

Abstract
Rational design of multivalent molecules represents a remarkable modem tool to transform weak non-covalent interactions into strong binding by creating multiple finely-tuned points of contact between multivalent ligands and their supposed multivalent targets. Here, we describe several prominent examples where the multivalent blockers were investigated for their ability to directly obstruct oligomeric channel-forming bacterial exotoxins, such as the pore-forming bacterial toxins and B component of the binary bacterial toxins. We address problems related to the blocker/target symmetry match and nature of the functional groups, as well as chemistry and length of the linkers connecting the functional groups to their multivalent scaffolds. Using the anthrax toxin and AB5 toxin case studies, we briefly review how the oligomeric toxin components can be successfully disabled by the multivalent non-channel-blocking inhibitors, which are based on a variety of multivalent scaffolds.
1. Introduction
1.1. Multivalency in Nature
A Burdock—clawed my Gown—
Not Burdock’s—blame—
But mine—
Who went too near
The Burdock’s Den
—Emily Dickinson
Many nature-lovers are familiar with the pesky burr, the plant seed-sacks that attach themselves to clothing or to animal fur in order to travel to wider fertile planting areas. The famous “Queen Recluse,” American legendary poet Emily Dickinson gave a dark poetic description of the burdock sticking to her gown. In a more practical way, Swiss electrical engineer George De Mestral examined the cockleburs, which clung to his clothes and his dog’s fur after his summer walks in the Alps, through a microscope. De Mestral discovered that the burrs were covered by multiple small hooks which were readily able to attach to the diminutive loops on the clothing and fur of those passing near the “Burdock’s Den.” In 1955 after more than eight years of experimenting with different types of material, from natural to synthetic, De Mestral invented “the zipperless zipper,” the product known today as Velcro®, the word that originates from French “vel ours” (velvet) and “cro chet” (hook). The product was made up of two strips of fabric, one covered by thousands of small hooks and the other with thousands of small loops designed to give an ideal and firm match. Increasing the material surface led to an increase in the mutual effect. Originally receiving skepticism from the fashion designers, today Velcro® production has become a multimillion dollar industry. Another fascinating manufacturing example is related to the U.S. military, developing special climbing gloves that would enable soldiers to climb vertical surfaces. The invention is inspired by the unique feet of the gecko species that are covered by an array of tiny long tubular filaments, the so-called setae with branched tips, each containing hundreds of miniature fibers. This exceptional surface architecture allows the lizards to cling equally effectively on various hydrophobic and hydrophilic surfaces due to dry adhesion of gecko setae by van der Waals forces (Autumn et al. 2002). With a single van der Waals interaction being lower than 2 kJ/mol, the numerous points of contact provide sizable amount of energy to secure tight but reversible binding. These types of multivalent interactions are ubiquitous in nature, where strong non-covalent binding is achieved by operation of multiple recognition events between multivalent ligands and receptors. Additional examples include adhesion of viruses to their target cell receptors, antibody-antigen interaction, intercellular recognition, and many others. Through the billions of years of evolution, nature has optimized these interactions ensuring that an array of weak molecular recognition events is acting in a powerful balanced concert (Mahon and Fulton 2014). Therefore, design and investigation of the synthetic multivalent compounds mimicking nature’s ability for multiple recognition events have tremendous prospects for the rational drug design goals (Fasting et al. 2012).
1.2. Multivalent Ligands to Combat Multivalent Targets
Multivalency is one of the best modern tools to transform relatively weak, non-covalent monovalent interactions into a strong binding by creating multiple points of contact between multivalent ligands and their receptor interfaces (Fasting et al. 2012). Traditionally, multivalent interaction is defined as specific simultaneous binding of multiple functional groups attached onto an inert platform, named scaffold, to multiple target receptor sites (Choi 2004). Numerous natural or designed multivalent molecules are built either on linear scaffolds or on preorganized, rigid, and conformationally defined scaffolds that allow for the suitable positioning and density of the functional group ligands. The proper display of the functional groups is frequently intended to match arrangement and distances between receptors on the complementary surface. The design and synthesis of the multivalent drug molecules involves covalent attachment of multiple copies of the previously identified functional groups onto a scaffold using molecular fragments, called linkers, or spacers. It is expected that the linker would serve as at least an “innocent observer” (Fasting et al. 2012) securing appropriate spacing between the functional groups. The linkers could also be designed to favorably contribute to the ligand/receptor interaction (Choi 2004). In addition, parameters such as linker length, conformational rigidity or flexibility, and chemical nature are frequently considered as important factors in the multivalent compound design (Mammen et al. 1998b; Shewmake et al. 2008; Krishnamurthy et al. 2007; Kane 2010). This chapter is written to discuss some of the recent advances in designing multivalent inhibitors of the channel-forming bacterial toxins. We also highlight several prominent examples of using the multivalent molecules in other aspects of ion channel research and provide examples of the multivalent inhibitors that counteract the channel-forming bacterial toxins without direct pore blockage. The multivalent inhibitors targeting non-pore-forming bacterial toxins will be described briefly in Sect. 3; for additional examples, we address the reader to the recent reviews published on the subject (Branson and Turnbull 2013; Weisman et al. 2015).
2. Multivalent Inhibitors of Channel-Forming Bacterial Exotoxins
Bacterial exotoxins are polypeptides or proteins secreted by pathogenic bacteria during invasion to destroy or damage host cells. Many of them oligomerize on the cell surface constituting the ideal symmetrical multivalent receptor targets for rational design of multivalent antitoxin ligands (Joshi et al. 2008; Ivarsson et al. 2012). Nowadays when biological origins of many diseases are investigated on a molecular level, with methods such as X-ray crystallography, cryo-EM, and all-atom molecular dynamic simulations, providing deep insight into structure/function relationships of the biomolecules, rational drug discovery strategies concurrently evolve. Below we discuss the recent efforts to rationally design multivalent antitoxins specifically focusing on ones tuned to interact with the multivalent targets made of oligomeric pore-forming bacterial toxins. In a way, this approach represents the development of an original idea related to attachment of an influenza virus to a target cell that suggests that multivalent attachment can be blocked effectively by an inhibitor that is itself multivalent (Mammen et al. 1998a).
2.1. Pore-Forming Bacterial Toxins as Targets for Multivalent Blocker Development
In 2001, Joseph E. Alouf had estimated that 35 % (115 out of 325) of bacterial exotoxins, identified by that time, attack mammalian cells damaging the cytoplasmic phospholipid bilayer membranes (Alouf 2001). Most of these exotoxins do so by forming transmembrane pores which allow the uncontrollable flow of ions down their electrochemical gradients leading to reduction in the membrane potential and eventually to the collapse of the plasma membrane barrier function (Bernheimer 1996). To reflect the mechanism of action of these toxins, they are often referred to as membrane-perforating or pore-forming bacterial toxins (PFTs). When added into model bilayer membrane bathing solutions, many PFTs spontaneously form large (nS conductance), stable (last for hours) oligomeric pores ideally suitable for molecular-sensing approaches that can be expanded to rational design of the effective multivalent pore blockers. One of the most striking examples is heptameric β-barrel α-hemolysin (αHL) of Staphylococcus aureus. The biosensing properties of this toxin, its stability, and structural robustness have determined its use for the stochastic resistive-pulse sensing of a variety of substrates, ranging from small molecules to polymers and biomacromolecules. Interestingly, even though these applications are not directly related to the toxic properties of αHL, they provide a deep insight into single molecule biophysics of the particle/channel binding reactions proved to be helpful for rational channel blocker design approaches.
Being secreted as a water-soluble 293-amino acid monomeric polypeptide, αHL forms heptameric pores in target cell membranes (reviewed in Berube and Bubeck Wardenburg 2013). In 1981, Oleg Krasilnikov and coauthors reconstituted αHL proteins into bilayer lipid membranes for the first time and recorded formation of large, stable, and slightly anion-selective channels of ~ 1 nS conductance in 1 M KCl solution (Krasilnikov et al. 1981). The channels remained in an open low-noise state for hours. The 1.9-Å resolution crystal structure of the heptamer revealed a hallow mushroom-like 100 Å × 100 Å (length by diameter) channel made of stem, cap, and rim domains with two narrow 0.9-nm and 0.6- to 0.7-nm constriction zones (Song et al. 1996). In the last twenty years after gaining momentum from the pioneering articles by Kasianowicz and Bezrukov (Bezrukov and Kasianowicz 1993; Kasianowicz and Bezrukov 1995), who suggested to employ αHL as a nanoscopic cuvette for reaction dynamic studies, this PFT was explored as a biological nanosensor to detect a wide variety of analytes. Among the multiple virulence factors secreted by S. aureus, in the context of this review, it is worth mentioning bicomponent octameric γ-hemolysin (Hlg), leukocidin (Luk), and Panton-Valentine leukocidin (PVL). These toxins are formed as a result of the interaction between two distinct polypeptides named F and S components (Kaneko and Kamio 2004; Alonzo and Torres 2014).
2.2. Binary Bacterial Toxins as Targets for Multivalent Blocker Development
In contrast to the PFTs damaging host cells by acting on their cell membranes, AB-type exotoxins employ a different mode of intoxication enzymatically modifying specific intracellular targets (Geny and Popoff 2006). These toxins are frequently secreted as single-chain polypeptides containing at least two functionally distinct domains, the binding B domain that docks to specific surface receptors and the active A domain that modifies certain substrates in the cytosol. Alternatively, the AB-type toxins, the so-called binary toxins, are made of two (or three) individual non-linked proteins with one acting as a binding/translocation B factor and another as an enzymatic A factor acting intracellularly (Barth et al. 2004). The B components of the binary toxins, after binding to their specific cell receptors, self-assemble to form ring-shaped oligomeric prepores able to bind several A components. These complexes are subsequently endocytosed and the oligomers are converted into the membrane-spanning ion-conductive pores reported to mediate the A component transport from the acidic endosomal environment into the cytosol. When incorporated into model lipid bilayers, B components of the binary toxins form robust and structurally stable β-barrel channels, apt for single-channel single molecule investigation (Blaustein et al. 1989; Schmid et al. 1994; Knapp et al. 2002). Therefore, for the purposes of this review, we will focus on the multivalent pore blockers designed to inhibit the 2nd group of the AB-type toxins.
The existing efforts to develop pore-forming bacterial toxin inhibitors mainly concentrate on targeting the anthrax toxin (Fig. 1a) and a family of related clostridial binary toxins, C2, iota, and CDT (Nestorovich et al. 2011; Roeder et al. 2014). Following intentional dissemination of Bacillus anthracis spores via the “anthrax letters” in Sept. 2001, quite remarkable developments were made in understanding molecular details of anthrax toxin uptake (Moayeri et al. 2015; Liu et al. 2015). Briefly, the tripartite anthrax toxin is released as three separate proteins, lethal factor (LF), edema factor (EF), and protective antigen (PA) that self-assemble at the host cell surface to form complexes contributing to the symptoms of anthrax. LF is a Zn-metalloprotease that cleaves MAP kinase kinases (Duesbery et al. 1998; Vitale et al. 2000) and Nlrp1 (Levinsohn et al. 2012). EF is a Ca2+ and calmodulin-activated adenylyl cyclase (Leppla 1982, 1984). PA, named this way for its ability to produce the protective antibodies in the anthrax vaccines, is a receptor-binding and translocation component that is important for the intracellular delivery of LF and EF. The anthrax toxin uptake occurs in several stages. After binding to the cellular CMG2 and TEM8 receptors and being proteolytically cleaved, PA oligomerizes to make heptameric (Petosa et al. 1997) and/or octameric (Kintzer et al. 2009, 2010) ring-shaped prepores. The oligomeric (PA63)7 prepore formation creates three (Mogridge et al. 2002) or four (Kintzer et al. 2009) LF- and EF-binding sites. The toxic AB complexes then undergo the receptor-mediated endocytosis (Pilpa et al. 2011). Subsequently, the acidic environment of the endosome causes substantial conformational changes of the PA oligomers leading to their insertion into endosomal membranes and formation of cation-selective channels (Blaustein et al. 1989). 2.9-Å cryo-EM imaging of the channel showed an elongated “flower-on-a-stem” heptamer with an external diameter ranging from ~27 to ~ 160 Å with 75-Å long bud and 105-Å long stem (Jiang et al. 2015) with channel radius varying from ~ 16 Å to as low as ~3.5 Å. The broadly accepted model of anthrax toxin translocation asserts that the PA oligomer acts as an effective translocase, capable of unfolding and translocating LF and EF into the cytosol using the proton gradient across endosomal membranes (pHendosome < pHcytosol) (Zhang et al. 2004a, 2004b; Krantz et al. 2006). The translocation was shown to be facilitated by a ring of phenylalanyl residues at position 427 of (PA63)7 (ϕ-clamp) (Krantz et al. 2005) and a substrate binding α-clamp (Brown et al. 2015; Feld et al. 2010). Kasianowicz and colleagues developed an alternative model showing that the anthrax toxin is able to catalyze the rupture of endosomal membranes allowing for the toxic complexes to be released into the cytosol (Nablo et al. 2013). Interestingly, PA63 was shown to deliver LF not only into the cytosol but also inside endosomal intraluminal vesicles that later fuse and release LF into the cytosol (Abrami et al. 2013).
Fig. 1.

Multivalent channel-blocking inhibitors of PA63 of anthrax toxin. a Illustration of the idea: a multivalent blocker as effective inhibitor of PA63 of tripartite anthrax toxin. b The most prominent examples of the multivalent blockers are based on cyclodextrin (Karginov et al. 2005) (top) and dendrimer (Forstner et al. 2014) (bottom) scaffolds
A group of clostridial binary channel-forming toxins (Knapp et al. 2015a): C2 (Ohishi and Odagiri 1984; Aktories et al. 1986; Simpson 1984), iota (Simpson et al. 1987; Stiles and Wilkins 1986), and CDT (Popoff et al. 1988) are related both structurally and functionally. They are made of two components where the A subunit acts through mono-ADP-ribosylation of G-actins (Aktories and Wegner 1989; Barth et al. 2015) and the B subunit binds and mediates delivery of the A subunit into the cytosol (Barth and Stiles 2008). PA and clostridial binary toxin B subunits have high degrees (from 27 to 38 %) of amino acid homology and are made of four distinct domains, each involved in the host receptor interaction, oligomerization, channel formation, and binding of the A subunits (Petosa et al. 1997; Schleberger et al. 2006). Similar to PA63, the proteolytically activated B subunits form ring-shaped heptamers, called prepores, on the host cell surface or in solution (Barth et al. 2000). The cell-bound AB complexes, C2I/C2IIa, Ia/Ib, and CDTa/CDTb are then internalized by receptor-mediated endocytosis (Blocker et al. 2001; Stiles et al. 2002; Nagahama et al. 2009; Pust et al. 2010) and enzymatic components translocate across the endosomal membranes into the cytosol. This process may involve use of the pores formed by binding/translocation components of the toxins as translocation corridors (Barth et al. 2000; Stiles et al. 2002; Bachmeyer et al. 2001; Blocker et al. 2003; Gibert et al. 2007). In mildly acidic conditions (pH < 6.6) in vitro, the B subunits of C2 and iota toxins form cation-selective ion channels (Schmid et al. 1994; Knapp et al. 2002). The phenylalanine clamp preserved in position 428 (C2IIa) and 454 (Ib) was reported to catalyze unfolding of C2I and Ia in the course of their transport across endosomal membrane (Knapp et al. 2015b; Lang et al. 2008; Neumeyer et al. 2008). Remarkably, PA63 is able to bind and translocate His-tagged A component of the C2 toxin (C2I). These similarities suggest that the universal approaches, for instance, those targeting the channel-forming components of the toxins, should be considered in rational design of broad-spectrum binary toxin inhibitors.
2.3. Cyclodextrin-Based Channel Blockers
When designing multivalent inhibitors of the oligomeric bacterial toxins which are frequently arranged into the centrosymmetric structures, one may consider settling ligands into a rigid cyclic scaffold (Fig. 1b) with a controlled number of preassembled attachment sites to achieve their proper positioning (Choi 2004). Cyclodextrins (CDs), cyclic oligomers of glucose which are typically composed of six (αCD), seven (βCD), and eight (γCD) subunits represent a well-studied example of the rigid scaffolds. Non-modified CDs are amphipathic molecules shaped like truncated cones. The CD’s exterior is hydrophilic due to presence of the outward-facing 2- and 3-OH groups on the wider rim and 6-OH group on the narrow rim. The hydroxyl groups can be easily derivatized by covalent attachment of different ligands (Choi 2004; Wenz 1994), which provides unlimited opportunities for their selective rational modifications (Khan et al. 1998). The hydroxyl groups at positions 2 and 3 form hydrogen bonds that are required to keep the molecule rigid, as a result, the hydroxyl groups at position 6 are frequently considered as a favorable site to introduce substitutions (Khan et al. 1998). Szejtli estimated that by 2004 over 15,000 αCD, βCD, and γCD derivatives had been reported and their properties were investigated in a number of diverse applications (Szejtli 2004). The hydrophobic interior of CD’s cavity defines its remarkable ability to form water-soluble “host-guest” inclusion complexes with otherwise poorly soluble small molecules and macromolecule fragments. This unique property has determined a long history of CD use in pharmaceutical, agrochemical, environmental, cosmetic, and food industries. In addition, CDs typically do not elicit immune responses and have relatively low toxicity in animals and humans (Davis and Brewster 2004). We specifically want to refer our readers to an excellent review on history of CDs that covers many different aspects of the 120 years of development of these “novel” compounds (Crini 2014).
The original idea (Karginov et al. 2005) to rationally modify CDs into effective antitoxins was based on the wealth of earlier research on stochastic sensing of organic analytes by an αHL channel containing a non-covalently bound βCD as a molecular adapter (Gu et al. 1999). The approach was designed to transform a water-filled lumen of αHL into a sensor element capable of detecting a number of small organic molecules that normally would not bind to this channel. To do so, Bayley and colleagues equipped an αHL incorporated into bilayer lipid membranes with CDs that were able to enter inside the pore lumen causing partial (~60 %) reversible blockages of the ion current. Because of the partial mode of the current blockage, the authors used the system to detect and quantify a variety of guest molecules, such as adamantanamine hydrochloride, adamantine carboxylic acid, promethazine, and imipramine, which presumably interact with the hydrophobic cavity of the βCD. The symmetry match between heptameric αHL pore and sevenfold symmetrical βCD secured the comfortable fit between the channel and adapter molecules. At the same time, sixfold symmetrical αCDs and eightfold symmetrical γCDs were also shown to reversibly interact with this heptameric channel (Gu et al. 1999).
The rationally modified cyclodextrin molecules were also investigated as potent channel-blocking antitoxins against αHL (Karginov et al. 2007; Ragle et al. 2010) and binary bacterial anthrax (Karginov et al. 2005), C2, iota (Nestorovich et al. 2011), and CDT toxins (Roeder et al. 2014). As channel-forming components of the binary bacterial toxins are known to be preferentially selective to cations (Blaustein et al. 1989), nearly any tested positively charged ligands were shown to reversibly block K+ current through PA63 (Krantz et al. 2005), C2IIa (Bachmeyer et al. 2003; Beitzinger et al. 2013; Bronnhuber et al. 2014), and Ib (Knapp et al. 2002) channels in a wide nM-mM range of effective concentrations. The significant increase in efficiency of the cationic compounds was achieved when multiple copies of positively charged ligands were covalently linked to a cyclodextrin scaffold (Karginov et al. 2005, 2006b). Thus, synthetic tailor-made sevenfold symmetrical βCDs (d ≈ 15 Å) carrying seven amino groups, each attached to the 6th position of the ring via a variety of hydrophobic linkers (Fig. 1b, top), effectively blocked PA63 current (Nestorovich et al. 2010) (Table 1). To optimize the 7+βCD compounds, the authors focused on a number of structural characteristics of these molecules, such as length and chemical nature of the linkers connecting functional groups to the CD scaffold, nature of functional groups, and ligand/blocker symmetry complementarity.
Table 1.
Multivalent positively charged cyclodextrin-based blockers of PA63 channel ion current and LT cytotoxicity
| No. | Cyclodextrin | Substituent | Inhibition of PA63 channels in model lipid membranes, IC50 (nM) | Inhibition of LT cytotoxicity, IC50 (μM) |
|---|---|---|---|---|
| Hepta-6-aminoalkyl β-cyclodextrin derivatives (Karginov et al. 2006a) | ||||
| 1 | β | -NH2 | 140 ± 90 | 20 ± 9 |
| 2 | β | -S(CH2)2NH2 | 3.5 ± 0.9 | 7.8 ± 2.4 |
| 3 | β | -S(CH2)3NH2 | 0.57 ± 0.39 | 2.9 ± 1.0 |
| 4 | β | -S(CH2LNH2 | 1.1 ± 0.5 | 5.1 ± 2.4 |
| 5 | β | -S(CH2)5NH2 | 3.8 ± 1.0 | 7.5 ± 2.4 |
| 7 | β | -S(CH2)6NH2 | 0.97 ± 0.38 | 0.6 ± 0.3 |
| 8 | β | -S(CH2)7NH2 | 4.6 ± 3.2 | 1.9 ± 1.1 |
| 9 | β | -S(CH2)8NH2 | 2.4 ± 0.95 | 0.3 ± 0.1 |
| 10 | β | -S(CH2)!0NH2 | 27.0 ± 17.0 | 2.6 ± 0.7 |
| Hepta-6-guanidinealkyl β-cyclodextrin derivatives (Karginov et al. 2006a) | ||||
| 11 | β | 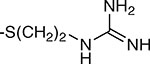 |
5.3 ± 3.2 | 8.9 ± 6.0 |
| 12 | β | 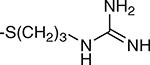 |
12.6 ± 9.0 | 12.2 ± 2.9 |
| Hepta-6-arylamine β-cyclodextrin derivative (Karginov et al. 2006a; Yannakopoulou et al. 2011) | ||||
| 13 | β | 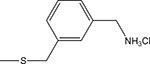 |
0.13 ± 0.10 | 0.8 ± 0.5 |
| Cationic α- and γ-cyclodextrin derivatives (Yannakopoulou et al. 2011) | ||||
| 14 | α | -NH2 | 1200 ± 300 | >100 |
| 15 | γ | -nh2 | 170 ± 50 | 12 ± 3 |
| 16 | α | 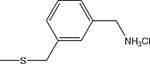 |
29 ± 5 | 45 ± 13 |
| 17 | γ | 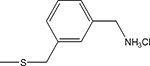 |
2.8 ± 1.3 | 5.4 ± 0.8 |
Length and chemical nature of linkers
Multivalent compounds are frequently designed by connecting multiple copies of a functional group to a scaffold with a linker (Choi 2004). These linkers are not only used as the covalent connectors for the ligands but also secure proper spacing to allow for the optimal fit between the multivalent target and the multivalent molecule. To determine an optimal length of the spacers, which tether the amino groups to the βCD, a group of hepta-6-thioaminoalkyl derivatives was investigated. By combining planar lipid bilayer measurements and cell assay studies, the authors reported that the alkyl linkers (3–8 CH2-linkers) were almost equally effective, with IC50 values from 0.6 to 4.6 nM in planar bilayers and from 0.3 to 7.5 μM in cell assays (Karginov et al. 2006a). Shorter spacers were less effective (Table 1), which is apparently related to the reduced size of these blockers and/or restricted mobility of the linkers. 7+βCDs with longer linkers (9–10 CH2-linkers) did show some evident channel blockage at low-nM concentrations but induced instability in the bilayer membranes and toxicity to the RAW cells. Positive linear correlation (R2 = 0.84) between the planar lipid bilayer and cell assay measurements supports the concept that 7+βCDs inhibit anthrax toxins by blocking the PA63 channel. The most significant increase in 7+βCD pore blocking activity was achieved when a single phenyl group was introduced into each thio-hydrocarbon linker (Karginov et al. 2006a). Three hepta-6-arylamine βCD derivatives (AMBnTβCDs) with the methylamino groups located in 2nd, 3rd, and 4th position of the aromatic ring were reported to protect RAW cells with IC50 values being as low as 0.5 ± 0.2 μM. When tested using the planar lipid bilayer technique, AMBnTβCD blocked PA63 channel conductance with Kd = 0.13 ± 0.1 nM. AMBnTβCD was found to completely protect Fisher F344 rats from LT. Moreover, in combination with ciprofloxacin, the antibiotic used to treat Bacillus anthracis infection, AMBnTβCD significantly increased mice survival in an infection mode of anthrax (Moayeri et al. 2008). AMBnTβCD represents an interesting example of a multivalent compound with linkers favorably contributing to a contact formed between target and ligand. The exact mechanism of phenyl group contribution to the blocking activity of AMBnTβCD is not clear at the moment. On one hand, the small-molecule cationic compounds containing aromatic fragments were reported to block PA63 more effectively than their aliphatic molecular counterparts (Krantz et al. 2005). This effect was explained by the stabilizing aromatic-aromatic, π-π or cation-π interactions between aromatic groups on the blockers and phenylalanyl residues of the ϕ-clamp. On the other hand, inclusion of the aromatic fragments can change conformation and flexibility of the linkers optimizing the binding. To understand the physical forces responsible for the channel/blocker interaction, the binding reaction was studied in solution with different electrolyte concentrations and under a wide range of applied transmembrane voltages (−100 to +200 mV) (Bezrukov et al. 2012). It was demonstrated that in the 7+βCD/PA63 binding reaction, salt concentration-independent short-range forces prevail. At relatively low salt concentrations, βCD residence time inside the channel was influenced by long-range Coulomb interactions. The existence of an additional electrostatic component was indicated by an increase in the blocker residence time as a function of transmembrane voltage.
Nature of functional groups
To determine if the nature of tethered positively charged groups is critical for effective binding, the aminoalkyl substituents were compared with a group of hepta-6-guanidine β-CD derivatives, in which positive charges were distributed between two nitrogens on the guanidine group (Table 1, compounds 11 and 12) (Karginov et al. 2006a). No increase in the blocker activity was detected; instead these compounds showed slightly lower PA63 channel-blocking and cell-protective activity.
Symmetry of the cyclodextrin scaffolds
The original idea behind design of the effective PA63 channel blockers was to build the sevenfold symmetrical blocker molecules to complement the heptameric structure of the channel. However, later it was demonstrated that this requirement is not strict because both 6+αCD and 8+γCD were able to inhibit channel conductance (Yannakopoulou et al. 2011). 6+αCD/PA63 binding was noticeably weaker (IC50 =1.2 μM in planar bilayers and >100 μM in cell assays), whereas 8+γCD/PA63 binding was comparable with that of 7+βCD/PA63 both in planar bilayers and in cell assays (Table 1, compounds 14–17). At the same time, the sevenfold symmetry of the blockers turned out to be fundamentally important when the symmetry match idea was extended to the cyclodextrin antitoxins designed to block ion current through αHL (Karginov et al. 2007). Thus, a seven positively charged hepta-6-substituted βCD derivative, IB201, effectively inhibited ion current through αHL in planar bilayers and protected rabbit red blood cells from αHL-triggered hemolysis. In a murine model of S. aureus infection, IB201 was reported to prevent the αHL-mediated alveolar epithelial cell lysis and mortality (Ragle et al. 2010). The sixfold and eightfold symmetrical IB201 analogues were not effective (Table 2). The molecular details of the αHL inhibition by IB201 were revealed using single-channel planar lipid bilayer experiments. Thus, application of IB201 blocker to the side of the membrane that corresponds to the cap side of the heptamer resulted in αHL switching to a weakly conductive state with a residual conductance ranging from 1 to 15 % of the open-channel conductance. The αHL/IB201 binding was reported to be significantly influenced by the transmembrane voltage, negative from the side opposite to the blocker addition. Moreover, within the time period of the measurements (several hours), IB201 binding to αHL was irreversible resembling the voltage-gating type of the channel closure. Interestingly, the symmetry complementarity was not required for the CD passive diffusion across the monomeric 14-stranded β-barrel bacterial porin, CymA, of Klebsiella oxytoca (Pajatsch et al. 1998, 1999). Recently, Winterhalter and colleagues suggested an interesting model to describe the passive diffusion of CDs through CymA (van den Berg et al. 2015).
Table 2.
α, β, and γCD blockers of αHL channel ion current and toxicity (Yannakopoulou et al. 2011)
| No. | Cyclodextrin | Substituent | Inhibition of a single αHL channel in model lipid membranes, IC50 (nM) | Inhibition of αHL toxicity, IC50 (μM) |
|---|---|---|---|---|
| 1 | α | >5000 | >100 | |
| 2 | β | *50 | 3.3 ± 2.3 | |
| 3 | γ | >5000 | >100 |
2.4. Dendrimer-Based Channel Blockers
Dendrimers are rigid highly divaricate starburst-shaped polymers, composed of regularly repeated branches originating from a central core. This architecture offers multiple unique opportunities for development of multivalent ligands (recently reviewed in Wu et al. 2015). Dendrimer synthesis processes are well established. The controlled production of monodispersed dendrimers, where each consecutive growing step results in a new “generation” with an increased diameter and doubled number of surface groups, determines dendrimer surface valence (Tomalia and Frechet 2002; Wijagkanalan et al. 2011). Dendrimer properties, such as their nanoscale dimensions, rigidity, stability, monodispersity, highly regulated multi-valency, and ability to form host-guest complexes, have resulted in numerous emergent medical and industrial applications, including their use as antibacterial, antiviral, and antiparasitic agents (Helms and Meijer 2006). The void interior of dendrimer molecules has been used to incase drugs for targeted delivery or biomarkers for imagining purposes. The outer shell is densely packed with a well-controlled number of terminals, which can be modified with a wide variety of functional groups to vary their biological activity and bioavailability. The starburst configuration makes dendrimers particularly useful for analyzing modes of multivalent compound interaction within confined objects, such as nanopores (Martin et al. 2007; Ficici et al. 2015; Forstner et al. 2014; Bustamante et al. 2000). Dendrimer-related studies on ion channels are limited. In 2007, Howorka’s group used sulfhydryl-reactive polyamido amine (PAMAM) dendrimers of generations 2, 3, and 5 with a mixed surface of terminal hydroxyl/amine groups to couple with αHL channels containing engineered cysteine residues (Martin et al. 2007). This type of modification was expected to change the stochastic-sensing properties of αHL under placement of charged and dense dendrimer polymers into their lumen. The authors reported that PAMAM dendrimers acted as both an ion-selectivity filter and a molecular sieve for the passage of small molecules and biopolymers. The extent of this modification was dependent on the PAMAM generation; the bigger G5 dendrimers did not enter the cis entrance, while G3 was able to couple inside the pore. Fluorescently labeled starburst amino-terminated dendrimers were used for rapid nuclear pore patch-clamp sizing, as an inert class of particles that can enter pores without irreversibly affecting their gating (Bustamante et al. 2000). Polypropylenimine dotriacontaamine G3 and G4 dendrimers were tested for their ability to block E. coli E69 pore-forming Wza K30 capsular polysaccharide transporter (Kong et al. 2013). However, no detectable interaction of the dendrimers with the Wza pore was detected. At the same time, rationally designed tetrameric G1 poly(propylene imine) dendrimer decorated with four arylpiperazine moieties (Cappelli et al. 2011) was shown to be effective in activating the pentameric serotonin 5-HT3 receptor (Paolino et al. 2014). 5-HT3 receptors are ligand-gated ion channels that open upon binding of serotonin, allowing for the influx of Na+ and K+ and creating an excitatory response in neurons. The group designed two homotetravalent compounds with shorter, TETRA-S and longer, TETRA-L linkers. Interestingly, the correct geometry of the bioactive moiety was only achieved with the longer TETRA-L. TETRA-L was shown to be more potent than the corresponding monovalent ligand (threefold difference) and the bivalent ligand (twofold difference). In another study, transport of dye-labeled amino-terminated PAMAM dendrimers through silica colloidal nanopores, as a function of the nanopore and dendrimer size, was investigated as a potential application in size-selective separations (Ignacio-de Leon and Zharov 2011).
Recently, commercially available amino-terminated PAMAM dendrimers of different generations and imperfect dendrimers, known as dendrons, were tested for their ability to block channel-forming components of binary anthrax and C2 toxins (Table 3, Fig. 1b, bottom) (Forstner et al. 2014). According to the manufacturer (Dendritech®), PAMAM dendrimers are synthesized based on an ethylene diamine core and amidoamine repeat branching structure and come in generations (G0-G10) with increasing molecular diameter (d = 15–135 Å) and doubled number of reactive surface sites with every subsequent generation (ranging from z = +4 to +4096). All but the smallest G0 dendrimers were reported to inhibit PA63 channels at low-nM and C2IIa channels at high-nM concentrations in the model bilayer membranes. Interestingly, the IC50 values (0.16 – 230 nM) for these commercially available dendrimers compare well with the ones for the very first rationally designed β-CD blocker AmPrβCD (0.55 nM), which was selected out of dozens of related compounds. At that, G1, G2, and G3 dendrimers were found to have higher per-functional group activity in inhibiting PA63 and C2IIa in comparison with G0 and G4, G8, and G10 dendrimers. Moreover, dendrimers of generation 2 and higher affected the morphology of the tested cells by their own. G0 and G1 did not interfere with cell morphology and viability but protected HeLa cells from intoxication with C2 toxin by inhibiting His-C2I delivery into the host cell cytosol.
Table 3.
Multivalent PAMAM dendrimer- and dendron-based blockers of PA63 channel ion current (Forstner et al. 2014)
| No. | Generation | Measured diameter (Å) | Number of surface NH2 groups | Inhibition of PA63 channels in model lipid membranes, IC50 (nM) |
|---|---|---|---|---|
| PAMAM-NH2 dendrimers | ||||
| 1 | 0 | 15 | 4 | 128 ± 44 |
| 2 | 1 | 22 | 8 | 5.3 ± 2.6 |
| 3 | 2 | 29 | 16 | 7.15 ± 4.7 |
| 4 | 3 | 36 | 32 | 5.0 ± 1.4 |
| 5 | 4 | 45 | 64 | 2.4 ± 1.3 |
| 6 | 8 | 97 | 1024 | 0.22 ± 0.08 |
| 7 | 10 | 135 | 4096 | 0.16 ± 0.07 |
| PAMAM-OH dendrimers | ||||
| 8 | 2 | n/a | 0 | 142 ± 36 |
| 9 | 3 | n/a | 0 | 45 ± 14 |
| PAMAM-SA dendrimers | ||||
| 10 | 2 | n/a | 0 | > 150000 |
| PAMAM-COONa dendrimers | ||||
| 11 | 0.5 | n/a | 0 | > 400000 |
| 75%OH/25%NH2 PAMAM dendrimers | ||||
| 12 | 2 | n/a | 4 (ave) | 122 ± 35 |
| PAMAM-NH2 dendrons | ||||
| 13 | 0 dendron | n/a | 2 | 26 ± 7 |
| 14 | 1 dendron | n/a | 4 | 4.9 ± 0.7 |
| 15 | 2 dendron | n/a | 8 | 4.2 ± 0.9 |
Based on the molecular modeling of PA63 oligomeric channel (Lee et al. 2012) and its negative-stain electron microscopy image (Katayama et al. 2008), the authors expected 15–45 Å G0-G4 dendrimers to enter and block PA63 channels, similarly to that observed with the 7+βCD inhibitors. However, not only smaller G0-G4 but also G8 (d = 97 Å) and G10 (d = 135 Å) dendrimers were shown to effectively inhibit both PA63 and C2IIa channels. This finding is in agreement with a recently published article addressing PAMAM dendrimer behavior inside the confined space of αHL channel (Ficici et al. 2015). Using single-channel measurements and molecular dynamics simulations, the authors show that PAMAM permeation inside the αHL lumen is not determined by the apparent hydrodynamic size of the dendrimers but rather by their generation-dependent conformational flexibility. Easily compressible G1 and G2 dendrimers were able to completely enter inside the stem opening of αHL, G3 did not fully enter the barrel. Small fragments of G4 and G5 dendrimers, for example, several branches, were still able to enter inside the pore.
To address the problem of toxicity, several modified dendrimers were investigated (Table 3). In particular, PAMAM G2 and G3 dendrimers functionalized with neutral hydroxyl (compounds 8 and 9), but not with anionic succinamate and carboxyl surface groups (compounds 10 and 11), inhibited PA63 conductance in planar lipid bilayers. Thus, G2-OH and G3-OH were 20 and 9 times less effective than the amino-terminated dendrimers of the same generation. This finding was explained by the positively charged tertiary amino groups at the branching points of the PAMAM core structure interacting with the negatively charged PA63 lumen. The favorable therapeutic window can sometimes be achieved by partial surface modification to lower the density of the attached ligands, or by degradation of the dendrimers to the “fractured” or “imperfect” dendrons (compounds 13–15) (Tang et al. 1996). The mixed surface 75%OH/25%NH2 G2 PAMAM dendrimer (compound 12) was ~ 17 times less active compared to its 16-positively charged G2-NH2 analogue (compound 3). However, activity of the G1-NH2 dendron, functionalized with 4 positively charged amines (compound 14), was ~26 times higher (IC50 = 4.9 ± 0.7) compared with that of the G0-NH2 dendrimer (compound 1), which also carries four positive charges (IC50 = 128 ± 44). The authors explain this effect either by increase in mobility of the surface primary amino groups of the dendrons or by improved access of the tertiary amino groups to the binding sites in the PA63 lumen.
2.5. Blocking Channel-Forming Bacterial Toxins Without Blocking the Channel
While the antitoxins discussed above were mostly design to block the toxin channels directly, the channel-forming bacterial toxins could also be inhibited by multivalent molecules that do not permeate into the channels. Kane’s group has successfully designed a number of ligands attached to several multivalent scaffolds (Vance et al. 2009) for disabling the channel-forming component of anthrax toxin either by blocking its cellular receptors (Basha et al. 2006) or by blocking PA63 interaction with LF and EF (Mourez et al. 2001). We focus here on the 2nd type of inhibitors (Fig. 2), because they are designed to directly bind to the PA63 oligomers. Using the peptide phage-display library, Mourez et al. identified an inhibitory peptide (HTSTYWWLDGAP) that was able to weakly (IC50 = 150 μM) bind the (PA63)7 prepores and inhibit their interaction with the enzymatic components of anthrax toxin (Mourez et al. 2001). When multiple copies of this peptide were covalently attached to an N-acryloyloxysuccinimide (pNAS) polymer backbone, the resulting polymer inhibited LF and EF binding to PA63 with a 7500-fold improvement in efficacy, and protected rats from anthrax toxin (Fig. 2a). The group has also used a reversible addition fragmentation chain transfer (RAFT) technique to optimize the synthesis process to get more control over the spacing of functional groups and the ability to copolymerize pNAS with non-active acrylamide (Gujraty et al. 2008). It was also reported that although multivalency can greatly enhance ligand potency, overcrowding of the binding sites can have the opposite effect (Gujraty et al. 2006). Instead of a pNAS backbone, Joshi et al. used activated poly-L-glutamic acid to conjugate HTSTYWWLDGAP peptide copies, and reported IC50 values of 20 nM per-peptide basis, which were comparable to the polyacrylamide-based inhibitors (Joshi et al. 2006). More recently instead of using the peptide decorated polymeric molecules, the group synthesized polypeptide repeats to interpose multiple instances of modified inhibitory HTSTYWWLDGAP (LIG) peptides with flexible peptide linkers in the sequence of SE[LIG-(SE)m]n to assure optimal linker length, flexibility, and ligand density (Patke et al. 2014). Guided by molecular dynamics simulation data, the authors created and tested a range of monodisperse candidate inhibitors with (H)10-SE[LIG(SE)5]7 compound made of decahistidine tag that aids in the purification of the polypeptides, five sequential repeats of serine and glutamic acid, and seven HTSTYWWLDGAP repeats. The IC50 value of this blocker was 4 ± 0.5 nM per inhibitory peptide, exceeding the corresponding monovalent ligands by fourfold. When seven copies of HTSTYWWLDGAP peptide were attached to a β-cyclodextrin core with polyethylene glycol (PEG) linkers (Fig. 2b), the resulting compound effectively neutralized anthrax lethal toxin, being 100,000-fold more effective than the monovalent peptide (Joshi et al. 2011). The authors varied the linker length to determine that PEGn allowed for an optimal fit, matching the 30-Å distance from the center of the PA63 oligomer to the peptide-binding faces.
Fig. 2.

Multivalent non-channel-blocking inhibitors of PA63 of anthrax toxin. a Peptide-based inhibitors. b Cyclodextrin-based inhibitor of anthrax toxin complexes assembly. c Liposome-based inhibitor of anthrax toxin complexes assembly. Reprinted with permission from Joshi et al. (2011), Rai et al. (2006), and Gujraty et al. (2005). © 2005, 2011. American Chemical Society. © 2006. Nature Publishing Group
Significant increase in the ligand activity was achieved when ~ 50-Å liposomes (Fig. 2C) were decorated with multiple copies of HTSTYWWLDGAP peptides (IC50 = 20 nM on a per-peptide basis) (Rai et al. 2006). This strategy was refined with design of liposome containing raft-like membrane microdomains to create regions of optimal ligand density (Rai et al. 2007). A major advancement of this approach was recently suggested in a study where artificial liposomes, containing higher than in vivo concentrations of cholesterol and sphingomyelin, were tailored to effectively compete with host cell membranes for toxin binding (Henry et al. 2015). These “membrane mimicking” nanoparticles efficiently sequestered a significant number of toxins secreted by a variety of staphylococcal and streptococcal pathogens and treated fatal invasive diseases in mouse models.
Calix[n]arenes (C[n]s), cone-shaped phenol-formaldehyde macrocycles, provide another easily adaptable scaffold for the design of high-affinity multivalent ligands (Varejao et al. 2013; Baldini et al. 2007). In a pioneering study, para-sulfonated calixarenes were found to effectively block outward rectifying chloride channels, showing subnanomolar inhibition constants and exceptionally long blockage times (Singh et al. 1995). Modified calix[4]arenes were also reported to effectively interact with tetrameric voltage-dependent potassium channels by binding to their surface in a reversible manner (Martos et al. 2009). Several p-sulfonato-C[n]s were effective in inhibiting staphylococcal pore-forming cytolysins, Hlg, Luk, and PVL (Laventie et al. 2013). These heterooctameric toxins are made via interaction of the two distinct class F and class S polypeptides. Even though the original compound selection was based on the potential ability of these compounds to block the upper ring of the Luk pores, the actual mechanism of toxin inhibition is probably different. The authors suggested that p-sulfonato-C[n]s act by preventing the binding of class S proteins to membranes and eventual formation of Luk pores.
3. Representative Examples of Multivalent Inhibitors of Other Bacterial Exotoxins
Multisubunit organization is inherent to a number of clinically relevant non-pore-forming bacterial exotoxins. One of the most prominent examples is represented by AB5 toxins of the cholera toxin family, which include cholera toxin, E. coli heat-labile enterotoxin, Shiga-like toxins (SLTI and SLTII), and pertussis toxin (Beddoe et al. 2010). AB5 toxins are made of a single catalytic A subunit, which disrupts essential host functions intracellularly, and a homopentameric B subunit, responsible for binding to glycan receptors on the target cell. Blocking these multivalent B subunit/receptor interactions would inhibit the toxin’s intracellular uptake. Extensive structural information on these proteins and their complexes with different small ligands made the AB5 family toxins an attractive model system for design of multivalent ligands (Fig. 3). These inhibitors act by blocking the initial cell host receptor-binding step during the AB5 toxins uptake (Fan and Merritt. 2002). A representative of this group is Shiga-like toxin of E. coli that binds to a specific glycolipid, globotriaosylceramide, Gb3 to enter host cells. Ling et al., resolving a 2.8 Å crystal structure of Shiga-like toxin I complexed with an analogue of the Gb3 trisaccharide, has shown that three trisaccharide molecules bind to each monomer of the B-pentamer complex resulting in 15 trisaccharides interacting with a single pentamer of the toxin (Ling et al. 1998). While the individual affinity of the carbohydrate-protein interaction was low, it was outdone by the multivalent toxin binding; the feature that required employment of the multivalent inhibitors “tailored” to the structure of the B-subunit pentamer (Kitov et al. 2000). The authors designed a decavalent water-soluble carbohydrate ligand, named STARFISH, with 1- to 10 million-fold higher in vitro inhibitory activity against Shiga toxin B-pentamer compared with that of monovalent ligands. Interestingly, crystallographic study of the complex formed between Shiga toxin I B-pentamer and STARFISH showed a binding pattern which was different from the one projected by the rational design of the compound. It turned out that STARFISH molecules bind to two B-subunit monomers from separate toxin molecules, forming a molecular sandwich between two pentamers, instead of bridging two sites on a single molecule. The 2:1 toxin/ligand complexes were formed even though STARFISH concentration was sufficient to form a 1:1 complex. However, solution-phase measurements predicted that 1:1 complexes could also form (Mulvey et al. 2003). Nevertheless, the study represents an example of unusually high efficacy (nM) for glycoconjugates of such size and valency protecting susceptible cells against prolonged exposure to SLTI and more clinically significant SLTII. The same group has extended the study designing a more active 2nd-generation multivalent inhibitor that was capable of simultaneously interacting with several binding sites on the same B component subunit (Mulvey et al. 2003). In vivo studies have shown only modest activity of the lead STARFISH inhibitor in protecting mice against Shiga toxin. A modified version of STARFISH, called Daisy, designed to simultaneously occupy binding sites 1 and 2 in the STLI and STLII subunits, was capable of protecting mice from the lethal effects of both SLTI and SLTII. Bundle’s group created a class of optimized inhibitors where polymeric scaffolds were used to display covalently preorganized heterobifunctional ligand pairs, designed to mediate formation of specific complexes of an endogenous multivalent protein with a multivalent target (Fig. 3a) (Kitov et al. 2008). The polymeric inhibitors/adaptors containing preordered heterobifunctional ligands that recognize Shiga toxin Type 1 and human serum amyloid P components have shown a significant increase in in vivo activity to protect mice against Shigotoxemia.
Fig. 3.

Multivalent inhibitors of AB5 bacterial toxins. a A schematic representation of polymeric preordered heterobifunctional ligands. b Conceptual design of pentavalent inhibitors based on symmetrical core, variable number of linker units, and monovalent fingers blocking the toxin receptor binding site. Reprinted with permission from Kitov et al. (2008) and Merritt et al. (2002). © 2008 National Academy of Sciences, USA. © 2002 American Chemical Society
Unlike the Shiga toxins, cholera toxin and E. coli heat-labile enterotoxin’s B-pentamers contain only one set of five host cell receptor binding sites. This property simplifies rational design of the effective multivalent inhibitors against these toxins. However, large, ~45 Å separation between the non-adjacent binding sites creates some challenges (Fan and Merritt 2002). First, a series of pentavalent 1-β-amidated D-galactose ligands attached to an acylated pantacyclen 5 core with a number of flexible linkers of different lengths was suggested (Fan et al. 2000). The compounds with an ideal match to the size of the B-pentamer were >100,000-fold more effective (0.56 μM vs. 58 mM) compared to the monovalent ligands. This approach was further refined when pentavalent inhibitors based on more effective monovalent ligands were constructed (Fig. 3b) (Merritt et al. 2002). Thus, the inhibitor carrying five copies of m-nitrophenyl-α-galactoside (MNPG) instead of β-D-galactose was >100-fold more effective (0.9 μM vs. 15 μM). Using dynamic light scattering and X-ray crystallography, the authors provided proof for a 1:1 toxin/ligand association that occurs through single-site binding interaction of the pentavalent inhibitor with the surface of the toxin B-pentamer. The MNPG moiety was later investigated as a substructure to anchor hydrophobic ring systems able to bind in a variety of modes within the receptor binding site of both cholera toxin and E. coli enterotoxin B-pentamers (Pickens et al. 2002). The group has later extended the study replacing the morpholine ring core of the blockers by cyclic peptide cores. This approach, together with manipulation of the linker length, allows for the controlled tuning of the multivalent inhibitor dimensions to obtain an optimal match with the target (Zhang et al. 2004c).
Recent advancements in search of multivalent AB5 toxin inhibitors include ganglioside GM1 oligosaccharide ligands attached on corannulene (IC50 = 5 nM) (Mattarella et al. 2013), calixarene (IC50 = 450 pM) (Garcia-Hartjes et al. 2013), and the inactive cholera toxin B subunits (104 pM) (Branson et al. 2014). It was shown that the multivalent GM1-inhibitors with mismatched valencies can show significant increase in potency compared to the monovalent ligand, apparently due to formation of higher-order structures (Sisu et al. 2009; Fu et al. 2015). About 3-orders of magnitude affinity increase was observed by structure-based design of heterobifunctional ligands that were able to dimerize two different multivalent proteins. In a particular example of cholera toxin, the bifunctional ligands allowed for dimerization of the B-pentamer with human serum amyloid P component (SAP) (Liu et al. 2005). Similarly, markedly enhanced activity of the multivalent ligands was achieved with creation of a multivalent heterofunctional inhibitor-adapter, called “BAIT” capable of capturing a Shiga toxin subunit by one ligand of a heterobivalent headgroup and an endogenous SAP trap proteins by the other ligand (Solomon et al. 2005).
4. Concluding Remarks
Despite the massive vaccination campaigns and the 1928 Fleming’s discovery of penicillin that revolutionized modern medicine, bacterial infections remain a significant cause of morbidity and mortality worldwide. The well-known microbial resistance problem is entangled by the ability of many pathogens to produce a wide variety of virulent factors, including the endotoxins that act outside the bacterial cell being not susceptible to the antibiotic action. Therefore, search for the novel antidotes counteracting toxin poisoning is an important complementary approach to development of new antibacterial therapies. If implemented successfully, the antitoxins would allow for more time and energy for the infected organisms to respond to the bacterial infection and build protective immunity. In the last two decades, the concept of multivalent constructs decorated with ligands that have proven affinity to the target bacterial toxin have advanced and developed (Weisman et al. 2015). Many bacterial toxins form radially symmetrical oligomeric structures. This structural property could be used for rational design of multivalent ligands exploring the inhibitor-toxin symmetry match idea, where the ligands would simultaneously occupy complementary symmetrical binding sites of the target toxin. In this chapter, we discussed several examples when this principle was successfully used in rational development of the multivalent blockers acting in the limited confinements of pore-forming bacterial toxins including the channel-forming component of the binary bacterial toxins. It was estimated that the most effective multivalent channel-blocking antitoxins compare well with the blockers of classical ion-selective channels of neurophysiology (Table 3 in Nestorovich and Bezrukov 2012). However, in contrast to the blocker of the ion-selective channels of excitable cells, which are designed to be highly channel-specific, the toxin pore blockers offer advantages of being universally active in inhibiting a number of structurally related proteins. Thus, the sevenfold symmetrical cationic AMBnTβCD blocker was protective against anthrax (Karginov et al. 2005), C2, iota (Nestorovich et al. 2011), and CDT toxins (Roeder et al. 2014). Another extensively studied family of bacterial toxins is represented by the AB5-type bacterial toxins, for which a great number of multivalent inhibitors design tactics was suggested. As we briefly discuss above, several smart tuning strategies focused on the available structural data has allowed for the design of multivalent AB5 toxin inhibitors that are over 10 million-fold more effective in the in vitro interaction with the toxin complexes compared to the monovalent ligands of the same type.
There is little doubt that the strategies for the multivalent molecule applications, already manifold, will continue to emerge stronger (Fasting et al. 2012; Badjic et al. 2005). The main caveat here is related to challenges that the developers of these compounds will face transferring them from laboratories to clinics. Traditions given by the widely used classical Lipinski’s “rule of five” (Lipinski et al. 1997), designed to predict the drug-likeness for orally available small-molecule compounds, automatically excludes many multivalent ligands from the pool of potential therapeutic agents. We hope the dogmatic drug discovery practices will become more flexible with many successful exceptions reported from this otherwise very informative rule (Zhang and Wilkinson 2007; Walters 2012; Doak et al. 2014). In our days of “the dawning era” of polymer therapeutics (Duncan 2003), advances in synthetic polymer chemistry have already led to considerable progress in changing the pharmaceutical community perceptions of polymer-based drug candidates (Duncan 2011, 2014; Duncan and Vicent 2013). Thus, two polymer-based drugs Neulasta®, used to prevent neutropenia, and Copaxone®, used to treat multiple sclerosis, were listed in the US top 10 selling therapeutic agents of 2013 (U.S. Pharmaceutical Sales.). Many other macromolecule-based compounds are being reviewed for a number of emergent applications in the field of nanomedicine (Duncan and Gaspar 2011). To summarize, we believe the rational strategies currently being developed will pave the road toward discovery and approval of effective multivalent drugs tuned to act against different targets, including those formed by bacterial exotoxins.
Acknowledgements
Our laboratory research is supported by the startup funds from The Catholic University of America and by NIAID of the NIH under award number 1R15AI099897-01A1.
References
- Abrami L, Brandi L, Moayeri M, Brown MJ, Krantz BA, Leppla SH et al. (2013) Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep 13(27):986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktories K, Wegner A (1989) ADP-ribosylation of actin by clostridial toxins. J Cell Biol 109(4 Pt 1):1385–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktories K, Barmann M, Ohishi I, Tsuyama S, Jakobs KH, Habermann E (1986) Botulinum C2 toxin ADP-ribosylates actin. Nature 322(6077):390–392 [DOI] [PubMed] [Google Scholar]
- Alonzo F 3rd, Torres VJ (2014) The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev 78(2):199–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alouf JE (2001) Pore-forming bacterial toxins: an overview In: Van der Goot G (ed) Pore-forming toxins. Springer, Berlin, pp 1–14 [PubMed] [Google Scholar]
- Autumn K, Sitti M, Liang YA, Peattie AM, Hansen WR, Sponberg S et al. (2002) Evidence for van der Waals adhesion in gecko setae. Proc Natl Acad Sci USA 99(19):12252–12256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmeyer C, Benz R, Barth H, Aktories K, Gilbert M, Popoff MR (2001) Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J 15(9):1658–1660 [DOI] [PubMed] [Google Scholar]
- Bachmeyer C, Orlik F, Barth H, Aktories K, Benz R (2003) Mechanism of C2-toxin inhibition by fluphenazine and related compounds: investigation of their binding kinetics to the C2II-channel using the current noise analysis. J Mol Biol 333(3):527–540 [DOI] [PubMed] [Google Scholar]
- Badjic JD, Nelson A, Cantrill SJ, Turnbull WB, Stoddart JF (2005) Multivalency and cooperativity in supramolecular chemistry. Acc Chem Res 38(9):723–732 [DOI] [PubMed] [Google Scholar]
- Baldini L, Casnati A, Sansone F, Ungaro R (2007) Calixarene-based multivalent ligands. Chem Soc Rev 36(2):254–266 [DOI] [PubMed] [Google Scholar]
- Barth H, Stiles BG (2008) Binary actin-ADP-ribosylating toxins and their use as molecular Trojan horses for drug delivery into eukaryotic cells. Curr Med Chem 15(5):459–469 [DOI] [PubMed] [Google Scholar]
- Barth H, Blocker D, Behlke J, Bergsma-Schutter W, Brisson A, Benz R et al. (2000) Cellular uptake of Clostridium botulinum C2 toxin requires oligomerization and acidification. J Biol Chem 275(25):18704–18711 [DOI] [PubMed] [Google Scholar]
- Barth H, Aktories K, Popoff MR, Stiles BG (2004) Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev 68 (3):373–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Stiles BG, Popoff MR (2015) ADP-ribosylating toxins modifying the actin cytoskeleton In: Alouf JA, Ladant D, Popoff MR (eds) The comprehensive sourcebook of bacterial protein toxins, 4th Edn. Elsevier, Amsterdam, pp 397–423. ISBN 9780128001882 [Google Scholar]
- Basha S, Rai P, Poon V, Saraph A, Gujraty K, Go MY et al. (2006) Polyvalent inhibitors of anthrax toxin that target host receptors. Proc Natl Acad Sci USA 103(36):13509–13513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddoe T, Paton AW, Le Nours J, Rossjohn J, Paton JC (2010) Structure, biological functions and applications of the AB5 toxins. Trends Biochem Sci 35(7):411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitzinger C, Bronnhuber A, Duscha K, Riedl Z, Huber-Lang M, Benz R, et al. (2013) Designed azolopyridinium salts block protective antigen pores in vitro and protect cells from anthrax toxin. PLoS One 8(6). doi: 10.1371/journal.pone.0066099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheimer AW (1996) Some aspects of the history of membrane-damaging toxins. Med Microbiol Immunol 185(2):59–63 [DOI] [PubMed] [Google Scholar]
- Berube BJ, Bubeck Wardenburg J (2013) Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel) 5(6):1140–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezrukov SM, Kasianowicz JJ (1993) Current noise reveals protonation kinetics and number of ionizable sites in an open protein ion channel. Phys Rev Lett 70(15):2352–2355 [DOI] [PubMed] [Google Scholar]
- Bezrukov SM, Liu X, Karginov VA, Wein AN, Leppla SH, Popoff MR et al. (2012) Interactions of high-affinity cationic blockers with the translocation pores of B. anthracis, C. botulinum, and C. perfringens binary toxins. Biophys J 103(6):1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein RO, Koehler TM, Collier RJ, Finkelstein A (1989) Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci USA 86 (7):2209–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker D, Behlke J, Aktories K, Barth H (2001) Cellular uptake of the clostridium perfringens binary iota-toxin. Infect Immun 69(5):2980–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker D, Pohlmann K, Haug G, Bachmeyer C, Benz R, Aktories K et al. (2003) Clostridium botulinum C2 toxin: low pH-induced pore formation is required for translocation of the enzyme component C2I into the cytosol of host cells. J Biol Chem 278(39):37360–37367 [DOI] [PubMed] [Google Scholar]
- Branson TR, Turnbull WB (2013) Bacterial toxin inhibitors based on multivalent scaffolds. Chem Soc Rev 42(11):4613–4622 [DOI] [PubMed] [Google Scholar]
- Branson TR, McAllister TE, Garcia-Hartjes J, Fascione MA, Ross JF, Warriner SL et al. (2014) A protein-based pentavalent inhibitor of the cholera toxin B-subunit. Angew Chem Int Ed Engl 53(32):8323–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronnhuber A, Maier E, Riedl Z, Hajos G, Benz R, Barth H (2014) Inhibitions of the translocation pore of Clostridium botulinum C2 toxin by tailored azolopyridinium salts protects human cells from intoxication. Toxicology 3(316C):25–33 [DOI] [PubMed] [Google Scholar]
- Brown MJ, Thoren KL, Krantz BA (2015) Role of the alpha clamp in the protein translocation mechanism of anthrax toxin. J Mol Biol 427(20):3340–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante JO, Michelette ER, Geibel JP, Hanover JA, McDonnell TJ, Dean DA (2000) Dendrimer-assisted patch-clamp sizing of nuclear pores. Pflugers Arch 439(6):829–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelli A, Manini M, Paolino M, Gallelli A, Anzini M, Mennuni L et al. (2011) Bivalent ligands for the serotonin 5-HT3 receptor. ACS Med Chem Lett 2(8):571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S (2004) Synthetic multivalent molecules. Wiley-Interscience, New York [Google Scholar]
- Crini G (2014) Review: a history of cyclodextrins. Chem Rev 114(21):10940–10975 [DOI] [PubMed] [Google Scholar]
- Davis ME, Brewster ME (2004) Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov 3(12):1023–1035 [DOI] [PubMed] [Google Scholar]
- Doak BC, Over B, Giordanetto F, Kihlberg J (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol 21(9):1115–1142 [DOI] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD et al. (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280 (5364):734–737 [DOI] [PubMed] [Google Scholar]
- Duncan R (2003) The dawning era of polymer therapeutics. Nat Rev Drug Discov 2(5):347–360 [DOI] [PubMed] [Google Scholar]
- Duncan R (2011) Polymer therapeutics as nanomedicines: new perspectives. Curr Opin Biotechnol 22(4):492–501 [DOI] [PubMed] [Google Scholar]
- Duncan R (2014) Polymer therapeutics: top 10 selling pharmaceuticals—what next? J Control Release 190:371–80 [DOI] [PubMed] [Google Scholar]
- Duncan R, Gaspar R (2011) Nanomedicine(s) under the microscope. Mol Pharm 8(6):2101–2141 [DOI] [PubMed] [Google Scholar]
- Duncan R, Vicent MJ (2013) Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev 65(1):60–70 [DOI] [PubMed] [Google Scholar]
- Fan E, Merritt EA (2002) Combating infectious diseases through multivalent design. Curr Drug Targets Infect Disord 2(2):161–167 [DOI] [PubMed] [Google Scholar]
- Fan E, Zhang Z, Minke WE, Hou Z, Verlinde CLMJ, Hol WGJ (2000) High-affinity pentavalent ligands of escherichia coli heat-labile enterotoxin by modular structure-based design. J Am Chem Soc 122(11):2663–2664 [Google Scholar]
- Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B et al. (2012) Multivalency as a chemical organization and action principle. Angew Chem Int Ed 51(42):10472–10498 [DOI] [PubMed] [Google Scholar]
- Feld GK, Thoren KL, Kintzer AF, Sterling HJ, Tang II, Greenberg SG et al. (2010) Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat Struct Mol Biol 17(11):1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficici E, Andricioaei I, Howorka S (2015) Dendrimers in nanoscale confinement: the interplay between conformational change and nanopore entrance. Nano Lett 15(7):4822–4828 [DOI] [PubMed] [Google Scholar]
- Forstner P, Bayer F, Kalu N, Felsen S, Fortsch C, Aloufi A et al. (2014) Cationic PAMAM dendrimers as pore-blocking binary toxin inhibitors. Biomacromolecules 15(7):2461–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu O, Pukin AV, van Ufford HC, Branson TR, Thies-Weesie DM, Turnbull WB et al. (2015) Tetra-versus pentavalent inhibitors of cholera toxin. ChemistryOpen 4(4):471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Hartjes J, Bernardi S, Weijers CA, Wennekes T, Gilbert M, Sansone F et al. (2013) Picomolar inhibition of cholera toxin by a pentavalent ganglioside GM1os-calix[5]arene. Org Biomol Chem 11(26):4340–4349 [DOI] [PubMed] [Google Scholar]
- Geny B, Popoff MR (2006) Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol Cell 98(11):667–678 [DOI] [PubMed] [Google Scholar]
- Gibert M, Marvaud JC, Pereira Y, Hale ML, Stiles BG, Boquet P et al. (2007) Differential requirement for the translocation of clostridial binary toxins: iota toxin requires a membrane potential gradient. FEBS Lett 581(7):1287–1296 [DOI] [PubMed] [Google Scholar]
- Gu LQ, Braha O, Conlan S, Cheley S, Bayley H (1999) Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 398(6729):686–690 [DOI] [PubMed] [Google Scholar]
- Gujraty K, Sadacharan S, Frost M, Poon V, Kane RS, Mogridge J (2005) Functional characterization of peptide-based anthrax toxin inhibitors. Mol Pharm 2(5):367–372 [DOI] [PubMed] [Google Scholar]
- Gujraty KV, Joshi A, Saraph A, Poon V, Mogridge J, Kane RS (2006) Synthesis of polyvalent inhibitors of controlled molecular weight: structure-activity relationship for inhibitors of anthrax toxin. Biomacromolecules 7(7):2082–2085 [DOI] [PubMed] [Google Scholar]
- Gujraty KV, Yanjarappa MJ, Saraph A, Joshi A, Mogridge J, Kane RS (2008) Synthesis of homopolymers and copolymers containing an active ester of acrylic acid by RAFT: scaffolds for controlling polyvalent ligand display. J Polym Sci A Polym Chem 46(21):7246–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms B, Meijer EW (2006) Chemistry. Dendrimers at work. Science 313(5789):929–930 [DOI] [PubMed] [Google Scholar]
- Henry BD, Neill DR, Becker KA, Gore S, Bricio-Moreno L, Ziobro R et al. (2015) Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat Biotechnol 33(1):81–88 [DOI] [PubMed] [Google Scholar]
- Ignacio-de Leon PA, Zharov I (2011) Size-selective molecular transport through silica colloidal nanopores. Chem Commun (Camb) 47(1):553–555 [DOI] [PubMed] [Google Scholar]
- Ivarsson ME, Leroux JC, Castagner B (2012) Targeting bacterial toxins. Angew Chem Int Ed Engl 22(51):4024. [DOI] [PubMed] [Google Scholar]
- Jiang J, Pentelute BL, Collier RJ, Zhou ZH (2015) Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 521(7553):545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Saraph A, Poon V, Mogridge J, Kane RS (2006) Synthesis of potent inhibitors of anthrax toxin based on poly-L-glutamic acid. Bioconjugate Chem 17(5):1265–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Vance D, Rai P, Thiyagarajan A, Kane RS (2008) The design of polyvalent therapeutics. Chem Eur J 14(26):7738–7747 [DOI] [PubMed] [Google Scholar]
- Joshi A, Kate S, Poon V, Mondal D, Boggara MB, Saraph A et al. (2011) Structure-based design of a heptavalent anthrax toxin inhibitor. Biomacromolecules 12(3):791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane RS (2010) Thermodynamics of multivalent interactions: influence of the linker. Langmuir 26 (11):8636–8640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko J, Kamio Y (2004) Bacterial two-component and hetero-heptameric pore-forming cytolytic toxins: structures, pore-forming mechanism, and organization of the genes. Biosci Biotechnol Biochem 68(5):981–1003 [DOI] [PubMed] [Google Scholar]
- Karginov VA, Nestorovich EM, Moayeri M, Leppla SH, Bezrukov SM (2005) Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc Natl Acad Sci USA 102(42):15075–15080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov VA, Nestorovich EM, Yohannes A, Robinson TM, Fahmi NE, Schmidtmann F et al. (2006a) Search for cyclodextrin-based inhibitors of anthrax toxins: synthesis, structural features, and relative activities. Antimicrob Agents Chemother 50(11):3740–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov VA, Yohannes A, Robinson TM, Fahmi NE, Alibek K, Hecht SM (2006b) Beta-cyclodextrin derivatives that inhibit anthrax lethal toxin. Bioorg Med Chem 14(1):33–40 [DOI] [PubMed] [Google Scholar]
- Karginov VA, Nestorovich EM, Schmidtmann F, Robinson TM, Yohannes A, Fahmi NE et al. (2007) Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg Med Chem 15(16):5424–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasianowicz JJ, Bezrukov SM (1995) Protonation dynamics of the alpha-toxin ion channel from spectral analysis of pH-dependent current fluctuations. Biophys J 69(1):94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP et al. (2008) GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol 15 (7):754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AR, Forgo P, Stine KJ, D’Souza VT (1998) Methods for selective modifications of cyclodextrins. Chem Rev 98(5):1977–1996 [DOI] [PubMed] [Google Scholar]
- Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II et al. (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol 392 (3):614–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer AF, Sterling HJ, Tang II, Abdul-Gader A, Miles AJ, Wallace BA et al. (2010) Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J Mol Biol 399(5):741–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS et al. (2000) Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403(6770):669–672 [DOI] [PubMed] [Google Scholar]
- Kitov PI, Mulvey GL, Griener TP, Lipinski T, Solomon D, Paszkiewicz E et al. (2008) In vivo supramolecular templating enhances the activity of multivalent ligands: a potential therapeutic against the Escherichia coli O157 AB5 toxins. Proc Natl Acad Sci USA 105(44):16837–16842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp O, Benz R, Gibert M, Marvaud JC, Popoff MR (2002) Interaction of Clostridium perfringens iota-toxin with lipid bilayer membranes. Demonstration of channel formation by the activated binding component Ib and channel block by the enzyme component Ia. J Biol Chem 277(8):6143–6152 [DOI] [PubMed] [Google Scholar]
- Knapp O, Benz R, Popoff MR (2015a) Pore-forming activity of clostridial binary toxins. Biochim Biophys Acta [DOI] [PubMed] [Google Scholar]
- Knapp O, Maier E, Waltenberger E, Mazuet C, Benz R, Popoff MR (2015b) Residues involved in the pore-forming activity of the Clostridium perfringens iota toxin. Cell Microbiol 17(2):288–302 [DOI] [PubMed] [Google Scholar]
- Kong L, Harrington L, Li Q, Cheley S, Davis BG, Bayley H (2013) Single-molecule interrogation of a bacterial sugar transporter allows the discovery of an extracellular inhibitor. Nat Chem 5 (8):651–659 [DOI] [PubMed] [Google Scholar]
- Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z et al. (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309(5735):777–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz BA, Finkelstein A, Collier RJ (2006) Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol 355(5):968–979 [DOI] [PubMed] [Google Scholar]
- Krasilnikov O, Ternovsky V, Tashmukhamedov B (1981) Properties of ion channels induced by alpha-staphylotoxin in bilayer lipid membranes. Biofisica 26:271–275 [Google Scholar]
- Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM (2007) Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J Am Chem Soc 129(5):1312–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Neumeyer T, Sun J, Collier RJ, Benz R, Aktories K (2008) Amino acid residues involved in membrane insertion and pore formation of Clostridium botulinum C2 toxin. Biochemistry 47(32):8406–8413 [DOI] [PubMed] [Google Scholar]
- Laventie BJ, Potrich C, Atmanene C, Saleh M, Joubert O, Viero G et al. (2013) p-Sulfonato-calix [n]arenes inhibit staphylococcal bicomponent leukotoxins by supramolecular interactions. Biochem J 450(3):559–571 [DOI] [PubMed] [Google Scholar]
- Lee KI, Jo S, Rui H, Egwolf B, Roux B, Pastor RW et al. (2012) Web interface for Brownian dynamics simulation of ion transport and its applications to beta-barrel pores. J Comput Chem 33(3):331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla SH (1982) Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA 79(10):3162–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla SH (1984) Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eucaryotic cells. Adv Cyclic Nucleotide Protein Phosphorylation Res 17:189–198 [PubMed] [Google Scholar]
- Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S et al. (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8(3): e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD, Brunton JL et al. (1998) Structure of the shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37(7):1777–1788 [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1–3):3–25 [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang Z, Tan X, Hol WG, Verlinde CL, Fan E (2005) Protein heterodimerization through ligand-bridged multivalent pre-organization: enhancing ligand binding toward both protein targets. J Am Chem Soc 127(7):2044–2045 [DOI] [PubMed] [Google Scholar]
- Liu S, Moayeri M, Pomerantsev AP, Leppla SH (2015) Bacillus anthracis toxins In: Alouf JA, Ladant D, Popoff MR (eds) The comprehensive sourcebook of bacterial protein toxins, 4th edn. Elsevier, Amsterdam, pp 361–396. ISBN 9780128001882 [Google Scholar]
- Mahon CS, Fulton DA (2014) Mimicking nature with synthetic macromolecules capable of recognition. Nat Chem 6(8):665–672 [DOI] [PubMed] [Google Scholar]
- Mammen M, Choi S, Whitesides GM (1998a) Polyvalent Interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed 37:2754–2794 [DOI] [PubMed] [Google Scholar]
- Mammen M, Shakhnovich EI, Whitesides GM (1998b) Using a convenient, quantitative model for torsional entropy to establish qualitative trends for molecular processes that restrict conformational freedom. JOrg Chem 63(10):3168–3175 [DOI] [PubMed] [Google Scholar]
- Martin H, Kinns H, Mitchell N, Astier Y, Madathil R, Howorka S (2007) Nanoscale protein pores modified with PAMAM dendrimers. J Am Chem Soc 129(31):9640–9649 [DOI] [PubMed] [Google Scholar]
- Martos V, Bell SC, Santos E, Isacoff EY, Trauner D, de Mendoza J (2009) Molecular recognition and self-assembly special feature: Calix[4]arene-based conical-shaped ligands for voltage-dependent potassium channels. Proc Natl Acad Sci USA 106(26):10482–10486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattarella M, Garcia-Hartjes J, Wennekes T, Zuilhof H, Siegel JS (2013) Nanomolar cholera toxin inhibitors based on symmetrical pentavalent ganglioside GM1os-sym-corannulenes. Org Biomol Chem 11(26):4333–4339 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Zhang Z, Pickens JC, Ahn M, Hol WG, Fan E (2002) Characterization and crystal structure of a high-affinity pentavalent receptor-binding inhibitor for cholera toxin and E. coli heat-labile enterotoxin. J Am Chem Soc 124(30):8818–8824 [DOI] [PubMed] [Google Scholar]
- Moayeri M, Robinson TM, Leppla SH, Karginov VA (2008) In vivo efficacy of beta-cyclodextrin derivatives against anthrax lethal toxin. Antimicrob Agents Chemother 52(6):2239–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri M, Leppla SH, Vrentas C, Pomerantsev A, Liu S (2015) Anthrax pathogenesis. Annu Rev Microbiol 16(69):185–208 [DOI] [PubMed] [Google Scholar]
- Mogridge J, Cunningham K, Collier RJ (2002) Stoichiometry of anthrax toxin complexes. Biochemistry 41(3):1079–1082 [DOI] [PubMed] [Google Scholar]
- Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR et al. (2001) Designing a polyvalent inhibitor of anthrax toxin. Nat Biotechnol 19(10):958–961 [DOI] [PubMed] [Google Scholar]
- Mulvey GL, Marcato P, Kitov PI, Sadowska J, Bundle DR, Armstrong GD (2003) Assessment in mice of the therapeutic potential of tailored, multivalent Shiga toxin carbohydrate ligands. J Infect Dis 187(4):640–649 [DOI] [PubMed] [Google Scholar]
- Nablo BJ, Panchal RG, Bavari S, Nguyen TL, Gussio R, Ribot W et al. (2013) Anthrax toxin-induced rupture of artificial lipid bilayer membranes. J Chem Phys 139(6). doi: 10.1063/1.4816467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama M, Hagiyama T, Kojima T, Aoyanagi K, Takahashi C, Oda M et al. (2009) Binding and internalization of Clostridium botulinum C2 toxin. Infect Immun 77(11):5139–5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorovich EM, Bezrukov SM (2012) Obstructing toxin pathways by targeted pore blockage. Chem Rev 11(112):6388–6430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorovich EM, Karginov VA, Berezhkovskii AM, Bezrukov SM (2010) Blockage of anthrax PA63 pore by a multicharged high-affinity toxin inhibitor. Biophys J 99(1):134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorovich EM, Karginov VA, Popoff MR, Bezrukov SM, Barth H (2011) Tailored ss-cyclodextrin blocks the translocation pores of binary exotoxins from C. botulinum and C. perfringens and protects cells from intoxication. PLoS One 6(8). doi: 10.1371/journal.pone.0023927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumeyer T, Schiffler B, Maier E, Lang AE, Aktories K, Benz R (2008) Clostridium botulinum C2 toxin. Identification of the binding site for chloroquine and related compounds and influence of the binding site on properties of the C2II channel. J Biol Chem 283(7):3904–3914 [DOI] [PubMed] [Google Scholar]
- Ohishi I, Odagiri Y (1984) Histopathological effect of botulinum C2 toxin on mouse intestines. Infect Immun 43(1):54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajatsch M, Gerhart M, Peist R, Horlacher R, Boos W, Bock A (1998) The periplasmic cyclodextrin binding protein CymE from Klebsiella oxytoca and its role in maltodextrin and cyclodextrin transport. J Bacteriol 180(10):2630–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajatsch M, Andersen C, Mathes A, Bock A, Benz R, Engelhardt H (1999) Properties of a cyclodextrin-specific, unusual porin from Klebsiella oxytoca. J Biol Chem 274(35):25159–25166 [DOI] [PubMed] [Google Scholar]
- Paolino M, Mennuni L, Giuliani G, Anzini M, Lanza M, Caselli G, Galimberti C, Menziani MC, Donati A, Cappelli A (2014) Dendrimeric tetravalent ligands for the serotonin-gated ion channel. Chem Commun (Camb) 50(62):8582–8585 [DOI] [PubMed] [Google Scholar]
- Patke S, Boggara M, Maheshwari R, Srivastava SK, Arha M, Douaisi M et al. (2014) Design of monodisperse and well-defined polypeptide-based polyvalent inhibitors of anthrax toxin. Angew Chem Int Ed Engl 53(31):8037–8040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC (1997) Crystal structure of the anthrax toxin protective antigen. Nature 385(6619):833–838 [DOI] [PubMed] [Google Scholar]
- Pickens JC, Merritt EA, Ahn M, Verlinde CL, Hol WG, Fan E (2002) Anchor-based design of improved cholera toxin and E. coli heat-labile enterotoxin receptor binding antagonists that display multiple binding modes. Chem Biol 9(2):215–224 [DOI] [PubMed] [Google Scholar]
- Pilpa RM, Bayrhuber M, Marlett JM, Riek R, Young JA (2011) A receptor-based switch that regulates anthrax toxin pore formation. PLoS Pathog. 7(12). doi: 10.1371/journal.ppat.1002354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff MR, Rubin EJ, Gill DM, Boquet P (1988) Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun 56(9):2299–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pust S, Barth H, Sandvig K (2010) Clostridium botulinum C2 toxin is internalized by clathrin- and Rho-dependent mechanisms. Cell Microbiol 12(12):1809–1820 [DOI] [PubMed] [Google Scholar]
- Ragle BE, Karginov VA, Bubeck Wardenburg J (2010) Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob Agents Chemother 54(1):298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S et al. (2006) Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat Biotechnol 24 (5):582–586 [DOI] [PubMed] [Google Scholar]
- Rai PR, Saraph A, Ashton R, Poon V, Mogridge J, Kane RS (2007) Raftlike polyvalent inhibitors of the anthrax toxin: modulating inhibitory potency by formation of lipid microdomains. Angew Chem Int Ed 46(13):2207–2209 [DOI] [PubMed] [Google Scholar]
- Roeder M, Nestorovich EM, Karginov VA, Schwan C, Aktories K, Barth H (2014) Tailored cyclodextrin pore blocker protects mammalian cells from clostridium difficile binary toxin CDT. Toxins (Basel) 6(7):2097–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleberger C, Hochmann H, Barth H, Aktories K, Schulz GE (2006) Structure and action of the binary C2 toxin from Clostridium botulinum. J Mol Biol 364(4):705–715 [DOI] [PubMed] [Google Scholar]
- Schmid A, Benz R, Just I, Aktories K (1994) Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J Biol Chem 269(24):16706–16711 [PubMed] [Google Scholar]
- Shewmake TA, Solis FJ, Gillies RJ, Caplan MR (2008) Effects of linker length and flexibility on multivalent targeting. Biomacromolecules 9(11):3057–3064 [DOI] [PubMed] [Google Scholar]
- Simpson LL (1984) Molecular basis for the pharmacological actions of Clostridium botulinum type C2 toxin. J Pharmacol Exp Ther 230(3):665–669 [PubMed] [Google Scholar]
- Simpson LL, Stiles BG, Zepeda HH, Wilkins TD (1987) Molecular basis for the pathological actions of Clostridium perfringens iota toxin. Infect Immun 55(1):118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Venglarik CJ, Bridges RJ (1995) Development of chloride channel modulators. Kidney Int 48(4):985–993 [DOI] [PubMed] [Google Scholar]
- Sisu C, Baron AJ, Branderhorst HM, Connell SD, Weijers CA, de Vries R et al. (2009) The influence of ligand valency on aggregation mechanisms for inhibiting bacterial toxins. ChemBioChem 10(2):329–337 [DOI] [PubMed] [Google Scholar]
- Solomon D, Kitov PI, Paszkiewicz E, Grant GA, Sadowska JM, Bundle DR (2005) Heterobifunctional multivalent inhibitor-adaptor mediates specific aggregation between Shiga toxin and a pentraxin. Org Lett 7(20):4369–4372 [DOI] [PubMed] [Google Scholar]
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274(5294):1859–1866 [DOI] [PubMed] [Google Scholar]
- Stiles BG, Wilkins TD (1986) Purification and characterization of Clostridium perfringens iota toxin: dependence on two nonlinked proteins for biological activity. Infect Immun 54(3):683–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles BG, Hale ML, Marvaud JC, Popoff MR (2002) Clostridium perfringens iota toxin: characterization of the cell-associated iota b complex. Biochem J 367(Pt 3):801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szejtli J (2004) Past, present, and future of cyclodextrin research. Pure Appl Chem 76(10):1825–1845 [Google Scholar]
- Tang MX, Redemann CT, Szoka FC Jr (1996) In vitro gene delivery by degraded polyamidoamine dendrimers. Bioconjug Chem 7(6):703–714 [DOI] [PubMed] [Google Scholar]
- Tomalia DA, Frechet JMJ (2002) Discovery of dendrimers and dendritic polymers: a brief historical perspective. J Polym Sci. Part A Polym Chem 40:2719–2728 [Google Scholar]
- U.S. Pharmaceutical Sales. Available at: http://www.drugs.com/stats/top100/sales
- van den Berg B, Prathyusha Bhamidimarri S, Dahyabhai Prajapati J, Kleinekathofer U, Winterhalter M (2015) Outer-membrane translocation of bulky small molecules by passive diffusion. Proc Natl Acad Sci USA 112(23):E2991–E2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance D, Martin J, Patke S, Kane RS (2009) The design of polyvalent scaffolds for targeted delivery. Adv Drug Deliv Rev 61(11):931–939 [DOI] [PubMed] [Google Scholar]
- Varejao EV, de Fatima A, Fernandes SA (2013) Calix[n]arenes as goldmines for the development of chemical entities of pharmaceutical interest. Curr Pharm Des 19(36):6507–6521 [DOI] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C (2000) Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 15(352):739–745 [PMC free article] [PubMed] [Google Scholar]
- Walters WP (2012) Going further than Lipinski’s rule in drug design. Expert Opin Drug Discov 7 (2):99–107 [DOI] [PubMed] [Google Scholar]
- Weisman A, Chou B, O’Brien J, Shea KJ (2015) Polymer antidotes for toxin sequestration. Adv Drug Deliv Rev 1(90):81–100 [DOI] [PubMed] [Google Scholar]
- Wenz G (1994) Cyclodextrins as building blocks for supramolecular structures and functional units. Angew Chem Int Ed Engl 33(8):803–822 [Google Scholar]
- Wijagkanalan W, Kawakami S, Hashida M (2011) Designing dendrimers for drug delivery and imaging: pharmacokinetic considerations. Pharm Res 28(7):1500–1519 [DOI] [PubMed] [Google Scholar]
- Wu LP, Ficker M, Christensen JB, Trohopoulos PN, Moghimi SM (2015) Dendrimers in medicine: therapeutic concepts and pharmaceutical challenges. Bioconjug Chem 26(7):1198–1211 [DOI] [PubMed] [Google Scholar]
- Yannakopoulou K, Jicsinszky L, Aggelidou C, Mourtzis N, Robinson TM, Yohannes A et al. (2011) Symmetry requirements for effective blocking of pore-forming toxins: comparative study with alpha-, beta-, and gamma-cyclodextrin derivatives. Antimicrob Agents Chemother 55(7):3594–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MQ, Wilkinson B (2007) Drug discovery beyond the ‘rule-of-five’. Curr Opin Biotechnol 18(6):478–488 [DOI] [PubMed] [Google Scholar]
- Zhang S, Finkelstein A, Collier RJ (2004a) Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc Natl Acad Sci USA 101(48):16756–16761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Udho E, Wu Z, Collier RJ, Finkelstein A (2004b) Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys J 87(6):3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu J, Verlinde CL, Hol WG, Fan E (2004c) Large cyclic peptides as cores of multivalent ligands: application to inhibitors of receptor binding by cholera toxin. J Org Chem 69 (22):7737–7740 [DOI] [PubMed] [Google Scholar]