

Abstract
Background
Sickle cell disease is a genetic haemoglobin disorder, which can cause severe pain, significant end‐organ damage, pulmonary complications, and premature death. Sickle cell disease is one of the most common severe monogenic disorders in the world, due to the inheritance of two abnormal haemoglobin (beta globin) genes. The two most common chronic chest complications due to sickle cell disease are pulmonary hypertension and chronic sickle lung disease. These complications can lead to morbidity (such as reduced exercise tolerance) and increased mortality.
This is an update of a Cochrane Review first published in 2011 and updated in 2014 and 2016.
Objectives
We wanted to determine whether trials involving people with sickle cell disease that compare regular long‐term blood transfusion regimens with standard care, hydroxycarbamide (hydroxyurea) any other drug treatment show differences in the following: mortality associated with chronic chest complications; severity of established chronic chest complications; development and progression of chronic chest complications; serious adverse events.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register. Date of the last search: 19 September 2019.
We also searched for randomised controlled trials in the Cochrane Central Register of Controlled Trials (CENTRAL) (the Cochrane Library, Issue 10, 14 November 2018), MEDLINE (from 1946), Embase (from 1974), CINAHL (from 1937), the Transfusion Evidence Library (from 1950), and ongoing trial databases to 14 November 2018.
Selection criteria
We included randomised controlled trials of people of any age with one of four common sickle cell disease genotypes, i.e. Hb SS, Sβº, SC, or Sβ+ that compared regular red blood cell transfusion regimens (either simple or exchange transfusions) to hydroxycarbamide, any other drug treatment, or to standard care that were aimed at reducing the development or progression of chronic chest complications (chronic sickle lung and pulmonary hypertension).
Data collection and analysis
We used the standard methodological procedures expected by Cochrane.
Main results
No studies matching the selection criteria were found.
Authors' conclusions
There is a need for randomised controlled trials looking at the role of long‐term transfusion therapy in pulmonary hypertension and chronic sickle lung disease. Due to the chronic nature of the conditions, such trials should aim to use a combination of objective and subjective measures to assess participants repeatedly before and after the intervention.
Plain language summary
The effect of long‐term red blood cell transfusions on chronic chest complications of sickle cell disease
Review question
We reviewed the evidence to see if regular long‐term red blood cell transfusions helped to reduce the occurrence or progression of chronic chest complications compared to hydroxycarbamide (hydroxyurea), any other treatment or standard care in people with sickle cell disease. This is an update of a previously published Cochrane Review.
Background
Oxygen is transported from our lungs to all parts of our body by haemoglobin, which is a major component of red blood cells. Sickle cell disease is an inherited disorder of haemoglobin. In people with sickle cell disease red blood cells become rigid once they have given up their oxygen and are often shaped like crescents. These rigid cells can block blood vessels, which causes problems throughout the body, including the lungs. The two most common chronic chest complications due to sickle cell disease are pulmonary hypertension and chronic sickle lung disease. Pulmonary hypertension is high blood pressure in the pulmonary arteries (the arteries that supply blood to the lungs). High blood pressure in these arteries are associated with an increased risk of death. Chronic sickle lung disease arises as a result of lung damage and loss of lung tissue.
Regular blood transfusions for people with sickle cell disease reduce the amount of the person's own sickled cells in their blood by replacing them with donated, non‐sickled cells. Regular transfusions have already been shown to reduce the risk of strokes in people with sickle cell disease. We aimed to find out if regular long‐term blood transfusions in people with this disease lead to a reduction in new chronic chest complications or slowed the progression of any chronic chest complications that have already developed. We also aimed to consider death rates due to chronic chest complications and any adverse effects of the transfusion programme.
Study characteristics
The evidence is current to 19 September 2019. We found no studies in this update of the review.
Key results
There are no results because we found no relevant randomised controlled trials. We would need to design a study with at least 946 participants to be able to detect a decrease in the number of people who died from 12 in 100 to six in 100.
Quality of the evidence
There is no evidence from randomised controlled trials to answer our review questions.
Background
Description of the condition
Sickle cell disease (SCD) is a genetic haemoglobin disorder, which can cause severe pain, significant end‐organ damage, pulmonary complications, and premature death (Chakravorty 2015). It is one of the most common severe monogenic disorders in the world, due to the inheritance of two abnormal haemoglobin (beta globin) genes (Rees 2010). Populations originating from sub‐Saharan Africa, Spanish‐speaking regions in the Western Hemisphere (South America, the Caribbean, and Central America), the Middle East, India and parts of the Mediterranean are predominantly affected. Reductions in infant and child mortality and increasing migration from highly affected countries have made this a worldwide problem (Piel 2012). Over 12,500 people in the United Kingdom and 100,000 in the United States of America suffer from the disease (NICE 2010; Pleasants 2014). A recent study estimated that approximately 305,800 babies were born with SCD in 2010, of which two thirds were born in Africa, and this could increase by 25% to approximately 404,200 by 2050 (Piel 2012).
The term 'sickle cell disease' refers to all genotypes that cause the clinical syndrome. There are three main types of SCD. Sickle cell anaemia is the most common form of the disease (up to 70% of cases of SCD in people of African origin) and is due to the inheritance of two beta globin S (βS) alleles (haemoglobin (Hb)SS). The second most common genotype (up to 30% of cases in people of African origin) is haemoglobin SC disease (HbSC disease) it is due to the co‐inheritance of the βS and βC alleles and tends to be a more moderate form of the disease. The third major type of SCD occurs when βS is inherited with a β‐thalassaemia allele, causing HbS/β‐thalassaemia (Rees 2010). People who have inherited a thalassaemia null mutation (HbSβº) have a disease that is clinically indistinguishable from sickle cell anaemia, whereas people with HbSβ+ thalassaemia have a milder disorder. In developed nations, people with SCD are expected to live into their 40's, 50's and beyond, whereas in low‐income countries including some African nations it is estimated that between 50% to 90% of children born with HbSS die before their fifth birthday (Gravitz 2014; Grosse 2011).
Key biochemical processes are understood to be red‐cell dehydration and increased adhesion of these erythrocytes to the endothelium (Hebbel 1997; Nagalla 2010). In SCD under conditions of low oxygen levels, acidity and cellular dehydration, the HbS molecules polymerise and begin to distort the red blood cells taking on the appearance of sickle‐shaped cells. The main determinant of disease severity is the rate and extent of this HbS polymerisation (Rees 2010).
The two most common chronic chest complications due to SCD are pulmonary hypertension and chronic sickle lung disease, though there is some overlap between the two conditions. These complications can lead to morbidity (such as reduced exercise tolerance) and increased mortality. Pulmonary complications leading to acute episodes of morbidity, such as the acute chest syndrome and asthma, have not been considered as part of this review. Blood transfusion as a potential treatment for acute chest syndrome has been considered in a separate review (Alhashimi 2010).
In the past all chronic chest complications due to SCD were considered together (Powars 1988); however, in this review we will consider pulmonary hypertension and chronic sickle lung disease separately.
Pulmonary hypertension is defined as having a resting mean pulmonary artery pressure of equal to or greater than 25 mm Hg determined by right heart catheterisation (Expert Panel Report 2014; Gordeuk 2016); it is thought to be multifactorial in origin and may involve haemolysis, impaired nitric oxide bioavailability, chronic hypoxaemia, thromboembolism, and parenchymal and vascular injury (Machado 2005;Vij 2010). Pulmonary hypertension is a common finding and affects approximately 10% of adults with SCD (Gordeuk 2016). Symptoms of pulmonary hypertension include shortness of breath during routine activities, fatigue, lethargy, chest pain, lower limb oedema. These symptoms need to be distinguished from similar symptoms that occur as a manifestation of SCD (Expert Panel Report 2014). People with SCD and pulmonary hypertension have an increased mortality rate compared to people with SCD and no pulmonary hypertension, with studies citing three‐year mortality rates between 12% and 37% (Gordeuk 2016).
It is estimated that approximately 90% of adults with SCD have abnormal pulmonary function, the most common abnormality is a mild restrictive defect (Klings 2006). Chronic sickle lung disease is used to describe lung fibrosis which may develop as a result of recurrent acute chest syndrome, but this is not always the case (Khoury 2011). Symptoms of chronic sickle lung disease include: difficulty breathing; episodes of chest pain; and exercise intolerance. Chronic lung disease is reported in approximately 5% of people with SCD. It tends to occur in individuals at an average age of 25 to 33 years and consists of perfusion and diffusion defects (defects in blood oxygenation), indicating that pulmonary fibrosis (scarring of the lungs) as well as vascular occlusion (blockage of the blood vessel) contribute to the disorder (Collins 1982; Powars 1988).
The diagnosis and assessment of chronic sickle lung disease include could include arterial blood gas sampling, pulse oximetry, spirometry, transfer factor, and high resolution computerized tomography (CT) lung scan. As with pulmonary hypertension it is also possible to assess the exercise tolerance of the individual.
Description of the intervention
In general, therapeutic options for chronic SCD complications are limited and consist of chronic red blood cell transfusions, hydroxycarbamide (hydroxyurea), and haematopoietic stem cell transplantation.
Recently, the Walk‐PHaSST study of sildenafil in people with sickle cell with tricuspid regurgitant velocity (TRV) at least 2.5 m/s was stopped early because of an increased incidence of painful crisis in the sildenafil arm (Gordeuk 2016; Machado 2009).
Red blood cell transfusions can be given to treat complications of SCD, e.g. acute chest syndrome (this often involves a single transfusion episode), or they can be part of a regular long‐term transfusion programme to prevent complications of SCD (Yawn 2014). People with SCD can be placed on a long‐term transfusion programme to prevent recurrence of a complication they have already experienced or to prevent the first episode of a complication e.g. stroke in children with abnormal transcranial dopplers (Adams 1998). Both a single transfusion episode or chronic transfusion programmes can use either a simple transfusion regime or an exchange transfusion regime (Josephson 2007). Red cell transfusions can be restrictive (to increase the total haemoglobin) or liberal (to decrease the haemoglobin S level below a specified percentage).
Red blood cell transfusions have reduced complications and improved the quality of life with SCD, however they can also cause adverse events that are sometimes serious (Josephson 2007). The benefits of transfusion therapy must be balanced against risks including infections, iron overload, alloimmunisation, acute or delayed haemolytic transfusion reactions, and increased complexity of compatibility testing (Chou 2013aChou 2013b; Porter 2013; Scheunemann 2010; Ubesie 2012).
How the intervention might work
Pulmonary hypertension occurs in a variety of chronic haemolytic anaemias including thalassaemia, hereditary spherocytosis as well as in SCD suggesting that pulmonary hypertension may be associated with haemolysis (Khoury 2011). Regular red blood cell transfusions might improve or stabilise existing chronic chest complications of SCD. Transfusions would be expected to reduce the baseline level of haemolysis, a contributor to pulmonary hypertension. They might improve the circulation of the lung and tissue oxygen delivery by reducing the amount of sickling in the pulmonary vasculature and the rate of haemolysis (Mehari 2016).
Sickled red blood cells increase blood viscosity (resistance to flow) through intrinsic properties of the sickled cells as well as through abnormal interactions of these cells with white cells, platelets, the vessel wall, and clotting factors. Transfusion of normal donor red blood cells is used to mitigate these effects (Yawn 2014) and several regimens are used in current clinical practice. These include 'simple' transfusion in which normal red cells are given to decrease anaemia without removal of the individual's blood. In people with SCD who do not have severe anaemia 'simple' blood transfusions can cause hyperviscosity syndrome because they raise the haemoglobin, but only marginally lower the HbS percentage (Schmalzer 1987).
Exchange transfusion involves removing some of the individual's own blood and transfusing allogeneic blood, thereby lowering the concentration of HbS through dilution. This reduces the effects of a given haemoglobin level on blood viscosity. A full exchange transfusion involves a full blood volume exchange by manual or automated apheresis, this allows for rapid lowering of the HbS level to 30 per cent or less, and correction of anaemia. A partial (limited) exchange transfusion refers to manual removal of some of the individual's own blood, this is less effective in lowering the HbS level but is more easily performed when automated exchange is not available. In order to lower the HbS below 30 per cent, repeat partial exchange transfusions may be necessary.
A restrictive (conservative) transfusion policy involves giving a simple transfusion to reach a pre‐specified target haemoglobin.
A liberal (aggressive) transfusion policy involves giving a transfusion to reduce HbS percentage below a pre‐specified threshold. In people with SCD with severe anaemia 'simple' blood transfusions can lead to a significant reduction in HbS percentage without the need for an exchange transfusion. In a trial of people with SCD due to have an operation, 36% of participants randomised to the 'aggressive' transfusion arm (to reduce HbS percentage to 30 or below) were treated with a 'simple' transfusion pre‐operatively (Vichinsky 1995).
Why it is important to do this review
Chronic chest complications are common in SCD. Some of the problems may have a degree of reversibility or could be stabilised to prevent further deterioration. These chronic complications are associated with significant morbidity and mortality. The optimal management of these conditions is unknown. Regular red blood cell transfusion is a widely available treatment that may improve outcomes in people with SCD who have chronic chest complications.
Regular transfusions may provide benefits but are associated with problems that can limit its use. Some common issues are:
iron overload with potential damage to the liver, the heart, and endocrine system; treatment of iron overload requires the use of chelating agents which have side effects of their own;
transfusion reactions;
alloimmunisation which can make it difficult to provide a compatible transfusion in the future. Studies of alloimmunisation have shown rates in transfused SS individuals of between 18% and 30% in the UK and the USA (Davies 1986; Rosse 1990; Vichinsky 1990).
transfusion transmitted infections such as HIV and hepatitis B.
This is an update of a previously published Cochrane Review (Cho 2011; Cho 2014; Estcourt 2016).
Objectives
To determine whether trials involving people with SCD that compare regular long‐term blood transfusion regimens with standard care, hydroxycarbamide or any other drug treatment show differences in the following:
mortality associated with chronic chest complications;
severity of established chronic chest complications;
development and progression of chronic chest complications;
serious adverse events.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs), irrespective of language or publication status (RCTs may be unpublished, or published as a peer‐reviewed article, letter, or abstract).
Types of participants
Males and females of any age and with one of four common SCD genotypes, i.e. Hb SS, Sβ0, SC, Sβ+ (proven by haemoglobin electrophoresis and sickle solubility test, with family studies or DNA tests as appropriate). Participants may or may not have a reported history of chronic or acute chest complications.
Types of interventions
Intervention
Regular red blood cell transfusion regimens (either simple or exchange transfusions)
Comparisons
Standard care (as defined in the studies)
Hydroxycarbamide
Any other drug treatments
Types of outcome measures
Primary outcomes
-
Number of people with new diagnoses of chronic chest complications
chronic sickle lung disease
pulmonary hypertension (using a cut off of TRV 2.5 m/s to define pulmonary hypertension)
-
Deaths
from any cause
from chronic chest complications (chronic sickle lung disease and pulmonary hypertension)
-
Transfusion‐related complications (up to 30 days post transfusion) including:
alloimmunisation
infection from blood products
procedural complications (such as device malfunction)
iron overload (measured by serum ferritin, liver iron, or non‐invasive imaging)
transfusion reactions
We will categorise diagnosis of chronic chest complications and deaths due to any cause according to short‐, medium‐, and long‐term outcomes. We will report the exact definition of these time frames over time periods that are common to as many studies as possible (for example, zero to five years, six to 10 years, over 10 years).
Secondary outcomes
-
Severity of pulmonary hypertension (assessment of treatment effects will compare baseline results with those at set time intervals: three months; one year; greater than one year (if applicable); and the end of treatment) as determined by:
participant exercise tolerance (for example six‐minute walk test);
pulmonary artery pressure by catheter study;
echocardiogram measurement of tricuspid regurgitant velocity (TRV);
stabilisation or improvement of symptoms (which might be based on subjective reports of changes or assessed in a formal way using a standardised scale).
-
Severity of chronic sickle lung disease (assessment of treatment effects will compare baseline results with those at set time intervals: three months; one year; greater than one year (if applicable); and the end of treatment) as determined by:
pulmonary function tests;
CT chest scan;
chest X‐ray appearance.
Quality of life
Any other adverse events
Search methods for identification of studies
Electronic searches
Potentially relevant trials were identified from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: (sickle cell OR (haemoglobinopathies AND general)) AND blood transfusion*.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (Clinical Trials) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Public Health Agency Annual Scientific Meeting (formerly the Caribbean Health Research Council Meeting); and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the last search of the Group's Haemoglobinopathies Trials Register: 19 September 2019.
We also searched for RCTs in the following databases: (seeAppendix 1 for search strategies).
CENTRAL & DARE (the Cochrane Library Issue 10, 14 November 2018)
MEDLINE (OvidSP, 1946 to 14 November 2018)
Embase (1974 to 14 November 2018)
CINAHL (EBSCOhost,1937 to 14 November 2018)
Pubmed (epublications only to 14 November 2018)
Transfusion Evidence Library (www.transfusionevidencelibrary.com) (1950 to 14 November 2018)
LILACS (BIREME/PAHO/WHO, 1982 to 14 November 2018)
KoreaMed (1997 to 14 November 2018)
IndMed (1986 to 14 November 2018)
PakMediNet (2001 to 14 November 2018)
Web of Science (Conference Proceedings Citation Index‐ Science (CPCI‐S) ‐ 1990 to 14 November 2018)
We combined searches in MEDLINE, Embase, and CINAHL with adaptations of the Cochrane RCT search filters, as detailed in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2011). We did not limit searches by language or publication status.
Databases of ongoing trials
We also searched ClinicalTrials.gov (clinicaltrials.gov/ct2/search), the WHO International Clinical Trials Registry (ICTRP) (apps.who.int/trialsearch/), the ISRCTN Register (www.controlledtrials.com/isrctn/), the EU Clinical Trials Register (www.clinicaltrialsregister.eu/ctr‐search), and the Hong Kong Clinical Trials Register (www.hkclinicaltrials.com/) in order to identify ongoing trials to 14 November 2018.
Searching other resources
We augmented database searching with the following.
Handsearching of reference lists
We checked references of all relevant review articles, and current treatment guidelines for further literature. We limited these searches to the 'first generation' reference lists.
Data collection and analysis
Selection of studies
We selected studies according to chapter 7 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). The Systematic Review Initiative's Information Specialist (CD) initially screened all search hits for relevance against the eligibility criteria and discarded all those that were clearly irrelevant. Thereafter, two review authors (PF, LE) independently screened all the remaining references for relevance against the full eligibility criteria. We retrieved full‐text articles for all references for which a decision on eligibility could not be made from title and abstract alone. The two review authors discussed the results of study selection and resolved any any disagreements on trial inclusions by consensus. We reported the results of study selection using a PRISMA flow diagram (Moher 2009).
Data extraction and management
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), two review authors (PF, LE) planned to independently extract data onto standardised forms. However, no completed study was included in this review.
We planned to extract the following information for each study.
Source: study ID; report ID; review author ID; date of extraction; ID of author checking extracted data; citation of paper; contact authors details.
General study information: publication type; study objectives; funding source; conflict of interest declared; other relevant study publication reviewed.
Study details and methods: location; country; setting; number of centres; total study duration; recruitment dates; length of follow‐up; power calculation; primary analysis (and definition); stopping rules; method of sequence generation; allocation concealment; blinding (of clinicians, participants and outcome assessors); and any concerns regarding bias.
Characteristics of interventions: number of study arms; description of experimental arm; description of control arm; type of blood transfusion regimen; hydroxycarbamide dosing; description of standard care or any other drug treatment used.
Characteristics of participants: age; gender; primary diagnosis; type of chest complication.
Participant flow: total number screened for inclusion; total number recruited; total number excluded; total number allocated to each study arm; total number analysed (for review outcomes); number of allocated participants who received planned treatment; number of drop‐outs with reasons (percentage in each arm); protocol violations; missing data.
Outcomes measured: number of people with new diagnoses of chronic chest complications, deaths, transfusion related complications, severity of pulmonary hypertension or sickle lung disease, quality of life, other adverse events.
Assessment of risk of bias in included studies
We planned to perform an assessment of all RCTs using the Cochrane 'Risk of bias' tool according to chapter eight of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). We planned to use Cochrane's tool for assessing risk of bias, which includes the following domains.
Selection bias (random sequence generation and allocation concealment)
Performance bias (blinding of participants and personnel)
Detection bias (blinding of outcome assessment)
Attrition bias (incomplete outcome data)
Reporting bias (selective reporting)
Other bias
However, no completed study was identified in this review and therefore we could not perform an assessment of risk of bias.
Measures of treatment effect
We did not perform any of the planned analyses because no completed study was included in this review. We planned to record the following data for this review:
continuous outcomes: mean, standard deviation (SD) and total number of participants in both the treatment and control groups;
dichotomous outcomes: number of events and total number of participants in both the treatment and control groups.
The following analyses were planned for this review and will be performed in future updates of this review:
for continuous outcomes using the same scale: analyses using the mean difference (MD) with 95% confidence intervals (CIs);
for continuous outcomes measured with different scales: analyses using the standardised mean difference (SMD);
extraction and reporting of hazard ratios (HRs) for mortality data or, if HRs were not available, every effort would be made to estimate the HR as accurately as possible using the available data and a purpose‐built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007);
for dichotomous outcomes: reporting the pooled risk ratio (RR) with a 95% CI. Where the number of observed events was small (less than 5% of sample per group), and where trials have balanced treatment groups, we planned to report the Peto's Odds Ratio (OR) with 95% CI (Deeks 2011).
If data allowed, we planned to undertake quantitative assessments using the Review Manager software (RevMan 2014).
Where appropriate, we planned to report the number needed to treat to benefit (NNTB) and the number needed to treat to harm (NNTH) with CIs.
If we could not report the available data in any of the formats described above, we planned to present a narrative report, and if appropriate we planned to present the data in tables.
Unit of analysis issues
We planned to treat any unit of analysis issues in accordance with the advice given in chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011d). However, no completed study was identified in this review and there were therefore no unit of analysis issues.
Dealing with missing data
We did not need to contact any study authors directly to enable us to make a decision on whether a study should be excluded.
Assessment of heterogeneity
We did not perform any of the planned analyses because no completed study was included in this review.
We had planned to combine the data to perform a meta‐analysis if the clinical and methodological characteristics of individual studies were sufficiently homogeneous. We planned to assess statistical heterogeneity of treatment effects between studies using a Chi² test with a significance level at P < 0.1. We planned to use the I² statistic to quantify the degree of potential heterogeneity, and classify it as moderate if the I² statistic was greater than 50%, or considerable if I² was greater than 80%. We perceived that we would identify at least moderate clinical and methodological heterogeneity within the studies selected for inclusion; in such cases, we planned to use the random‐effects model. If statistical heterogeneity was considerable, we planned not to report the overall summary statistic. We planned to assess potential causes of heterogeneity by sensitivity and subgroup analyses (Deeks 2011).
Assessment of reporting biases
We did not perform a formal assessment of potential publication bias (small trial bias) by generating a funnel plot and statistically test using a linear regression test (Sterne 2011), because there were no completed trials within this review.
Data synthesis
We planned to perform analyses according to the recommendations of chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions, using aggregated data for analysis (Deeks 2011).
We did not perform any of the planned analyses because no completed study was included in this review.
Summary of findings table
We planned to use the GRADE approach to create 'Summary of findings' tables as suggested in chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). We planned to use the GRADE approach to rate the quality of the evidence as 'high', 'moderate', 'low', or 'very low' using the five GRADE considerations.
Risk of bias: serious or very serious
Inconsistency: serious or very serious
Indirectness: serious or very serious
Imprecision: serious or very serious
Publication bias: likely or very likely
The outcomes we planned to include are listed below.
Number of people with new diagnoses of chronic chest complications
Death from any cause
Severity of pulmonary hypertension
Severity of chronic sickle lung disease
Transfusion‐related complications
Quality of life
Subgroup analysis and investigation of heterogeneity
If adequate data were available, we planned to perform subgroup analyses according to Cochrane recommendations for each of the following outcomes in order to assess the effect on heterogeneity (Deeks 2011).
Age of participants: neonate, child (one to 15 years), adult (16 years and older)
Genotype (homozygous sickle cell disease (SS), sickle beta thalassaemia (Sβ0 and Sβ+) and sickle haemoglobin C
disease (SC))
Severe SCD complications (strokes, acute chest syndrome, painful crisis, priapism)
Follow‐up duration: longer‐term RCTs (one year or longer) versus shorter term RCTs (less than one year). The authors chose this time stratification as chronic lung fibrosis will not change rapidly. Although pulmonary artery pressure can change quickly, it is the effect of the treatment over an extended period that is of most interest;
Participants with and without a reported history of chronic or acute chest complications
However, no completed study was identified in this review and therefore we could not perform any subgroup analyses.
Sensitivity analysis
We planned to assess the robustness of our findings by performing the following sensitivity analyses according to the recommendations of Cochrane (Deeks 2011) where appropriate.
Including only those studies with a ‘low risk of bias’ (for example, RCTs with methods assessed as low risk for random sequence generation and concealment of treatment allocation).
Including only those studies with less than a 20% dropout rate.
However, no completed study was identified in this review and therefore we could not perform any sensitivity analyses.
Results
Description of studies
See Characteristics of excluded studies.
Results of the search
The original review and first update identified a total of 114 potentially relevant records. Two review authors (GC, IH) excluded all 114 records on the basis of the abstract (Cho 2011; Cho 2014). The search updated in 2016 identified a total of 971 potentially‐relevant records. There were 511 records after duplicates were removed. Two review authors (LE; PF) excluded 507 records on the basis of the abstract. Eight full text articles were retrieved for assessment by the same two review authors. All eight studies were excluded (Estcourt 2016).
The latest update of the search (conducted 14 September 2019) identified a total of 1620 potentially‐relevant records. There were 306 records after duplicates were removed. Two review authors (LE; PF) excluded 305 records on the basis of the abstract (Figure 1), the other record was a secondary citation of a previously excluded study (Atilla 2011).
1.
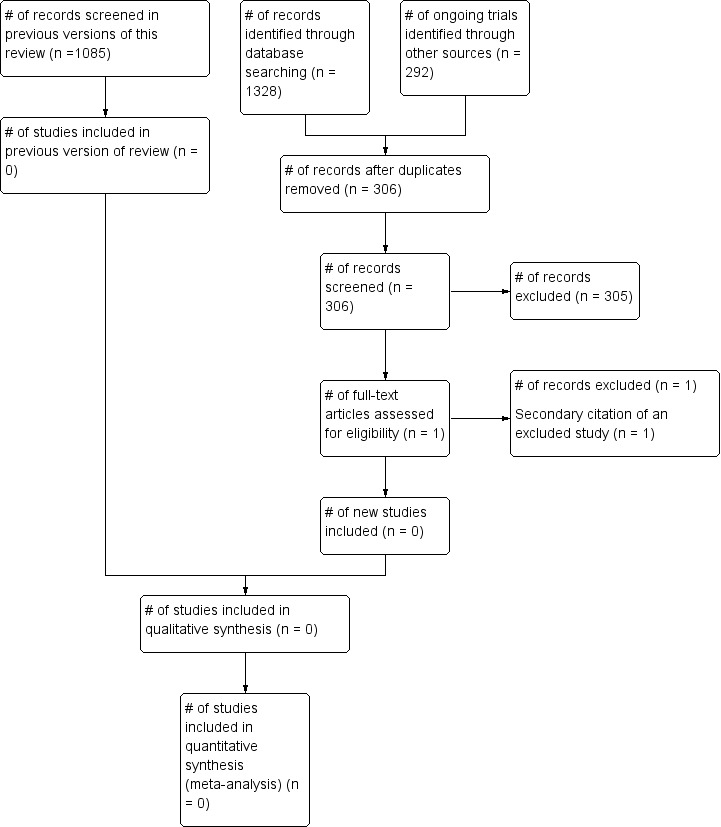
˜Study flow diagram.
Included studies
No completed or ongoing trials were included in this review. No randomised control trials that fit the criteria were found.
Excluded studies
See Characteristics of excluded studies for further details.
Three studies were not RCTs (Detterickh 2015; Lunt 2016; NCT00023296 )
Two studies did not include people with sickle cell disease (Atilla 2011; Chueamuangphan 2014)
Two studies did not include people with pulmonary hypertension or chronic sickle lung disease (Styles 2006; Styles 2012 )
One study was using a different intervention and did not include a blood transfusion arm (Barst 2010)
Risk of bias in included studies
No trials were identified for inclusion in the review.
Effects of interventions
No trials were identified for inclusion in the review.
Discussion
Summary of main results
There were no completed or ongoing RCTs that were relevant to this review.
Overall completeness and applicability of evidence
This review did not identify any completed RCTs and therefore there is no evidence that can be assessed.
If we assumed that death due to any cause occurred in 12 out of 100 people who had pulmonary hypertension (based on lower estimate of all‐cause mortality Gordeuk 2016) and this was reduced to six out of 100 with treatment we would need to design a study with at least 946 participants to detect this difference with 90% power and 5% significance. If there is 10% non‐compliance to the protocol in both study arms this will increase to 1480 participants.
Quality of the evidence
This review did not identify any completed RCTs and therefore there is no evidence that can be assessed.
Potential biases in the review process
To our knowledge, our review process is free from bias. We conducted a comprehensive search, searching data sources (including multiple databases, and clinical trial registries) to ensure that all relevant trials would be captured. The relevance of each paper identified was carefully assessed and all screening and data extractions were performed in duplicate.
We prespecified all outcomes and subgroups prior to analysis.
Agreements and disagreements with other studies or reviews
The Evidence‐Based Management of Sickle Cell Disease Report (Expert Panel Report 2014), included RCTs, non‐randomised intervention studies, and observational studies, also case reports and small case series when outcomes involved harm or rare complications in their reviews of the literature. They reported that most chronic complications in SCD were not the subject of RCTs or high quality observational studies, leaving few areas where strong recommendations were possible. The Panel made a strong recommendation to refer persons who have symptoms or signs suggestive of pulmonary hypertension for echocardiography based on moderate quality evidence, but no treatment recommendations were made (Yawn 2014).
The report included two RCTs and seven observational studies all dealing with pulmonary hypertension. None of the studies looked at red blood cell transfusion. There were no studies examining sickle lung disease and no recommendations were made for this indication. The report further highlights the paucity of evidence for management and treatment of chronic chest complications in SCD as identified in this systematic review.
Authors' conclusions
Implications for practice.
As no trials were found for inclusion in the review, it is not possible to comment on the evidence for or against the efficacy of long‐term red blood cell transfusions for managing chronic chest complications in people with sickle cell disease.
Implications for research.
There is a need for randomised controlled trials (RCTs) looking at the effect of long‐term transfusion therapy on pulmonary hypertension and chronic sickle lung disease. The most likely starting point for any series of trials will be the effect of transfusion on existing pulmonary hypertension. The effect of transfusion on disease incidence and mortality would require trials with longer‐term follow‐on, making them more costly and conceptually more difficult. The definition of chronic sickle lung disease include is not agreed by consensus and this is a stumbling block for further studies in this area.
New trials could consider using a combination of objective and subjective outcome measures. Effectiveness could be measured objectively, for example, through echocardiogram or pulmonary function testing, or subjectively by measuring symptoms such as chest pain on a standardised scale. Such trials might provide useful information on the rate of deterioration in chronic chest complications. Given the chronic nature of the condition, trials could consider measuring pre‐intervention 'severity' using an extended baseline 'steady state' period.
It should be remembered that transfusions may reduce symptoms such as breathlessness by increasing the haemoglobin level rather than having any beneficial effect per se on the chronic chest complication. Future RCTs in this area should have clear protocols for the aims of transfusion (such as a target haemoglobin level, or target sickle haemoglobin percentage) and how the long‐term transfusion programme is to be carried out, for example, by simple or exchange transfusion.
Possible transfusion complications are a key concern, and it would be important to collect information on the complications arising from long‐term transfusion therapy in trial participants.
What's new
| Date | Event | Description |
|---|---|---|
| 19 September 2019 | New citation required but conclusions have not changed | New searches have been undertaken for this update and minor changes have been made throughout the review. |
| 19 September 2019 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register identified 44 references, 43 references were excluded once duplicates had been removed, 1 was a secondary citation of an excluded study. An additional search was performed that identified 1,282 references plus 292 ongoing trials, which were reduced to 872 references plus 189 ongoing trials once duplicates had been removed, and to 254 references plus 23 ongoing trials once previously screened references had been removed. These were all excluded on the basis of the abstract. |
History
Protocol first published: Issue 2, 2010 Review first published: Issue 9, 2011
| Date | Event | Description |
|---|---|---|
| 16 May 2016 | New citation required but conclusions have not changed | The review was updated according to the new MECIR guidelines. New review authors have produced this updated version of the review which now conforms to the current Cochrane standards. |
| 16 May 2016 | New search has been performed | We updated the search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register but no new trials were identified. We expanded and updated additional searches for trials (Appendix 1); however, we did not identify any that were eligible for inclusion. Eight further studies have been added to the 'Excluded studies' section (Atilla 2011; Barst 2010; Chueamuangphan 2014; Detterickh 2015; Lunt 2016; NCT00023296; Styles 2006; Styles 2012). |
| 24 October 2013 | New citation required but conclusions have not changed | No new trials have been included in the review update, only minor changes have been made throughout the review. |
| 24 October 2013 | New search has been performed | Updated searches did not identify any eligible trials. |
Acknowledgements
We thank the editorial base of the Cochrane Cystic Fibrosis and Genetic Disorders Group for their help and support during the course of this review.
We thank Patricia Fortin who was involve in the searching; selection of trials; eligibility assessment; and review content update and development on the previous version of this review (Estcourt 2016). Patricia Fortin also helped in screening of the additional updated search of this review.
We thank the National Institute for Health Research (NIHR). This review is part of a series of reviews that have been partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. This research was also supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Appendices
Appendix 1. Search Strategies
CENTRAL
#1 MeSH descriptor: [Anemia, Sickle Cell] explode all trees
#2 ("hemoglobin s" or "haemoglobin s" or "hemoglobin sc" or "haemoglobin sc" or "hemoglobin se" or "haemoglobin se" or "hemoglobin ss" or "haemoglobin ss" or "hemoglobin c" or "haemoglobin c" or "hemoglobin d" or "haemoglobin d" or "Hb s" or "Hb sc" or "Hb se" or "Hb ss" or "Hb c" or "Hb d" or "sc disease" or "sc diseases" or sickle or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*)
#3 #1 or #2
#4 MeSH descriptor: [Hypertension, Pulmonary] this term only
#5 (hypertens* near/6 (lung* or pulmon*))
#6 MeSH descriptor: [Lung] explode all trees and with qualifier(s): [Physiopathology ‐ PP]
#7 MeSH descriptor: [Lung Diseases] this term only
#8 MeSH descriptor: [Lung Injury] this term only
#9 ((chest* or lung* or pulmon*) near/6 (disease* or damag* or dysfunction* or complication* or chronic* or persist* or recur* or permanent*))
#10 ((chronic* or persist* or recur* or permanent*) N5 (asthma* or dyspne* or dyspnoe*))
#11 MeSH descriptor: [Asthma] this term only
#12 MeSH descriptor: [Bronchoconstriction] this term only
#13 MeSH descriptor: [Dyspnea] explode all trees
#14 MeSH descriptor: [Sleep Apnea Syndromes] explode all trees
#15 MeSH descriptor: [Anoxia] explode all trees
#16 ("sleep disordered breathing" or apnea or apnoea or hypopnea or hypopnoea)
#17 (nocturnal hypox* or nocturnal hypoventilation or nocturnal anoxia)
#18 (pneumosclerosis or pneumofibrosis)
#19 MeSH descriptor: [Pulmonary Embolism] explode all trees
#20 MeSH descriptor: [Pulmonary Fibrosis] this term only
#21 MeSH descriptor: [Pulmonary Veno‐Occlusive Disease] this term only
#22 ((lung* or pulmon*) near/6 (fibrosis or sclerosis or embolism* or thrombosis or thromboembolism* or vasculopath* or function*))
#23 ("chronic respiratory dysfunction" or "chronic respiratory disease" or "chronic respiratory diseases" or bronchoconstrict*)
#24 #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23
#25 #3 and #24
MEDLINE
1. exp Anemia, Sickle Cell/
2. (h?emoglobin s or h?emoglobin sc or h?emoglobin se or h?emoglobin ss or h?emoglobin c or h?emoglobin d or Hb s or Hb sc or Hb se or Hb ss or Hb c or Hb d or sc disease*).tw,kf.
3. (sickle cell or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*).tw,kf.
4. or/1‐3
5. exp Hypertension, Pulmonary/
6. (hypertens* adj6 (lung* or pulmon*)).tw,kf.
7. Lung/pp
8. Lung Diseases/
9. Lung Injury/
10. ((chest* or lung* or pulmon*) adj6 (disease* or damag* or dysfunction* or complication* or chronic* or persist* or recurr* or permanent*)).tw,kf.
11. ((chronic* or persist* or recurr* or permanent*) adj5 (asthma* or dyspn?e*)).tw,kf.
12. Asthma/
13. Bronchoconstriction/
14. Dyspnea/
15. exp Sleep Apnea Syndromes/
16. Anoxia/
17. (sleep disordered breathing or apn?ea or hypopn?ea).tw,kf.
18. (nocturnal hypox* or nocturnal hypoventilation or nocturnal anoxia).tw,kf.
19. (pneumosclerosis or pneumofibrosis).tw,kf.
20. Pulmonary Embolism/
21. Pulmonary Fibrosis/
22. Pulmonary Veno‐occlusive Disease/
23. ((lung* or pulmon*) adj6 (fibrosis or sclerosis or embolism* or thrombosis or thromboembolism* or vasculopath* or function*)).tw,kf.
24. (chronic respiratory dysfunction* or chronic respiratory disease* or bronchoconstrict*).tw,kf.
25. or/5‐24
26. 4 and 25
Embase
1. exp Sickle Cell Anemia/
2. (h?emoglobin s or h?emoglobin sc or h?emoglobin se or h?emoglobin ss or h?emoglobin c or h?emoglobin d or Hb s or Hb sc or Hb se or Hb ss or Hb c or Hb d or sc disease*).tw.
3. (sickle cell or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*).tw.
4. or/1‐3
5. pulmonary hypertension/ or chronic thromboembolic pulmonary hypertension/ or persistent pulmonary hypertension/ or portopulmonary hypertension/ or pulmonary capillary hemangiomatosis/ or pulmonary vascular obstructive disease/ or pulmonary veno‐occlusive disease/
6. (hypertens* adj6 (lung* or pulmon*)).tw.
7. lung disease/ or chronic lung disease/ or lung injury/
8. ((chest* or lung* or pulmon*) adj6 (disease* or damag* or dysfunction* or complication* or chronic* or persist* or recur* or permanent*)).tw.
9. dyspnea/
10. hypoxia/ or hypoxemia/ or chronic intermittent hypoxia/ or hypoxic lung vasoconstriction/ or anoxia/
11. sleep disordered breathing/
12. asthma/ or severe persistent asthma/
13. ((chronic* or persist* or recur* or permanent*) adj5 (asthma* or dyspn?e*)).tw.
14. (sleep disordered breathing or apn?ea or hypopn?ea).tw.
15. (nocturnal hypox* or nocturnal hypoventilation or nocturnal anoxia).tw.
16. lung fibrosis/
17. (pneumosclerosis or pneumofibrosis).tw.
18. lung embolism/
19. ((lung* or pulmon*) adj6 (fibrosis or sclerosis or embolism* or thrombosis or thromboembolism* or vasculopath* or function*)).tw.
20. bronchoconstriction/
21. (chronic respiratory dysfunction* or chronic respiratory disease* or bronchoconstrict*).tw.
22. or/5‐21
23. 4 and 22
CINAHL
S1 (MH "Anemia, Sickle Cell+")
S2 TX ("hemoglobin s" or "haemoglobin s" or "hemoglobin sc" or "haemoglobin sc" or "hemoglobin se" or "haemoglobin se" or "hemoglobin ss" or "haemoglobin ss" or "hemoglobin c" or "haemoglobin c" or "hemoglobin d" or "haemoglobin d" or "Hb s" or "Hb sc" or "Hb se" or "Hb ss" or "Hb c" or "Hb d" or "sc disease" or "sc diseases" or sickle or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*)
S3 S1 OR S2
S4 (MH "Hypertension, Pulmonary") OR (MH "Pulmonary Arterial Hypertension")
S5 TX (hypertens* N6 (lung* or pulmon*))
S6 (MH "Lung+/PP")
S7 (MH "Lung Diseases")
S8 (MH "Lung Injury")
S9 TX ((chest* or lung* or pulmon*) N6 (disease* or damag* or dysfunction* or complication* or chronic* or persist* or recur* or permanent*))
S10 TX ((chronic* or persist* or recur* or permanent*) N5 (asthma* or dyspne* or dyspnoe*))
S11 (MH "Asthma")
S12 (MH "Bronchoconstriction")
S13 (MH "Dyspnea+")
S14 (MH "Sleep Apnea Syndromes+")
S15 (MH "Anoxia+")
S16 TX ("sleep disordered breathing" or apnea or apnoea or hypopnea or hypopnoea)
S17 TX (nocturnal hypox* or nocturnal hypoventilation or nocturnal anoxia)
S18 TX (pneumosclerosis or pneumofibrosis)
S19 (MH "Pulmonary Embolism")
S20 (MH "Pulmonary Fibrosis")
S21 TX ((lung* or pulmon*) N6 (fibrosis or sclerosis or embolism* or thrombosis or thromboembolism* or vasculopath* or function*))
S22 TX (chronic respiratory dysfunction* or chronic respiratory disease* or bronchoconstrict*)
S23 S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22
S24 S3 AND S23
PubMed
#1 ("hemoglobin s" OR "haemoglobin s" OR "hemoglobin sc" OR "haemoglobin sc" OR "hemoglobin se" OR "haemoglobin se" OR "hemoglobin ss" OR "haemoglobin ss" OR "hemoglobin c" OR "haemoglobin c" OR "hemoglobin d" OR "haemoglobin d" OR "Hb s" OR "Hb sc" OR "Hb se" OR "Hb ss" OR "Hb c" OR "Hb d" OR "sc disease" OR "sc diseases" OR sickle OR sicklemia OR sickled OR sickling OR meniscocyt* OR drepanocyt*)
#2 (hypertens* AND (lung OR lungs OR pulmon*))
#3 ((chest OR lung OR lungs OR pulmon*) AND (disease* OR damag* OR dysfunction* OR complication* OR chronic* OR persist* OR recurr* OR permanent*))
#4 ((chronic* OR persist* OR recurr* OR permanent*) AND (asthma* OR dyspne* OR dyspnoe*))
#5 ("sleep disordered breathing" OR apnea OR apnoea OR hypopnea OR hypopnoea)
#6 (nocturnal hypox* OR nocturnal hypoventilation OR nocturnal anoxia OR pneumosclerosis OR pneumofibrosis)
#7 ((lung OR lungs OR pulmon*) AND (fibrosis OR sclerosis OR embolism* OR thrombosis OR thromboembolism* OR vasculopath* OR function*))
#8 ("chronic respiratory dysfunction" OR "chronic respiratory disease" OR "chronic respiratory diseases" OR bronchoconstrict*)
#9 #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8
#10 #1 AND #9
Transfusion Evidence Library, LILACS, PakMediNet
sickle OR sicklemia OR sickled OR sickling
KoreaMed
sickl* [ALL] AND random* [ALL]
IndMed
(sickle OR sickled OR sickling OR sicklemia) AND (chest OR lung OR lungs OR pulmonary OR asthma OR apnea OR apnoea) AND (random OR randomly OR randomized OR randomised OR systematic OR trial OR group OR groups)
Web of Science CPCI‐S
TOPIC: (("hemoglobin s" OR "haemoglobin s" OR "hemoglobin sc" OR "haemoglobin sc" OR "hemoglobin se" OR "haemoglobin se" OR "hemoglobin ss" OR "haemoglobin ss" OR "hemoglobin c" OR "haemoglobin c" OR "hemoglobin d" OR "haemoglobin d" OR "Hb s" OR "Hb sc" OR "Hb se" OR "Hb ss" OR "Hb c" OR "Hb d" OR "sc disease" OR "sc diseases" OR sickle OR sicklemia OR sickled OR sickling OR meniscocyt* OR drepanocyt*)) AND TOPIC: ((lung OR lungs OR pulmon* OR asthma* OR dyspne* OR dyspnoe* OR apnea OR apnoea OR hypopnea OR hypopnoea)) AND TOPIC: ((systematic* OR random* OR blind* OR trial* OR control* OR group*))
ClinicalTrials.gov
Search Terms: chest OR lung OR pulmonary OR asthma OR dyspnea OR anoxia OR apnea
Study Type: Interventional Studies
Condition: sickle
WHO ICTRP
Title: chest OR lung OR pulmonary OR asthma OR dyspnea OR anoxia OR apnea OR apnoea OR hypopnea OR hypopnoea
Recruitment Status: ALL
Condition: sickle
ISRCTN
Text: chest OR lung OR pulmonary OR asthma OR dyspnea OR anoxia OR apnoea OR hypopnoea
Condition: sickle
EUCTR & HKUCTR
sickle
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Atilla 2011 | Wrong participant population |
| Barst 2010 | Wrong intervention |
| Chueamuangphan 2014 | Wrong participant population |
| Detterickh 2015 | Wrong study design |
| Lunt 2016 | Wrong study design |
| NCT00023296 | Wrong study design |
| Styles 2006 | Wrong indication |
| Styles 2012 | Wrong indication |
Differences between protocol and review
Changes from previous versions of this review
There are several differences between the protocol (Cho 2010) and this version, and the 2016 version, of the review (Estcourt 2016).
Selection of studies
We are not including quasi‐randomised studies within this review.
Outcomes
We added 'Quality of life' and 'Any other adverse events' to the list of secondary outcomes.
Search strategies
We changed the search strategies and increased the number of databases searched (Appendix 1).
Measures of treatment effect
We planned to extract and report hazard ratios (HRs) for mortality data or, if HRs were not available, every effort would be made to estimate the HR as accurately as possible using the available data and a purpose‐built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007).
For dichotomous outcomes: reporting the pooled risk ratio (RR) with a 95% CI. Where the number of observed events was small (less than 5% of sample per group), and where trials have balanced treatment groups, we planned to report the Peto odds ratio (OR) with 95% CI (Deeks 2011).
Study flow diagram
We created a study flow diagram and based it on guidance adapted from the PRISMA flow diagram and recommended for updates to Cochrane reviews (Stovold 2014).
Summary of findings
We planned to use the GRADE approach to build a 'Summary of Findings' table as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). We planned to use the GRADE approach to rate the quality of the evidence as 'high', 'moderate', 'low', or 'very low' using the five GRADE considerations. This had not been done in the previous versions of the review.
Subgroups
We extended the subgroup analyses to include: age of participant (neonate, child (one to 15 years), adult (16 years and older)); and severe SCD complications (strokes, acute chest syndrome, painful crisis, priapism).
Aspects of the review that were not implemented due to lack of data
There are several differences between the protocol (Cho 2010) and this review due to lack of data. There were no completed studies and therefore we could not: • report on any of the primary or secondary outcomes of the review; • perform a ’Risk of bias’ assessment; • assess the quality of the evidence; • assess publication bias; • perform any analyses or subgroup analyses.
Contributions of authors
The original review and first update were authored by Gavin Cho and Ian Hambleton.
In this update all listed authors contributed to the review.
Lise Estcourt: searching; selection of trials; eligibility assessment; content expert, and review content update and development.
Sally Hopewell: methodological expert and review development.
Marialena Trivella: statistical and methodological expert and review development
Ian R Hambleton: protocol development, review development and statistical expert.
Gavin Cho: protocol development, review development and content expert.
Sources of support
Internal sources
-
NHS Blood and Transplant, Research and Development, UK.
To fund the work of the Systematic Review Initiative (SRI)
External sources
-
National Institute for Health Research (NIHR) Cochrane Programme Grant, UK.
To provide funding for systematic reviewers and methodological support from the Centre for Statistics in Medicine, Oxford
Declarations of interest
Lise Estcourt: none declared. Sally Hopewell: none declared. Marialena Trivella: none declared. Ian Hambleton: none declared. Gavin Cho: none declared.
New search for studies and content updated (no change to conclusions)
References
References to studies excluded from this review
Atilla 2011 {published data only}
- Atilla E, Topcuoglu P, Yavasoglu S, Karakaya A, Gencturk C, Bozdag S, Arslan O. A randomized comparison of hemoglobin content‐based vs standard (unit‐based) RBC transfusion policy efficiencies. Vox Sanguinis 2011; Vol. 101, issue S2:121. Abstract No. P‐303.
- Atilla E, Toprak SK, Civriz Bozdag S, Topcuoglu P, Arslan O. A randomized comparison of hemoglobin content‐based versus standard (unit‐based) red blood cell transfusion policy [Hemoglobin icerigine dayali transfuzyon ile standart eritrosit suspansiyonu transfuzyon etkinliginin karsilastirilmasi]. Turk J Haematol 2017 Aug 2;34(3):244‐9. [CENTRAL: CN‐01403554; CRS: 6688743; EMBASE: 617660321; PUBMED: 27149653] [DOI] [PMC free article] [PubMed] [Google Scholar]
Barst 2010 {published data only}
- Barst RJ, Mubarak KK, Machado RF, Ataga KI, Benza L, Castro O, Naeije R, Sood N, Swerdlow PS, Hildesheim M, Gladwin MT, on behalf of the ASSET study group. Exercise capacity and hemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: rResults of the ASSET studies. British Journal of Haematology 2010;149(3):426‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chueamuangphan 2014 {published data only}
- Chueamuangphan N, Patumanond J, Wongtheptien W, Nawarawong W, Sukonthasarn A, Chuncharunee S, et al. Benefits of chronic blood transfusion in hemoglobin E/β thalassemia with pulmonary arterial hypertension. International Journal of General Medicine 2014;7:411‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Detterickh 2015 {published data only}
- Detterich JA, Kato RM, Rabai M, Meiselman HJ, Coates TD, Wood JC. Chronic transfusion therapy improves but does not normalize systemic and pulmonary vasculopathy in sickle cell disease. Blood 2015;126(6):703‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lunt 2016 {published data only}
- Lunt A, McGheea E, Robinsona P, Rees D, Height S, Greenougha A. Lung function, transfusion, pulmonary capillary blood volume and sickle cell disease. Respiratory Physiology & Neurobiology 2016;222:6‐10. [DOI] [PubMed] [Google Scholar]
NCT00023296 {published data only}
- NCT00023296. Nitric oxide and transfusion therapy for sickle cell patients with pulmonary hypertension. ClinicalTrials.gov NCT00023296 (accessed 28 April 2016).
Styles 2006 {published data only}
- Styles LA, Abboud M, Larkin S, Lo, Kuypers FA1. Transfusion prevents acute chest syndrome predicted by elevated secretory phospholipase A2. British Journal of Haematology 2006;136(2):343‐44. [DOI] [PubMed] [Google Scholar]
Styles 2012 {published data only}
- Styles L, Wager CG, Labotka RJ, Smith‐Whitley K, Thompson AA, Lane PA, et al. Refining the value of secretory phospholipase A2 as a predictor of acute chest syndrome in sickle cell disease: results of a feasibility study (PROACTIVE). British Journal of Haematology 2012;157(5):627‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Adams 1998
- Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Controlled Clinical Trials 1998;19(1):110‐29. [PUBMED: 9492971] [DOI] [PubMed] [Google Scholar]
Alhashimi 2010
- Alhashimi D, Fedorowicz Z, Alhashimi F, Dastgiri S. Blood transfusions for treating acute chest syndrome in people with sickle cell disease. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD007843.pub2] [DOI] [PubMed] [Google Scholar]
Chakravorty 2015
- Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Archives of Disease in Childhood 2015;100(1):48‐53. [10.1136/ archdischild‐2013‐303773] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chou 2013a
- Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology 2013;2013:439‐46. [DOI] [PubMed] [Google Scholar]
Chou 2013b
- Chou ST, Jackson T, Vege S, Smith‐Whitley K, Friedman DF, Whesthoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh‐matched minority donors. Blood 2013;122(6):1062‐71. [DOI] [PubMed] [Google Scholar]
Collins 1982
- Collins FS, Orringer EP. Pulmonary hypertension and cor pulmonale in the sickle hemoglobinopathies. American Journal of Medicine 1982;73(6):814‐21. [PUBMED: 7148875] [DOI] [PubMed] [Google Scholar]
Davies 1986
- Davies SC, McWilliam AC, Hewitt PE, Devenish A, Brozovic M. Red cell alloimmunization in sickle cell disease. British Journal of Haematology 1986;63(2):241‐5. [PUBMED: 3087406] [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG, on behalf of the Cochrane Statistical Methods Group. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S, editor(s). Cochrane handbook for systematic reviews of interventions Version. 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Expert Panel Report 2014
- US Department of Health and Human Services National Institutes of Health National Heart Lung and Blood Institute Expert Panel Report 2014. Evidence‐based management of sickle cell disease: Expert Panel Report, 2014. http://www.nhlbi.nih.gov/health‐pro/guidelines/sickle‐cell‐disease‐guidelines (accessed 01 August 2015).
Gordeuk 2016
- Gordeuk VR, Castro OL, Machado R. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016;127(7):820‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gravitz 2014
- Gravitz L, Pincock S. Sickle‐cell disease. Nature 2014;515:S1. [DOI] [PubMed] [Google Scholar]
Grosse 2011
- Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa. A neglected cause of early childhood mortality. American Journal of Preventive Medicine 2011;41(6S4):S398‐S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hebbel 1997
- Hebbel RP. Perspectives series: cell adhesion in vascular biology. Adhesive interactions of sickle erythrocytes with endothelium. Journal of Clinical Investigation 1997;99(11):2561‐4. [PUBMED: 9169483] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JP, Altman DG, Sterne JA, on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaborartion, 2011. Available from handbook.cochrane.org.
Higgins 2011d
- Higgins JP, Deeks JJ, Altman DG, on behalf of the Cochrane Statistical Methods Group, editor(s). Chapter 16: Special topics in statistics. In: Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Josephson 2007
- Josephson Cassandra D, Su Leon L, Hillyer Krista L, Hillyer Christopher D. Transfusion in the patient with sickle cell disease: a critical review of the literature and transfusion guidelines. Transfusion Medicine Reviews 2007;21:118‐32. [DOI] [PubMed] [Google Scholar]
Khoury 2011
- Khoury RA, Musallam KM, Mroueh S, Abboud MR. Pulmonary complications of sickle cell disease. Hemoglobin 2011;35(5‐6):625‐35. [DOI] [PubMed] [Google Scholar]
Klings 2006
- Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. American Journal of Respiratory and Critical Care Medicine 2006;173(11):1264‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Machado 2005
- Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. British Journal of Haematology 2005;130(3):445‐53. [PUBMED: 16042696] [DOI] [PMC free article] [PubMed] [Google Scholar]
Machado 2009
- Machado RF, Barst RJ, Yovetich NA, Hassell KL, Goldsmith JC, Woolson R, et al. Safety and efficacy of sildenafil therapy for doppler‐defined pulmonary hypertension in patients with sickle cell disease: preliminary results of the walk‐PHaSST clinical trial. 51st American Society of Hematology Annual Meeting and Exposition. 2009.
Mehari 2016
- Mehari A, Klings ES. Chronic pulmonary complications of sickle cell disease. Chest 2016; Vol. 149, issue 5:1313‐24. [DOI] [PMC free article] [PubMed]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐Analyses:the PRISMA statement. Annals of Internal Medicine 2009;151(4):264‐9. [DOI] [PubMed] [Google Scholar]
Nagalla 2010
- Nagalla S, Ballas SK. Drugs for preventing red blood cell dehydration in people with sickle cell disease. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD003426.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2010
- NICE (National Institute for Health and Care Excellence). Sickle cell disease. http://cks.nice.org.uk/sickle‐cell‐disease (accessed 01 August 2015).
Parmar 1998
- Parmar M, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Piel 2012
- Piel F B, Patil A P, Howes R E, Nyangiri O A, Gething P W, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. Lancet 2012;381:142‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pleasants 2014
- Pleasants S. Epidemiology: a moving target. Nature 2014;515:S2‐3. [DOI] [PubMed] [Google Scholar]
Porter 2013
- Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. ASH Education Program Book 2013;1:447‐56. [DOI] [PubMed] [Google Scholar]
Powars 1988
- Powars D, Weidman JA, Odom‐Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine (Baltimore) 1988;67(1):66‐76. [PUBMED: 3336282] [PubMed] [Google Scholar]
Rees 2010
- Rees D C, Williams T N, Gladwin M T. Sickle‐cell disease. Lancet 2010;376:2018‐31. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rosse 1990
- Rosse WF, Gallagher D, Kinney TR, Castro O, Dosik H, Moohr J, et al. Transfusion and alloimmunization in sickle cell disease. The Cooperative Study of Sickle Cell Disease. Blood 1990;76(7):1431‐7. [PUBMED: 2207318] [PubMed] [Google Scholar]
Scheunemann 2010
- Scheunemann L P, Ataga K I. Delayed hemolytic transfusion reaction in sickle cell disease. The American Journal of the Medical Sciences 2010;339:266‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmalzer 1987
- Schmalzer E A, Lee J O, Brown A K, Usami S, Chien S. Viscosity of mixtures of sickle and normal red cells at varying hematocrit levels. Implications for transfusion. Transfusion 1987;27:228‐33. [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH on behalf of the Cochrane Applicability and Recommendations Methods Group and the Cochrane Statistical Methods Group. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S (editors). Cochrane handbook for systematic reviews of interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration 2011. Available from www.cochranehandbook.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al on behalf of the Cochrane Applicability and Recommendations Methods Group and the Cochrane Statistical Methods Group. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors). Cochrane handbook for systematic reviews of interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. The Cochrane Collaboration.
Sterne 2011
- Sterne JA, Egger M, Moher D, on behalf of the Cochrane Bias Methods Group, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Stovold 2014
- Stovold E, Beecher D, Foxlee R, Noel‐Storr A. Study flow diagrams in Cochrane systematic review updates: an adapted PRISMA flow diagram. Systematic Reviews 2014;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8(16):[DOI: 10.1186/1745‐6215‐8‐16]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ubesie 2012
- Ubesie A, Emodi I, Ikefuna A, Ilechukwu G, Ilechukwu G. Prevalence of human immunodeficiency virus transmission among transfused children with sickle cell anemia in Enugu Nigeria. Annals of Medical and Health Sciences Research 2012;2:109‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vichinsky 1990
- Vichinsky EP, Earles A, Johnson RA, Hoag MS, Williams A, Lubin B. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. New England Journal of Medicine 1990;322(23):1617‐21. [DOI] [PubMed] [Google Scholar]
Vichinsky 1995
- Vichinsky EP, CM Haberkern, L Neumayr, AN Earles, D Black, M Koshy, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. New England Journal of Medicine 1995;333:206‐13. [DOI] [PubMed] [Google Scholar]
Vij 2010
- Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest 2010;138(4):973‐83. [DOI] [PubMed] [Google Scholar]
Yawn 2014
- Yawn BP, Buchanan GR, Afenyi‐Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence‐based report by expert panel members. JAMA 2014;312(10):1033‐48. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cho 2010
- Cho G, Hambleton IR. Regular long‐term red blood cell transfusions for managing chronic chest complications in sickle cell disease. Cochrane Database of Systematic Reviews 2010, Issue 2. [DOI: 10.1002/14651858.CD008360] [DOI] [PubMed] [Google Scholar]
Cho 2011
- Cho G, Hambleton IR. Regular long‐term red blood cell transfusions for managing chronic chest complications in sickle cell disease. Cochrane Database of Systematic Reviews 2011, Issue 9. [DOI: 10.1002/14651858.CD008360.pub2] [DOI] [PubMed] [Google Scholar]
Cho 2014
- Cho G, Hambleton IR. Regular long‐term red blood cell transfusions for managing chronic chest complications in sickle cell disease. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD008360.pub3] [DOI] [PubMed] [Google Scholar]
Estcourt 2016
- Estcourt LJ, Fortin PM, Hopewell S, Trivella M, Hambleton IR, Cho G. Regular long‐term red blood cell transfusions for managing chronic chest complications in sickle cell disease. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD008360.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]