

Abstract
Myelofibrosis (MF), a rare disorder characterized by bone marrow fibrosis, has been implicated as a cause of pulmonary hypertension (PH). To date, studies examining this association have not looked at the impact of PH on survival in MF. In this single center, retrospective chart review we identified 65 patients with biopsy proven primary and secondary myelofibrosis of whom 31 underwent transthoracic echocardiography. After accounting for chronic obstructive pulmonary disease and left sided or valvular heart dysfunction which excluded 6 patients, we identified 14 patients (56%) who had echocardiographic evidence of Group 5 PH (Pulmonary Hypertension Due to Unclear or Multifactorial Mechanisms), 8 with primary myelofibrosis (PMF) and 6 with secondary MF. Myelofibrosis patients with PH trended toward being predominantly female, older, and less often had constitutional symptoms compared to the non-PH cohort. There was no effect of the presence of PH on overall survival in the entire MF cohort or in any subgroup analyzed including PMF versus secondary MF and PMF intermediate risk patients. We conclude that given the high prevalence of MF associated PH, there may be a larger role for routine screening using echocardiography in MF patients. Further, the underlying association between PH and MF may signify an endothelial plasticity or increased telomerase activity as part of the pathogenesis of MF.
Keywords: myelofibrosis, pulmonary hypertension, prevalence, overall survival, endothelial mesenchymal transition, telomerase
Introduction
Myelofibrosis (MF) is a clonal defect of hematopoietic stems cells in the bone marrow resulting in a release of cytokines that induce a reactive fibrosis of the marrow leading to extramedullary hematopoiesis and splenomegaly (1). It is more broadly classified as one of the chronic myeloproliferative neoplasms (MPNs), a heterogeneous group of disorders that also includes polycythemia vera (PV) and essential thrombocytosis (ET) (2).
Pulmonary hypertension (PH) is a disease characterized by a sustained elevation in pulmonary pressure which may be secondary to pulmonary vascular remodeling (3). Pulmonary hypertension due to MPNs is clinically classified by the World Health Organization as Group 5 (4) “Pulmonary Hypertension Due to Unclear or Multifactorial Mechanisms” based on published case reports and small series (5–10). The largest of these case series studied a population of 36 patients with myelofibrosis, 13 of whom (36%) had PH documented by echocardiogram (7).
Despite this relationship, there have been no larger studies quantifying this association. Furthermore, while there is suggestion that PH portends a poor prognosis (6) there are no studies directly comparing survival of MF patients with and without with PH. Herein, we examine the relationship between MF and PH by echocardiogram (ECHO) using a retrospective patient database and examine the influence of PH on overall survival.
Methods
In this single center retrospective chart review we identified patients with biopsy proven primary and secondary myelofibrosis within the Lifespan hospital system from 2000-2015. Only those patients with an echocardiogram (ECHO) available for review were included in the PH cohorts. The study was approved by the Institutional Review Board.
Baseline demographics, blood counts, cytogenetic testing, JAK2 testing, bone marrow assessment including degree of fibrosis, and ECHO reports were reviewed. Data regarding CALR and MPL mutational status was not collected as many of the patients included in this study predate the discovery of these gene mutations. Data regarding the use of cytoreductive therapy was also not readily available in the medical record. The Dynamic International Prognostic Scoring (DIPSS) was used to risk stratify patients with primary myelofibrosis (PMF) into low, intermediate-1, intermediate-2, and high-risk categories (11).
The diagnosis of Group 5 PH was made noninvasively using measurements from Doppler transthoracic echocardiography (TTE) and considered present if the right ventricular systolic pressure (RVSP) on echocardiogram was ≥ 36mmHg. Patients with an elevated RVSP but with a history of chronic obstructive pulmonary disease (COPD), a left ventricular ejection fraction (LVEF) < 50% or those with severe aortic/mitral valve disease were excluded from incidence and survival analysis.
Overall survival (OS) was defined as the time in months from initial diagnosis of MF by bone marrow biopsy until death. Fisher’s exact test and chi square analysis was used to determine differences in categorical outcomes while Wilcoxon rank-sum tests were used for differences in continuous variables. OS curves were estimated using the Kaplan-Meier method and compared using the log-rank test (12). Due to a limited number of patients meeting criteria for low and high risk PMF, OS curves based on PH were separately performed for patients with DIPSS Intermediate-1 and Intermediate-2 PMF.
Results
A total of 65 biopsy proven myelofibrosis patients were identified from 2000 to 2015, 46 with primary myelofibrosis (PMF) and 19 secondary to polycythemia vera (PV) or essential thrombocytosis (ET). Baseline clinical and laboratory characteristics of the entire MF population are presented in Table 1, subdivided based on primary or secondary MF. Patients with PMF had lower WBC at the time of diagnosis and were more often JAK2 negative than those with secondary myelofibrosis. PMF patients also trended toward a greater need for transfusions. Within the entire MF cohort, 31 (47.7%) patients underwent transthoracic echocardiography. Six patients met criteria for PH but either had COPD or LVEF < 50% and were excluded from analysis. Of the remaining twenty-five patients, fourteen (56%), 8 with primary myelofibrosis (PMF) and 6 with secondary MF, had echocardiographic evidence of group 5 PH.
Table 1:
Baseline clinical and laboratory characteristics for entire myelofibrosis cohort, subdivided based on primary and secondary myelofibrosis
| Primary Myelofibrosis n=46 (71%) | Secondary Myelofibrosis n=19 (29%) | P-value | |
|---|---|---|---|
| Age | 70 | 65.5 | 0.35 |
| Sex | |||
| Male | 29 | 11 | 0.70 |
| Female | 17 | 8 | |
| Group 5 PH | 8 | 6 | 0.21 |
| WBC | 4.7 | 13.3 | 0.001 |
| Hgb | 9.9 | 9.9 | 0.79 |
| Platelets | 159 | 209 | 0.27 |
| Constitutional Symptomsa | 4 (n=38) | 4 (n=16) | 0.17 |
| Transfusion Requirement | 15 (n=39) | 2 (n=16) | 0.06 |
| Circulating Blast Cells ≥1%a | 7 (n=40) | 6 (n=17) | 0.14 |
| JAK 2 Statusb | 14( (n=35) | 12 (n=15) | 0.009 |
Sample size (n) limited by incomplete data entry in the electronic medical record
Sample size limited based on availability of ordered test and/or results
In analysis of all patients who underwent echocardiography, we compare baseline clinical and laboratory characteristics for those with and without PH (Table 2) and further limit this comparison to only patients with PMF (Table 3). Amongst all patients and in the PMF only group, the PH cohort trended toward being older, predominantly female, and less likely to have constitutional symptoms compared to the non-PH group. Otherwise there were no statistical trends nor differences between cohorts.
Table 2:
Baseline clinical and laboratory characteristics for myelofibrosis patients who had echocardiograms. Patients are divided into with and without PH cohorts.
| MF without PH n=11 (44%) | MF with Group 5 PH n=14 (56%) | P-value | |
|---|---|---|---|
| Age | 66 | 70.5 | 0.11 |
| Sex | |||
| Male | 8 | 5 | 0.07 |
| Female | 3 | 9 | |
| Primary MF | 8 | 8 | 0.42 |
| WBC | 4.6 | 8.7 | 0.13 |
| Hgb | 9.8 | 9.2 | 0.91 |
| Platelets | 121 | 261 | 0.35 |
| Constitutional Symptomsa | 4(40)% (n=10) | 1(7.7%) (n=13) | 0.06 |
| Transfusion Requirement | 3 | 3 | 0.71 |
| Circulating Blast Cells ≥1%a | 2(20%) (n=10) | 5(36%) (n=14) | 0.4 |
| JAK 2 Statusb | 5(63)% (n=8) | 4(36%) (n=11) | 0.26 |
Sample size (n) limited by incomplete data entry in the electronic medical record
Sample size limited based on availability of ordered test and/or results
Table 3:
Baseline clinical and laboratory characteristics for patients with primary myelofibrosis with and without pulmonary hypertension
| PMF without PH n=8 (50%) | PMF with Group 5 PH n=8 (50%) | P-value | |
|---|---|---|---|
| Age | 65 | 74 | 0.07 |
| Sex | |||
| Male | 6 | 2 | 0.13 |
| Female | 2 | 6 | |
| WBC | 3.7 | 4.6 | 0.38 |
| Hgb | 10.4 | 9.0 | 0.95 |
| Platelets | 97 | 261 | 0.56 |
| Constitutional Symptomsa | 2(29)% (n=7) | 0 (n=8) | 0.2 |
| Transfusion Requirement | 3 | 3 | 1.0 |
| Circulating Blast Cells ≥1%a | 2(29%) (n=7) | 2(25%) (n=8) | 1.0 |
| JAK 2 Positiveb | 2(40)% (n=5) | 1(17%) (n=6) | 0.55 |
| Median DIPSS Risk | 2 | 2.5 | 0.37 |
Sample size (n) limited by incomplete data entry in the electronic medical record
Sample size limited based on availability of ordered test and/or results
We initially examined OS in all primary myelofibrosis patients based on their respective DIPSS risk category (Figure 1) which showed a clear difference in OS (p=0.008). The median OS for Low, Intermediate-1, Intermediate-2, and High Risk PMF patients was 36, 57, 23, and 1 month, respectively. 10 patients without PH and 12 patients with Group 5 PH had sufficient survival data available for analysis. OS for all MF patients with an ECHO based on the presence or absence of PH was not different (Figure 2, p=0.51) with a median OS of 12.5 months for those without PH and 30 months for those with PH.
Figure 1:
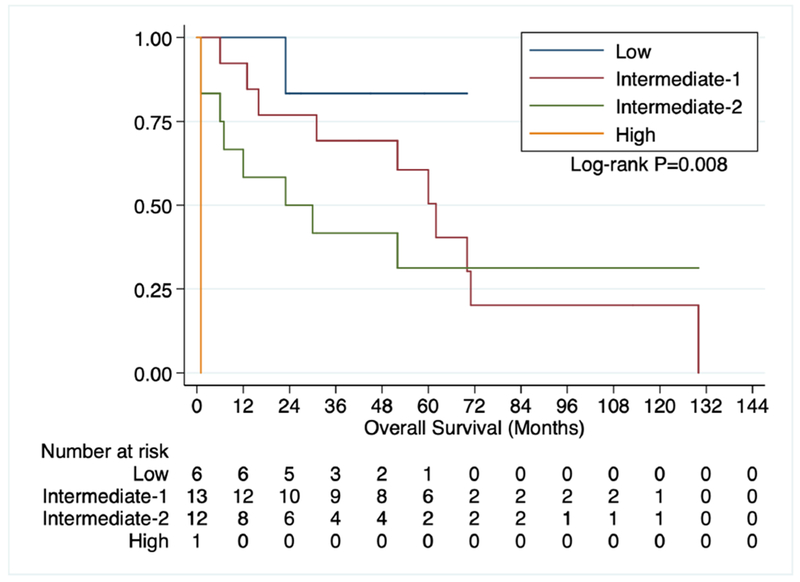
OS of primary myelofibrosis patients based on DIPSS.
Figure 2:
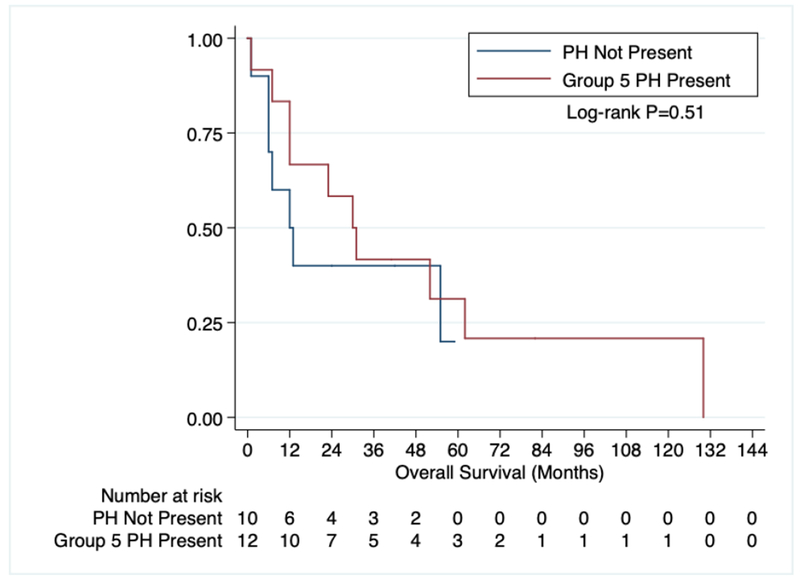
OS of all myelofibrosis patients with an ECHO based on the presence of Group 5 pulmonary hypertension.
In order to determine the influence of DIPSS risk category and PH on OS, we evaluated those PMF patients with DIPSS Intermediate-1 and Intermediate-2 risk disease. Low and high-risk patients were excluded from analysis because of low numbers (2 patients and 1 patient, respectively) and an absence of Group 5 PH in each group. Similar to all MF patients there was no difference in survival amongst intermediate risk PMF patients with (8 patients, median OS 30.5 months) and without (3 patients, median OS 13 months) Group 5 PH (Figure 3).
Figure 3:
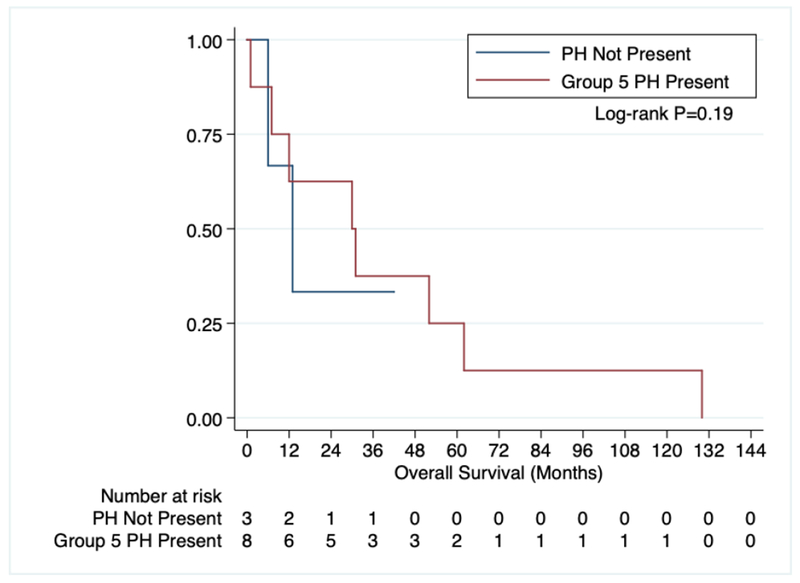
OS of primary myelofibrosis patients with intermediate-1 and intermediate-2 DIPSS based on the presence of Group 5 pulmonary hypertension.
Discussion
The association of PH with MF has been reported in a number of case series, but studies have been limited due to sample size. This retrospective study represents the largest evaluation to our knowledge to better quantify the exact prevalence of MF and PH. Using non-invasive echocardiography, we show that overall prevalence of group 5 PH in our entire MF cohort of 65 patients to be 21.5%, consistent with prior retrospective studies (6, 7, 9). However, if we consider only the cohort of patients who underwent an ECHO and exclude those patients with other potential causes of PH we see a much higher prevalence of 56%. There appears to be very little difference in clinical and baseline laboratory characteristics for MF patients with and without Group 5 PH except possibly for age, sex, and presence of constitutional symptoms. Older patients may either have more comorbid disease contributing to or a longer disease course leading to PH. Female sex is a known risk factor for idiopathic and associated forms of pulmonary arterial hypertension (PAH) and sex steroids have been implicated in PAH pathobiology, but a sex bias has not been previously described in WHO Group 5 PH (13).
While this study suggests and supports an ongoing notion that incidence of PH is increased in myelofibrosis, we are limited by its retrospective nature. Given limitations in EMR documentation, it was often unclear why echocardiograms were obtained, and while we assume that it was for cardiopulmonary evaluation, it is not known if patients were symptomatic at time of diagnosis (considered more likely) or if investigators were specifically screening for PH (considered less likely). In addition, a large proportion of the MF cohort (53%) did not undergo an echocardiogram. While their PH status is unknown, the absence of an ECHO implies that at the very least if PH was present it was likely not clinically significant.
This data is additionally limited by the lack of right heart catheterization data which is the required diagnostic test for PH, although we attempted to control for this by excluding those patients with COPD, LVEF < 50% or those with severe aortic/mitral valve disease. This may ultimately lead an overestimation of the true prevalence of group 5 PH in MF and may be especially relevant given those with PH tended to be older with more comorbid disease. Given these limitations, in the absence of routine ECHO or R-heart catheterization data in all MF patients, the exact incidence of PH in MF remains unknown.
There is a suggestion in the literature that the presence of PH portends a poor prognosis in MF (6), although the data to support this claim is scant. Figure 1 serves as an internal validation tool for our patient population, and in accordance with prior data (11), survival in MF worsens as DIPSS scores increase. Group 5 PH, however, appeared to have no effect on survival amongst all MF patients and in those PMF patients that were Intermediate Risk-1 and 2 at diagnosis. The lack of worsened prognosis of Group 5 PH in our MF patients may be a reflection of the relatively small sample sizes, particularly when considering our subgroup analysis. Whether or not a larger patient population that has been more thoroughly screened for Group 5 PH by ECHO would detect a survival difference is unknown but worth future consideration.
A number of studies have indicated a role for bone marrow derived endothelial progenitor cells (EPCs) in the development of PH. Farha et al. reported that non-affected family members of patients with heritable PH displayed elevated circulating concentrations of CD34+CD133+ progenitor cells, which were comparable to their affected relatives with PH, and had significant increase in marrow fibrosis compared to healthy unrelated controls. It was hypothesized that a subclinical myeloproliferative process appears intrinsic to PH, with myeloid abnormalities promoting pathologic vascular remodeling in the lung (14). It was also observed that in lung tissue from patients with PH, there was marked infiltration of c-kit+ hematopoietic progenitor cells in the perivascular compartment of remodeled pulmonary arteries (15), and the therapeutic targeting of c-kit+ cells prevent hypoxia-induced PH in mice (16,17). In transplant studies on hypoxic and monocrotaline treated rats with PH, labeled donor marrow derived cells were observed in vascular regions, including CD45+ hematopoietic cells, smooth muscle alpha-actin expressing myofibroblasts and endothelial-like cells in the adventitia (18–20). Moreover, to further underscore the association of PH with the bone marrow, BM-derived CD133+ progenitor cells from patients with PH induce PH when transplanted into immune deficient mice (21).
Recent studies on mice with monocrotaline induced PH have directly indicated a major role for endothelial progenitors. Initially it was demonstrated that extracellular vesicles from the lungs of monocrotaline treated mice could induce PH in normal mice. Furthermore, the whole bone marrow from these mice could induce PH and in fact, the PH inducing marrow cells were EPCs (22). Lastly, these changes were shown to be preventable and reversible by extracellular vesicles derived from a separate population of marrow derived, mesenchymal stem cells (23).
These same marrow derived EPCs implicated in the pathogenesis of PH undergo an endothelial-to-mesenchymal transition in MF, a process in which cells lose their endothelial cell phenotype and gain a mesenchymal or smooth muscle phenotype, contributing to bone marrow fibrosis (24–27). In addition, endothelial to hematopoietic transition is also thought to play a role in the pathogenesis of pulmonary vascular disease (28). Both PH and MPNs show a close inter-dependence between hematopoietic disease and endothelial cell/EPC dysfunction, suggesting a common hematopoietic-endothelial bi-lineage progenitor may be involved in both diseases that is supported by our finding of an association between MF and PH.
Another potential unifying theory of the link between PH and MF lies in telomerase activity. Increased telomerase reverse transcriptase (TERT) has been found in the lungs of patients with idiopathic PH and in the pulmonary artery smooth muscle of a murine PH model. Furthermore, the TERT inhibitor imetelstat is able to decrease pulmonary artery smooth muscle growth and reduce PH in these murine models (29). Telomerase has also been shown to be active in cancer cells with inhibition of telomerase by imetelstat having been shown to not only exert activity but to actually result in durable disease modifying complete and partial remissions for approximately 20% of patients with myelofibrosis (30).
Overall, our data continues to support the notion that PH is an under-recognized complication of MF and given the high prevalence there is a role for standardized screening. Given the limitations associated with echocardiography, alternative screening tools such as cardiopulmonary exercise testing (CPET), with a recent study identifying pulmonary perfusion defects in asymptomatic PMF patients with no alteration in baseline echocardiogram (31), and incorporating the measurement of plasmatic biomarkers associated with PH, including BNP, NR-proBNP (32) may be used in the future. For now, however, the widespread availability and relative ease of echocardiography continues to make it a useful screening tool for PH, especially early in its disease course when appropriate therapies may be available. Moreover, the high prevalence of Group 5 PH in MF further underscores a potential role of endothelial to mesenchymal cell transition and/or telomerase activity in the development of myelofibrosis. Additional research into these processes is needed to further knowledge on myelofibrosis development.
Acknowledgements
Dr. Olin Liang reports receiving grant support from the following: NIH/NIGMS P20 GM119943, NIH/NHLBI R01 HL112860, NIH/NHLBI R01 HL123965, and NIH/NIDDK R01 DK112808. Drs. Austin, Quesenberry, and Reagan designed the research study. Dr. Austin and Reagan collected and analyzed the data. All authors wrote the paper and approved the final draft.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tefferi A Myelofibrosis with Myeloid Metaplasia. New England Journal of Medicine. 2000;342(17):1255–1265. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 3.Jeffery TK, and Morrell NW. Molecular and Cellular Basis of Pulmonary Vascular Remodeling in Pulmonary Hypertension. Progress in Cardiovascular Diseases. 2002; 45(3):173–202. [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, et al. Updated Clinical Classification of Pulmonary Hypertension. Journal of the American College of Cardiology. 2013;62(25) [DOI] [PubMed] [Google Scholar]
- 5.Marvin KS, and Spellberg RD. Pulmonary Hypertension Secondary to Thrombocytosis in a Patient with Myeloid Metaplasia. Chest. 1993;103(2):642–644. [DOI] [PubMed] [Google Scholar]
- 6.Dingli D, et al. Unexplained Pulmonary Hypertension in Chronic Myeloproliferative Disorders.” Chest. 2001;120(3):801–808. [DOI] [PubMed] [Google Scholar]
- 7.Cortelezzi A, et al. Pulmonary Arterial Hypertension in Primary Myelofibrosis Is Common and Associated with an Altered Angiogenic Status. Leukemia. 2007;22(3):646–649. [DOI] [PubMed] [Google Scholar]
- 8.Perrone C, et al. Pulmonary Hypertension Diagnosed by Echocardiography during Idiopathic Myelofibrosis. A Case Report and a Brief Review of the Literature. Multidisciplinary Respiratory Medicine. 2010;5(4):267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roach EC, et al. Impaired Right Ventricular-Pulmonary Vascular Function in Myeloproliferative Neoplasms. The Journal of Heart and Lung Transplantation. 2015;34(3):390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Manero G, et al. Pulmonary Hypertension in Patients with Myelofibrosis Secondary to Myeloproliferative Diseases. American Journal of Hematology. 1999;60(2):130–135. [DOI] [PubMed] [Google Scholar]
- 11.Passamonti F, et al. A Dynamic Prognostic Model to Predict Survival in Primary Myelofibrosis: a Study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2009;115(9):1703–1708. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan EL, and Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association. 1958;53(282):457. [Google Scholar]
- 13.Foderaro A, and Ventetuolo CE. Pulmonary Arterial Hypertension and the Sex Hormone Paradox. Current Hypertension Reports. 2016;18(11). [DOI] [PubMed] [Google Scholar]
- 14.Farha S, et al. Hypoxia-Inducible Factors in Human Pulmonary Arterial Hypertension: a Link to the Intrinsic Myeloid Abnormalities. Blood. 2011;117(13):3485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montani D, et al. C-Kit Positive Cells Accumulate in Remodeled Vessels of Idiopathic Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine. 2011; 184(1) 2011:116–123. [DOI] [PubMed] [Google Scholar]
- 16.Farha S, et al. Imatinib in Pulmonary Arterial Hypertension: C-Kit Inhibition. Pulmonary Circulation. 2014;4(3):452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gambaryan N, et al. Targeting of c-Kit Haematopoietic Progenitor Cells Prevents Hypoxic Pulmonary Hypertension. European Respiratory Journal. 2010;37(6):1392–1399. [DOI] [PubMed] [Google Scholar]
- 18.Hayashida K, et al. Bone Marrow-Derived Cells Contribute to Pulmonary Vascular Remodeling in Hypoxia-Induced Pulmonary Hypertension. Chest. 2005;127(5):1793–1798. [DOI] [PubMed] [Google Scholar]
- 19.Spees JL, et al. Bone Marrow Progenitor Cells Contribute to Repair and Remodeling of the Lung and Heart in a Rat Model of Progressive Pulmonary Hypertension. The FASEB Journal. 2008;22(4):1226–1236. [DOI] [PubMed] [Google Scholar]
- 20.Marsboom G, et al. Sustained Endothelial Progenitor Cell Dysfunction After Chronic Hypoxia-Induced Pulmonary Hypertension. Stem Cells. 2008;26(4):1017–1026. [DOI] [PubMed] [Google Scholar]
- 21.Asosingh K, et al. Pulmonary Vascular Disease in Mice Xenografted with Human BM Progenitors from Patients with Pulmonary Arterial Hypertension. Blood. 2012;120(6):1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aliotta JM, et al. Induction of Pulmonary Hypertensive Changes by Extracellular Vesicles from Monocrotaline-Treated Mice. Cardiovascular Research. 2013;100(3):354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aliotta JM, et al. Exosomes Induce and Reverse Monocrotaline-Induced Pulmonary Hypertension in Mice. Cardiovascular Research. 2016;110(3):319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erba B, et al. Endothelial-to-Mesenchymal Transition in Bone Marrow and Spleen of Primary Myelofibrosis. The American Journal of Pathology. 2017;187(8):1879–1892. [DOI] [PubMed] [Google Scholar]
- 25.Ranchoux B, et al. Endothelial-to-Mesenchymal Transition in Pulmonary Hypertension. Circulation. 2015; 131(11):1006–1018. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, et al. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2018;314(1):L118–L126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takagi K, et al. IL-13 enhances mesenchymal transition of pulmonary artery endothelial cells via down-regulation of miR-424/503 in vitro. Cellular Signalling. 2018;42:270–280. [DOI] [PubMed] [Google Scholar]
- 28.Liang O, et al. Endothelial to haematopoietic transition contributes to pulmonary arterial hypertension. Cardiovascular Research. 2017;113(13):1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouraret N, Houssaini A, Abid S, et al. Role for telomerase in pulmonary hypertension. Circulation. 2015;131 (8):742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tefferi A, Lasho TL, Begna KH, et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N Engl J Med. 2015;373(10):908–919. [DOI] [PubMed] [Google Scholar]
- 31.Sciumè M, et al. Early Detection of Pulmonary Hypertension in Primary Myelofibrosis: The Role of Echocardiography, Cardiopulmonary Exercise Testing, and Biomarkers. American Journal of Hematology. 2015;92(4). [DOI] [PubMed] [Google Scholar]
- 32.Pezzuto B, et al. Circulating Biomarkers in Pulmonary Arterial Hypertension: Update and Future Direction. The Journal of Heart and Lung Transplantation. 2015;34(3):282–305. [DOI] [PubMed] [Google Scholar]