit. Nat. Commun. 2014, 5, 5304. 10.1038/ncomms6304. [DOI] [PubMed] [Google Scholar]
- Godfrin C.; Ferhat A.; Ballou R.; Klyatskaya S.; Ruben M.; Wernsdorfer W.; Balestro F. Operating quantum states in single magnetic molecules: Implementation of Grover’s quantum algorithm. Phys. Rev. Lett. 2017, 119, 187702. 10.1103/PhysRevLett.119.187702. [DOI] [PubMed] [Google Scholar]
- Nakazawa S.; Nishida S.; Ise T.; Yoshino T.; Mori N.; Rahimi R. D.; Sato K.; Morita Y.; Toyota K.; Shiomi D.; Kitagawa M.; Hara H.; Carl P.; Hofer P.; Takui T. A synthetic two-spin quantum bit: g-engineered exchange-coupling biradical designed for controlled-NOT gate operations. Angew. Chem., Int. Ed. 2012, 51, 9860–9864. 10.1002/anie.201204489. [DOI] [PubMed] [Google Scholar]
- Aguila D.; Barrios L. A.; Velasco V.; Roubeau O.; Repolles A.; Alonso P. J.; Sese J.; Teat S. J.; Luis F.; Aromi G. Heterodimetallic [LnLn’] lanthanide complexes: Toward a chemical design of two-qubit molecular spin quantum gates. J. Am. Chem. Soc. 2014, 136, 14215–14222. 10.1021/ja507809w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzori M.; Chiesa A.; Morra E.; Chiesa M.; Sorace L.; Carretta S.; Sessoli R. A two-qubit molecular architecture for electron-mediated nuclear quantum simulation. Chem. Sci. 2018, 9, 6183–6192. 10.1039/C8SC01695J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant S. R.; Jevric M.; Morton J. J. L.; Ardavan A.; Khlobystov A. N.; Briggs G. A. D.; Profyrakis K. A two-step approach to the synthesis of N@C60 fullerene dimers for molecular qubits. Chem. Sci. 2013, 4, 2971–2975. 10.1039/c3sc50395j. [DOI] [Google Scholar]
- Morita Y.; Yakiyama Y.; Nakazawa S.; Murata T.; Ise T.; Hashizume D.; Shiomi D.; Sato K.; Kitagawa M.; Nakasuji K.; Takui T. Triple-stranded metallo-helicates addressable as Lloyd’s electron spin qubits. J. Am. Chem. Soc. 2010, 132, 6944–6946. 10.1021/ja102030w. [DOI] [PubMed] [Google Scholar]
- Fernandez A.; Moreno Pineda E.; Muryn C. A.; Sproules S.; Moro F.; Timco G. A.; McInnes E. J. L.; Winpenny R. E. P. g-Engineering in hybrid rotaxanes to create AB and AB2 electron spin systems: EPR spectroscopic studies of weak interactions between dissimilar electron spin qubits. Angew. Chem., Int. Ed. 2015, 54, 10858–10861. 10.1002/anie.201504487. [DOI] [PubMed] [Google Scholar]
- Larsen F. K.; McInnes E. J. L.; El Mkami H.; Overgaard J.; Piligkos S.; Rajaraman G.; Rentschler E.; Smith A. A.; Smith G. M.; Boote V.; Jennings M.; Timco G. A.; Winpenny R. E. P. Synthesis and characterization of heterometallic {Cr7Ni} wheels. Angew. Chem. 2003, 115, 105–109. 10.1002/ange.200390002. [DOI] [PubMed] [Google Scholar]
- McInnes E. J. L.; Timco G. A.; Whitehead G. F. S.; Winpenny R. E. P. Heterometallic rings: Their physics and use as supramolecular building blocks. Angew. Chem., Int. Ed. 2015, 54, 14244–14269. 10.1002/anie.201502730. [DOI] [PubMed] [Google Scholar]
- Piligkos S.; Weihe H.; Bill E.; Neese F.; El Mkami H.; Smith G. M.; Collison D.; Rajaraman G.; Timco G. A.; Winpenny R. E. P.; McInnes E. J. L. EPR spectroscopy of a family of CrIII7MII (M= Cd, Zn, Mn, Ni) “wheels“: Studies of isostructural compounds with differenet spin ground states. Chem. - Eur. J. 2009, 15, 3152–3167. 10.1002/chem.200801895. [DOI] [PubMed] [Google Scholar]
- Ardavan A.; Rival O.; Morton J. J. L.; Blundell S. J.; Tyryshkin A. M.; Timco G. A.; Winpenny R. E. P. Will spin-relaxation times in molecular magnets permit quantum information process?. Phys. Rev. Lett. 2007, 98, 057201. 10.1103/PhysRevLett.98.057201. [DOI] [PubMed] [Google Scholar]
- Wedge C. J.; Timco G. A.; Spielberg E. T.; George R. G.; Tuna F.; Rigby S.; McInnes E. J. L.; Winpenny R. E. P.; Blundell S. J.; Ardavan A. Chemical engineering of molecular qubits. Phys. Rev. Lett. 2012, 108, 107204. 10.1103/PhysRevLett.108.107204. [DOI] [PubMed] [Google Scholar]
- Ferrando-Soria J.; Fernandez A.; Moreno Pineda E.; Varey S. A.; Adams R. W.; Vitorica-Yrezabal I. J.; Tuna F.; Timco G. A.; Muryn C. A.; Winpenny R. E. P. Controlled synthesis of nanoscopic metal cages. J. Am. Chem. Soc. 2015, 137, 7644–7647. 10.1021/jacs.5b04664. [DOI] [PubMed] [Google Scholar]
- Ballesteros B.; Faust T. B.; Lee C.; Leigh D. A.; Muryn C. A.; Pritchard G. A.; Schultz D.; Teat S. J.; Timco G. A.; Winpenny R. E. P. Synthesis, structure and dynamic properties of hybrid organic-inorganic rotaxanes. J. Am. Chem. Soc. 2010, 132, 15435–15444. 10.1021/ja1074773. [DOI] [PubMed] [Google Scholar]
- Ardavan A.; Bowen A. M.; Fernandez A.; Fielding A. J.; Kaminski D.; Moro F.; Muryn C. A.; Wise M. D.; Ruggi A.; McInnes E. J. L.; Severin K.; Timco G. A.; Timmel C. R.; Tuna F.; Whitehead G. F. S.; Winpenny R. E. P. Engineering coherent interaction in molecular nanomagnet dimers. npj Quantum Inf. 2015, 1, 15012. 10.1038/npjqi.2015.12. [DOI] [Google Scholar]
- Fernandez A.; Ferrando-Soria J.; Pineda E. M.; Tuna F.; Vitorica-Yrezabal I. J.; Knappke C.; Ujma J.; Muryn C. A.; Timco G. A.; Barran P. E.; Ardavan A.; Winpenny R. E. P. Making hybrid [n]-rotaxanes as supramolecular arrays of molecular electron spin qubits. Nat. Commun. 2016, 7, 10240. 10.1038/ncomms10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead G. F. S.; Cross B.; Carthy L.; Milway V. A.; Harapriya R.; Fernandez A.; Heath S. L.; Muryn C. A.; Pritchard R. P.; Teat S. J.; Timco G. A.; Winpenny R. E. P. Rings and threads as linkers in metal-organic frameworks and poly-rotaxanes. Chem. Commun. 2013, 49, 7195–7197. 10.1039/c3cc42300j. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Burchfield J. M.; Du J. L.; More K. M.; Eaton S. S.; Eaton G. R. Enhancement of electron spin relaxation rates of metalloporphyrins due to interaction with a faster relaxing metal bound to an appended bipyridyl. Inorg. Chim. Acta 1997, 263, 23–33. 10.1016/S0020-1693(97)05590-4. [DOI] [Google Scholar]
- Fielding A. J.; Fox S.; Millhauser G. L.; Chattopadhyay M.; Kroneck M. H.; Fritz G.; Eaton G. R.; Eaton S. S. Electron spin relaxation of copper(II) complexes in glassy solution between 10 and 120 K. J. Magn. Reson. 2006, 179, 92–104. 10.1016/j.jmr.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H.; Kathirvelu V.; Spagnol G.; Rajca S.; Rajca R.; Eaton S. S.; Eaton G. R. Impact of electron-electron spin interaction on electron spin relaxation of nitroxide diradicals and tetraradical in glassy solbents between 10 and 300 K. J. Phys. Chem. B 2008, 112, 2818–2828. 10.1021/jp073600u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowsky M. H.; More K. M.; Kulikov A. V.; Eaton G. R.; Eaton S. S. Time domain electron paramagnetic resonance as a a probe of electron-electron spin-spin interaction in spin-labelled low-spin iron porphyrins. J. Am. Chem. Soc. 1995, 117, 2049–2057. 10.1021/ja00112a019. [DOI] [Google Scholar]
- Eaton G. R.; Eaton S. S. EPR studies of long-range intramolecular electron-electron exchange interaction. Acc. Chem. Res. 1988, 21, 107–113. 10.1021/ar00147a003. [DOI] [Google Scholar]
- Chilton N. F.; Anderson R. P.; Turner L. D.; Soncini A.; Murray K. S. PHI: a powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f- block complexes. J. Comput. Chem. 2013, 34, 1164–1175. 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- Casadei C. M.; Bordonali L.; Furukawa Y.; Borsa F.; Garlatti E.; Lascialfari A.; Carretta S.; Sanna S.; Timco G.; Winpenny R. E. P. Local spin density in the Cr7Ni antiferromagnetic molecular ring and 53Cr-NMR. J. Phys.: Condens. Matter 2012, 24, 406002. 10.1088/0953-8984/24/40/406002. [DOI] [PubMed] [Google Scholar]
- Timco G. A.; Carretta S.; Troiani F.; Tuna F.; Pritchard R. J.; Muryn C. A.; McInnes E. J. L.; Ghirri A.; Candini A.; Santini P.; Amoretti G.; Affronte M.; Winpenny R. E. P. Engineering the coupling between molecular spin qubits by coordination chemistry. Nat. Nanotechnol. 2009, 4, 173–178. 10.1038/nnano.2008.404. [DOI] [PubMed] [Google Scholar]
- Richardson D. E.; Taube H. Electronic interactions in mixed-valence molecules as mediated by organic bridging groups. J. Am. Chem. Soc. 1983, 105, 40–51. 10.1021/ja00339a009. [DOI] [Google Scholar]
- Cargill Thompson A. M. W.; Gatteschi D.; McCleverty J. A.; Navas J. A.; Rentschler E.; Ward M. D. Effects of systematic variations in bridging ligand structure on the electrochemical and magnetic properties of a series of dinuclear molybdenum complexes. Inorg. Chem. 1996, 35, 2701–2703. 10.1021/ic9515057. [DOI] [PubMed] [Google Scholar]
- Faust T. B.; Bellini V.; Candini A.; Carretta S.; Lorusso G.; Allan D. R.; Carthy L.; Collison D.; Docherty R. J.; Kenyon J.; Machin J.; McInnes E. J. L.; Muryn C. A.; Nowell H.; Pritchard R. G.; Teat S. J.; Timco G. A.; Tuna F.; Whitehead G. F. S.; Wernsdorfer W.; Affronte M.; Winpenny R. E. P. Chemical control of spin propagation between heterometallic rings. Chem. - Eur. J. 2011, 17, 14020–14030. 10.1002/chem.201101785. [DOI] [PubMed] [Google Scholar]
- Boulon M.-E.; Fernandez A.; Moreno Pineda E.; Chilton N. F.; Timco G.; Fielding A. J.; Winpenny R. E. P. Measuring spin···spin interactions between heterospins in a hybrid [2]rotaxane. Angew. Chem., Int. Ed. 2017, 56, 3876–3879. 10.1002/anie.201612249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G. Dipolar spectroscopy – Double-resonance methods. eMagRes. 2016, 5, 1459–1476. 10.1002/9780470034590.emrstm1518. [DOI] [Google Scholar]
- Banham J. E.; Baker C. M.; Ceola S.; Day I. J.; Grant G. H.; Groenen E. J. J.; Rodgers C. T.; Jeschke G.; Timmel C. R. Distance measurements in the borderline region of applicability of CW EPR and DEER: A model study on a homologous series of spin-labelled peptides. J. Magn. Reson. 2008, 191, 202–218. 10.1016/j.jmr.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Spindler P. E.; Schöps P.; Bowen A. M.; Endeward B.; Prisner T. H. Shaped pulses in EPR. eMagRes. 2016, 5, 1477–1492. 10.1002/9780470034590.emrstm1520. [DOI] [Google Scholar]
- Timco G. A.; Fernandez A.; Kostopoulos A. A.; Charlton J. F.; Lockyer S. J.; Hailes T. R.; Adams R. W.; McInnes E. J. L.; Tuna F.; Vitorica-Yrezabal I. J.; Whitehead G. F. S.; Winpenny R. E. P. Hybrid organic-inorganic rotaxanes, including a hetero-hybrid [3]rotaxanes featuring two distinct heterometallic rings and a molecular shuttle. Angew. Chem., Int. Ed. 2018, 57, 10919–10922. 10.1002/anie.201805439. [DOI] [PubMed] [Google Scholar]
- Moro F.; Kaminski D.; Tuna F.; Whitehead G. F. S.; Timco G. A.; Collison D.; Winpenny R. E. P. Coherent electron spin manipulation in a dilute oriented ensemble of molecular nanomagnets: pulsed EPR on doped single crystals. Chem. Commun. 2014, 50, 91–93. 10.1039/C3CC46326E. [DOI] [PubMed] [Google Scholar]
- Atzori M.; Serpe A.; Deplano P.; Schlueter J. A.; Mercuri M. L. Tailoring magnetic properties of molecular materials through non-covalent interactions. Inorg. Chem. Front. 2015, 2, 108–115. 10.1039/C4QI00179F. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.