

Abstract
Chronic use of prescription opioids can exacerbate risk and severity of ischemic stroke. Annually, 6 million people die from stroke worldwide and there are no neuroprotective or neurorestorative agents to improve stroke outcomes and promote recovery. Prescribed opioids such as morphine have been shown to alter tight junction protein expression, resulting in the disruption of the blood brain barrier (BBB), ultimately leading to stroke pathogenesis. Consequently, protection of the of BBB has been proposed as a therapeutic strategy for ischemic stroke. This perspective addresses the deficiency in stroke pharmacological options and examines a novel application and repurposing of FDA-approved opioid antagonists as a prospective neuroprotective therapeutic strategy to minimize BBB damage, reduce stroke severity, and promote neural recovery. Future directions discuss potential drug design and delivery methods to enhance these novel therapeutic targets.
Keywords: Ischemic stroke, Opioid antagonist, Blood brain barrier, Neuroprotection, Naloxone, Naltrexone
Graphical abstract
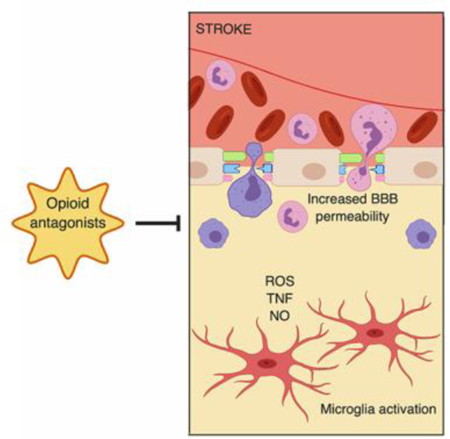
1. INTRODUCTION
As of 2017, the US government declared the opioid epidemic as a public health emergency that is linked to a number of serious health issues, including an increase in cerebrovascular events such as stroke (1, 2). Chronic prescription opioid use exacerbates risk and severity of ischemic stroke. Simultaneously, stroke is the fifth overall cause of death in the US and costing the US health care system over $30 billion annually (3–7). Despite this, treatment options for ischemic stroke remain limited. Currently, there are no FDA-approved treatments for the resulting pathological damage to the blood brain barrier (BBB) that arises from an ischemic stroke and there is a need for novel drugs to promote stroke recovery as there are no approved neuroprotective or neurorestorative treatments for stroke (8). While substantial research for novel treatments for the protection of the brain from damage after a stroke has been conducted in the past decade, success has been limited, and many neuroprotective treatments have failed in safety or efficacy in clinical trials. BBB disruption is a pathological hallmark in ischemic stroke, thus suggesting that protection of the BBB as a therapeutic strategy during stroke and for stroke recovery is of critical importance. Simultaneously, inflammatory responses are activated during ischemic injury. A potential therapeutic strategy is to modulate resulting microglia and macrophage activation in the ischemic region to reduce neuroinflammation and prevent secondary neurodegeneration resulting from phagocytosis of viable neurons. In this perspective, we survey the current state of stroke recovery interventions centered on neuroprotective agents for stroke recovery, specifically, opioid antagonists. As several reviews focusing on neuroprotection for ischemic stroke have already been published, this paper focuses on using FDA-approved opioid antagonists as novel drug repurposing for promising neuroprotective pharmacological options for ischemic stroke. While the exact mechanism of action of opioid antagonists is not fully understood, this class of drugs provides an attractive therapeutic option for treating ischemic stroke due to their anti-inflammatory properties, reduction of secondary neuronal loss, and minimization of BBB perturbations through suppression of microglial activation and reduction of cytokines, ultimately, proposing a potential recycling of FDA-approved therapeutics for treatment of prescription opioid induced stroke. An examination and critical review of promising work involving the use of opioid antagonists as prospective stroke therapeutics, and their respective efficacy in primitive human studies and later animal models is discussed.
2. BACKGROUND
2.1. Ischemic stroke
Stroke is the 5th leading causes of death in US, and attributes to 1 of every 20 deaths (9, 10). An ischemic stroke accounts for 87% of all strokes and occurs when there is an obstruction in the blood vessel, such as a blood clot, and fresh blood can no longer reach the brain (5). When a blockage occurs, the brain lacks the oxygen and nutrients needed for cellular energy, resulting in necrosis (11). During an ischemic stroke, the BBB is disrupted (12–15).
In ischemic stroke, intracellular tight junctions (TJs) are disrupted, resulting in compromised BBB integrity and increased permeability and poor regulation of transfer of molecules and ions across the BBB (Figure 1). Often, when BBB integrity is disturbed, neuronal dysfunction, neuroinflammation, and neurodegeneration may occur (12, 16, 17). During an ischemic stroke, the affected area suffers oxidative stress, in turn challenging the integrity of the BBB and resulting in breakdown (Figure 1) (18–20). Oxidative stress is indicative of an increase in reactive oxygen species (ROS) which aid in TJ protein dysregulation (19, 21, 22). Much of the vascular and tissue damage in stroke is attributed to neuroinflammation and oxidative stress, and oxidative stress may be one of the underlying mechanisms of BBB disruption in ischemic stroke (12, 14, 15, 23, 24).
Figure 1. Blood-brain barrier (BBB) disruption during ischemic stroke.
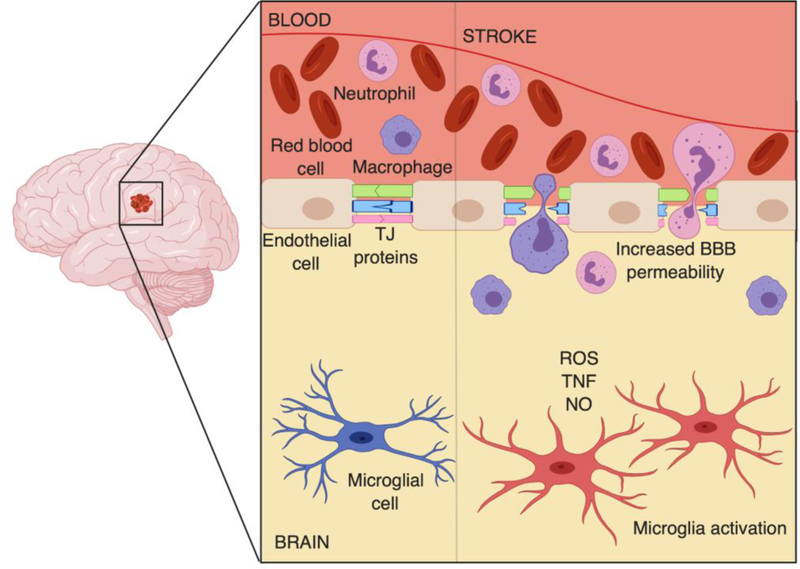
Ischemia, caused by restricted blood flow, results in activation of microglia, leading to release of reactive oxidative species (ROS), nitric oxide (NO), and inflammatory cytokines, such as TNF-alpha, in turn compromising the integrity of BBB. Tight junction (TJ) proteins, such as occludin, junctional adhesion molecule (JAM), and zonula occludens (ZO), become also disrupted, further contributing to dysfunction of the BBB. Dysregulation of TJ proteins results in increased BBB permeability and entry of blood-borne substances and cells, such as macrophages and neutrophils, into the infarct zone and brain parenchyma.
Down-regulation or dysregulation of TJ proteins such as occludin and claudin-5 is frequently observed in ischemic stroke (23, 25). TJ proteins such as occludin, junctional adhesion molecule (JAM), and submembranous zonula occludens (ZO) proteins are crucial to the cytoskeleton of the BBB as they regulate cellular traffic into the central nervous system (CNS) (23, 25–27). Dysregulation of these proteins can promote the migration of inflammatory cells across the BBB, resulting in neuroinflammation. BBB dysfunction following ischemic stroke has been suggested to be progressive or biphasic, and the time-course of the post stroke BBB opening is not clearly understood (28). Several studies have reported opposing data indicating that is unclear if post stroke damage may occur progressively following the stroke or if BBB disruption is exceptional during the first 3 h after stroke, or BBB permeability is biphasic such that significant damage is observed at 4–6 h and then again at 24 or 72 h following a stroke (29–32). Disruption to the BBB results in increased barrier permeability to blood-borne substances, including leakage of blood proteins (i.e., albumin) as well as monocytes and neutrophils into the CNS, ultimately challenging the homeostasis of the brain microenvironment that is necessary for proper neural functioning (33–35). This sequence of events has been observed in numerous clinical studies and confirmed in experimental models of a widely used rodent model of ischemic stroke, transient middle cerebral artery occlusion (MCAO) (3, 14, 36, 37).
2.2. Opioids and stroke
Pain management is critical in the effective care of patients after surgery, as well as patients with cancer, and severe acute and chronic diseases (38, 39). For example, opioids have been a basis of cancer pain treatment regimen, and morphine and its derivatives are the most used opioid drugs (40–42). The action of these opioids is mediated primarily through activation of the μ opioid receptor. As the principle target for opioids, the μ-opioid receptor is a G protein coupled receptor (GPCR) on brain endothelial cells with high affinity and specific binding towards commonly clinically used opioids such as morphine. While the molecular basis of the μ-opioid receptors is not clearly understood, the μ-opioid receptor and mediates the effects of morphine through activation of downstream G-proteins and stimulation of various signaling pathways such as mitogen-activated protein kinase (MAPK)-pathway (43–46). Morphine is the ultimate analgesic, but, unfortunately, is also highly addictive (42, 47, 48). Long-term pain management with opioids present severe side effects, including addiction, abuse, and neurovascular complications, such as ischemic stroke (48–50). Chronic use of prescription opioids induces mitochondrial dysfunction and oxidative stress, which are critical factors in stimulating neuroimmune activation. As a result, these painkillers are now linked to higher risk for stroke by compressing the carotid artery or causing cardio-embolism, hypoxia, or hypoperfusion (6, 51–54). Pathologically, chronic opioid use is also shown to alter the BBB integrity (55, 56). Morphine contributes to the breakdown of BBB by disrupting the expression of TJ proteins (56). Exposure to morphine results in a significant increase in the transendothelial migration of peripheral blood mononuclear blood cells (PBMC). In addition, increased JAM-2 expression, decreased ZO-1 and occludin gene expression are observed, thus compromising the integrity of the BBB (56). For example, prostate cancer patients receiving intense morphine had approximately a 3-fold higher risk for ischemic stroke in comparison to non-morphine users. This risk was found to also be enhanced with increased morphine dosage (6).
In addition to opioids, opium has been linked to stroke in several clinical studies (51, 52, 57). Nearly half of a cohort of 35 ischemic stroke consisting of 14 men and 21 women that expressed co-morbidity with muscle weakness were observed to have suffered from opium abuse. Consequently, opium abuse was the most common risk factor for ischemic stroke in this study (58). Similarly, nearly 40% of a sample of 97 ischemic stroke patients that also experienced large vessel involvement such as a large artery stenosis, were found to be dependent on opium (52). The relationship between stroke and opium dependence was also studied in a case-control study of 105 stroke and 105 control patients (51). Patients were diagnosed with a stroke by clinical diagnosis and CT scan and opium dependency was confirmed by patients’ medical history and DSM-IV-TR diagnosis. Analysis of the results indicated statistical significance, therefore opium dependency was suggested as a plausible independent risk factor for stroke (51).
Opioids have also been linked to an increased prevalence of atrial fibrillation, which is a significant risk factor for stroke (59–61). The prevalence of atrial fibrillation has been observed to be significantly higher in hydrocodone, propoxyphene, and tramadol users in comparison to non-opioid users (12.5% vs 7.6%; p < 0.001) in a cross-sectional association between prescription opioid use and atrial fibrillation using data collected from 30,239 participants (59). This association between opioids and atrial fibrillation may be explained by the down-regulation of opioid receptors (59). As chronic opioid use leads to tolerance, a decrease in opioid receptor signaling is observed, indicative of an opioid receptor desensitization (60, 62). This mechanism was proposed in rats that were chronically exposed to morphine and μ-opioid receptor (MOR) activity was reduced compared to animals that did not receive morphine (60). Conventionally, during ischemia, endogenous opioids can exhibit cardioprotective effects by opening mitochondrial K+ ATP channels, as a protective mechanism against oxidative stress. However, this protective mechanism may be lost with chronic opioid use, causing damage to atrial myocytes and eventually leading to atrial fibrillation (59).
2.3. Current stroke treatment
Recent advances have been made in preventing the occurrence of stroke, however there are only few therapeutic agents for treatment of ischemic stroke. Currently, there is only one FDA approved drug for stroke treatment: tissue-type plasminogen activator (tPA) (3, 63). Recombinant tPA (r-tPA) is a thrombolytic protein that was approved in 1996 as an acute stroke treatment to dissolve the blood clot and restore blood flow to the brain (8, 64). However, there are many limitation to this drug including a narrow therapeutic window, thus the patient must receive tPA between 3–4.5 h after their stroke onset (8, 64–66). As less than 15% of patients arrive to the hospital within this window, and, in addition, patients with certain medical conditions are excluded from receiving tPA, only 3% of ischemic patients are eligible to receive this treatment (4, 8). An impaired BBB, such as that exhibited in stroke, also limits the uses of tPA by increasing likelihood of a hemorrhagic transformation (HT) (67, 68). Further, tPA has no apparent neuroprotective or neurological recovery effects.
Unlike tPA that target the thrombus, neuroprotective agents are potential stroke therapeutics that aim to minimize BBB damage and secondary neural damage before and after ischemic injury. Neuroprotective treatments intend to restore or reverse the injury that has occurred to the ischemic region, subsequently to prevent greater or irreversible injury to the ischemic brain (Table 1) (69). Next, we will draw attention to FDA-approved opioid antagonists and their novel use as prospective neuroprotective agents for stroke.
Table 1. Therapeutic agents undergoing clinical trials for stroke treatment.
Updated from Small DL et al., 2002 (4).
| Drug Class | Drug |
|---|---|
| Drugs for improving blood flow | |
| Antithrombotic | Heparin, Nadroparin, Tinzaparin, Danaparoid |
| Anti-platelet | Aspirin, Abciximab |
| Fibrinogen depleting | Ancrod |
| Improve capillary flow | Pentoxifylline |
| Thrombolytics | Pro-urokinase, Tissue plasminogen activator, Streptokinase, Urokinase |
| Drugs to protect brain tissue (neuroprotective agents) | |
| Calcium channel blockers | Nimodipine, Flunarizine |
| Free radical scavengers-antioxidants | Ebselen, Tirilazad, NYP-059 |
| GABA agonists | Clomethiazole |
| AMPA antagonists | GYKI 52466, NBQX, YM90K, YM872, ZK-200775 (MPQX) |
| Kainate antagonist | SYM 2081 |
| Competitive NMDA antagonists | CGS 19755 (Selfotel) |
| NMDA channel blockers | Aptiganel (Cerestat), Dextrorphan, Dextromethorphan |
| Magnesium | Memantine, MK-801, NPS 1506, AR-R15896AR, HU-211, Remacemide |
| Glycine site antagonists | ACEA 1021, GV 150526 |
| Polyamine site antagonists | Eliprodil, Ifenprodil |
| Growth factors | Fibroblast Growth factor (bFGF) |
| Leukocyte adhesion inhibitor | Anti-ICAM antibody (Enlimonab), Hu23F2G |
| Nitric oxide inhibitor | Lubeluzole |
| Opioid antagonists | Naloxone, Nalmefene |
| Phosphatidylcholine precursor | Citicoline (CDP-choline) |
| Serotonin agonists | Bay × 3072 |
| Sodium channel blockers | Fosphenytoin, Lubeluzole, 619C89 |
| Potassium channel opener | BMS-204352 |
3. NOVEL THERAPEUTIC STRATEGIES FOR ISCHEMIC STROKE
Various therapeutic agents are being tested in clinical trials for stroke including antithrombotics, antiplatelet agents, and thrombolytics (Table 1). Nonetheless, their uses are limited to dissolving the blood clot and restoring blood flow (4). As protection of the BBB has been suggested as a therapeutic strategy for ischemic stroke, we surveyed neuroprotective agents for stroke recovery, specifically opioid antagonists, as other neuroprotective agents such as NMDA antagonists and GABA agonists have been previously extensively reviewed (Table 1) (4, 8, 55). Protection of the BBB should be prioritized during a stroke and developed as a therapeutic tool for stroke recovery (8, 70). Combining therapeutic agents with tPA can help to minimize BBB perturbations and appears to be an attractive therapeutic objective (71). For example, a study based on an opioid use in a mouse model observed that a small dose of an opioid antagonist, naloxone, significantly reduced the effects of morphine on BBB permeability, suggesting that naloxone may have neuroprotective effects (55). Novel therapeutic agents, in conjunction with tPA, that are aimed to minimize BBB perturbation may also minimize the risk for hemorrhagic transformation and increase the therapeutic window of tPA, in turn, increasing the applicability of the drug for a larger number of stroke patients (8).
3.1. Naloxone as a potential therapeutic for ischemic stroke
Naloxone ((–)-naloxone) is an FDA-approved opioid overdose treatment and is administered as a nasal spray or injection (72–74) (Figure 2). It functions as a competitive antagonist by quickly occupying opioid receptors, preventing opiates from binding and activating the receptors (75). Initial dosing is one spray (0.4 mg/mL) intranasally or an injection of 0.4 mg/mL for opioid overdose (76, 77). Although commonly used as opioid abuse medication, naloxone treatment has also been proposed as a promising treatment for ischemic stroke. Naloxone was first suggested as a therapeutic agent for cerebral ischemia in 1981, and its respective neuroprotective effects was initially observed in in humans (78). In an initial study, repeated intravenous naloxone was concluded to reverse secondary cerebral ischemia neurological deficits, such as hemiplegia in two human patients (78). In another clinical study, the potential neurorestorative effects of naloxone was observed in thirteen patients with acute stroke that presented neurologic deficits. More than half of these patients returned to their pre-stroke neurological state by the end of their hospital stay after intravenous administration of naloxone (79). Naloxone was also shown to reduce neurologic deficits in opioid use animal models of ischemic stroke, specifically MCAO ischemic stroke in gerbils that received morphine sulfate (80). Intraperitoneal injection of naloxone at 1 mg/kg was found to reverse signs of stroke within minutes of administration, albeit the effect lasted for only 30 min (80). While limited to a small sample size and/or experimental stroke models, these primitive human and animal studies indicate that naloxone administration may be an effective neurorestorative therapeutic to reverse neurologic deficits in acute stroke models (78–80) (Table 2).
Figure 2.
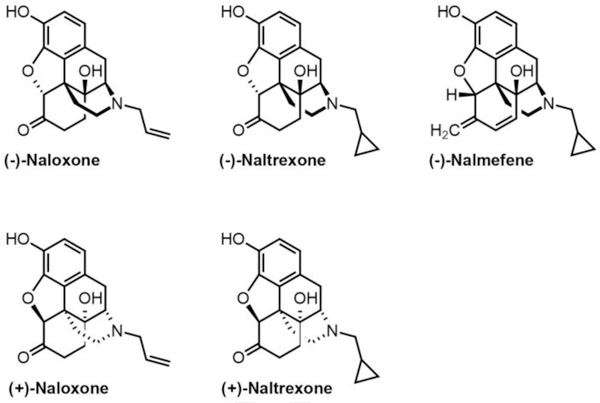
Chemical structures of surveyed opioid antagonists.
Table 2.
Survey of opioid antagonists for promoting stroke recovery
| Opioid antagonist |
Dose | Frequency | Organism | Disease | Reference |
|---|---|---|---|---|---|
| Naloxone | |||||
| (-)-Naloxone | 0.4 mg intravenous injection | Repeated as needed | Humans | Ischemic stroke | 79 |
| (-)-Naloxone | 1 mg/kg intraperitoneal injection | Repeated as needed | Gerbils | MCAO ischemic stroke | 80 |
| (-)-Naloxone | 0. 4 mg-1.2 mg intravenous injection | 2–3 doses | Humans | Ischemic stroke | 77 |
| (-)-Naloxone | 82.5 nmol intracerebroventricular infusion (i.c.v) | Every 4 h | Rats | MCAO ischemic stroke | 81 |
| (-)-Naloxone | 1 mg/mL or 10 mg/mL intracerebroventricular infusion (i.c.v) | Every 4 h | Rats | MCAO ischemic stroke | 82 |
| (-)-Naloxone | 0.32 mg/kg intranasally | Twice a day for 7 days | Rats | MCAO ischemic stroke | 84 |
| (-)-Naloxone | 10 mg/kg initial intraperitoneal injection, 5 mg/kg/h subcutaneously | Continuous | Feline | MCAO ischemic stroke | 87 |
| Naloxone enantiomer | |||||
| (+)-Naloxone | 0.32 mg/kg - 0.8 mg/kg intranasally | Twice a day for 7 days | Rats | MCAO ischemic stroke | 84 |
| Naltrexone | |||||
| (-)-Naltrexone | 10 mg/kg initial intraperitoneal injection, 1 mg/kg/h subcutaneously | Continuous | Feline | MCAO ischemic stroke | 87 |
| Naltrexone enantiomer | |||||
| (+)- Naltrexone | 3 mg/kg or 6 mg/kg intraperitoneal injection | Twice a day for 2 days | Mouse | Cardiac arrest | 83 |
| Nalmefene | |||||
| Nalmefene | 0.05 mg/kg initial dose intravenously, then 0.01 mg/kg | 24 h | Humans | Ischemic stroke | 91 |
| Nalmefene | 0.2 mg intravenous injection | Twice a day for 10 days | Humans | Large cerebral infarction | 93 |
The neuroprotective mechanisms of naloxone are not clearly understood (Figure 3). While many studies suggest that this neuroprotection occurs via blocking opioid receptor activation, other reports have shown that the neuroprotective effects are independent of opioid receptors (Figure 3). In a study observing the neuroprotective impact of naloxone against ischemic injury in rats, blockage of opioid receptor activation was suggested as a method for decreasing extent of ischemic injury (81). To test if opioid receptors are involved in the neuroprotective role of naloxone, (–)-naloxone was compared to its enantiomer, (+)-naloxone, an inactive form of the drug that is not a competitive antagonist for opiates and binding to opioid receptors (Figure 2). Results found that intracerebroventricular infusion of (–)-naloxone significantly reduced the extent of infarct volume in comparison to enantiomer (+)-naloxone that was ineffective (81). As a result, naloxone’s neuroprotective role was concluded to involve an opioid receptor mechanism via blocking ^-opioid receptor properties (Figure 3A) (Table 2). Naloxone was also found to significantly decrease inflammatory cell accumulation as quantified by myeloperoxidase (MPO) activity. Blocking μ-opioid receptor activation by an opioid antagonist was observed to be protective against ischemic injury as brain infarction and neutrophil accumulation were conclusively reduced with naloxone treatment in rat models of ischemic injury (Figure 3A) (81). Similarly, treatment with (–)-naloxone (1 mg/mL or 10 mg/mL) prior to cerebral ischemic injury significantly reduced the extent of the ischemic brain injury in MCAO rats (82). Accumulation of inflammatory cells such as neutrophils, macrophages, leukocytes, and microglia is also a hallmark of ischemic injury as a consequence of compromised BBB integrity and increased barrier permeability (Figure 1). Simultaneously, as MPO activity in the ischemic area is increased within 24 h of injury, pre-treatment with naloxone was found to attenuate this event. These findings not only suggest that naloxone may reduce ischemic neuronal loss and cell infiltration by reducing microglia activation in rats with ischemic brain injury, but also qualify naloxone as a promising effective neuroprotective agent for reducing ischemic injuries (82) (Table 2).
Figure 3. Suggested neuroprotective mechanisms for opioid antagonists.
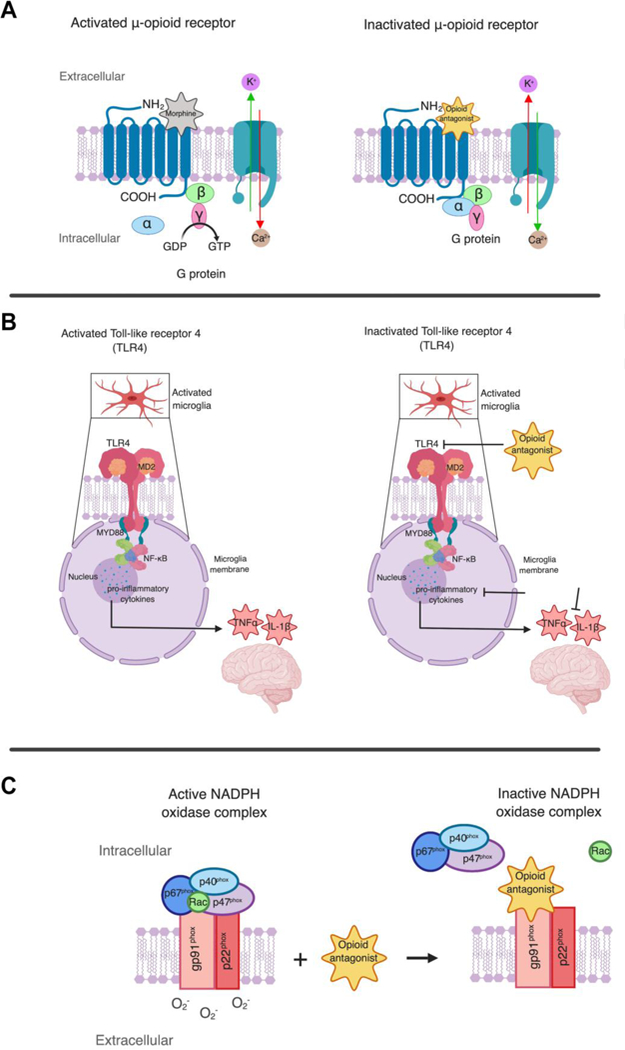
While the neuroprotective mechanisms of opioid antagonists are not clearly understood, the following mechanisms are being considered. A. μ-opioid receptors are 7 transmembrane spanning that activate G proteins composed of α, β and γ subunits which convert GDP to GTP. When activated, μ-opioid receptors exhibit inhibition of Ca2+ influx and activation of K+ channels. Opioid antagonists block μ-opioid receptor activation by competitive binding. B. TLR4 signaling pathway is activated in microgliosis. As a result, neurotoxic mediators such as TNFα and IL-1β are released. Opioid antagonists are suggested to block TLR4 signaling, leading to inhibition of pro-inflammatory cytokine production of TNFα and IL-1β. C. NADPH (dihydronicotinamide adenine dinucleotide phosphate) oxidase is an enzyme complex involved in the induction of oxidative stress that consists a membrane bound gp91phox subunit and p22phox as well as three cytosolic proteins (p40phox, p47phox, and p67phox). During an ischemic stroke, the NADPH complex is activated as and the cytosolic components are translocated to plasma membrane to interact with the membrane bound gp91phox subunit and p22phox to assemble an active NADPH oxidase enzyme complex stimulating increased superoxide O2- generation. Opioid antagonists inhibit enzymatic activity of NADPH oxidase by binding to the gp91phox subunit and induce a conformational change of the NADPH protein complex affecting the binding affinity of the cytosolic subunits p40phox, p47phox, p67phox. As a result, oxidative stress that compromises BBB integrity is reduced
Microgliosis occurs as a response to ischemia and results in a neurotoxic environment (83). During an ischemic injury, stressed cells release danger-associated molecular pattern molecules that are agonists for TLR4 which induces microgliosis (Figure 3B). As a result, neurotoxic mediators such as TNFα and IL-1β are released (83). Naloxone’s anti-inflammatory properties and its respective suppression of microglial activation were studied in the MCAO rat model (84). Activation of microglia was the most pronounced on day 7 post-ischemic stroke and neuronal loss was observed in the thalamus 14 days after MCAO. (–)-Naloxone and its enantiomer, (+)-naloxone, were synthesized and intranasally administered, evaluating if difference in affinity to opioid receptor antagonist by naloxone isoforms may result in varying neuroprotective effects and behavioral recovery. One day after MCAO, (+)-naloxone was administered to rats at a dose of 0.32 mg/kg every 12 hours for 7 days. On days 10 and 14, body asymmetry and neurological deficits were all significantly reduced in the ischemic rats. By day 14, measured locomotor activity was significantly improved. (+)-Naloxone (0.32–0.8 mg/kg) administered post-stroke also significantly reduced infarction size on day 14 post-stroke and prevented delayed neuronal death. (–)-Naloxone (0.32 mg/kg, administered intranasally) was shown to reduce body asymmetry on days 10 and 14 following stroke. Findings from this study indicated that post-stroke intranasal administration of naloxone to MCAO rat models of ischemic stroke reduces neuroinflammation and promotes behavioral recovery, suggesting that targeting microglia/macrophage activation within the regions of ischemia may be a potential target for stroke therapeutic agents (84) (Table 2). Therefore, it is also suggested that the efficacy of (+)- naloxone in reducing stroke symptoms and its’ respective anti-inflammatory and neuroprotective effects may be independent of opioid receptors.
3.2. Naltrexone as a potential therapeutic for ischemic stroke
As ischemic injury leads to microglia and macrophage activation, which in turn results in neuroinflammation and neuronal loss, the neuroprotective role of naltrexone has been considered. (–)-Naltrexone is an FDA-approved opioid antagonist for opioid addiction that may also be neuroprotective following an ischemic injury (83, 85, 86) (Figure 2; Table 2). The neuroprotective capacity of (+)-naltrexone, an enantiomer of naltrexone, was observed in reducing microgliosis, neuronal injury, and neuronal death after cardiac arrest (CA) and cardiopulmonary resuscitation (CPR) in mice (83) (Table 2). CA was induced in mice by injecting cold KCl into the jugular catheter, and confirmed by EKG. CPR was given after 8 min of CA by epinephrine injection, chest compressions and oxygen ventilation. CA/CPR leads to microglial activation and therefore an increase in pro-inflammatory cytokines such as TNF and IL-1β is observed. (+)-Naltrexone intraperitoneal injection was administered at either 3 mg/kg or 6 mg/kg doses to mice twice a day for two days 30 min after CA. (+)-Naltrexone was used in place of its stereoisomer (–)-naltrexone as it blocks TLR4 signaling and does not bind opioid receptors. Both doses of (+)-naltrexone were shown to significantly protect against ischemic cell death, while the 6 mg/kg dose showed greater neuron protection (Table 2). (+)-Naltrexone was also observed to significantly attenuate production of inflammatory cytokines by microglia and lymphocyte cell infiltration in the mice which is common during BBB disruption. Conclusively, (+)-naltrexone was suggested to be beneficial for reducing neuronal death and neurotoxicity by blocking TLR4 activation (Figure 3B) (83).
Acute and long-term effects of continuous naloxone and naltrexone administration were shown to improve motor function after an ischemic stroke in a feline model of cerebral ischemia generated using MCAO (87). Naloxone or naltrexone intraperitoneal injection, both administered at an initial dose of 10 mg/kg and then transferred to a lower continuous dose for 24 h, significantly improved motor function and prolonged survival of cats with MCAO compared to controls receiving the saline (control) injection. Moreover, a significant improvement in motor function was observed with naloxone and naltrexone administration, and cats regained normal walking abilities. These results suggest that naloxone and naltrexone opiate antagonists may have neurorestorative neurologic effects and may be useful in treating ischemic neurologic deficits (87) (Table 2).
Conclusively, the studies above highlight the prospective neuroprotective and neurorestorative properties of opioid receptor antagonists, naloxone and naltrexone (Table 2). As previously stated however, the mechanisms by which these antagonists elicit their effects are not fully understood. Additionally, naloxone and naltrexone have been suggested to inhibit NADPH (dihydronicotinamide adenine dinucleotide phosphate) oxidase (NOX2), an enzyme complex responsible for oxidative stress (Figure 3C) (88, 89). Above, we described the increase in oxidative stress as a result of microglial activation in the pathogenesis of ischemic stroke and comprised BBB integrity. In order to inhibit the increase in oxidative stress and stroke progression, blockage of NADPH oxidase (or NOX2) has been suggested (90). This enzyme complex consists of membrane bound gp91phox subunit and p22phox, as well as three cytosolic proteins (p40phox, p47phox, and p67phox). Upon cell activation, these cytosolic components are translocated to plasma membrane to interact with the membrane bound gp91phox subunit and p22phox to assemble an active NADPH oxidase enzyme complex resulting in superoxide O2− generation (Figure 3C). Naloxone and naltrexone may function by inhibiting enzymatic activity of NADPH oxidase by binding to the gp91phox subunit and inducing a conformational change of the NADPH protein complex, affecting the binding affinity of the cytosolic subunits, p40phox, p47phox, and p67phox. Consequently, pro-inflammatory cytokine production, ROS, and NO that compromise BBB integrity are reduced as suggested by in vitro studies (24, 89). Naloxone inhibition of superoxide production is suggested to be independent of opioid receptors as superoxide production induced by LPS (lipopolysaccharide) was significantly and dose-dependently inhibited by (–) and (+)- naloxone isomers (78). Direct targeting of NOX2 and suppression of superoxide generation by naloxone was studied using blood neutrophils due to their abundance of NOX2. Neutrophils were treated with PMA (phorbol myristate acetate), a commonly used agent for superoxide production to stimulate NOX2. Naloxone was found to inhibit NADPH-dependent superoxide generation by PMA-stimulated neutrophil membranes, indicating a direct inhibitory effect of naloxone on NOX2 (Figure 3C) (78).
3.3. Nalmefene as a potential therapeutic for ischemic stroke
Nalmefene is an opioid receptor antagonist that has also been studied for improved stroke recovery and its neuroprotective effects (Figure 2, Table 2). As the k receptor has been shown to be dysfunctional following a CNS injury, studies have employed nalmefene hydrochloride for acute ischemic stroke treatment due to its k opioid receptor antagonist properties (91). To date, the effects of nalmefene, commercially sold as Cervene, is not fully understood in human ischemic stroke patients. In a pilot study, the efficacy of Cervene was compared to placebo in a randomized double-blind clinical trial. Specifically, 34 ischemic stroke patients received 0.05 mg/kg of Cervene intravenously for 15 min and then were transferred to a dosage of 0.01 mg/kg for 24 h. A control group of 10 ischemic stroke patients that received placebo was maintained as well. Cervene efficacy was assessed by comparing the patient’s National Institutes of Health Stroke Scale Score (NIHSS) at baseline to scores 7 days after treatment. Glasgow Coma Scale (GCS), which is a measure of recovery from brain injuries, were obtained 3 month after as a secondary efficacy measure (92). Results indicated that while statistically significant efficacy of Cervene cannot be deduced from this small scale study, this opioid antagonist is safe and tolerable, and may be a beneficial stroke treatment for neurological recovery and improved functional recovery (91) (Table 2). Another study observed the neuroprotective effects of Nalmefene in patients with cerebral infarctions as large cerebral infarctions often lead to hypoxia, ischemia, and necrosis (93). Specifically, 236 patients with middle cerebral artery trunk infarction were randomly divided into two groups: a control group receiving conventional treatment and an experimental group receiving 0.2 mg of intravenous Nalmefene hydrochloride injections twice per day for 10 days (93). Patient treated with nalmefene had significantly low NIHSS scores in comparison to control group patients with large cerebral infarction. Similarly, there was a statistically significant difference between GCS scores of patients in the nalmefene treatment group in comparison to those in the control group (93). However, the long-term therapeutic efficacy of Nalmefene was not studied and cannot be concluded from this study. Indeed, as only few clinical studies with Nalmefene have been conducted, the therapeutic efficacy and ability to restore neurologic function remain largely unknown.
4. FUTURE DIRECTIONS
Current stroke treatment is restricted to only one FDA-approved drug, tPA. Efficient tPA use is limited to 3% of patients and has no apparent neuroprotective or neurological recovery effects. There is a need for novel drugs and drug delivery to promote stroke recovery through protection of the BBB. While further human and animal studies need to be conducted to evaluate therapeutic efficacy and more clearly understand mechanism, the use of opioid antagonists as a potential therapeutic agent for ischemic stroke suggests a novel repurposing of FDA-approved opioid antagonists that should be further explored.
Future work should study the mechanism by which these opioid antagonists are inducing neuroprotective and neurorestoration effects. By better understanding these drugs mechanistically, drugs with similar mechanism of actions may also be explored for their protective effects. Simultaneously, to date, there are there are no in vitro studies observing the effects of naloxone, naltrexone, and nalmefene on an in vitro model of ischemic stroke. Therefore, in addition to further in vivo studies evaluating the mechanism and further evaluating the therapeutic efficacy of these opioid antagonists as agents for stroke, future studies should also include in vitro studies as additional studies that may shed light on the mechanism by which these drugs are inducing their neuroprotective or neurorestorative effects. Similarly, while naloxone, naltrexone, and nalmefene are the only FDA-approved centrally activated opioid antagonists, it may also be beneficial to explore peripherally activated opioid antagonists for any neuroprotective effects. Nevertheless, long-term studies should be conducted to not only ensure the efficacy of these drugs in their neuroprotective properties, but to also ensure that these drugs have no negative side-effects, including toxicity, with long term use. Various treatments regimes and dosage should also be evaluated to determine the most effective treatment plans.
As opioid antagonists should be further studied for their potential as stroke therapeutics, it is also important to draw attention to the need for enhancing the deliveries of these opioid antagonists through use of novel drug delivery strategies and state of the art drug designs. Many promising neuroprotective agents have failed in clinical trials due to safety or efficacy (94). Drugs are most commonly administered via oral delivery or as an injection. As a result, the drug may have off target effects by affecting healthy cells and organs as well (95). Simultaneously, drug efficacy is lost as the majority of the drug may be metabolized by other organs such as the liver, with a small dose reaching the organ of interest (96). Consequently, a higher dose of the drug is needed to make up for the low bioavailability of injections or oral delivery. One major hurdle for targeting a drug to the brain is the highly restrictive BBB, especially for non-invasive transport of drug to the brain. Oral delivery of drugs poses many issues including low bioavailability, slow absorption, hepatic first-pass metabolism, and GI side effects (97). Many of the current drug delivery strategies utilized in the above studies to enhance drug permeability through the BBB are invasive including intraventricular or intracerebral infusion of the drug. These techniques are high risk and can have many dangerous complications for the patient (98).
One way to overcome the need for very invasive drug delivery, such as intracerebral drug infusion is to enhance the design of stroke therapeutics, i.e. the prospective opioid antagonists, for more effective passage across the BBB. Nanotechnology is an innovative form of drug development that can be used to enhance the delivery of opioid antagonists for stroke therapeutics through optimization of various characteristics of the drug molecule shape and size to achieve a nanoparticle formulation of the opioid antagonists that is lipid soluble, has a low molecular weight, and is small in size, in turn enhancing the delivery of the drug across the BBB (99).
Nanoparticles are solid colloidal particles that can be controlled to be very small in size to freely cross the BBB while not disturbing BBB integrity (94, 95). The goal of developing a drug into nanoparticles is to ensure release of drug at a specific rate, dose, and site (100). Nanotechnology based drug delivery offers localized, controlled, and sustained drug delivery, in turn increasing the therapeutic efficiency of the drug, reducing dosage and frequency of doses, as well as reducing off target effects to other organs and cells (95). Due to the reduced particle size and decreased diffusion distance, nanoparticles offer faster and more effective drug absorption. The small particle size provides increased contact area, allowing for increased drug adhesiveness to the cell surface, in turn, increasing drug bioavailability (101). Nanoparticles preserve the innate therapeutic and non-toxic properties of original drugs while increasing bioavailability in comparison to traditional drug delivery forms. Therefore, dosage and frequency of dosage is decreased (101). Simultaneously, the therapeutic effects of the original drug are preserved.
Composition of nanoparticle surface has been studied to be critical when targeting the brain. Nanoparticles fabricated with nonionic surfactants have been shown to exhibit increased uptake by the brain and more successful passage through the BBB. Other strategies such as use of viral vectors and exosomes have also been studied for brain drug delivery, however may not be effective strategies for ischemic stroke brain drug delivery (99). Viral vectors, for example, are beneficial for transfecting genes to patients that cannot normally cross the BBB. However, they have many limitation including patient safety and production costs as well as invasive administration routes such as injection into the cerebrospinal fluid (99). Exosomes are another drug delivery technique that involves the use of cell vesicles as a carrier for brain drugs delivery (99). Exosomes have often been studied for brain gene delivery, transporting proteins and nucleic acids across the BBB. As exosomes are non-immunogenic, they allow for enhanced circulation of the drug or protein of interest. However, exosomes also have many limitations including selection of the exosome carrier cell and vesicle loading (99). Further toxicity studies also need to be conducted with exosomes.
Nanotechnology has the potential to enhance potential stroke recovery therapeutics, such as opioid antagonists, and their respective passage of the treatment across the BBB to achieve a more direct delivery to the brain. As a result, a significantly invasive delivery (i.e. cerebral infusion) will not be necessary for successful drug administration (102, 103). Concurrently, nanoparticles offer many advantages to traditional drug delivery systems, including increased drug solubility, bioavailability, and therapeutic efficacy, nanoparticles may be a plausible future development in drug delivery methods of opioid antagonists, with the goal to ultimately improve patient outcomes.
Future human and animal studies should generate new knowledge to further understand the therapeutic efficacy and cellular and molecular mechanisms underlying the effectiveness of opioid antagonists for their potential in attenuating stroke severity, promoting recovery, and protecting the BBB against opioid-associated cerebrovascular complications. Progress in drug delivery methods to enhance these prospective stroke recovery treatments is suggested, with the ultimate goal of improving the lives of patients and their recovery from ischemic stroke.
Highlights.
Prescription opioids exacerbate risk and severity of ischemic stroke.
This perspective addresses a novel application and repurposing of FDA-approved opioid antagonists to minimize BBB damage, reduce stroke severity, and promote neural recovery.
Future directions discuss potential drug design and delivery methods to enhance these novel therapeutic targets.
Acknowledgments
This work was supported by the National Institutes of Health (R01GM114321, R01GM127706, R01MH104656, R01MH110415, HL126559, DA039576, DA040537, and DA044579) and the National Science Foundation (CHE-1506740, CBET-1841419). SD thanks the Miller School of Medicine of the University of Miami for the Lucille P. Markey Chair in Biochemistry and Molecular Biology.
Footnotes
Competing interest declaration
The authors’ state that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE LIST
- 1.Services UDoHaH. What is the U.S. Opioid Epidemic? 2018. Available from: https://www.hhs.gov/opioids/about-the-epidemic/index.html.
- 2.Update CH. Rising Numbers of Deaths Involving Fentanyl and Fentanyl Analogs, Including Carfentanil, and Increased Usage and Mixing with Non-opioids. CDC Health Alert Network; 2018. [Google Scholar]
- 3.Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445–54. Epub 2015/07/15. doi: 10.2147/DDDT.S56071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Small DL, Morley P, Buchan AM. Current and experimental treatment of stroke Neuropsychopharmacology: The Fifth Generation of Progress 2002. [Google Scholar]
- 5.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C, Stroke Statistics S. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135(10):e146–e603. Epub 2017/01/27. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee CW, Muo CH, Liang JA, Sung FC, Kao CH. Association of intensive morphine treatment and increased stroke incidence in prostate cancer patients: a population-based nested case-control study. Jpn J Clin Oncol. 2013;43(8):776–81. Epub 2013/06/26. doi: 10.1093/jjco/hyt080. [DOI] [PubMed] [Google Scholar]
- 7.LoCasale R, Kern DM, Chevalier P, Zhou S, Chavoshi S, Sostek M. Description of cardiovascular event rates in patients initiating chronic opioid therapy for noncancer pain in observational cohort studies in the US, UK, and Germany. Adv Ther. 2014;31(7):708–23. Epub 2014/07/19. doi: 10.1007/s12325-014-0131-y. [DOI] [PubMed] [Google Scholar]
- 8.Sifat AE, Vaidya B, Abbruscato TJ. Blood-Brain Barrier Protection as a Therapeutic Strategy for Acute Ischemic Stroke. AAPS J. 2017;19(4):957–72. Epub 2017/05/10. doi: 10.1208/s12248-017-0091-7. [DOI] [PubMed] [Google Scholar]
- 9.Yang Q, Tong X, Schieb L, Vaughan A, Gillespie C, Wiltz JL, King SC, Odom E, Merritt R, Hong Y, George MG. Vital Signs: Recent Trends in Stroke Death Rates - United States, 2000–2015. MMWR Morb Mortal Wkly Rep. 2017;66(35):933–9. Epub 2017/09/08. doi: 10.15585/mmwr.mm6635e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stroke Rodgers H.. Handb Clin Neurol. 2013;110:427–33. Epub 2013/01/15. doi: 10.1016/B978-0-444-52901-5.00036-8. [DOI] [PubMed] [Google Scholar]
- 11.Ferdinand P, Roffe C. Hypoxia after stroke: a review of experimental and clinical evidence. Exp Transl Stroke Med. 2016;8:9 Epub 2016/12/17. doi: 10.1186/s13231-016-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96. Epub 2013/12/07. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. 2012;79(13 Suppl 1):S52–7. Epub 2012/10/04. doi: 10.1212/WNL.0b013e3182697e70. [DOI] [PubMed] [Google Scholar]
- 14.Bertrand L, Meroth F, Tournebize M, Leda AR, Sun E, Toborek M. Targeting the HIV- infected brain to improve ischemic stroke outcome. Nat Commun. 2019;10(1):2009 Epub 2019/05/03. doi: 10.1038/s41467-019-10046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertrand L, Dygert L, Toborek M. Antiretroviral Treatment with Efavirenz Disrupts the Blood-Brain Barrier Integrity and Increases Stroke Severity. Sci Rep. 2016;6:39738 Epub 2016/12/23. doi: 10.1038/srep39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Disdier C, Chalansonnet M, Gagnaire F, Gate L, Cosnier F, Devoy J, Saba W, Lund AK, Brun E, Mabondzo A. Brain Inflammation, Blood Brain Barrier dysfunction and Neuronal Synaptophysin Decrease after Inhalation Exposure to Titanium Dioxide Nano-aerosol in Aging Rats. Sci Rep. 2017;7(1):12196 Epub 2017/09/25. doi: 10.1038/s41598-017-12404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvey PM, Hendey B, Monahan AJ. The blood-brain barrier in neurodegenerative disease: a rhetorical perspective. J Neurochem. 2009;111(2):291–314. Epub 2009/08/08. doi: 10.1111/j.1471-4159.2009.06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shirley R, Ord EN, Work LM. Oxidative Stress and the Use of Antioxidants in Stroke. Antioxidants (Basel). 2014;3(3):472–501. Epub 2014/01/01. doi: 10.3390/antiox3030472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12(5):698–714. [DOI] [PubMed] [Google Scholar]
- 20.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–70. Epub 2009/11/26. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 21.Rao R Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci. 2008;13:7210–26. Epub 2008/05/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, Blasig IE, Dijkstra CD, Ronken E, de Vries HE. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21(13):3666–76. Epub 2007/06/26. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 23.Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9(1):23 Epub 2012/11/13. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehner C, Gehwolf R, Tempfer H, Krizbai I, Hennig B, Bauer HC, Bauer H. Oxidative stress and blood-brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid Redox Signal. 2011;15(5):1305–23. Epub 2011/02/08. doi: 10.1089/ars.2011.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummins PM. Occludin: one protein, many forms. Mol Cell Biol. 2012;32(2):242–50. Epub 2011/11/16. doi: 10.1128/MCB.06029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balbuena P, Li W, Ehrich M. Assessments of tight junction proteins occludin, claudin 5 and scaffold proteins ZO1 and ZO2 in endothelial cells of the rat blood-brain barrier: cellular responses to neurotoxicants malathion and lead acetate. Neurotoxicology. 2011;32(1):58–67. Epub 2010/10/26. doi: 10.1016/j.neuro.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol. 2008;6(3):179–92. Epub 2009/06/10. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hone EA, Hu H, Sprowls SA, Farooqi I, Grasmick K, Lockman PR, Simpkins JW, Ren X. Biphasic Blood-Brain Barrier Openings after Stroke. Neurological Disorders and Stroke International. 2018;1(2). [Google Scholar]
- 29.Giraud M, Cho TH, Nighoghossian N, Maucort-Boulch D, Deiana G, Ostergaard L, Baron JC, Fiehler J, Pedraza S, Derex L, Berthezene Y. Early Blood Brain Barrier Changes in Acute Ischemic Stroke: A Sequential MRI Study. J Neuroimaging. 2015;25(6):959–63. Epub 2015/02/24. doi: 10.1111/jon.12225. [DOI] [PubMed] [Google Scholar]
- 30.Hjort N, Wu O, Ashkanian M, Solling C, Mouridsen K, Christensen S, Gyldensted C, Andersen G, Ostergaard L. MRI detection of early blood-brain barrier disruption: parenchymal enhancement predicts focal hemorrhagic transformation after thrombolysis. Stroke. 2008;39(3):1025–8. Epub 2008/02/09. doi: 10.1161/STROKEAHA.107.497719. [DOI] [PubMed] [Google Scholar]
- 31.Kuntz M, Mysiorek C, Petrault O, Petrault M, Uzbekov R, Bordet R, Fenart L, Cecchelli R, Berezowski V. Stroke-induced brain parenchymal injury drives blood-brain barrier early leakage kinetics: a combined in vivo/in vitro study. J Cereb Blood Flow Metab. 2014;34(1):95–107. Epub 2013/10/03. doi: 10.1038/jcbfm.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Li T, Zhao Y, Qian Y, Li X, Dai X, Huang D, Pan T, Zhou L. Platelet glycoprotein receptor Ib blockade ameliorates experimental cerebral ischemia-reperfusion injury by strengthening the blood-brain barrier function and anti-thrombo-inflammatory property. Brain Behav Immun. 2018;69:255–63. Epub 2017/12/03. doi: 10.1016/j.bbi.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 33.Lulit Price CW, and Gerald Grant. Chapter 4 Blood-Brain Barrier Pathophysiology following Traumatic Brain Injury In: Laskowitz DGG, editor. Translational Research in Traumatic Brain Injury; 2016. [Google Scholar]
- 34.Patel JP, Frey BN. Disruption in the Blood-Brain Barrier: The Missing Link between Brain and Body Inflammation in Bipolar Disorder? Neural Plast. 2015;2015:708306. Epub 2015/06/16. doi: 10.1155/2015/708306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wunder A, Schoknecht K, Stanimirovic DB, Prager O, Chassidim Y. Imaging blood-brain barrier dysfunction in animal disease models. Epilepsia. 2012;53 Suppl 6:14–21. Epub 2013/01/03. doi: 10.1111/j.1528-1167.2012.03698.x. [DOI] [PubMed] [Google Scholar]
- 36.Chiang T, Messing RO, Chou WH. Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011(48). Epub 2011/03/05. doi: 10.3791/2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saunders NR, Dziegielewska KM, Mollgard K, Habgood MD. Markers for blood-brain barrier integrity: how appropriate is Evans blue in the twenty-first century and what are the alternatives? Front Neurosci. 2015;9:385 Epub 2015/11/19. doi: 10.3389/fnins.2015.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell K Portenoy M, Zankhana Mehta, Ebtesam Ahmed. Cancer pain management: General principles and risk management for patients receiving opioids 2018. [Google Scholar]
- 39.Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, Ripamonti CI, Committee EG. Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2018;29(Supplement_4):iv166–iv91. Epub 2018/07/28. doi: 10.1093/annonc/mdy152. [DOI] [PubMed] [Google Scholar]
- 40.Nersesyan H, Slavin KV. Current aproach to cancer pain management: Availability and implications of different treatment options. Ther Clin Risk Manag. 2007;3(3):381–400. Epub 2008/05/20. [PMC free article] [PubMed] [Google Scholar]
- 41.Wiffen PJ, Wee B, Moore RA. Oral morphine for cancer pain. Cochrane Database Syst Rev. 2016;4:CD003868. Epub 2016/04/23. doi: 10.1002/14651858.CD003868.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simmons CP, Macleod N, Laird BJ. Clinical management of pain in advanced lung cancer. Clin Med Insights Oncol. 2012;6:331–46. Epub 2012/11/02. doi: 10.4137/CMO.S8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Y, He X, Yang Y, Chao D, Lazarus LH, Xia Y. Current research on opioid receptor function. Curr Drug Targets. 2012;13(2):230–46. Epub 2011/12/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Wang H, Shah K, Karamyan VT, Abbruscato TJ. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011;1383:307–16. Epub 2011/02/02. doi: 10.1016/j.brainres.2011.01.083. [DOI] [PubMed] [Google Scholar]
- 45.Chaves C, Remiao F, Cisternino S, Decleves X. Opioids and the Blood-Brain Barrier: A Dynamic Interaction with Consequences on Drug Disposition in Brain. Curr Neuropharmacol. 2017;15(8):1156–73. Epub 2017/05/06. doi: 10.2174/1570159X15666170504095823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leo S, Nuydens R, Meert TF. Opioid-induced proliferation of vascular endothelial cells. J Pain Res. 2009;2:59–66. Epub 2009/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan MM, Christie MJ. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br J Pharmacol. 2011;164(4):1322–34. Epub 2011/03/26. doi: 10.1111/j.1476-5381.2011.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woller SA, Moreno GL, Hart N, Wellman PJ, Grau JW, Hook MA. Analgesia or addiction?: implications for morphine use after spinal cord injury. J Neurotrauma. 2012;29(8):1650–62. Epub 2012/01/05. doi: 10.1089/neu.2011.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fields HL. The doctor’s dilemma: opiate analgesics and chronic pain. Neuron. 2011; 69(4):591–4. Epub 2011/02/23. doi: 10.1016/j.neuron.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenblum A, Marsch LA, Joseph H, Portenoy RK. Opioids and the treatment of chronic pain: controversies, current status, and future directions. Exp Clin Psychopharmacol. 2008;16(5):405–16. Epub 2008/10/08. doi: 10.1037/a0013628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamzei Moqaddam A, Ahmadi Musavi SM, Khademizadeh K. Relationship of opium dependency and stroke. Addict Health. 2009;1(1):6–10. Epub 2009/07/01. [PMC free article] [PubMed] [Google Scholar]
- 52.Hamzei-Moghaddam A, Shafa MA, Khanjani N, Farahat R. Frequency of Opium Addiction in Patients with Ischemic Stroke and Comparing their Cerebrovascular Doppler Ultrasound Changes to Non-Addicts. Addict Health. 2013;5(3–4):95–101. Epub 2014/02/05. [PMC free article] [PubMed] [Google Scholar]
- 53.Yu YP, Tan L. The Vulnerability of Vessels Involved in the Role of Embolism and Hypoperfusion in the Mechanisms of Ischemic Cerebrovascular Diseases. Biomed Res Int. 2016;2016:8531958. Epub 2016/06/18. doi: 10.1155/2016/8531958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baldini A, Von Korff M, Lin EH. A Review of Potential Adverse Effects of Long-Term Opioid Therapy: A Practitioner’s Guide. Prim Care Companion CNS Disord. 2012;14(3). Epub 2012/10/30. doi: 10.4088/PCC.11m01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baba M, Oishi R, Saeki K. Enhancement of blood-brain barrier permeability to sodium fluorescein by stimulation of mu opioid receptors in mice. Naunyn Schmiedebergs Arch Pharmacol. 1988;337(4):423–8. Epub 1988/04/01. [DOI] [PubMed] [Google Scholar]
- 56.Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, Chawda R, Shanahan TC, Schwartz SA. Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J Clin Immunol. 2008;28(5):528–41. Epub 2008/06/25. doi: 10.1007/s10875-008-9208-1. [DOI] [PubMed] [Google Scholar]
- 57.Ebrahimi H, Haghjoo Javanmard S, Asgary S, Dehghani L, Amiri M, Saadatnia M. Opium Addiction and Ischemic Stroke in Isfahan, Iran: A Case-Control Study. Eur Neurol. 2018;79(1–2):82–5. Epub 2017/12/25. doi: 10.1159/000485098. [DOI] [PubMed] [Google Scholar]
- 58.Iranmanesh F Prognostic value of electrocardiography and electroencephalography in patients with ischemic stroke. Acta Neurol Taiwan. 2008;17(4):228–32. Epub 2009/03/14. [PubMed] [Google Scholar]
- 59.Qureshi WT, O’Neal WT, Khodneva Y, Judd S, Safford MM, Muntner P, Soliman EZ. Association Between Opioid Use and Atrial Fibrillation: The Reasons for Geographic and Racial Differences in Stroke (REGARDS) Study. JAMA Intern Med. 2015;175(6):1058–60. Epub 2015/04/29. doi: 10.1001/jamainternmed.2015.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allouche S, Noble F, Marie N. Opioid receptor desensitization: mechanisms and its link to tolerance. Front Pharmacol. 2014;5:280 Epub 2015/01/08. doi: 10.3389/fphar.2014.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolf PA AR, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;8:983–8; [DOI] [PubMed] [Google Scholar]
- 62.Rivat C, Ballantyne J. The dark side of opioids in pain management: basic science explains clinical observation. Pain Rep. 2016;1(2):e570 Epub 2016/09/08. doi: 10.1097/PR9.0000000000000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bansal S, Sangha KS, Khatri P. Drug treatment of acute ischemic stroke. Am J Cardiovasc Drugs. 2013;13(1):57–69. Epub 2013/02/06. doi: 10.1007/s40256-013-0007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu S, Feng X, Jin R, Li G. Tissue plasminogen activator-based nanothrombolysis for ischemic stroke. Expert Opin Drug Deliv. 2018;15(2):173–84. Epub 2017/09/26. doi: 10.1080/17425247.2018.1384464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gladstone DJ, Black SE. Update on intravenous tissue plasminogen activator for acute stroke: from clinical trials to clinical practice. CMAJ. 2001;165(3):311–7. Epub 2001/08/24. [PMC free article] [PubMed] [Google Scholar]
- 66.Dela Pena IC, Yang S, Shen G, Fang Liang H, Solak S, Borlongan CV. Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke. Int J Mol Sci. 2018;19(6). Epub 2018/06/03. doi: 10.3390/ijms19061635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32 Epub 2013/04/09. doi: 10.3389/fneur.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan Z, Lucke-Wold BP, Logsdon AF, Turner RC, Tan C, Li X, Hongpaison J, Alkon DL, Simpkins JW, Rosen CL, Huber JD. Bryostatin extends tPA time window to 6 h following middle cerebral artery occlusion in aged female rats. Eur J Pharmacol. 2015;764:404–12. Epub 2015/07/21. doi: 10.1016/j.ejphar.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lutsep HL, Clark WM. Current status of neuroprotective agents in the treatment of acute ischemic stroke. Curr Neurol Neurosci Rep. 2001; 1(1):13–8. Epub 2002/03/20. [DOI] [PubMed] [Google Scholar]
- 70.Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. 2018;315(3):C343–C56. Epub 2018/06/28. doi: 10.1152/ajpcell.00095.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gravanis I, Tsirka SE. Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opin Ther Targets. 2008;12(2):159–70. Epub 2008/01/23. doi: 10.1517/14728222.12.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Services UDoHaH. Narcan (naloxone nasal spray) Approved to Reverse Opioid Overdose 2018. Available from: https://www.fda.gov/Drugs/DrugSafety/ucm472958.htm.
- 73.Lewis CR, Vo HT, Fishman M. Intranasal naloxone and related strategies for opioid overdose intervention by nonmedical personnel: a review. Subst Abuse Rehabil. 2017;8:79–95. Epub 2017/10/27. doi: 10.2147/SAR.S101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryan SA, Dunne RB. Pharmacokinetic properties of intranasal and injectable formulations of naloxone for community use: a systematic review. Pain Manag. 2018;8(3):231–45. Epub 2018/04/24. doi: 10.2217/pmt-2017-0060. [DOI] [PubMed] [Google Scholar]
- 75.Helm S, Trescot AM, Colson J, Sehgal N, Silverman S. Opioid antagonists, partial agonists, and agonists/antagonists: the role of office-based detoxification. Pain Physician. 2008;11 (2):225–35. Epub 2008/03/21. [PubMed] [Google Scholar]
- 76.Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9(1):63–88. Epub 2018/01/11. doi: 10.1177/2042098617744161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reference PD. naloxone hydrochloride - Drug Summary. Available from: http://www.pdr.net/drug-summary/Narcan-naloxone-hydrochloride-3837.
- 78.Baskin DS, Hosobuchi Y. Naloxone reversal of ischaemic neurological deficits in man. Lancet. 1981;2(8241):272–5. Epub 1981/08/08. [DOI] [PubMed] [Google Scholar]
- 79.Jabaily J, Davis JN. Naloxone administration to patients with acute stroke. Stroke. 1984;15(1):36–9. Epub 1984/01/01. [DOI] [PubMed] [Google Scholar]
- 80.Hosobuchi Y, Baskin DS, Woo SK. Reversal of induced ischemic neurologic deficit in gerbils by the opiate antagonist naloxone. Science. 1982;215(4528):69–71. Epub 1982/01/01. [DOI] [PubMed] [Google Scholar]
- 81.Liao SL, Chen WY, Raung SL, Chen CJ. Neuroprotection of naloxone against ischemic injury in rats: role of mu receptor antagonism. Neurosci Lett. 2003;345(3):169–72. Epub 2003/07/05. [DOI] [PubMed] [Google Scholar]
- 82.Chen CJ, Liao SL, Chen WY, Hong JS, Kuo JS. Cerebral ischemia/reperfusion injury in rat brain: effects of naloxone. Neuroreport. 2001;12(6):1245–9. Epub 2001/05/08. [DOI] [PubMed] [Google Scholar]
- 83.Grace PM, Shimizu K, Strand KA, Rice KC, Deng G, Watkins LR, Herson PS. (+)- Naltrexone is neuroprotective and promotes alternative activation in the mouse hippocampus after cardiac arrest/cardiopulmonary resuscitation. Brain Behav Immun. 2015;48:115–22. Epub 2015/03/17. doi: 10.1016/j.bbi.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anttila JE, Albert K, Wires ES, Matlik K, Loram LC, Watkins LR, Rice KC, Wang Y, Harvey BK, Airavaara M. Post-stroke Intranasal (+)-Naloxone Delivery Reduces Microglial Activation and Improves Behavioral Recovery from Ischemic Injury. eNeuro. 2018;5(2). Epub 2018/05/17. doi: 10.1523/ENEURO.0395-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goonoo N, Bhaw-Luximon A, Ujoodha R, Jhugroo A, Hulse GK, Jhurry D. Naltrexone: a review of existing sustained drug delivery systems and emerging nano-based systems. J Control Release. 2014;183:154–66. Epub 2014/04/08. doi: 10.1016/j.jconrel.2014.03.046. [DOI] [PubMed] [Google Scholar]
- 86.Liu JC, Ma JD, Morello CM, Atayee RS, Best BM. Naltrexone metabolism and concomitant drug concentrations in chronic pain patients. J Anal Toxicol. 2014;38(4):212–7. Epub 2014/03/25. doi: 10.1093/jat/bku019. [DOI] [PubMed] [Google Scholar]
- 87.Baskin DS, Kuroda H, Hosobuchi Y, Lee NM. Treatment of stroke with opiate antagonists--effects of exogenous antagonists and dynorphin 1–13. Neuropeptides. 1985;5(4–6):307–10. Epub 1985/02/01. [DOI] [PubMed] [Google Scholar]
- 88.Younger J, Parkitny L, McLain D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin Rheumatol. 2014;33(4):451–9. Epub 2014/02/15. doi: 10.1007/s10067-014-2517-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, Hong JS. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012;9:32 Epub 2012/02/22. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qin L, Block ML, Liu Y, Bienstock RJ, Pei Z, Zhang W, Wu X, Wilson B, Burka T, Hong JS. Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J. 2005;19(6):550–7. Epub 2005/03/26. doi: 10.1096/fj.04-2857com. [DOI] [PubMed] [Google Scholar]
- 91.Clark WM, Coull BM, Karukin M, Hendin B, Kelley R, Rosing H, Zachariah S, Winograd M, Raps E, Walshe T, Singer S, Mettinger KL. Randomized trial of Cervene, a kappa receptor-selective opioid antagonist, in acute ischemic stroke. J Stroke Cerebrovasc Dis. 1996;6(1):35–40. Epub 1996/09/01. [DOI] [PubMed] [Google Scholar]
- 92.McMillan T, Wilson L, Ponsford J, Levin H, Teasdale G, Bond M. The Glasgow Outcome Scale - 40 years of application and refinement. Nat Rev Neurol. 2016;12(8):477–85. Epub 2016/07/16. doi: 10.1038/nrneurol.2016.89. [DOI] [PubMed] [Google Scholar]
- 93.Li X HW, Song L. Nalmefene improves prognosis in patients with a large cerebral infarction: study protocol and preliminary results of a randomized, controlled, prospective trial. Clin Trials Degener Dis. 2017;2(4):101–7. [Google Scholar]
- 94.Panagiotou S, Saha S. Therapeutic benefits of nanoparticles in stroke. Front Neurosci. 2015;9:182 Epub 2015/06/05. doi: 10.3389/fnins.2015.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singh S, Pandey VK, Tewari RP, Agarwal V. Nanoparticle Based Drug Delivery System: Advantages and Applications. Indian Journal of Science & Technology 2011; 4(3). [Google Scholar]
- 96.Gavhane YN, Yadav AV. Loss of orally administered drugs in GI tract. Saudi Pharm J. 2012;20(4):331–44. Epub 2013/08/21. doi: 10.1016/j.jsps.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Salatin S, Barar J, Barzegar-Jalali M, Adibkia K, Milani MA, Jelvehgari M. Hydrogel nanoparticles and nanocomposites for nasal drug/vaccine delivery. Arch Pharm Res. 2016;39(9):1181–92. Epub 2016/06/29. doi: 10.1007/s12272-016-0782-0. [DOI] [PubMed] [Google Scholar]
- 98.Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood-brain barrier by nanoparticles. J Control Release. 2012;161(2):264–73. Epub 2011/08/30. doi: 10.1016/j.jconrel.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 99.Dong X Current Strategies for Brain Drug Delivery. Theranostics. 2018;8(6):1481–93. Epub 2018/03/21. doi: 10.7150/thno.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Singh R, Lillard JW Jr Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–23. Epub 2009/02/03. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Junyaprasert VB, Morakul B. Nanocrystals for enhancement of oralbioavailability of poorly water-soluble drugs. Asian Journal of Pharmaceutical Sciences 2014:13–23. [Google Scholar]
- 102.Saraiva C, Praca C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34–47. Epub 2016/05/22. doi: 10.1016/j.jconrel.2016.05.044. [DOI] [PubMed] [Google Scholar]
- 103.Chen L, Gao X. The application of nanoparticles for neuroprotection in acute ischemic stroke. Ther Deliv. 2017;8(10):915–28. Epub 2017/09/26. doi: 10.4155/tde-2017-0023. [DOI] [PubMed] [Google Scholar]