

Abstract
Homologous recombination deficiency conferred by alterations in BRCA1 or BRCA2 are common in breast tumors and can drive sensitivity to platinum chemotherapy and PARP inhibitors. Alterations in nucleotide excision repair (NER) activity can also impact sensitivity to DNA damaging agents, but NER activity in breast cancer has been poorly characterized. Here, we apply a novel immunofluorescence-based cellular NER assay to screen a large panel of breast epithelial and cancer cell lines. Although the majority of breast cancer models are NER proficient, we identify an example of a breast cancer cell line with profound NER deficiency. We show that NER deficiency in this model is driven by epigenetic silencing of the ERCC4 gene, leading to lack of expression of the NER nuclease XPF, and that ERCC4 methylation is also strongly correlated with ERCC4 mRNA and XPF protein expression in primary breast tumors. Re-expression of XPF in the ERCC4-deficient breast cancer rescues NER deficiency and cisplatin sensitivity, but does not impact PARP inhibitor sensitivity. These findings demonstrate the potential to use functional assays to identify novel mechanisms of DNA repair deficiency and nominate NER deficiency as a platinum sensitivity biomarker in breast cancer.
INTRODUCTION
DNA repair pathway alterations are common in tumors and can have important implications for therapy selection. In breast cancer, alterations in homologous repair (HR) pathway genes – particularly BRCA1 and BRCA2 – have been associated with increased sensitivity to platinum agents as well as poly(ADP ribose)polymerase inhibitors (PARPi’s).[1, 2] However, the frequency and clinical relevance of alterations in DNA repair pathways beyond HR have been poorly characterized.
The nucleotide excision repair (NER) pathway is a highly conserved DNA repair pathway that corrects intrastrand crosslinks created by carcinogens such as ultraviolet (UV) light and tobacco byproducts.[3, 4] In addition, the NER pathway repairs damage created by several classes of chemotherapy agents, including platinum drugs such as cisplatin and carboplatin. NER-mediated repair of DNA crosslinks begins with lesion recognition: the transcription-coupled NER (TC-NER) sub-pathway specifically recognizes lesions that block RNA polymerase II-mediated gene transcription while the global genome NER (GG-NER) sub-pathway can recognize and bind lesions across the genome. Following lesion recognition via either sub-pathway, the transcription factor II H (TFIIH) complex is assembled, the DNA helix is unwound, and a single-stranded DNA fragment containing the damaged base is excised. The resulting single-stranded gap is then filled by a DNA polymerase, resulting in error-free repair of the lesion.[4]
Given the widespread use of platinum-based chemotherapies in a variety of tumor contexts, tumor alterations in NER pathway genes have been investigated as a potential biomarker of platinum response.[5] Our group and others have demonstrated an association between somatic missense mutations in the NER gene ERCC2 and improved response to cisplatin-based chemotherapy in muscle-invasive bladder cancer.[6, 7] Functional studies revealed that the observed mutations failed to rescue the NER deficiency of an ERCC2-deficient cell line, suggesting that an ERCC2 mutation is sufficient to drive cisplatin sensitivity in this context.[8] Similarly, in multiple myeloma cell lines and patient samples, low ERCC3 transcript levels were found to correlate with sensitivity to alkylating chemotherapy.[9]
There is renewed interest in the use of platinum-based chemotherapy in breast cancer, particularly among triple negative tumors (i.e., tumors that lack immunohistochemical staining of estrogen receptor [ER] and progesterone receptor [PR], and do not have HER2/neu amplification).[2] Germline or somatic alterations in BRCA1 or BRCA2 are present in up to 25% of triple negative tumors and these tumors have high response rates to platinum drugs.[10] BRCA1/2 alterations are also common in high-grade serous ovarian cancer (HGSOC) and are associated with improved response and prolonged survival following platinum-based therapy.[11] In addition, we recently showed that a subset of HGSOCs possess NER pathway mutations and that these NER alterations are associated with cisplatin sensitivity and improved clinical outcomes independent of BRCA1/2 status.[12]
Despite these advances in other tumor settings, the impact of NER pathway dysfunction in breast cancer remains poorly understood. An inherited truncating mutation in ERCC3 was recently shown to confer increased risk of breast cancer, and functional studies confirmed that the mutant ERCC3 was unable to support normal cellular NER.[13] Furthermore, studies using unscheduled DNA synthesis to infer NER activity suggest higher rates of relative NER deficiency among early-stage breast tumors compared to primary breast and ovarian epithelial cells, but the underlying mechanisms are unknown.[14, 15]
To characterize NER pathway activity in breast cancer, we functionally profiled a panel of breast cancer cell lines using a novel NER assay that relies on specific binding of a DDB2 proteo-probe to UV-induced 6,4-photoproducts.[16] We find that nearly all breast cancer cell lines are NER proficient; however, we identify one cell line – MDA-MB-468 – with a severe defect in NER and corresponding sensitivity to UV and cisplatin. Genomic analysis revealed promoter methylation of the NER gene ERCC4, resulting in lack of expression of the ERCC4 (XPF) nuclease. Accordingly, NER function was restored and cisplatin sensitivity was decreased following expression of wild-type ERCC4. This study highlights the ability of functional profiling to identify novel mechanisms of tumor DNA repair pathway deficiency and also nominates the NER pathway as a mediator of cisplatin sensitivity in breast cancer.
RESULTS
The DDB2 proteo-probe assay detects cellular NER deficiency
To functionally assess cellular NER capacity, we applied a novel fluorescence-based microscopy assay previously developed in our laboratory.[16] The assay utilizes a purified DDB2 (Damage-specific DNA binding protein 2) proteo-probe that binds specifically to UV-induced (6,4)-photoproducts (6,4-PP’s).[8, 9, 16, 17] We used the DDB2 proteo-probe to measure NER capacity across a panel of fibroblast cell lines derived from patients with xeroderma pigmentosum (XP), Cockayne syndrome (CS), or trichothiodystrophy (TTD)(Supplementary Fig. 1). Biallelic germline mutations in the NER pathway can cause XP, an autosomal recessive disease characterized by extreme sensitivity to UV light and increased risk of skin cancer.[18, 19] In addition, several NER proteins are also components of the transcription factor IIH (TFIIH) complex, and biallelic mutations in one of these genes can result in Cockayne syndrome (CS) or trichothiodystrophy (TTD), diseases characterized by altered transcription as well as a DNA repair defect in some cases.[20] DDB2 (XPE) is a member of the GG-NER sub-pathway; therefore the DDB2 proteo-probe assay measures activity of the GG-NER and shared NER pathway.
In cell lines with loss of a canonical XP gene (XPA-XPG), the extent of residual DNA damage two hours following UV treatment was significantly higher than in a fibroblast cell line (BJ1) lacking any known NER pathway alterations (p-values all <0.0001; Fig. 1, Supplementary Fig. 1). Residual damage at two hours ranged from 58% in XPG-mutated fibroblasts to 193% percent in XPC-mutated fibroblasts. The kinetics of damage recognition and NER are known to be impacted by sequence-specific and epigenetic factors[21], and an increase in DDB2 foci from five minutes to two hours (i.e., values >100%) in NER deficient lines may be due to chromatin remodeling following DNA damage, which allows presentation of additional (6,4)-PP’s to the DDB2 proteo-probe.[22, 23]
Figure 1:
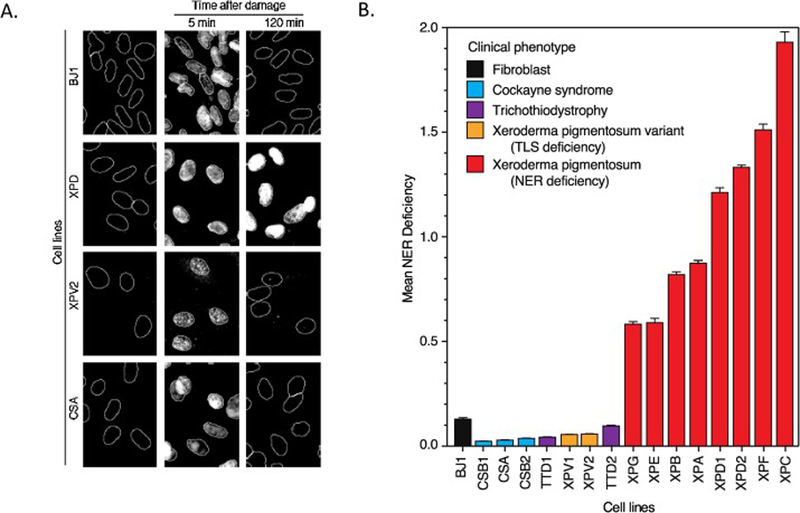
The DDB2 proteo-probe assay detects GG-NER defects in XP cell lines, but not CS or TTD cell lines. A. Cells harvested before, 5 minutes after, or 120 minutes after UV exposure are treated with the purified DDB2 proteo-probe. In all cell lines, DDB2 foci are absent prior to UV exposure but are present 5 minutes after UV exposure. In cells with intact GG-NER, DDB2 foci resolve by 120 minutes, indicating repair, whereas foci persist in cells lacking NER. B. The DDB2 assay was performed on a panel of fibroblast cell lines established from patients with XP, CS, or TTD. Mean GG-NER deficiency is calculated by dividing the DDB2 proteo-probe signal present 2 hours following UV exposure by the signal present 5 minutes after exposure. XP cell lines (red) are GG-NER deficient while CS, TTD, and XP variant cell lines have GG-NER capacity similar to a control fibroblast cell line (BJ1).
Unlike cell lines from patients with canonical XP gene mutations (XPA-XPG), cell lines from CS and TTD patients repaired UV-induced DDB2 foci, and there were no significant differences in repair capacity between these lines and the NER-proficient BJ1 fibroblast line (p-values 0.14 to 0.99; Fig. 1). These results are consistent with loss of TC-NER – but preserved GG-NER – in CS patients. Together, these results demonstrate the utility of the DDB2 proteo-probe in identifying cell lines with functional deficiency of the GG-NER or shared NER pathway.
Functional profiling of NER in breast cancer cell lines
We next analyzed NER activity in a panel of 43 breast cell lines, including five non-transformed mammary epithelial and thirty-eight tumor cell lines comprising all major breast cancer subtypes as defined by estrogen receptor (ER), progesterone receptor (PR), and HER2 (ERBB2) status (Supplementary Fig. 2).
All five non-transformed mammary epithelial cell lines were NER proficient (Fig. 2). Similarly, the majority of breast cancer cell lines were also NER proficient. However, there was one clear exception: the MDA-MB-468 cell line, which was initially established from a malignant pleural effusion from a patient with a high-grade triple negative (ER negative, PR negative, HER2 non-amplified) breast adenocarcinoma. MDA-MB-468 cells showed a markedly higher ratio of unrepaired DDB2 foci than any other breast epithelial or tumor cell line (0.89 for MDA-MB-468 vs <0.2 for all other cell lines; Fig. 2). Remarkably, the MDA-MB-468 appeared to have an NER deficiency as profound as many of the XP cell lines.
Figure 2:

NER profiling of breast epithelial cancer cell lines using the DDB2 proteo-probe assay. Mean NER deficiency is defined as the DDB2 proteo-probe signal remaining 2 hours after UV exposure divided by the signal detected 5 minutes after exposure. TNBC, triple negative breast cancer (defined as estrogen receptor [ER] negative, progesterone receptor [PR] negative, HER2/neu non-amplified).
NER deficiency correlates with cisplatin and UV sensitivity
To validate and extend our findings from the DDB2 proteo-probe assay, we measured sensitivity of our breast cell line panel to UV irradiation and cisplatin, two sources of DNA damage that are typically repaired by the NER pathway. In agreement with the DDB2 proteo-probe assay, MDA-MB-468 was the most sensitive breast cell line to both cisplatin and UV irradiation (Fig. 3A, B). Across the remaining breast cancer cell lines, there was no correlation between the small variations in NER activity measured by the DDB2 proteo-probe assay and changes in sensitivity to UV or cisplatin.
Figure 3:
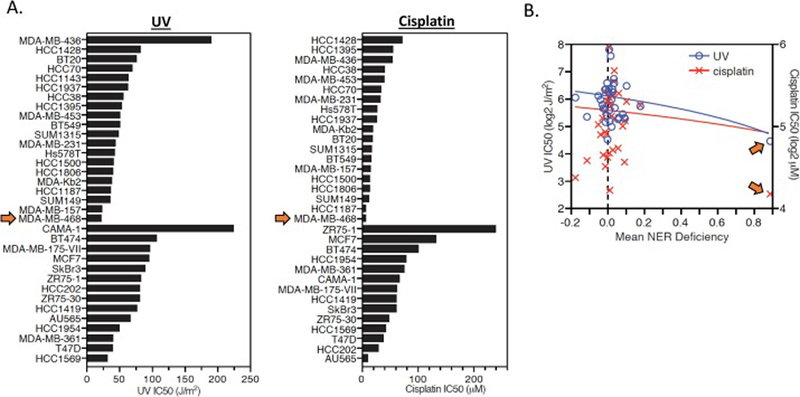
UV and cisplatin sensitivity of breast epithelial and cancer cell lines. A. UV (left) and cisplatin (right) sensitivity is plotted for each mammary epithelial and breast cancer cell line. The MDA-MB-468 cell line (denoted by orange arrows) was the most sensitive cell line to both UV and cisplatin. B. UV and cisplatin IC50 values are plotted against mean NER deficiency (from the DDB2 proteo-probe assay) for each cell line. The MDA-MB-468 cell line (denoted by orange arrows) has the highest mean NER deficiency and lowest UV and cisplatin IC50 of all cell lines.
ERCC4 promoter methylation results in lack of XPF expression
To investigate the mechanism underlying its apparent NER deficiency, we performed whole exome sequencing (WES) of the MDA-MB-468 cell line (Methods). In agreement with published sequencing analysis of this cell line[24, 25], we did not identify non-synonymous mutations in any of the 33 genes comprising our NER gene set list (Supplementary Fig. 3).
We next performed gene expression analysis in MDA-MB-468 using available gene expression data. For each of 110 NER-related genes[26], we compared expression in MDA-MB-468 to expression in a panel of 27 other breast cancer cell lines (Methods). This analysis revealed that ERCC4 was the most significantly under-expressed NER gene in MDA-MB-468 compared to other breast cancer cell lines (Fig. 4A). Indeed, across 877 cancer cell lines representing 36 tumor types from the Cancer Cell Line Encyclopedia (CCLE), MD-MBA-468 had the lowest ERCC4 expression among breast cancer cell lines and the second lowest ERCC4 expression overall (Supplementary Fig. 4).
Figure 4:
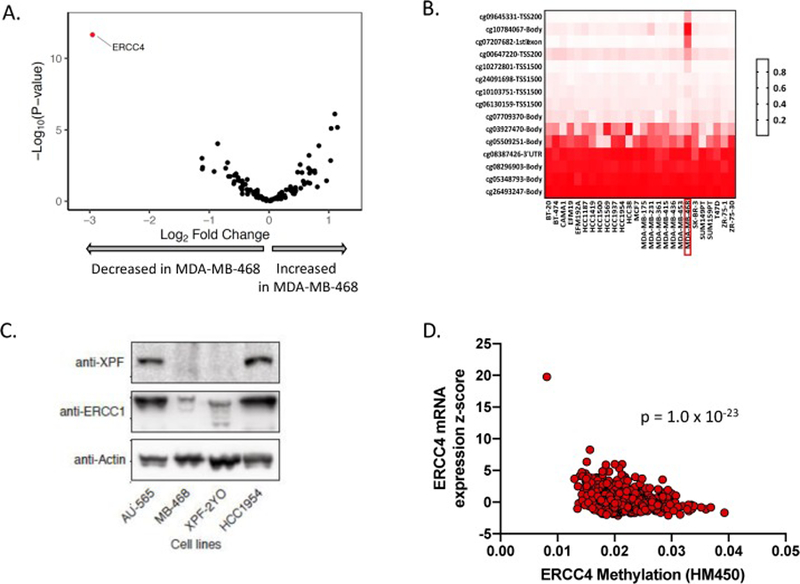
MDA-MB-468 cells have high ERCC4 methylation levels and low ERCC4 mRNA and XPF protein levels. A. Across 110 NER-related genes, ERCC4 was the most under-expressed gene in MDA-MB-468 compared to a panel of 27 other breast cancer cell lines. B. ERCC4 promoter and CpG island methylation of the ERCC4 gene was increased in MDA-MB-468 compared to other breast cancer cell lines. C. Immunoblot showing XPF (encoded by ERCC4 gene), ERCC1 (XPF binding partner), and actin (loading control) protein levels for 4 cells lines. XPF and ERCC1 protein levels are lower in MDA-MB-468 compared to AU-565 and HCC1954 breast cancer cell lines, and instead resemble XPF/ERCC1 levels from XPF-2YO, a fibroblast cell line from a XP complementation group F (XPF−/−) patient. D. ERCC4 methylation is strongly correlated with ERCC4 mRNA levels in the TCGA breast cancer cohort.
To determine if low ERCC4 expression in MDA-MB-468 was driven by an epigenetic mechanism, we analyzed published DNA methylation data from breast cancer cell lines. Across 27 breast cancer cell lines, MDA-MB-468 had the highest levels of ERCC4 promoter and CpG island methylation (Fig. 4B; Methods). A similar analysis of cell lines from the CCLE also showed increased ERCC4 methylation in MDA-MB-468 compared to other breast cancer cell lines (Supplementary Fig. 5).
To determine whether the observed ERCC4 gene methylation correlated with decreased expression of XPF, the protein encoded by the ERCC4 gene, we compared XPF levels in MDA-MB-468 cells to the NER-proficient breast cancer cell lines AU565 and HCC1954 as well as to the ERCC4-deficient fibroblast cell line 2YO (Fig. 4C). XPF was expressed in the AU565 and HCC1954 cell lines, but no expression was detected in MDA-MB-468 or 2YO cells. XPF forms a stable heterodimer with ERCC1, and ERCC1 levels were also lower in MDA-MB-468 and 2YO cells than in the NER proficient AU565 and HCC1954 cell lines. In the TCGA breast cancer cohort, ERCC4 methylation was strongly associated with ERCC4 mRNA (p=1.9e-23; n=785) and XPF protein (p=0.0156; n=28) levels (Fig. 4D, Supplementary Fig. 6), suggesting that ERCC4 methylation status drives ERCC4 mRNA and XPF protein levels and may therefore impact NER activity in breast tumors.
ERCC4 re-expression in MDA-MB-468 rescues cisplatin and UV sensitivity
To test if the lack of XPF contributes to NER dysfunction and cisplatin sensitivity in MDA-MB-468 cells, we expressed wild-type (WT) XPF in MDA-MB-468 cells and measured the effect on NER pathway function and cisplatin sensitivity. Stable expression of XPF nearly completely rescues the NER deficiency of the MDA-MB-468 cell line (84.8% vs 2.3% repair at 2 hours, p=0.0089), and significantly decreases cisplatin sensitivity (Fig. 5A–C). However, re-expression of XPF did not impact sensitivity to the PARP inhibitor olaparib (Fig. 5D). Taken together, these findings demonstrate that the NER deficiency and cisplatin sensitivity of the MDA-MB-468 cell line is driven by lack of XPF protein expression, but that NER deficiency does not impact sensitivity to PARP inhibition in this setting.
Figure 5:
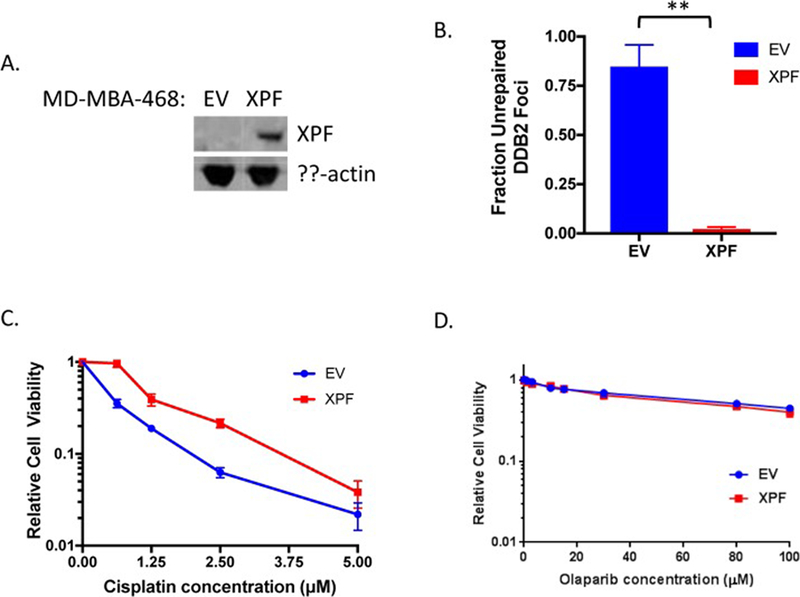
Exogenous XPF expression in MDA-MB-468 cells rescues NER deficiency and cisplatin sensitivity. A. Immunoblot showing stable expression of wild-type XPF in MDA-MB-468 cells. B. XPF expression rescues NER deficiency in the DDB2 proteo-probe assay (**p<0.01). C. XPF expression rescues cisplatin sensitivity of the MDA-MB-468 cell line. D. XPF expression does not impact sensitivity to the PARP inhibitor olaparib. EV: empty vector.
DISCUSSION
The NER pathway plays a central role in repairing genotoxic lesions created by environmental and chemotherapeutic agents. Many aspects of the genetics and biochemistry of the NER pathway were elucidated from studies of germline NER pathway mutations in patients with xeroderma pigmentosum (XP), Cockayne syndrome (CS), or trichothiodystrophy (TTD). However, recent large-scale genomic studies have also revealed that alterations in the NER pathway occur in several tumor types. Approximately 15% of muscle-invasive bladder tumors have a somatic missense mutation in ERCC2, and these ERCC2 mutations have been shown to abrogate NER function and are associated with improved response to platinum-based chemotherapy.[6–8] A similar association between somatic NER alterations and platinum response has also been observed in ovarian cancer.[12] In multiple myeloma, transcript levels of ERCC3, the gene encoding the NER helicase XPB, correlate with NER activity and sensitivity to alkylating agents.[9] These studies suggest that differences in NER function across tumors may have important therapeutic implications in specific clinical contexts.
In breast cancer, HR deficiency driven by germline or somatic alterations in BRCA1/2 can have important implications for clinical management; however, the frequency and clinical relevance of alterations in DNA repair pathways beyond HR are less well characterized. Several recent studies have shown that a small subset of breast tumors possess microsatellite instability (MSI)[27, 28], indicative of a defect in the mismatch repair (MMR) pathway and sensitivity to immune checkpoint blockade.[29]
Although no NER genes are recurrently mutated in breast cancer, we hypothesized that a subset of breast tumors may be functionally NER deficient. To obtain an unbiased view of NER pathway activity, we screened a large panel of genomically diverse breast epithelial and tumor cell lines using the DDB2 proteo-probe assay, a reliable and efficient method for directly quantifying NER-mediated repair in cells.[9, 16] Our results demonstrate that most breast cancer cell lines are NER proficient. Interestingly, differences in NER activity among cell lines was much smaller than differences in cisplatin or UV sensitivity among cell lines, highlighting that differences in clinical response to cross-linking agents such as cisplatin are multi-factorial and are driven by the integrated function (or dysfunction) of multiple DNA repair and cell signaling pathways.
Despite the similarities in NER capacity across most breast cancer cell lines, the MDA-MB-468 cell line was a clear outlier in our functional analysis and exhibited significantly lower NER activity and higher cisplatin and UV sensitivity than any other cell line tested. Although whole exome sequencing did not reveal mutations in any NER genes, we found that methylation of the ERCC4 gene was associated with low ERCC4 mRNA levels and lack of XPF protein expression. A similar relationship among ERCC4 promoter methylation, ERCC4 mRNA levels, and XPF protein levels was also observed in primary breast tumors from the TCGA cohort, suggesting that epigenetic silencing of ERCC4 may modulate NER activity in primary breast tumors. Importantly, re-expression of XPF was sufficient to restore NER activity and decrease cisplatin sensitivity of the MDA-MB-468 cell line.
The identification of ERCC4 methylation as a mechanism of NER loss in breast cancer may have therapeutic implications. Promoter methylation of the BRCA1 gene is one of the most frequently observed mechanisms of HR loss among breast and ovarian tumors. However, epigenetic silencing of ERCC4 (or other NER genes) has not been previously reported as a mechanism of functional NER deficiency. Whereas HR deficiency driven by genetic or epigenetic mechanisms is often associated with sensitivity to both cisplatin and PARP inhibitors, NER deficiency conferred by mutation or epigenetic silencing is associated with cisplatin sensitivity but not PARP inhibitor sensitivity (Fig. 5D).[12] Therefore, using platinum sensitivity to predict PARP inhibitor sensitivity is only appropriate for cases in which the platinum sensitivity is driven by HR deficiency.
Although assays to profile DNA repair capacity in clinical breast specimens and other tumor types have been described[30, 31], challenges with tissue collection and processing as well as assay reproducibility and cost considerations have limited widespread clinical incorporation. Because the DDB2 proteo-probe assay provides a direct readout of NER activity, the assay can be completed in approximately 3 hours. In addition, because the assay relies on detection of a repair factor rather than a specific DNA lesion, the DDB2 proteo-probe assay may be useful in measuring GG-NER activity in response to a variety of DNA damaging agents.
In summary, we applied a novel NER functional assay to identify a previously unappreciated mechanism of NER loss in breast cancer. Taken together, our data highlight the utility of functional assays in identifying novel mechanisms of DNA repair deficiency beyond and suggest that NER loss may be a platinum sensitivity biomarker in breast cancer.
MATERIALS AND METHODS
Cell culture
Mammary epithelial and breast cancer cell lines used in this study were purchased from ATCC (Manassas, VA) with the exception of SUM149PT, which was a gift from Dr. Daniel Silver. Cell lines were grown in RPMI supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C, 100% humidity and 5% CO2. Breast cancer cell lines were characterized by hormone receptor status as defined by Neve et al[32] or the ATCC database.
Cell lines from patients with CS, TTD, or XP were purchased from Coriell Cell Repository (Camden, NJ) and grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C, 100% humidity and 5% CO2.
The wild-type ERCC4 lentiviral plasmid[12] transfected with viral packaging vectors into 293T cells. Supernatant containing virus was harvested after 48 hours, filtered using a 0.45 μm syringe filter, and then used to infect MDA-MB-468 cells. After 24 hours, puromycin was used to select a polyclonal cell population stably expressing XPF.
DDB2 proteo-probe assay
The DDB2 proteo-probe assay was performed as previously described.[16] Briefly, the purified HA-tagged DDB2 protein complex was used as a probe in an immunofluorescence-based assay. Cells were plated on 12 mm coverslips and exposed to 20 J/m2 UV-C at 254 nm using a StrataLinker 2400 irradiator (Stratagene, Agilent Technologies). Five or 120 minutes following UV exposure, cells were fixed with ice cold methanol for 10 minutes at room temperature. After serial rehydration with phosphate-buffered saline (PBS), non-specific binding sites were blocked by incubation with PBA-BSA (PBS, 0.3% bovine serum albumin, 0.1% sodium azide). DDB2 proteo-probe was diluted in PBS-BSA and added to the fixed cells for 30 minutes at 37°C. Cells were then washed twice with PBS and mouse anti-HA antibody (diluted 1:200 in PBS-BSA) was used to label the hybridized DDB2 proteo-probe. Cells were then washed again with PBS and goat anti-mouse antibody coupled to Alexa fluor488 fluorochrome (diluted 1:300 in PBS-BSA) was added. Following two final washes in PBS and one in purified water, coverslips were mounted in Vectashield medium containing DAPI (Vector Laboratories). All immunofluorescence experiments were performed in duplicate.
The DDB2 proteo-probe assay was also performed in MDA-MB-468 cells following stable expression of Myc-tagged XPF. Cells were incubated with both mouse anti-HA antibody and rabbit anti-Myc antibody (Cell Signaling Technology) 1:1000 in PBS-BSA for one hour at 37°C, followed by secondary antibody incubation with goat anti-mouse antibody coupled to Alexa fluor488 fluorochrome (as described above) and concurrent goat anti-rabbit antibody coupled to Alexa fluor594 (Thermo-Fisher) diluted 1:300 in PBS-BSA.
Imaging was performed as previously described with a Imager.M2 Zeiss microscope with AxioCam MRM camera.[16] Images were processed to remove non-specific signal with the “Projection” module of CellProfiler.[33] To calculate NER deficiency, CellProfiler was used to calculate the average fluorescence signal both inside and outside nuclei in the processed images. Background fluorescence (defined as the average fluorescence of the area outside nuclei) was subtracted from the average fluorescence of nuclei for each image, and the average fluorescence of nuclei in non-irradiated cells was then subtracted from the average fluorescence of nuclei in cells fixed at 5 or 120 minutes following UV exposure. Finally, NER deficiency was defined as the ratio of fluorescence at 120 minutes compared to 5 minutes. Data management and graphing were performed using Microsoft Excel and GraphPad Prism 6.0.
UV and cisplatin sensitivity assays
To measure cisplatin sensitivity, cells were seeded in triplicate in 96-well plates at a density of approximately 5,000 cells per well. The following day, cisplatin (Sigma) dissolved in cell media was added to the wells to final concentrations ranging from 0 to 80 μM. Forty-eight hours later, cells were incubated with MTS reagent (Promega) for 2 hours. Cell viability was measured as per manufacturer instructions. Normalized values were fit using nonlinear regression in GraphPad Prism to calculate IC50 values.
To assess sensitivity to UV-C radiation, cells were seeded at 1,000 cells per well using the DropArray system (Curiox Biosystems Inc). The following day, media was aspirated and cells were exposed to nine doses of UV-C light ranging from 0 to 75 J/m2. Forty-eight hours after UV-C exposure, the MTS assay was performed.
Correlations among NER deficiency, cisplatin IC50, and UV IC50 were calculated using a multifactorial linear regression model constructed in R. Best linear fit models were generated using GraphPad Prism.
Immunoblotting
Soluble cell lysates were heated at 70°C for 10 minutes and combined with NuPAGE sample loading buffer. Proteins were separated by electrophoresis on a NuPAGE Bis-Tris 4–12% gradient gel (Thermo Fisher) and then transferred to a 0.45 um pore nitrocellulose membrane (Bio Rad).
For immunoblotting, the following primary antibodies were used: rabbit anti-XPF (clone D3G8C, 1:500; Cell Signaling), rabbit anti-ERCC1 (clone D61F5, 1:500; Cell Signaling), and mouse anti-actin (1:5000; Sigma). Secondary antibodies were goat anti-mouse and goat anti-rabbit conjugated to HRP (1:2000; Cell Signaling). All antibodies were diluted in PBS with 0.05% Tween. HRP was activated by a three-minute incubation with SuperSignal West Pico Reagent (Thermo Fisher) and visualized by chemiluminescence with a LAS-4000 Luminescent Image Analyzer (Fujifilm).
Genomic analyses
Whole exome sequencing of the MDA-MB-468 cell line used in the DDB2 analyses was performed by Personal Genome Diagnostics (Baltimore, MD) to a mean depth of 224x. No coding mutations were observed in any of the NER genes listed in Supplementary Figure 3.
ERCC4 methylation data was downloaded from the Gene Expression Omnibus (GEO) dataset GPL13534. Promoter and CpG island methylation sites assigned to ERCC4 were queried across 27 breast cancer cell lines. Methylation sites were ranked and ordered based on the median value calculated across the cohort for each site. Methylation data were plotted and analyzed using Prism 6 (GraphPad Software, Inc; La Jolla, CA).
Patient-level mutation data was downloaded from The Cancer Genome Atlas Research Network (https://portal.gdc.cancer.gov/). Microarray datasets were downloaded from GEO using the GEOquery package. The Limma package was used to determine differentially expressed genes between the sample MB-468 and other breast cancer cell lines. The data were stratified by study and differential gene expression analysis was performed in R.
Statistical analyses
When not otherwise specified, statistical tests and graphing were performed using GraphPad Prism. For comparison of NER activity by DDB2 assay (Figure 1), p-values were calculated using a one-way ANOVA with Dunnett test for multiple comparisons.
Supplementary Material
Highlights.
nucleotide excision repair (NER) function is poorly characterized in breast cancer
we developed a novel DDB2 proteo-probe assay to measure cellular NER
NER function is interrogated across a panel of breast epithelial and tumor cell lines
epigenetic silencing of ERCC4 is identified as a driver of NER deficiency
re-expression of ERCC4 (XPF) rescues NER deficiency and UV/cisplatin sensitivity
Footnotes
No conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Robson M, et al. , Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med, 2017. 377(6): p. 523–533. [DOI] [PubMed] [Google Scholar]
- 2.Silver DP, et al. , Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol, 2010. 28(7): p. 1145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marteijn JA, et al. , Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol, 2014. 15(7): p. 465–81. [DOI] [PubMed] [Google Scholar]
- 4.Scharer OD, Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol, 2013. 5(10): p. a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mouw KW, D’Andrea AD, and Konstantinopoulos PA, Nucleotide excision repair (NER) alterations as evolving biomarkers and therapeutic targets in epithelial cancers. Oncoscience, 2015. 2(12): p. 942–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu D, et al. , Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol, 2016. 2(8): p. 1094–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Allen EM, et al. , Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov, 2014. 4(10): p. 1140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, et al. , ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin Cancer Res, 2019. 25(3): p. 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szalat R, et al. , Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia, 2017. [DOI] [PMC free article] [PubMed]
- 10.von Minckwitz G, et al. , Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol, 2014. 15(7): p. 747–56. [DOI] [PubMed] [Google Scholar]
- 11.Yang D, et al. , Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA, 2011. 306 (14): p. 1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceccaldi R, et al. , A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res, 2015. 75(4): p. 628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vijai J, et al. , A Recurrent ERCC3 Truncating Mutation Confers Moderate Risk for Breast Cancer. Cancer Discov, 2016. 6(11): p. 1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latimer JJ, et al. , Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer. Proc Natl Acad Sci U S A, 2010. 107(50): p. 21725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latimer JJ, et al. , Cell-type-specific level of DNA nucleotide excision repair in primary human mammary and ovarian epithelial cell cultures. Cell Tissue Res, 2008. 333(3): p. 461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dreze M, et al. , Monitoring repair of UV-induced 6–4-photoproducts with a purified DDB2 protein complex. PLoS One, 2014. 9(1): p. e85896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scrima A, et al. , Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell, 2008. 135(7): p. 1213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cleaver JE, Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer, 2005. 5(7): p. 564–73. [DOI] [PubMed] [Google Scholar]
- 19.Bradford PT, et al. , Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet, 2011. 48(3): p. 168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleaver JE, Lam ET, and Revet I, Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet, 2009. 10(11): p. 756–68. [DOI] [PubMed] [Google Scholar]
- 21.Hu J, et al. , Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci U S A, 2017. 114(26): p. 6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fei J, et al. , Regulation of nucleotide excision repair by UV-DDB: prioritization of damage recognition to internucleosomal DNA. PLoS Biol, 2011. 9(10): p. e1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luijsterburg MS, et al. , Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci, 2007. 120(Pt 15): p. 2706–16. [DOI] [PubMed] [Google Scholar]
- 24.Abaan OD, et al. , The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res, 2013. 73(14): p. 4372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barretina J, et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fabregat A, et al. , The Reactome Pathway Knowledgebase. Nucleic Acids Res, 2018. 46(D1): p. D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nik-Zainal S, et al. , Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature, 2016. 534(7605): p. 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maruvka YE, et al. , Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat Biotechnol, 2017. 35(10): p. 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le DT, et al. , PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med, 2015. 372(26): p. 2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willers H, et al. , Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res, 2009. 7(8): p. 1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill SJ, et al. , Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov, 2018. 8(11): p. 1404–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neve RM, et al. , A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell, 2006. 10(6): p. 515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamentsky L, et al. , Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics, 2011. 27(8): p. 1179–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.