

SUMMARY
The protein deacetylase SIRT1 (Sirtuin 1) regulates many cellular processes, including cell-cycle progression, DNA damage response, and metabolism. Although the centrosome is a key regulator of cell-cycle progression and genome stability, little is known concerning SIRT1 controlled centrosome-associated events. Here we report that the centrosome protein Plk2 is acetylated and undergoes deacetylation by SIRT1. Acetylation protects Plk2 from ubiquitination, and SIRT1-mediated deacetylation promotes ubiquitindependent degradation of Plk2. SIRT1 controls centriole duplication by temporally modulating centrosomal Plk2 levels. AURKA phosphorylates SIRT1 and promotes the SIRT1-Plk2 interaction in mitosis. In early-mid G1, phosphorylated SIRT1 deacetylates and promotes Plk2 degradation. In late G1, SIRT1 is hypophosphorylated and its affinity to Plk2 is decreased, resulting in a rapid accumulation of centrosomal Plk2, which contributes to the timely initiation of centriole duplication. Collectively, our findings uncover a critical role of SIRT1 in centriole duplication and provide a mechanistic insight into SIRT1-mediated centrosome-associated functions.
Graphical Abstract
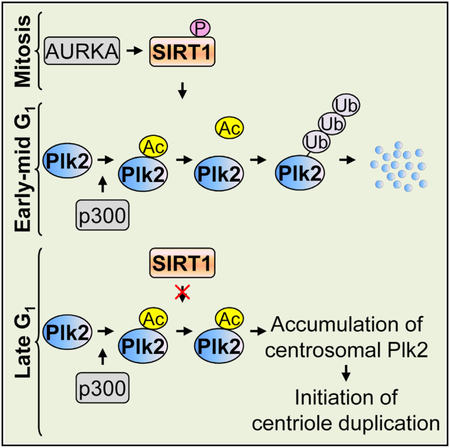
In Brief
Ling et al. demonstrate that SIRT1 deacetylates and thereby destabilizes Plk2, which in turn results in the suppression of centriole duplication.
INTRODUCTION
The centrosome is the major microtubule-organizing center in animal cells. A mature centrosome contains a pair of centrioles and surrounding amorphous pericentriolar material, which direct the formation of bipolar spindles in mitosis and ensure the equal distribution of chromosomes to daughter cells. Centrosome (as well as centriole) duplication occurs once in each cell cycle. After mitosis, a daughter cell inherits one centrosome (two centrioles) from the mother cell. The centrosome initiates duplication in late G1 and early S phases, in which each centriole inherited from the mother cell serves as a template for procentriole production (Doxsey, 2001; Hinchcliffe and Sluder, 2001, 2002). Defects in the regulatory mechanism for centrosome/centriole duplication may lead to multiple rounds of duplication (reduplication) in a single cell cycle, resulting in the generation of centrosome (>2) or centriole (>4) amplification and, in turn, aberrant mitoses and chromosomal segregation errors. Centrosome amplification is common in various types of cancer and is one of the major causes of chromosomal instability (Winey, 1996; D’Assoro et al., 2002; Fukasawa, 2005).
Many proteins participate in the regulation of centrosome duplication, and their activities are mainly controlled by posttranslational modifications. Among various modifications, past research efforts have primarily focused on phosphorylation. Indeed, many kinases and phosphatases have been identified as regulators of centrosome duplication (Fukasawa, 2007). Acetylation that occurs on the s-amino group of lysine (Lys) residues is another common modification, which is catalyzed by histone acetyltransferases (HATs) and reversed by histone deacetylases (HDACs). Acetylation eliminates positive charges and potentially affects the protein interactions, activity, and localization of the target substrates. The acetylation and deacetylation reactions are also known to cross-talk with other modifications. For instance, acetylation often antagonizes ubiquitination, hence stabilizing the target protein, either directly by competing for the same Lys residues or indirectly by altering the overall structure of the target proteins. Reversible (de)acetylation and its modifiers, HDACs and HATs, have been thus implicated in many biological processes (Caron et al., 2005; Yang and Seto, 2008). However, their roles in centrosome duplication have not been closely studied.
Previously, in an effort to systematically examine the effects of all identified human HDACs, including 11 classical HDACs (HDAC1–11) and 7 Sirtuins (SIRT1–7), on centrosome duplication, we found that SIRT1, the founding member of class III HDACs (Sirtuins), can localize to centrosomes and suppress centrosome duplication in cycling cells and centrosome reduplication in arrested cells (Ling et al., 2012). Here, we further report that SIRT1 deacetylates polo-like kinase 2 (Plk2) to control centriole duplication. Plk2 is an important centrosome duplication regulator (Warnke et al., 2004; Cizmecioglu et al., 2008; Ling et al., 2015), which drives centrosome duplication through phosphorylating several centrosome targets (Krause and Hoffmann, 2010; Chang et al., 2010; Cizmecioglu et al., 2012). We found that Plk2 is acetylated primarily by p300, which protects Plk2 from being ubiquitinated, whereas SIRT1-mediated deacetylation promotes ubiquitination, and hence degradation, of Plk2. Moreover, phosphorylation of SIRT1 by aurora kinase A (AURKA), which occurs during mitosis of the mother cell, upregulates the binding affinity of SIRT1 to Plk2, and is critical for the timely accumulation of centrosomal Plk2 and the initiation of centrosome duplication in daughter cells. We thus delineated a series of events involving SIRT1, p300, Plk2, AURKA, and ubiquitin (Ub)dependent proteolytic machineries, which contribute to the timely initiation of centrosome duplication.
RESULTS
Plk2 Is an Acetylated Centrosomal Protein
U2OS human osteosarcoma cells were co-immunostained for acetyl-lysine (AcK) and γ-tubulin, as well as SAS-6. The anti-AcK antibody-reactive signals were readily detected at both unduplicated and duplicated centrosomes (Figure 1A). Immuno- blotting of the centrosome-enriched fraction with the anti-AcK antibody (Figure 1B, fraction 3) showed that multiple centrosomal proteins are acetylated in human cells. Mass spectrometry (MS) analysis was performed to identify potential acetylated centrosomal proteins (Figure S1), of which we focused on Plk2, a protein that has been implicated in the regulation of centriole and centrosome duplication and reduplication.
Figure 1. Plk2 Is Acetylated by p300.

(A) U2OS cells were co-immunostained with anti-AcK (green) and anti-γ-tubulin or anti-SAS-6 antibodies (red). The arrows point to centrosomes. The insets show the magnified images. Scale bars: 10 μm.
(B) Fractionated samples of U2OS cells for centrosome enrichment were blotted with anti-AcK and anti-γ-tubulin antibodies.
(C) HEK293T cells were co-transfected with GFP-Plk2 and HA-p300, HA-Tip60, FLAG-HBO1, or FLAG-PCAF expression plasmids. Lysates were immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies.
(D) HEK293T cells were co-transfected with GFP-Plk2 expression plasmid and increasing amounts (0, 0.5, and 1 μg) of plasmids expressing Myc-tagged p300 WT, as well as acetyltransferase-deficient mutant (D1399Y). The lysates were immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies.
(E) p300-depleted and control cells were transfected with GFP-Plk2 expression plasmid. The lysates were then immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies.
(F) Bacterially purified GST-Plk2 was subjected to an in vitro acetylation assay using recombinant p300 HAT domain and PCAF protein, and blotted with an anti-Ack antibody.
(G) HEK293T cells were transfected with GFP-Plk2 expression plasmid and treated with 100 ng/mL TSA, 20 μM NIA, or both for 16 hr. The lysates were immunoprecipitated with an anti-GFP antibody and were blotted with anti-AcK and anti-GFP antibodies.
(H) Mapping of acetylated Lys residues of Plk2.
(I) HEK293T cells were transfected with GFP-Plk2/WT or KQ expression plasmids and were treated with NIAfor16 hr.The lysates were immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies.
To identify the HAT(s) responsible for acetylating Plk2, several major HATs (PCAF, HBO1, p300, Tip60) were co-expressed with GFP-Plk2, and GFP-Plk2 acetylation was examined. Expression of p300, but not other HATs, resulted in a significant increase in the acetylation level of GFP-Plk2 (Figure 1C). Moreover, the acetylation level of GFP-Plk2 increased proportionally with the level of p300 protein, but not the acetylase-deficient p300/DY mutant (Figure 1D). When p300 was depleted, the acetylation level of GFP-Plk2 was correspondingly reduced (Figure 1E). Lastly, Plk2 was efficiently acetylated by p300 in vitro (Figure 1F). Thus, p300 appears to be the main HAT that catalyzes Plk2 acetylation.
The acetylation states of proteins are also controlled by deacetylases (HDACs). To gain insight into which deacetylase(s) targets Plk2, we examined the acetylation state of Plk2 in the presence of deacetylase inhibitors. Cells expressing GFP-Plk2 were treated with either Trichostatin A (TSA; a classical HDACs inhibitor) or nicotinamide (NIA; Sirtuins inhibitor), and the acetylation level of GFP-Plk2 was determined (Figure 1G). When Sirtuins were inhibited by NIA, the acetylation level of Plk2 was markedly increased, indicating that one or several Sirtuins, the class III NAD+-dependent HDACs, are responsible for the deacetylation of Plk2.
Through MS analysis of Plk2 immunopurified from NIA-treated cells overexpressing Plk2 and p300, we identified a total of 12 potentially acetylated Lys residues on Plk2 (Figure 1H). When these residues were replaced with Gln (Plk2/2KQ, 4KQ, 6KQ, and 12KQ), acetylation of Plk2 was decreased (Figure 1I), confirming that Plk2 is acetylated on some or all of these identified Lys sites.
SIRT1 Binds to and Deacetylates Plk2
To identify which SIRT(s) deacetylates Plk2, we tested the ability of SIRT1–7 to bind to Plk2. FLAG-SIRTs and GFP-Plk2 were coexpressed in cells, followed by co-immunoprecipitation assays. Among all SIRTs, GFP-Plk2 was co-immunoprecipitated exclusively with SIRT1 (Figure 2A), and FLAG-SIRT1 was co-immunoprecipitated with GFP-Plk2 (Figure 2B). Plk2 lacking either the kinase or PB domains resulted in a marked decrease in SIRT1-Plk2 interaction. Particularly, the linker domain of Plk2 is required for Plk2 to bind to SIRT1 (Figure 2C). We also tested the physical interaction between endogenous SIRT1 and endogenous Plk2. Plk2 readily co-immunoprecipitated with SIRT1 (Figure 2D, left) and vice versa (Figure 2D, right), confirming that SIRT1 binds to Plk2 under normal physiological conditions.
Figure 2. SIRT1 Interacts with and Deacetylates Plk2.

(A) HEK293T cells were co-transfected with GFP-Plk2 and FLAG-SIRT1-7 expression plasmids. The lysates were immunoprecipitated with an anti-FLAG antibody and blotted with anti-GFP and anti-FLAG antibodies.
(B) Lysates prepared from HEK293T cells expressing GFP-Plk2 and FLAG-SIRT1 were immunoprecipitated with an anti-GFP antibody and blotted with anti-GFP and anti-FLAG antibodies.
(C) HEK293T cells were co-transfected with plasmids expressing Myc-SIRT1 and GFP-tagged Plk2 deletion mutants, and lysates were immunoprecipitated with an anti-GFP antibody and then blotted with anti-Myc and anti-GFP antibodies.
(D) U2OS cell lysates were immunoprecipitated with either an anti-SIRT1 antibody(left) oranti-Plk2 antibody(right) and blotted with antibodies to Plk2 and SIRT1.
(E) HEK293T cells were transfected with GFP-Plk2 and FLAG-SIRT1-7 expression plasmids. The lysateswere immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies.
(F) GFP-Plk2 immunoprecipitated from HEK293T cells expressing GFP-Plk2 after NIA treatment for 16 hr was subjected to an in vitro deacetylation assay with purified GST-SIRT1. The reaction mixtures were blotted with anti-AcK, anti-GFP, and anti-GST antibodies.
(G) SIRT1-depleted and control cells were transfected with GFP-Plk2 expression plasmid. The lysates were immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti-GFP antibodies
We next tested SIRT1–7’s ability to deacetylate Plk2. GFP-Plk2 was immunoprecipitated from cells co-transfected with FLAG-SIRTs and GFP-Plk2 expression plasmids, and then was examined for acetylation status (Figure 2E). Introduction of SIRT1, but not other SIRTs, resulted in a reduction in the acetylation level of Plk2. Consistent with this observation, SIRT1 efficiently deacetylated Plk2 in vitro (Figure 2F). Moreover, when SIRT1 was depleted in cells, the acetylation level of GFP-Plk2 was markedly increased (Figure 2G). Based on these findings, we conclude that SIRT1 is the chief HDAC responsible for deace- tylating Plk2.
SIRT1-Mediated Deacetylation Promotes Ubiquitin-Dependent Degradation of Plk2
The ubiquitin-proteasome-dependent protein degradation plays a role in the regulation of many cellular processes, including centrosome duplication. For example, the SCF-F-box protein Ub ligases control centrosome duplication and reduplication by modulating the stability of key centrosomal proteins: SCF/Slimb targets Plk4 (Rogers et al., 2009; Cunha-Ferreira et al., 2009; Korzeniewski et al., 2009), and SCF/FBXL1 and HEK293T cells were co-transfected with GFP-Plk2 expression plasmid and increasing amounts (0, 0.5, and 1 μg) of plasmids expressing Myc-tagged p300 WT, aswell as acetyltransferase-deficient mutant (D1399Y). The lysateswere immunoprecipitated with an anti-GFP antibody and blotted with anti-AcK and anti- GFP antibodies. SCF/FBXW7 target cyclin E and/or p27 (Nakayama et al., 2000; Cizmecioglu et al., 2012). The majority of proteins that are subjected to ubiquitin-proteasome-dependent degradation require the initial ubiquitination and polyubiquitination on Lys residues. Because both acetylation and ubiquitination occur on Lys residues, acetylation often counteracts ubiquitination, hence stabilizing the target protein (Caron et al., 2005). We asked whether acetylation affects the stability of Plk2. The acetylated Lys residues of Plk2 are clustered in the polo-like kinase domain (PKD) and linker domains (Figures 1H and 1I). We tested the effect of acetylation mimetic mutations (Plk2/2KQ, 4KQ, 6KQ, and 12KQ) on the stability of the Plk2 protein. The Plk2/wild-type (WT) and mutant expression plasmids were transfected into cells, and their protein levels were examined (Figure 3A). The levels of Plk2/2KQ and Plk2/4KQ were similar to that of Plk2/ WT, indicating that the acetylation of Lys residues in the N terminus and kinase domain is not involved in Plk2 stability. In contrast, the levels of Plk2/6KQ and Plk2/12KQ were 2-fold higher than that of Plk2/WT, implying that acetylation of Lys residues in the linker domain stabilizes Plk2. To extend this finding, cells were transfected with plasmids expressing GFP-Plk2/WT, GFP-Plk2/6KQ acetylation mimetic mutant, or GFP-Plk2/6KR (Lys to Arg) non-acetylatable mutant, and exposed to cycloheximide (CHX) for 8 hr, to determine the half-life (t1/2) of the respective GFP-Plk2 (Figure 3B). The t1/2 of GFP-Plk2/WT was ~3 hr, whereas the t1/2 of both Plk2/6KQ and Plk2/6KR mutants were extended to more than 8 hr, implying that the six potentially acetylated Lys residues in the linker domain are critical determinants for the stability of Plk2. To test the possibility that the six potentially acetylated Lys residues may be ubiquitination targets, and that acetylation and ubiquitination may compete for the same Lys residues, cells were transfected with GFP-Plk2/WT, GFP- Plk2/6KQ, or GFP-Plk2/6KR together with hemagglutinin (HA)-Ub expression plasmids. After treatment with the proteasome inhibitor MG132, GFP-Plk2 was examined for ubiquitination by immunoblotting with an anti-HA antibody (Figure 3C). Plk2/WT was heavily ubiquitinated, whereas the ubiquitination levels of both Plk2/6KQ and Plk2/6KR were reduced to less than 20%. Thus, the target Lys residue(s) of ubiquitination for Plk2 degradation within these six Lys residues, as well as the acetylation of these residues, blocks ubiquitination.
Figure 3. SIRT1 Destabilizes Plk2.

(A) Lysates prepared from HEK293T cells transfected with the indicated expression plasmids were blotted with antibodies to GFP and β-actin.
(B) Cells transfected with indicated plasmids were exposed to CHX (100 μM) for 8 hr. At every 2 hr, the lysates were blotted with anti-GFP and anti-α-tubulin antibodies (upper panel). The quantification of the GFP-Plk2 level (the GFP-Plk2 level at 0 hr is set to 1) is shown (lower panel).
(C) Cells were co-transfected with HA-Ub and GFP-Plk2/WT, GFP-Plk2/6KQ, or GFP-Plk2/6KR and then were treated with MG132 for 5 hr. The lysates were immunoprecipitated with an anti-GFP antibody and blotted with an anti-HA antibody (upper panel). The levels of ubiquitination were quantified (the level of GFP-Plk2/WT is set to 1) (lower panel).
(D) Cells were co-transfected with GFP-Plk2, HA-Ub, and either Myc-SIRT1/WT or Myc-SIRT1/H363Y expression plasmids. Cells were then treated with MG132 for 5 hr. The lysates were immunoprecipitated with an anti-GFP antibody and blotted with an anti-HA antibody. The lysates were separately blotted with anti-Myc and anti-β-actin antibodies. The quantification of the ubiquitination level (the level of GFP-Plk2 in the control is set to 1) is shown in the graph (lower panel).
(E) Cells were co-transfected with GFP-Plk2 and Myc-SIRT1 expression plasmids. At 30 hr post-transfection, cells were exposed to CHX for 8 hr. At every 2 hr, the lysates were blotted with anti-GFP, anti-Myc, and anti-b-actin antibodies. For reference, cells transfected with GFP-Plk2 were treated with NIA for 6 hr prior to CHX treatment. NIA-pretreated and untreated cells were then treated with CHX for 8 hr in the presence of NIA (upper panel). The quantification of the GFP-Plk2 levels is shown in the graph (the GFP-Plk2 level at 0 hr is set to 1) (lower panel).
(F) U2OS cells were transfected with either FLAG-SIRT1 or SIRT1 small interfering RNA (siRNA) expression plasmids and then were exposed to CHX for 3 hr. Lysates prepared from cells harvested every hour were blotted with anti-Plk2, anti-SIRT1, and anti-β-actin antibodies (left panel). Quantification of the Plk2 levels (Plk2 level at 0 hr is set to 1) is shown in the graph (right panel).
We then asked whether SIRT1-mediated deacetylation promotes the ubiquitination of Plk2 by examining the ubiquitination levels of Plk2 upon co-expression with either WT SIRT1 or a deacetylase-dead mutant (SIRT1/H363Y) (Figure 3D). The Plk2 ubiquitination level in SIRT1/WT-transfected cells was ~2.5-fold higher than that in the control cells, whereas co-expression of the SIRT1/H363Y mutant led to a decrease in the ubiquitination level of Plk2, which likely reflects the dominant-negative nature of this mutant (Luo et al., 2001). Thus, SIRT1 promotes the ubiquitination of Plk2 in a deacetylase activity-dependent manner.
We next tested whether ectopic expression of SIRT1 and NIA-mediated inhibition of SIRTs affect the stability of Plk2. Cells were either transfected with GFP-Plk2 or co-transfected with GFP-Plk2 and Myc-SIRT1/WT expression plasmids, and were exposed to CHX for 8 hr. Two groups of the GFP-Plk2-transfectants were prepared for NIA treatment: one pre-treated with NIA for 6 hr (and continual treatment thereafter), and another treated with NIA only during CHX exposure. GFP-Plk2 protein levels were then determined with western blots (Figure 3E). In control cells, the t1/2 of Plk2 was ~3 hr. In the SIRT1-overexpressing cells, the t1/2 of Plk2 was shortened to 1.5 hr. In contrast, in the NIA-treated cells, the t1/2 of Plk2 was extended to ~7 hr, and when cells were pre-treated with NIA, it was further extended to more than 8 hr. Taken together, we conclude that SIRT1 de- acetylates and in turn destabilizes Plk2. We also examined the stability of endogenous Plk2 in SIRT1-overexpressing cells, as well as SIRT1-depleted cells (Figure 3F). Compared with the GFP-tagged Plk2, the endogenous Plk2 was found to be less stable, with a t1/2 of ~2 hr. However, consistent with the findings from ectopically expressed GFP-Plk2, Plk2 was stabilized in the SIRT1-depleted cells (t1/2 > 3 hr) and was destabilized in the SIRT1-overexpressing cells (t1/2 < 1 hr). These findings confirm that SIRT1 controls the stability of endogenous Plk2 by deacetylating and promoting Ub-dependent degradation of Plk2.
SIRT1 Suppresses Centriole Reduplication by Targeting Plk2
Plk2 is an important centrosome duplication regulator. Its expression level is critical for Plk2 to trigger centrosome and centriole duplication and reduplication; induced expression of Plk2 promotes, but knockdown of Plk2 suppresses, centriole and centrosome duplication and reduplication (Warnke et al., 2004; Cizmecioglu et al., 2008; Chang et al., 2010; Krause and Hoffmann, 2010; Cizmecioglu et al., 2012; Ling et al., 2015). We have preliminarily shown that SIRT1 suppresses centrosome duplication and reduplication (Ling et al., 2012). We thus asked whether SIRT1 targets Plk2 to control centriole duplication and reduplication.
First, we tested a direct co-localization of SIRT1 and Plk2 at the centrosomes. We previously reported that SIRT1 can localize to centrosomes (Ling et al., 2012. We revisited this finding by testing the anti-SIRT1 antibody in SIRT1-depleted cells. The anti-SIRT1 antibody detected dot signals overlapping the centrosome in the control cells, but not in the SIRT1-depleted cells (Figure 4A). We confirmed centrosomal localization of Plk2, as previously shown (Warnke et al., 2004) (Figure 4B). When cells were co-immunostained with anti-SIRT1 and anti-Plk2 antibodies, the dot signals detected by these antibodies likely co-localized at the centrosomes (Figure 4B).
Figure 4. SIRT1 Suppresses Centriole Reduplication via Targeting Plk2.

(A) SIRT1-depleted and control U2OS cells were co-immunostained with anti-γ-tubulin (green) and anti-SIRT1 (red) antibodies and were stained with DAPI (blue). Scale bars: 10 μm.
(B) U2OS cells were also co-immunostained with anti-Plk2 (green) and anti-γ-tubulin (red) or anti-SIRT1 (red) antibodies, and then were stained with DAPI (blue). The arrows point to centrosomes. The insets show the magnified images. Scale bars: 10 μm.
(C) U2OS cells were transfected with small interfering RNA (siRNA) targeting Plk2, SIRT1, or in combination. The lysates were blotted with anti-Plk2, anti-SIRT1, and anti-β-actin antibodies (upper left). The transfected cells were treated with HU for 72 hr, and their centriole profiles were determined by immunostaining centrin (red), as well as γ-tubulin (green). The results are shown in the graph as means ± SEM (lower left). **p < 0.01. The experiments were carried out in triplicate. For each experiment, >150 cells were examined. The represented immunostaining images are shown (right panel). The arrows point to centrioles and centrosomes. Scale bars: 10 μm.
(D) U2OS cells were transfected with either GFP-tagged Plk2/WT or Plk2/6KQ together with or without FLAG-SIRT1 expression plasmids. The lysates were blotted with anti-GFP, anti-FLAG, and anti-β-actin antibodies. Quantification of the Plk2 levels (the level of Plk2 WT without SIRT1 co-expression is set to 1) is shown on the bottom of the images (left panel). The transfected cells were treated with HU for 72 hr, and their centriole profiles were determined. The results are shown in the graph as means ± SEM (right). **p < 0.01. The experiments were carried out in triplicate. For each experiment, >150 cells were examined.
We next examined the functional relationship between SIRT1 and Plk2 in the regulation of centriole duplication. U2OS cells were depleted of SIRT1 and/or Plk2 and subjected to a centriole reduplication assay. In this assay, cells are arrested by exposure to the DNA synthesis inhibitor hydroxyurea (HU). Although DNA synthesis is inhibited in these cells, centrioles continue to duplicate, resulting in the generation of reduplicated centrioles (>4). Consistent with previous studies (Warnke et al., 2004; Ling et al., 2012), Plk2 depletion resulted in an almost complete inhibition of centriole reduplication, whereas SIRT1 depletion resulted in an increased frequency of centriole reduplication. Plk2 depletion completely abolished SIRT1 depletion-induced promotion of centriole reduplication (Figure 4C); thus, SIRT1 suppresses centriole duplication in a manner dependent on Plk2.
We then tested this finding using an overexpression approach. Cells were transfected with FLAG-SIRT1, GFP-Plk2/WT, or GFP-Plk2/6KQ expression plasmids individually or in combination. The level of GFP-Plk2/WT, but not GFP-Plk2/6KQ, was expectedly decreased (to ~51.5% and 83.1%, respectively, compared to without SIRT1 co-expression) when SIRT1 was co-expressed (Figure 4D, left). The transfected cells were subjected to the centriole reduplication assay (Figure 4D, right). As previously shown (Cizmecioglu et al., 2008; Ling et al., 2012), SIRT1 overexpression resulted in the suppression of centriole reduplication, whereas overexpression of Plk2/WT and Plk2/6KQ resulted in the promotion of centriole reduplication. When co-expressed with SIRT1, both Plk2/WT and Plk2/6KQ could overcome SIRT1 to promote centriole reduplication. Therefore, Plk2 functions downstream of SIRT1 in the regulation of centriole reduplication. Importantly, despite SIRT1 co-expression, Plk2/6KQ induced a higher frequency of centriole reduplication than Plk2/WT (Figure 4D, right), which is consistent with their expression levels in cells (Figure 4D, left), confirming that the expression level of the Plk2 protein is indeed critical for centriole reduplication. Because SIRT1 deacetylates and promotes Ub-dependent degradation of Plk2, SIRT1 is likely to suppress centriole reduplication, at least partly, via targeting Plk2, and most probably by deacetylating and destabilizing Plk2. It is noteworthy that, although only the level of Plk2/WT (but not Plk2/6KQ) was significantly decreased, when SIRT1 was co-expressed, either Plk2/ WT- or 6KQ-induced promotion of centriole reduplication was slightly reduced. Because it usually causes a noticeable increase in the expression level of target proteins, the overexpression assay could determine the relationship of SIRT1 and Plk2 in the regulation of centriole reduplication in our experiment, but it might not be able to exactly reflect the role of SIRT1-mediated deacetylation of Plk2. Thus, dissecting the effect of SIRT1 on the level of endogenous Plk2 is critical to better understand the function of the SIRT1-Plk2 axis in centriole duplication.
SIRT1 Controls the Timely Accumulation of Plk2 at Centrosomes and Initiation of Centriole Duplication
Centrosome localization is critical for Plk2 to drive centrosome and centriole duplication (Cizmecioglu et al., 2008). We examined the changes in the levels of centrosomal Plk2 upon overexpression and depletion of SIRT1 by immunocytochemical quantification. When SIRT1 was overexpressed (Figure 5A, left), the level of Plk2 at centrosomes was markedly reduced (Figure 5A, middle and right). In contrast, when SIRT1 was depleted (Figure 5B, left), the level of Plk2 at centrosomes increased (Figure 5B, middle and right). Therefore, SIRT1 negatively controls the level of Plk2 at centrosomes.
Figure 5. SIRT1 Modulates the Level of Centrosomal Plk2 and Centriole Duplication.

(A and B) U2OS cells were transfected with plasmids expressing (A) FLAG-SIRT1 or (B) SIRT1 small interfering RNA (siRNA), and lysates were blotted with anti-FLAG, or anti-SIRT1 and anti-β-actin antibodies (left panels). Transfectants were synchronized in S phase, co-immunostained with anti-Plk2 (green) and anti-γ-tubulin (red) antibodies, and stained with DAPI (blue). The fluorescence intensities of centrosomal Plk2 were quantified under a microscope. The representative immunostaining images and corresponding quantification graphs of the fluorescence intensity are presented in the middle panels. The quantification results are shown as means ± SD (right panels). **p < 0.01. Scale bars: 10 μm. For each experiment, >30 cells were examined.
(C) Cell-cycle synchronization scheme using Noc arrest (upper left). The SIRT1-depleted and control cells were arrested with Noc and released from arrest by the removal of Noc. The centrioles and centrosome were stained with anti-CP110 (green) and anti-γ-tubulin (red) antibodies. The representative images for cells with un-duplicated and duplicated centrioles are shown (lower left). Scale bars: 10 μm. The rates of centriole duplication are graphed as means ± SEM (upper right). The arrow points to the time at which the majority of cells initiated centriole duplication. The cells were also co-immunostained with anti-Plk2 and anti-γ-tubulin antibodies, and the fluorescence intensity of centrosomal Plk2 was quantified and graphed. The fluorescence intensities at 3 hr were set as 1 (lower right). The arrow points to the time at which the majority of unduplicated centrioles show an increased level of Plk2
We next tested whether SIRT1 suppresses centriole duplication in cycling cells via targeting Plk2. To this end, control and SIRT1-depleted cells were first arrested at mitosis by nocoda- zole (Noc) treatment, then were allowed to re-enter the cell cycle by removal of Noc. Cells were then examined for the rate of centriole duplication (the experimental scheme is shown in Figure 5C, upper left). The representative images for cells with unduplicated (two CP110-positive foci) and duplicated (four CP110-positive foci) centrioles are shown in Figure 5C (lower left). Most of the control cells had initiated centriole duplication by 9 hr after release. In contrast, most of the SIRT1-depleted cells had initiated centriole duplication by 6 hr, indicating that the depletion of SIRT1 accelerates the initiation of centriole duplication (Figure 5C, upper right).
We next examined the changes in the levels of centrosomally localized Plk2 during the chase period (Figure 5C, lower right). In control cells, centrosomal accumulation of Plk2 was detected at 9 hr. In the SIRT1-depleted cells, centrosomal accumulation of Plk2 was detected at 6 hr, indicating that SIRT1 depletion facilitates the centrosomal accumulation of Plk2. Importantly, in both the control and SIRT1-depleted cells, the time that Plk2 accumulates at centrosomes coincides with the time of initiation of centriole duplication, supporting the notion that centro- somal accumulation of Plk2 is an important event for centriole duplication (Cizmecioglu et al., 2008). Together, these findings suggest that SIRT1 suppresses the initiation of centriole duplication by preventing centrosomal accumulation of Plk2. We also examined the levels of Plk2 mRNA and centrosomally localized SIRT1 during the chase period, and found that they are constant in both the control and SIRT1-depleted cells (Figure S2). Thus, centrosomal accumulation of Plk2 is not caused at the transcriptional level or by the association or dissociation of SIRT1 to or from centrosomes. Instead, accumulation of Plk2 at centrosomes in late G1 is likely due to changes in SIRT1 activity.
AURKA Phosphorylation of SIRT1 Is Critical for SIRT1-Plk2 Interaction and Timely Initiation of Centriole Duplication
SIRT1 is phosphorylated on multiple Ser and Thr residues (Conrad et al., 2016; Guo et al., 2012; Lau et al., 2014; Sasaki et al., 2008). We wondered whether phosphorylation is involved in SIRT1-associated regulation of centriole duplication. Because centriole duplication is initiated in late G1, SIRT1 must prevent centrosomal accumulation of Plk2 in early-mid G1. Hence, SIRT1 transmitted from a mother cell must be highly active in deacetylating Plk2. Because mitotic kinase(s)-mediated phosphorylation is likely the last modification of SIRT1 prior to cytokinesis, we examined whether AURKA, a key centrosome- associated mitotic kinase that is highly activated in mitosis but inactivated in late G1 and early S phases (Gopalan et al., 1997; Stenoien et al., 2003), participates in the phosphorylation of SIRT1. AURKA interacts with and phosphorylates SIRT1 in vitro (Figures 6A and 6B). Using MS/MS analysis, we further identified that AURKA phosphorylates SIRT1 on Thr344 (Figure 6C), as well as on several other Ser residues (data not shown). Previous studies showed that the deacetylase activity of SIRT1 is negatively regulated by Deleted in Breast Cancer-1 (DBC1) protein; DBC1 binds tightly to SIRT1 and suppresses its deacetylase activity (Kim et al., 2008; Zhao et al., 2008). The AMPK-mediated Thr344 phosphorylation, however, releases SIRT1 from the sequestration of DBC1, leading to the activation of SIRT1’s deacetylase activity (Lau et al., 2014). This raises the possibility that AURKA-mediated Thr344 phosphorylation of SIRT1 might enhance SIRT1’s affinity to Plk2 in mitosis and early G1, which in turn attenuates the accumulation of centrosomal Plk2. To test this, we co-transfected cells with HA-Plk2 and either FLAG-SIRT1/WT, T344A, or T344E mutants (non-phosphorylation and phosphorylation mimetic mutants, respectively), together with Myc-DBC1. The lysates were immunoprecipitated with an anti-FLAG antibody and blotted with antiHA, anti-Myc, and anti-FLAG antibodies. Myc-DBC1 was co-immunoprecipitated with FLAG-SIRT1/WT and FLAG-SIRT1/ T344A, but not with FLAG-SIRT1/T344E mutant (Figure 6D). In contrast, the amount of HA-Plk2 co-immunoprecipitated with FLAG-SIRT1/T344E mutant was markedly increased (Figure 6D), indicating that Thr344 phosphorylation upregulates SIRT1’s binding affinity to Plk2. Moreover, co-expression of SIRT1/ T344E led to lower levels of centrosomal Plk2 than SIRT1/WT and SIRT1/T344A (Figure 6E). Taken together, Thr344 phosphorylation by AURKA is critical for SIRT1 to effectively bind to and deacetylate Plk2, which in turn keeps centrosomal Plk2 at a low level.
Figure 6. AURKA Phosphorylates SIRT1 and Increases Its Binding Affinity to Plk2.

(A) SIRT1 interacts with AURKA. U2OS cells were transfected with HA-AURKA expression plasmid. Lysates were immunoprecipitated with anti-SIRT1, anti-HA, or control antibodies, and blotted with anti-HA and anti-SIRT1 antibodies.
(B) AURKA phosphorylatesSIRT1 in vitro. Bacterially purified GST-SIRT1 was incubated with AURKA or control. The products were separated by SDS-PAGE and blotted with anti-phospho-Ser and anti-Thr antibodies.
(C) In vitro phosphorylated SIRT1 was subjected to tandem mass spectrum analysis, and the phosphorylated Thr344-containing peptide (NYT344QN I DTLEQVAGI QR) from AURKA-phosphorylated SIRT1 protein was identified. The inset shows the peptide measurement in the survey scan.
(D) Cells were co-transfected with Myc-DBC1 and HA-Plk2, together with FLAG-SIRT1/WT, FLAG-SIRT1/T344A, and FLAG-SIRT1/T344E expression plasmids or control vector. Lysates were immunoprecipitated with an anti-FLAG antibody and blotted with anti-Myc, anti-HA, and anti-FLAG antibodies. The lysates were also blotted with anti-Myc, anti-HA, anti-FLAG, and anti-β-actin antibodies.
(E) SIRT1-depleted cells were transfected with control, SIRT1/T344A, and T344E mutants, together with DBC1 expression plasmids. Transfected cells were arrested by Noc and released for 9 hr. The fluorescence intensity of centrosomal Plk2 was quantitated, and the results are shown in the graph as means ± SD. **p < 0.01. For each experiment, >30 cells were examined.
(F) Transfected cells were subjected to the centriole duplication assay. Cells were synchronized and released. The rate of centriole duplication was determined, and the results are shown in the graph as means ± SEM. **p < 0.01. The experiments were carried out in triplicate. For each experiment, >150 cells were examined.
We next tested the effect of AURKA-mediated phosphorylation of SIRT1 on centriole duplication. The SIRT1-depleted U2OS cells were transfected with plasmids expressing SIRT1/T344A or SIRT1/T344E, together with DBC1. The transfected cells were subjected to the centriole duplication assay using the synchronization scheme described in Figure 5C. Compared with SIRT1/T344A, the activity of SIRT1/T344E to suppress centriole duplication was markedly increased (Figure 6F), indicating that Thr344 phosphorylation by AURKA is important for SIRT1 to suppress the initiation of centriole duplication.
It has been reported that SIRT1 is phosphorylated by CDK1- cyclin B at Thr530 and Ser540 during mitosis (Sasaki et al., 2008). We therefore examined the potential role of Thr530/ Ser540 phosphorylation in SIRT1-associated regulation of centriole duplication. Thr530/Ser540 phosphorylation does not affect the centrosome localization of SIRT1; there is no difference in the centrosome localization patterns between SIRT1/ WT and the SIRT1/2A (SIRT/T530A, S540A) mutant (Figure S3A). However, similar to Thr344, the phosphorylation of Thr530/ Ser540 seems essential for maintaining a lower level of centroso- mally localized Plk2 (Figure S3B). Compared with WT SIRT1, the activity of SIRT1/2A to suppress centriole duplication was markedly reduced (Figure S3C), indicating that the Thr530/Ser540 phosphorylation by CDK1-cyclin B is also critical for SIRT1- mediated suppression of centriole duplication.
p300 Regulates Centriole Duplication and Reduplication via Targeting Plk2
Because p300 acetylates Plk2, we asked whether p300 plays a role in the regulation of centriole duplication. We first tested whether p300 localizes to centrosomes by co-immunostaining U2OS cells for γ-tubulin and p300. p300 could be detected at centrosomes of control, but not p300-depleted, cells (Figure 7A), demonstrating the high specificity of the anti-p300 antibody. We also tested HA-tagged p300 for its localization to centrosomes. Like endogenous p300, HA-p300 localized to centrosomes (Figure 7B).
Figure 7. p300 Regulates Centriole Duplication and Reduplication via Targeting Plk2.

(A) Control and p300-depleted U2OS cells were co-immunostained with anti-γ-tubulin (red) and anti-p300 (green) antibodies, and then were stained with DAPI (blue). Scale bars: 10 μm.
(B) U2OS cells transfected with a control or HA-p300 expression plasmid were immunostained with anti-γ-tubulin (red) and anti-HA (green) antibodies, and then were stained with DAPI (blue). Scale bars: 10 μm.
(C) U2OS cells were transfected with either p300 small interfering RNA (siRNA) or control siRNA. The lysates were blotted with anti-p300, anti-Plk2, and anti-Vinculin antibodies (left). The transfected cells were co-immunostained with anti-Plk2 and anti-γ-tubulin antibodies. The fluorescence intensity of centrosomal Plk2 was quantified, and the results are shown in the graph (right) as means ± SD. **p < 0.01. For each experiment, >30 cells were examined.
(D) The p300-depleted and control cells were subjected to a centriole duplication assay after synchronization and release. The rate of centriole duplication was determined, and the results are shown in the graph as means ± SEM. **p < 0.01. The experiments were carried out in triplicate. For each experiment, >150 cells were examined.
(E) Transfected cells were also subjected to a centriole reduplication assay (72-hr HU exposure). Results are shown in the graph as means ± SEM. *p < 0.05; **p < 0.01. The experiments were carried out in triplicate. For each experiment, >150 cells were examined
(F) SIRT1 has little effect on the auto-acetylation level of p300. Cells were co-transfected with plasmids expressing HA-p300 and FLAG-SIRT1 or control. HA-p300 was immunopurified with an anti-HA antibody and blotted with an anti-AcK antibody. The lysates were blotted with an anti-FLAG antibody (left). The immunopurified HA-p300 was incubated with GST-SIRT1 or control in deacetylation buffer in the presence of 5 mM NAD+ and 60 mM curcumin at 30°C for 1 hr. The reaction mixtures were blotted with an anti-AcK antibody (right).
Because SIRT1-mediated deacetylation promotes ubiquitina- tion and degradation of Plk2, depletion of p300 (i.e., loss of acetylation) may have a similar effect on Plk2. We thus examined the p300-depleted cells (Figure 7C, left) for centrosomal Plk2 levels (Figure 7C, right). As expected, p300 depletion led to a reduction of Plk2 at centrosomes.
The control and p300-depleted cells were subjected to the centriole duplication assay as described above. As with the overexpression of SIRT1, the depletion of p300 resulted in the delayed initiation of centriole duplication (Figure 7D). In the centriole reduplication assay, p300-depleted cells reduplicated centrioles less efficiently than the control cells (Figure 7E). Thus, p300 plays a role in the timely initiation of centriole duplication in cycling cells and induction of centriole reduplication in arrested cells. Importantly, overexpression of Plk2/WT could rescue the suppression of centriole reduplication caused by p300 depletion, and the frequency of centriole duplication was further increased when the acetylation-mimetic and ubiquitina- tion-attenuated Plk2 (Plk2/6KQ) was introduced (Figure 7E), indicating that p300 controls centriole duplication and reduplication by targeting Plk2.
We show that SIRT1 and p300 have antagonistic activities in controlling centriole duplication and reduplication through targeting Plk2. However, one issue remains to be addressed. It is known that autoacetylation of p300 influences its activity (Thompson et al., 2004; Black et al., 2006), and thus deacetylation of the acetylated residues negatively affects p300. Several deacetylases, including SIRT1, SIRT2, and non-SIRT HDACs, have been implicated in the deacetylation of p300 (Bouras et al., 2005; Stiehl et al., 2007; Black et al., 2008). We therefore tested for a possible interaction between SIRT1 and p300. Cells were co-transfected with plasmids expressing HA-p300 and FLAG-SIRT1 or control. HA-p300 was immunopurified, and the acetylation levels were examined by blotting with anti-AcK antibody (Figure 7F, left). Immunopurified HA-p300 was also subjected to an in vitro deacetylation assay by incubating HA- p300 with bacterially purified GST-SIRT1 (Figure 7F, right). In both cases, we failed to detect a decrease in the acetylation level of p300 caused by SIRT1. Thus, SIRT1 does not seem to affect the acetylation state of p300 in our experimental system.
DISCUSSION
SIRT1 is the founding member of class III HDACs (Sirtuins), which catalyzes the removal of the acetyl group from Lys residues in an NAD+-dependent manner. Besides histones, SIRT1 has been found to profoundly affect the function of many nonhistone substrates, including transcriptional co-activators and repressors, regulators of DNA damage responses, and metabolism, among others (Vaziri et al., 2001; Langley et al., 2002, Yuan et al., 2007; Lan et al., 2008; Peng et al., 2011; Limagne et al., 2017; Zaini et al., 2018). Here, we found that SIRT1 targets Plk2 to control centriole duplication. Plk2 was acetylated primarily by p300 on 12 Lys residues, of which acetylation at the 6 Lys residues in the linker domain profoundly affects the stability of Plk2. Replacing the Lys residues in the linker domain to non-ubiquitylatable residues in the Plk2 mutant increases its stability, suggesting that one or more of the six Lys residues are likely targeted by ubiquitination for degradation. Thus, acetylation stabilizes Plk2 at least in part by directly competing for the same Lys residue(s). We found that SIRT1 is responsible for the deacetylation of Plk2, and consequently, Plk2 is stabilized when SIRT1 is either depleted or inhibited. We have previously shown that SIRT1 suppresses centrosome duplication in cycling cells and reduplication in arrested cells in a deacetylase activity-dependent manner (Ling et al., 2012). Our present observation further implies that SIRT1 suppresses centriole duplication and reduplication through deacetylating and in turn destabilizing Plk2.
We further reveal that SIRT1 controls the initiation of centriole duplication through regulating the timely accumulation of Plk2 at centrosomes. Plk2 has been known to localize to the mother centriole in early-mid G1 and accumulates at the daughter centriole prior to centrosome duplication in late G1 (Cizmecioglu et al., 2008). Our analysis of the changes in the centrosomal level of Plk2 during the cell cycle revealed that the amount of Plk2 at centrosomes remains relatively low in early-mid G1 and rapidly increases in late G1, which at least partly reflects its accumulation at the daughter centrioles, as previously reported. We further found the critical involvement of SIRT1 and p300 in the late G1-specific accumulation of Plk2 at centrosomes. For instance, the depletion of SIRT1 results in an accelerated accumulation of Plk2 at centrosomes, whereas the depletion of p300 leads to a reduction of Plk2 at centrosomes. Thus, it is reasonable to predict the following scenario: in early-mid G1, SIRT1 effectively de- acetylates Plk2, leading to its centrosomal degradation. In late G1, SIRT1’s activity is downregulated, which favors p300-mediated acetylation of Plk2, leading to the stabilization and centro- somal accumulation of Plk2. If this is the case, SIRT1 inherited from the mother cells upon cytokinesis must be highly active in deacetylating Plk2. We found here that SIRT1 can be phosphor- ylated by mitotic centrosomal kinase AURKA. Phosphorylation of SIRT1 increases its deacetylase activity by freeing it from the sequestration of its inhibitor, DBC1, and enhances the binding affinity of SIRT1 to Plk2. Thus, SIRT1 transmitted from the mother cells effectively binds to and deacetylates Plk2 in early- mid G1, keeping centrosomal Plk2 at low levels. It is known that CDK1-cyclin B phosphorylates SIRT1 at Thr530/Ser540 in mitosis (Sasaki et al., 2008), which, we found here, led to less accumulation of Plk2 at centrosomes and the suppression of centriole duplication. Therefore, CDK1-cyclin B plays a role in the regulation of SIRT1-mediated suppression of centriole duplication too. We also tested the possible association of SIRT1’s function with another mitotic centrosomal kinase polo-like kinase 1 (Plk1), but the direct phosphorylation of SIRT1 by Plk1 was not detected in our MS analysis. However, we cannot exclude the possibility that multiple mitotic kinases phosphory- late SIRT1 in concert or separately to modulate SIRT1-associ- ated regulation in centriole duplication.
Taken together, we propose a model for the role of SIRT1, as well as p300, in the regulation of centriole and centrosome duplication. During mitosis of the mother cell, SIRT1 is phosphory- lated on Thr344 by AURKA, which results in the acquisition of a high binding affinity to Plk2. Thus, the phosphorylated SIRT1 transmitted to daughter cells effectively deacetylates and promotes Ub-dependent degradation of Plk2, thereby suppressing centrosomal accumulation of Plk2 in early-mid G1. In late G1, AURKA is inactivated, resulting in a decrease in SIRT1’s phosphorylation and its affinity to Plk2. This favors acetylation of Plk2 by p300, which protects Plk2 from being ubiquitinated, leading to the rapid accumulation of Plk2 at centrosomes. Together with other positive regulators of centriole and centro- some duplication (e.g., CDK2-cyclin E, Plk4, ROCKII, etc.), Plk2 accumulation at centrosomes triggers the timely initiation of centriole and centrosome duplication. Our present studies reveal how SIRT1, as well as p300, regulates centriole and centrosome duplication through deacetylating or acetylating the centrosome protein Plk2.
STAR ★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Edward Seto (seto@gwu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T and U2OS cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin. Transfection was performed using FuGENE HD (Promega) and Lipofectamine 2000 (Invitrogen).
METHOD DETAILS
Generation of plasmids
GFP-, HA- and GST-tagged Plk2 expression plasmids were generated by subcloning Plk2 cDNA into pEGFP-C3, pCDNA3-HA and pGEX4T-1 vectors. Plasmids expressing Plk2 and SIRT1 mutants were generated by a PCR-based method. To generate the plasmids containing siRNAs, the sequences targeting following genes were placed into pSuper vector (OligoEngine). Human SIRT1: 5′-GTTGGATGATATGACACTG-3′, human Plk2: 5′-GGACATGGCTGTGAATCAG-3′, human p300: 5′-GCGGCCTAAACTCTCATCT-3′, and luciferase (negative control): 5′-CATCACGTACGCGGAATAC-3′.
Immunoblot and immunoprecipitation
For immunoblot analysis, cells were lysed in lysis buffer [1% SDS, 1% NP-40, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, protease inhibitors]. The lysates were boiled for 5 min and cleared by centrifugation at 4°C. The samples were denatured in sample buffer [2% SDS, 10% glycerol, 60 mM Tris-HCl (pH 6.8), 5% β-mercaptoethanol, 0.01% bromophenol blue], resolved by SDS-PAGE, and transferred onto a membrane. The blots were incubated in blocking buffer [5% (w/v) nonfat dry milk in TBS + Tween 20 (TBS-T)] for 1 h, and incubated with primary antibodies for 16 h at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h at 25°C. The antibody-antigen complex was visualized by ECL chemiluminescence and was quantified by Quantity One (BioRad) or Image Studio Lite 5.2 (LI-COR).
For immunoprecipitation, cells were lysed in NP-40 lysis buffer [50 mM Tris-HCl (pH7.5), 50 mM β-Glycerophosphate, 150 mM NaCl, 5 mM MgCl2, 10 mM EGTA, 5 mM NaF, 1% Na deoxycholate, 1% NP-40, protease inhibitors]. The pre-cleared lysates were incubated with antibodies overnight at 4°C. The immunocomplexes were collected by protein G or A agarose beads, boiled in sample buffer, and resolved by SDS-PAGE.
Indirect immunofluorescence
Cells were either pre-extracted or untreated, then fixed with either 10% formalin at 25°C or cold methanol for 20 min, permeabilized, and blocked by 10% normal goat serum in PBS for 1 h. Cells were then incubated with primary antibodies for 1 h, and with either Alexa Fluor 488- or 594-conjugated secondary antibodies for 1 h. All immunostaining experiments in this study were accompanied with DAPI staining.
Quantitation of the fluorescence intensity of the immunostained Plk2 and SIRT1 at the centrosome was performed using NIS- Element AR 3.10 (Nikon Corp., Japan) or AxioVision 4.7 (Carl Zeiss Inc., Germany).
In vitro deacetylation, acetylation, and phosphorylation assays
In vitro deacetylation assays were performed by incubating deacetylase (SIRT1) and substrates in deacetylation buffer [10 mM Tris- HCl (pH 8.0), 150 mM NaCl, and 10% glycerol] containing 0.5 or 5 mM NAD+ at 30°C for 1 h.
For in vitro acetylation assays, the substrate was incubated with a recombinant p300 HAT domain, or PCAF protein in acetylation buffer [50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM dithiothreitol (DTT), and 10% glycerol] containing 20 mM acetyl-CoA at 30°C for 2 h.
For the non-radioactive in vitro phosphorylation assay, bacterially purified substrate protein was incubated with AURKA in kinase buffer [25mM Tris-HCl (pH7.5), 5 mM β-glycerophosphate, 0.1mM Na3VO4, 10 mM MgCl2 and 2 mM DTT] containing 200 mM ATP at 30°C for 1h.
Preparation of centrosome-enriched fraction
The centrosome-enriched fraction was prepared from U2OS cells as described previously (Okuda et al., 2000). Briefly, cytoplasmic extracts prepared by osmotic lysis and the removal of nuclei were subjected to discontinuous sucrose gradient centrifugation. The fractions were collected from the bottom of the tube.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using GraphPad Prism. For graphs, all data are shown as means ± SEM unless otherwise noted. Comparisons between two groups were performed with two tailed unpaired t test (Figures 4, 5, 6, 7, and S3). p < 0.05 was considered significant. The number of cells or replicates for each experiment is provided in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | CAT#T9026; RRID: AB_477593 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | CAT#A1978; RRID: AB_476692 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | CAT# F3165; RRID: AB_259529 |
| Mouse monoclonal anti-HA | Sigma-Aldrich | CAT#H3663; RRID: AB_262051 |
| Mouse monoclonal anti-γ-tubulin | Sigma-Aldrich | CAT#T6557; RRID: AB_477584 |
| Mouse monoclonal anti-γ-tubulin | Santa Cruz | CAT#sc-51715; RRID: AB_630410 |
| Mouse monoclonal anti-SAS-6 | Santa Cruz | CAT#sc-81431; RRID: AB_1128357 |
| Mouse monoclonal anti-SIRT1 | Santa Cruz | CAT#sc-74504; RRID: AB_2188348 |
| Mouse monoclonal anti-GFP | Roche | CAT#11814460001; RRID: AB_390913 |
| Mouse monoclonal anti-HA | Roche | CAT#11583816001; RRID: AB_514505 |
| Mouse monoclonal anti-centrin | Sanders and Salisbury, 1994 | N/A |
| Mouse monoclonal anti-Myc tag | Cell signaling | CAT#2276s; RRID: AB_331783 |
| Rabbit anti acetylated-Lysine | Cell Signaling | CAT#9441s; RRID: AB_331805 |
| Rabbit anti acetylated-Lysine | Calbiochem | CAT#ST1027; RRID: AB_10682447 |
| Rabbit polyclonal anti-FLAG | Sigma-Aldrich | CAT#F7425; RRID: AB_439687 |
| Rabbit polyclonal anti-HA | Santa Cruz | CAT#sc-805; RRID: AB_631618 |
| Rabbit polyclonal anti-SIRT1 | Santa Cruz | CAT#sc-15404; RRID: AB_2188346 |
| Rabbit polyclonal anti-p300 | Santa Cruz | CAT#sc-584; RRID: AB_2293429 |
| Rabbit polyclonal anti-Plk2 | Santa Cruz | CAT#sc-25421; RRID: AB_2167745 |
| Rabbit polyclonal anti-GFP | Santa Cruz | CAT#sc-8334; RRID: AB_641123 |
| Rabbit polyclonal anti-GST | Santa Cruz | CAT#sc-459; RRID: AB_631586 |
| Rabbit polyclonal anti-γ-tubulin | Shinmura et al., 2005 | N/A |
| Rabbit polyclonal anti-CP110 | Proteintech | CAT#12780-1-AP; RRID: AB_10638480 |
| Rabbit polyclonal anti-Phosphoserine | Invitrogen | CAT#61-8100; RRID: AB_2533940 |
| Rabbit polyclonal anti-Phosphothreonine | Invitrogen | CAT#71-8200; RRID: AB_2534000 |
| Rabbit monoclonal anti-Plk2 | Abcam | CAT#Ab154794 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trichostatin A | ApexBio | CAT#A8183 |
| nicotinamide | Sigma-Aldrich | CAT#N3376 |
| curcumin | Sigma-Aldrich | CAT#C1386 |
| Proteasome inhibitor MG132 | EMD Millipore | CAT#474790 |
| cycloheximide | Sigma-Aldrich | CAT#C1988 |
| nocodazole | Sigma-Aldrich | CAT# M1404 |
| p300 HAT domain | Upstate Bio | CAT#14-418 |
| PCAF protein | Upstate Bio | CAT#14-309 |
| Experimental Models:Cell Lines | ||
| Human HEK293T | ATCC | CAT#CRL-11268G-1 |
| Human U2OS | ATCC | CAT# HTB96 |
| Oligonucleotides | ||
| Oligonucleotides for the site-directed mutagenesis | Table S1 | N/A |
| shRNA SIRT1-F (from 5′ to 3′): GATCCCCGTTGGATGATATGACACTGTTCAAGAGACAGTGTCATATCATCCAACTTTTTA | Integrated DNA technologies | N/A |
| shRNA SIRT1-R (from 5′ to 3′): AGCTTAAAAAGTTGGATGATATGACACTGTCTCTTGAACAGTGTCATATCATCCAACGGG | Integrated DNA technologies | N/A |
| shRNA Plk2-F (from 5′ to 3′): GATCCCCGGACATGGCTGTGAATCAGTTCAAGAGACTGATTCACAGCCATGTCCTTTTTA | Integrated DNA technologies | N/A |
| shRNA Plk2-R (from 5′ to 3′): AGCTTAAAAAGGACATGGCTGTGAATCAGTCTCTTGAACTGATTCACAGCCATGTCCGGG | Integrated DNA technologies | N/A |
| shRNA p300-F (from 5′ to 3′): GATCCCCGCGGCCTAAACTCTCATCTTTCAAGAGAAGATGAGAGTTTAGGCCGCTTTTTA | Integrated DNA technologies | N/A |
| shRNA p300-R (from 5′ to 3′): AGCTTAAAAAGCGGCCTAAACTCTCATCTTCTCTTGAAAGATGAGAGTTTAGGCCGCGGG | Integrated DNA technologies | N/A |
| shRNA Control-F (from 5′ to 3′): GATCCCCCATCACGTACGCGGAATACTTCAAGAGAGTATTCCGCGTACGTGATGTTTTTA | Integrated DNA technologies | N/A |
| shRNA Control-R (from 5′ to 3′): AGCTTAAAAACATCACGTACGCGGAATAC TCTCTTGAAGTATTCCGCGTACGTGATGGGG | Integrated DNA technologies | N/A |
| Recombinant DNA | ||
| pEGFP-C3-Plk2 | This paper | N/A |
| pCDNA3-HA-Plk2 | This paper | N/A |
| pGEX4T1-GST-Plk2 | This paper | N/A |
| pCDNA3-HA-AURKA | This paper | N/A |
| pCDNA3-HA-p300 | This paper | N/A |
| pCMVβ-p300-Myc/D1399Y | This paper | N/A |
| pCDNA3-Flag-HBO1 | This paper | N/A |
| pCDNA-Myc-DBC1 | Hiraike et al., 2010 | Addgene Plasmid #35096 |
| pCMVβ-p300-Myc | Tso-Pang Yao lab | Addgene Plasmid #30489 |
| pCDCN3-HA-Ub | Kamitani et al., 1997 | Addgene Plasmid #18712 |
| pCX-Falg-PCAF | Yang et al., 1996 | N/A |
| Flag-SIRT1-7 | Yuan et al., 2007 | N/A |
| pGEX2TK-GST-SIRT1 | Langley et al., 2002 | N/A |
| pCDNA3.1B-Myc-SIRT1 | Langley et al., 2002 | N/A |
| PCDNA3.1B-Myc-SIRT1/H363Y | Langley et al.. 2002 | N/A |
Highlights.
Plk2 is an acetylated centrosomal protein
SIRT1 deacetylates and destabilizes Plk2
SIRT1 controls the initiation of centriole duplication via targeting Plk2
AURKA phosphorylates SIRT1 and promotes SIRT1 deacetylation of Plk2
ACKNOWLEDGMENTS
We thank W. El-Deiry at Fox Chase Cancer Center for the Plk2 expression plasmid. Additionally, we thank Huadong Pei and Sonali Bahl for discussions and comments that greatly improved the manuscript. This research was supported bygrantsfrom the NIH. Some preliminaryexperimentsdescribed inthis paper were performed at the Moffitt Cancer Center Core Facility.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.025.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Black JC, Choi JE, Lombardo SR, and Carey M (2006). A mechanism for coordinating chromatin modification and preinitiation complex assembly. Mol. Cell 23, 809–818. [DOI] [PubMed] [Google Scholar]
- Black JC, Mosley A, Kitada T, Washburn M, and Carey M (2008). The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 32, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, and Pestell RG (2005). SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280, 10264–10276. [DOI] [PubMed] [Google Scholar]
- Caron C, Boyault C, and Khochbin S (2005). Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. BioEssays 27, 408–415. [DOI] [PubMed] [Google Scholar]
- Chang J, Cizmecioglu O, Hoffmann I, and Rhee K (2010). PLK2 phosphor¬ylation is critical for CPAP function in procentriole formation during the centro- some cycle. EMBO J. 29, 2395–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O, Warnke S, Arnold M, Duensing S, and Hoffmann I (2008). Plk2 regulated centriole duplication is dependent on its localization to the cen- trioles and a functional polo-box domain. Cell Cycle 7, 3548–3555. [DOI] [PubMed] [Google Scholar]
- Cizmecioglu O, Krause A, Bahtz R, Ehret L, Malek N, and Hoffmann I (2012). Plk2 regulatescentrioleduplicationthrough phosphorylation-mediated degradation of Fbxw7 (human Cdc4). J. Cell Sci. 125, 981–992. [DOI] [PubMed] [Google Scholar]
- Conrad E, Polonio-Vallon T, Meister M, Matt S, Bitomsky N, Herbel C, Liebl M, Greiner V, Kriznik B, Schumacher S, et al. (2016). HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 23, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM, and Bettencourt-Dias M (2009). The SCF/ Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr. Biol. 19, 43–49. [DOI] [PubMed] [Google Scholar]
- D’Assoro AB, Lingle WL, and Salisbury JL (2002). Centrosome amplifica¬tion and the development of cancer. Oncogene 21, 6146–6153. [DOI] [PubMed] [Google Scholar]
- Doxsey S (2001). Re-evaluating centrosomefunction. Nat. Rev. Mol. Cell Biol. 2, 688–698. [DOI] [PubMed] [Google Scholar]
- Fukasawa K (2005). Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 230, 6–19. [DOI] [PubMed] [Google Scholar]
- Fukasawa K (2007). Oncogenes and tumour suppressors take on centro- somes. Nat. Rev. Cancer 7, 911–924. [DOI] [PubMed] [Google Scholar]
- Gopalan G, Chan CS, and Donovan PJ (1997). A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J. Cell Biol. 138, 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Kesimer M, Tolun G, Zheng X, Xu Q, Lu J, Sheehan JK, Griffith JD, and Li X (2012). The NAD(+)-dependent protein deacetylase activity of SIRT1 is regulated by its oligomeric status. Sci. Rep. 2, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, and Sluder G (2001). “Ittakestwo totango”: understanding howcentrosomeduplication is regulated throughoutthecell cycle. Genes Dev. 15, 1167–1181. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, and Sluder G (2002). Two for two: Cdk2 and its role in centrosome doubling. Oncogene 21, 6154–6160. [DOI] [PubMed] [Google Scholar]
- Hiraike H, Wada-Hiraike O, Nakagawa S, Koyama S, Miyamoto Y, Sone K, Tanikawa M, Tsuruga T, Nagasaka K, Matsumoto Y, et al. (2010). Identification of DBC1 asatranscriptional repressorfor BRCA1. Br. J. Cancer 102, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, and Yeh ET (1997). Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, and Lou Z (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586. [DOI] [PubMed] [Google Scholar]
- Korzeniewski N, Zheng L, Cuevas R, Parry J, Chatterjee P, Anderton B, Duensing A, Munger K, and Duensing S (2009). Cullin 1 functions as a cen- trosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 69, 6668–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause A, and Hoffmann I (2010). Polo-like kinase 2-dependent phosphor¬ylation of NPM/B23 on serine 4 triggers centriole duplication. PLoS ONE 5, e9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Cacicedo JM, Ruderman N, and Ido Y (2008). SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, and Kouzarides T (2002). Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AW, Liu P, Inuzuka H, and Gao D (2014). SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. Am. J. Cancer Res. 4, 245–255. [PMC free article] [PubMed] [Google Scholar]
- Limagne E, Thibaudin M, Euvrard R, Berger H, Chalons P, Vegan F, Humblin E, Boidot R, Rebe C, Derangfbre V, et al. (2017). Sirtuin-1 activation controls tumor growth by impeding Th17 differentiation via STAT3 deacetylation. Cell Rep. 19, 746–759. [DOI] [PubMed] [Google Scholar]
- Ling H, Peng L, Seto E, and Fukasawa K (2012). Suppression of centro- some duplication and amplification by deacetylases. Cell Cycle 11, 3779–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Hanashiro K, Luong TH, Benavides L, and Fukasawa K (2015). Functional relationship among PLK2, PLK4 and ROCK2 to induce centrosome amplification. Cell Cycle 14, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, and Gu W (2001). Negative control of p53 by Sir2a promotes cell survival under stress. Cell 107, 137–148. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, et al. (2000). Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 19, 2069–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, and Fukasawa K (2000). Nucleophosmin/B23 is a target ofCDK2/cyclin E in centrosome duplication. Cell 103, 127–140. [DOI] [PubMed] [Google Scholar]
- Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N, Koomen J, Chen J, Lane WS, and Seto E (2011). SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell. Biol. 31, 4720–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GC, Rusan NM, Roberts DM, Peifer M, and Rogers SL (2009). The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MA, and Salisbury JL (1994). Centrin plays an essential role in microtubule severing during flagellar excision in Chlamydomonas reinhardtii. J. Cell Biol. 124, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, Minor W, and Scrable H (2008). Phosphorylation regulates SIRT1 function. PLoS ONE 3, e4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinmura K, Tarapore P, Tokuyama Y, George KR, and Fukasawa K (2005). Characterization of centrosomal association of nucleophosmin/B23 linked to Crm1 activity. FEBS Lett. 579, 6621–6634. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Sen S, Mancini MA, and Brinkley BR (2003). Dynamic association of a tumor amplified kinase, Aurora-A, with the centrosome and mitotic spindle. Cell Motil. Cytoskeleton 55, 134–146. [DOI] [PubMed] [Google Scholar]
- Stiehl DP, Fath DM, Liang D, Jiang Y, and Sang N (2007). Histone deacetylase inhibitors synergize p300 autoacetylation that regulates its transactivation activity and complex formation. Cancer Res. 67, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, et al. (2004). Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol 11, 308–315. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, and Weinberg RA (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Warnke S, Kemmler S, Hames RS, Tsai HL, Hoffmann-Rohrer U, Fry AM, and Hoffmann I (2004). Polo-like kinase-2 is required for centriole duplication in mammalian cells. Curr. Biol. 14, 1200–1207. [DOI] [PubMed] [Google Scholar]
- Winey M (1996). Keeping the centrosome cycle on track. Genome stability. Curr. Biol. 6, 962–964. [DOI] [PubMed] [Google Scholar]
- Yang XJ, and Seto E (2008). Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, and Nakatani Y (1996). A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382, 319–324. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Zhang X, Sengupta N, Lane WS, and Seto E (2007). SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaini MA, Müller C, de Jong TV, Ackermann T, Hartleben G, Kortman G, Gührs KH, Fusetti F, Krämer OH, Guryev V, and Calkhoven CF (2018). A p300 and SIRT1 regulated acetylation switch of C/EBPα controls mitochondrial function. Cell Rep. 22, 497–511. [DOI] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, and Gu W (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.