

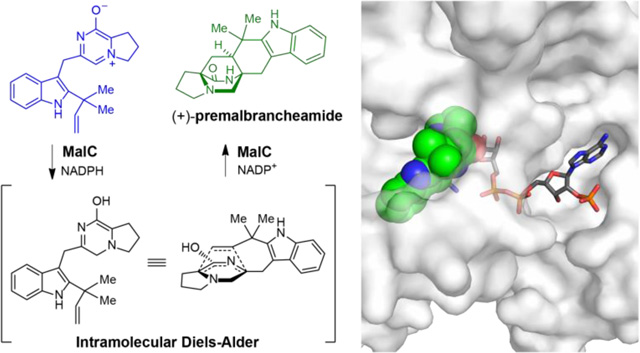
Abstract
Prenylated indole alkaloids, such as the calmodulin inhibitory malbrancheamides and anthelmintic paraherquamides possess great structural diversity and pharmaceutical utility. Here we report complete elucidation of the malbrancheamide biosynthetic pathway accomplished through complementary approaches. These include a biomimetic total synthesis to access the natural alkaloid and biosynthetic intermediates in racemic form, and in vitro enzymatic reconstitution that provides access to the natural antipode (+)-malbrancheamide. Reductive cleavage of a l-Pro-l-Trp dipeptide from the MalG nonribosomal peptide synthetase (NRPS) followed by reverse prenylation and a cascade of post-NRPS reactions culminates in an intramolecular [4+2] hetero-Diels-Alder (IMDA) cyclization to furnish the bicyclo[2.2.2]diazaoctane scaffold. Enzymatic assembly of optically pure (+)-premalbrancheamide involves an unexpected zwitterionic intermediate where MalC catalyzes enantioselective cycloaddition as a bifunctional NADPH-dependent reductase/Diels-Alderase. Crystal structures of substrate and product complexes together with site-directed mutagenesis and molecular dynamics simulations demonstrate how MalC and PhqE, its homolog from the paraherquamide pathway, catalyze diastereo- and enantioselective cyclization in the construction of this important class of secondary metabolites.
Graphical Abstract
Introduction
Prenylated indole alkaloids comprised of the bicyclo[2.2.2]diazaoctane core have attracted considerable interest due to their wide spectrum of biological activities and offer compelling targets for chemical synthesis and biosynthetic studies.1–3 Among them, 2-deoxy-paraherquamide A (derquantel) is a commercial therapeutic agent for treating parasitic nematodes in sheep.4–6 It is now clear that in various genera of fungi, two distinct families containing a bicyclo[2.2.2]diazaoctane system have evolved: (1) the diketopiperazines (DKPs) such as the anti-cancer stephacidins, insecticidal brevianamides and cytotoxic notoamides, and (2) the monoketopiperazines (MKPs), including the anthelmintic paraherquamides, asperparalines and calmodulin-inhibitory malbrancheamides (Fig. 1a, 1-5). In addition, the citrinadins represent another related series of alkaloids that are thought to be derived by deconstruction of monoketopiperazine progenitors containing the [2.2.2] ring system7,8 (Supplementary Fig. 1).
Figure 1. Fungal bicyclo[2.2.2]diazaoctane indole alkaloids and biosynthesis.

a. Representative natural products with the bicyclo[2.2.2]diazaoctane group colored in red. b. Scheme of malbrancheamide biosynthesis: the MalG NRPS performs reductive offloading of coupled natural substrates l-proline and l-tryptophan to produce the terminal aldehyde 6. This compound undergoes spontaneous cyclization and dehydration to give dienamine 8, which is reverse prenylated by MalE. Compound 9 undergoes spontaneous oxidation to prenylated zwitterion 11. MalC is a bifunctional enzyme which performs reduction and stereoselective [4+2] cycloaddition to furnish (+)-1. The final product malbrancheamide (+)-2 is generated by the MalA halogenase. The products from key biosynthetic steps are colored differently. Proteins are indicated by spheres; MalG domains are adenylation (A1 and A2), thiolation (T1 and T2), condensation (C) and reductase (R).
The bicyclo[2.2.2]diazaoctane core of these metabolites was first proposed in 1970 to arise from an intramolecular [4+2] Diels-Alder (IMDA) reaction.9 A long-held hypothesis assumed that both the DKP and MKP families shared a common biogenesis, with the tryptophan carbonyl of the latter family involved in a net four-electron reduction subsequent to a putative Diels-Alder construction.3,10 Based on initial genetic studies,11 and experimental corroboration described in this report, we have discovered that Nature employs divergent biogenetic pathways and biochemical mechanisms to generate the bicyclo[2.2.2]diazaoctane nucleus in these two distinct families of alkaloids11–15 (Supplementary Fig. 1). In the DKP-producing systems, the first enzyme is a nonribosomal peptide synthetase (NRPS) with a terminal condensation domain, responsible for production of a cyclo-Pro-Trp DKP. In contrast, analysis of the malbrancheamide and paraherquamide MKP biosynthetic gene clusters suggested that the NRPS terminal domain is a reductase that catalyzes off-loading of a reduced Pro-Trp leading to a monoketopiperazine intermediate11,15 (Fig. 1b, 6-12). Next, the monoketopiperazine intermediate is reverse prenylated, with a new carbon-carbon bond formed between Trp C2 and the C3 atom of the prenyl group. We hypothesized that an intramolecular [4+2] Diels-Alder reaction follows, producing the bicyclo[2.2.2]diazaoctane ring system. However, annotation of the mal and phq gene clusters11 failed to reveal a candidate enzyme for the IMDA reaction. The putative cycloaddition is stereospecific based on the syn- or anti- configuration of C12a (labeling in premalbrancheamide (1), Fig. 1a) and the relative position of the diene and the dienophile. Antipodal bicyclo[2.2.2]diazaoctanes have been isolated from different fungal strains producing either of the indole alkaloid families,16 while only (+)-malbrancheamide ((+)-2) has been isolated from Malbranchea aurantiaca,17 indicating strict diastereo- and enantioselectivity of the biosynthetic IMDA. Thus, the identification and characterization of this presumed catalytic step is fundamental for understanding the formation of these structurally diverse molecules.
Reports of Diels-Alderases remain rare, with few examples over the past decade.18–24 Among them, five crystal structures have been reported including, 1) the S-adenosyl-l-methionine (SAM)-dependent methyltransferase SpnF18,25 and LepI,22,29 2) the β-barrel protein PyrI426 and its homolog AbyU,27 and 3) the flavin-dependent enzyme PyrE3.28 A common theme in these Diels-Alderases is their apparent evolution from divergent ancestors, with evident active site reconfiguration. Accordingly, in all reported cases these enzymes have lost ancestral function and the sole remaining activity facilitates a spontaneous [4+2] pericyclic reaction with regio- and stereoselectivity. Cofactors, if present, do not serve their canonical catalytic role for the Diels-Alder cycloaddition, but rather play a structural role in maintaining the active site in a catalytically productive conformation. Similarly, distinct catalytic residues that abolished the enzymatic function were not identified in any previously characterized Diels-Alderase, suggesting that catalysis is achieved primarily through substrate positioning in the protein scaffold. The malbrancheamide and paraherquamide gene clusters lack homologous genes that encode known Diels-Alderases, which indicated the existence of a novel class of biocatalysts. In this article, we reveal the molecular basis for stereocontrolled construction of the monoketopiperazine bicyclic core in the malbrancheamide and paraherquamide biosynthetic pathways. These genetically homologous systems proceed through a bifunctional reductase and Diels-Alderase that evolved from an ancestral short-chain dehydrogenase (SDR) and is also encoded in several other fungal natural product biosynthetic gene clusters.
Results and Discussion
Biomimetic Synthesis of Premalbrancheamide, Malbrancheamide and Spiromalbramide
Early considerations regarding biogenesis of the bicyclo[2.2.2]diazaoctane core envisioned a biosynthetic Diels-Alder reaction. In order to chemically validate the sequence of events in malbrancheamide biosynthesis, we prepared the C2 reverse prenylated proposed biosynthetic intermediate, dipeptide aldehyde (17),11 and found that this substance undergoes the cascade of ring closure, dehydration, tautomerization and intramolecular cycloaddition, upon deprotection to give premalbrancheamide (1) (Fig. 2). This strategy was applied to two additional natural products, malbrancheamide (2) and spiromalbramide (4) (Supplementary Fig. 2), underscoring the utility of the biomimetic paradigm. The key, Fmoc-protected dipeptide aldehyde 17 was prepared through the peptide coupling of N-Fmoc proline (14) with the C2 reverse prenylated tryptophan methyl ester 13 through the agency of HATU in acetonitrile in 85% yield. Reduction of the methyl ester with sodium borohydride (82% to 15) followed by a Parikh-Doering oxidation, furnished the N-Fmoc aldehyde 17 in 72% yield. Removal of the N-Fmoc residue with diethylamine under anaerobic conditions provided the di-enamine 9, which could be isolated and characterized. Treatment of this substance with TFA in THF at temperatures between 0 °C and 50 °C, resulted in formation of (±)–1. The observed modest yield is possibly due to unfavorable tautomerization of 9 to 12. Under aerobic conditions, 9 spontaneously and rapidly oxidized to aromatic zwitterion 11 (Fig. 1b), which we initially reasoned to be a non-physiological byproduct. We later determined that 11 could be chemically reduced by NAD(P)H to 12, resulting in the spontaneous formation of racemic premalbrancheamide in 76% yield. This discovery suggested two possible biosynthetic routes ‒ aerobic vs. anaerobic ‒ and the possibility that 11 is an authentic pathway intermediate depending on the intracellular conditions during fungal biosynthesis. Significantly, both routes lead to only the syn-diastereomers upon cyclization as the corresponding anti-isomers was not detected in even trace amounts from the cycloaddition reactions. This finding agrees with density functional theory calculations that the azadiene 12 has a calculated relative transition state energy difference of about 2.6 kcal/mol favoring the syncycloadduct.30 The anti-pathway experiences unfavorable steric interactions between the pyrrolidine ring and the prenyl group (Supplementary Fig. 3). Our biomimetic synthesis of syn-malbrancheamides gave rise to the (+)- and (−)- enantiomers, raising the intriguing question regarding how optically pure (+)-premalbrancheamide is formed by Malbranchea aurantiaca. This further indicated the likely presence of an enzyme-directed cyclization in vivo, and motivated us to explore the biosynthetic origins of premalbrancheamide by in vitro pathway reconstitution.
Figure 2. Biomimetic synthesis of premalbrancheamide.

The biomimetic synthesis proceeded through a spontaneous intramolecular [4+2] Diels-Alder reaction from key azadiene intermediate 12 to produce a racemic mixture of syn-premalbrancheamides (1). Zwitterion 11 arises from spontaneous oxidation of 9 and was initially reasoned to be a non-physiological by-product. After discovering that 11 was the preferred substrate of MalC, non-enzymatic chemical reduction by NADH was explored providing 1 in 76% yield from 11. Only optically pure (+)-1 has been isolated from Malbranchea aurantiaca. See SI for complete methods.
In Vitro Reconstitution of the Malbrancheamide Biosynthetic Pathway
We aimed to reconstitute the biosynthesis of malbrancheamide as a representative monoketopiperazine alkaloid in a multi-component in vitro reaction (Supplementary Fig. 4). We hypothesized the first step of malbrancheamide biosynthesis involves coupling of l-proline and l-tryptophan by MalG, a dimodular NRPS containing six domains (A1-T1-C-A2-T2-R, Fig. 1b), to produce l-Pro-l-Trp aldehyde 6 through reductive off-loading. Since the full-length NRPS protein could not be produced in soluble form, we identified domain boundaries in MalG, developed expression constructs for the excised A1-T1, C, T2 and R domains (Supplementary Fig. 5), and loaded the putative amino acid substrates onto the MalG T1 and T2 domains (Supplementary Fig. 6). Phosphopantetheinylated MalG A1-T1 was loaded with l-proline in the presence of ATP and Mg2+, consistent with our functional annotation. With no access to soluble MalG A2, l-tryptophan was loaded onto MalG T2 using Sfp,31 a nonspecific 4’-phosphopantetheinyl transferase, and l-Trp-coenzyme A (CoA). l-Pro A1-T1 and l-Trp T2 were incubated with the MalG C domain and R domain with the presumed NADPH cofactor. Product formation was determined by LC/MS and comparison with authentic standards. Instead of the proposed dipeptidyl aldehyde-derived product 8, we identified aromatic zwitterion 10 as the main product (Fig. 1b and 3a). We hypothesized that 10 was produced from spontaneous oxidation of 8. This was confirmed by chemical synthesis of 8, which spontaneously and irreversibly converted to 10. This transformation was suppressed under anaerobic conditions, leading to the conclusion that the malbrancheamide NRPS product rapidly cyclized and dehydrated to 8 and subsequently spontaneously oxidized to 10 under aerobic conditions.
Figure 3. In vitro enzymatic reconstitution of malbrancheamide biosynthesis.

Reactions were monitored by LC/MS. Extracted ion counts (EIC) for key molecules in reaction mixtures are compared to authentic synthetic standards. a. MalG NRPS (excised A1-T1, C, T2, R domains) produced zwitterion 10 by spontaneous oxidation of 8. b – c. Addition of MalE or MalB prenyltransferase formed three products: a prenylated zwitterion 11, and (±)-1. d. MalC Diels-Alderase addition disabled formation of 11 and (−)-1 (see panel f). e. Malbrancheamide 2, the final pathway product, was produced by MalA halogenation of (+)-1. f. Chiral separation of (±)-1 indicates that MalC is an intramolecular [4+2] Diels-Alderase, while neither MalE nor MalB provide enantioselectivity for the spontaneous IMDA reaction. g. MalC-catalyzed reactions under aerobic (11 + MalC) or anaerobic (9 + MalC) conditions. The aerobic route with 11 as the pathway intermediate was more efficient than the anaerobic route from 9. h. Effect of cofactor on the enantiomeric excess of the MalC-catalyzed Diels-Alder reaction. MalC provided limited enantioselectivity when NADH was used as cofactor. EIC traces are colored by compound as in Figure 1b, authentic standards are in purple or pink. For panel g and h, all data represent the average of triplicate independent experiments (center values, mean; error bars, SD; n = 3).
To further test the hypothesis that the MalG terminal R-domain catalyzes an NADPH-dependent two-electron reductive release to produce an aldehyde (as opposed to a 4-electron reduction leading to an alcohol), we synthesized a dipeptidyl-CoA analog 23, in which the prolyl-N-atom was replaced with an O-atom to prevent nucleophilic addition of the prolyl-N-atom to the CoA thioester, and loaded 23 onto MalG T2 via Sfp (Supplementary Fig. 6). Product standards of aldehyde 24 and alcohol 25 were synthesized chemically. Compound 25 was nonreactive in methanol, while 24 epimerized and reacted to produce the hemiacetal 26 (Supplementary Fig. 7). Assays with 23-loaded T2 and MalG R yielded product 26, confirming that MalG generates an aldehyde product. NADPH was the preferred cofactor in this reaction (Supplementary Fig. 7). MalG R is an SDR reductase with catalytic Tyr and Lys amino acids, as demonstrated in the 2.6-Å crystal structure of an NADPH complex of PhqB R, the MalG R homolog of paraherquamide biosynthesis (Supplementary Fig. 8–10). The essential role of Tyr was confirmed with MalG R/Y2132F, which was incapable of reductive release (Supplementary Fig. 10).
We propose that the NRPS product 8 would undergo a reverse prenylation as the next biosynthetic step, thereby installing the dienophile for the IMDA reaction. Two genes, malE and malB (from the mal gene cluster) encode putative prenyltransferases. We incubated MalB or MalE with substrate-loaded MalG domains, NADPH and dimethylallyl pyrophosphate (DMAPP), the prenyl donor. MalE readily catalyzed a C2 reverse prenyl transfer reaction to produce zwitterion 11 (Fig. 3b), whereas MalB displayed modest activity, suggesting that malB may be a redundant gene in the pathway (Fig. 3c and Supplementary Fig. 14). Because we could not distinguish in this assay whether 8 or 10 was the MalE substrate, synthetic 8 was produced under anaerobic conditions by UV irradiation of an O-nitrobenzyl (ONB) photo-protected dipeptide aldehyde 30 and subjected to the prenyltransferase assay (Supplementary Fig. 11). This substrate was rapidly prenylated by MalE in contrast to synthetic 10, which showed low levels of conversion with the enzyme, indicating that 8 is the native substrate for MalE (Supplementary Fig. 12). This raised the question regarding how MalE accesses substrate 8 prior to its oxidation to 10. Thus, we considered the possibility that C2 reverse prenylation occurs with the substrate tethered to the NRPS T2 domain. To address this question, we tested whether MalE or MalB could prenylate l-Trp, l-Trp-loaded MalG T2, or 23-loaded MalG T2 (Supplementary Fig. 13). In all cases, no product was detected, confirming that the prenyl transfer reaction occurs on free substrate following the NRPS-catalyzed reaction. It is evident that some process is employed in vivo to ensure access to the reduced substrate for prenylation.
We noticed low levels of premalbrancheamide in the reconstitution assays with MalG and MalE or MalB. Chiral LC/MS analysis revealed a 1:1 racemic mixture of (±)-1 (Fig. 3f), in agreement with the biomimetic synthesis described above (Fig. 2). Further investigation using synthetic 11 revealed that racemic premalbrancheamide arose through non-enzymatic reduction of 11 by NADPH to azadiene 12, which undergoes spontaneous cycloaddition in the reaction buffer, thereby explaining the background accumulation of the Diels-Alder products (±)-1 from in vitro assays. From these studies we ascertained that MalG and MalE are the minimal components required for premalbrancheamide biosynthesis, albeit lacking stereocontrol in the IMDA reaction.
Premalbrancheamide isolated from Malbranchea aurantiaca is optically pure (+)-1, which strongly implicates enzymatic control in the IMDA reaction. Known Diels-Alderases have diverse origins, but the annotated mal and phq gene clusters did not contain an evident candidate biosynthetic enzyme. Nonetheless, we tested whether MalC, annotated as a short-chain dehydrogenase/reductase (SDR), could function as the presumed Diels-Alderase. When MalC was incubated with substrate-loaded MalG and MalE (NADPH and DMAPP included), neither aromatic zwitterion intermediate, 10 or 11, was detected; instead the sole product was (+)-1, confirming that MalC functions as an intramolecular [4+2] Diels-Alderase (Fig. 3d and 3f). To our surprise, when MalC was added to the reaction mixture after significant amounts of 11 had accumulated, the oxidized intermediate was converted to (+)-1, indicating that MalC possessed the ability to reduce the zwitterion 11 to the reactive azadiene 12 prior to conducting the diastereo- and enantio-controlled cycloaddition reaction. This unexpected reactivity of MalC was confirmed using synthetic 11 and NADPH (Supplementary Fig. 15). To our knowledge, this is a unique example where reduction regenerates the biosynthetic substrate from an oxidized (aromatic) intermediate to provide a productive mode for cycloaddition. The fact that 11 is a MalC substrate indicates that it is an authentic pathway intermediate, and motivated us to address whether an aerobic or anaerobic biosynthetic route is operative in vivo. This question was interrogated in two ways: first by performing MalC assays under anaerobic conditions with synthetic 9, which was generated by photo-deprotection of ONB prenyl dipeptidyl aldehyde 33 (Supplementary Fig. 11). Conversion to (+)-1 was observed only in the presence of MalC and NADP+ (Fig. 3g). However, the efficiency of this reaction was attenuated compared to the MalC-catalyzed conversion of 11 to (+)-1 under aerobic conditions, indicating that the dienamine tautomer 9 is not optimally recognized by MalC. It is unknown whether MalC can play a role in tautomerization of 9; notably, background conversion of 9 to racemic 1 was not detected under these conditions. Second, gene disruption of the malC homolog phqE was conducted in the paraherquamide-producing strain Penicillium simplicissimum using a CRISPR-Cas9 system.32 Extracts from the phqE mutant strain grown on CYA medium showed the presence of the expected (methyl-Pro-Trp prenyl) zwitterion intermediate 38 (Supplementary Fig. 16) by LC/MS analysis and co-injection with a synthetic standard, confirming the accumulation of this oxidized metabolite in vivo (Supplementary Fig. 17). Taken together, these data demonstrate that 11 is the native substrate for MalC en route to (+)-1.
For the MalC-catalyzed reduction of 11, either NADH or NADPH is effective as the cofactor. However, NADPH is required for strict stereocontrol of the IMDA reaction, as MalC produced a 63:37 mixture of (+)-1 and (−)-1 when using NADH (Fig. 3h). This is consistent with the anaerobic experiment in which NADP+ was required to generate (+)-1, and further indicates that NADPH plays an important role in the IMDA stereocontrol. The Michaelis-Menten kinetic constants for NADPH and NADH in reactions with MalC and 11 revealed a 10-fold greater catalytic efficiency (kcat/KM) with NADPH compared to NADH (Supplementary Fig. 15). A 6-fold greater KM with NADH also suggests that proper cofactor binding is a required component to achieve stereocontrol. Enzymatic rate enhancement of (+)-1 formation is evident under both assay conditions: aerobically through substrate 11 (post-reduction), or anaerobically through substrate 9 (post-tautomerization) (Fig. 3g). The dramatic shift in enantiomeric excess for the enzymatic reaction (from 0% to 96%) is indicative of enzymatic catalysis for the IMDA reaction.
To complete the biosynthetic pathway, flavin-dependent halogenase MalA was employed to add chlorine atoms on C8 and C9 of premalbrancheamide (+)-1 to provide malbrancheamide (+)-2.33 We incubated MalA with its pathway partners (l-Pro and l-Trp MalG, MalE and MalC, NADPH, DMAPP, NaCl and FADH2) and identified (+)-2 as the final product (Fig. 3e). We also found that MalA is stereoselective: when incubated with racemic mixture of 1, MalA chlorinated only the natural (+) enantiomer (Supplementary Fig. 18).
Probing the Catalytic Mechanism of the Bifunctional Diels-Alderase
To gain further insight into the function of the SDR-derived Diels-Alderase, the crystal structure of ligand-free MalC was solved at 1.6-Å resolution, revealing a classical SDR fold with a nucleotide-binding subdomain that contains an invariant “TGX3GXG” motif (P-loop), and a C-terminal substrate binding region that is less conserved and largely hydrophobic.34,35 The closest structural homologs are a group of SDRs including RasADH (2.3 Å Cα R.M.S.D., 27% overall sequence identity),36 which uses NAD(P) to catalyze reversible oxidation of secondary alcohols to aldehydes. In RasADH, a catalytic Tyr serves as the proton donor and a catalytic Lys facilitates proton transfer. Unexpectedly, MalC lacks the characteristic Tyr and Lys catalytic amino acids, and also the essential Asn and Ser residues of typical SDRs,34,35 suggesting that the active site is reconfigured to fit its unique catalytic roles (Supplementary Fig. 24).
Neither cofactor nor substrate was captured in complex with MalC due to crystal lattice constraints in which the active site was occluded by a neighboring tetramer. Thus, we turned to the homologous paraherquamide biosynthetic pathway. PhqE is a MalC homolog (54% identity) and catalyzed formation of (+)-premalbrancheamide using the zwitterionic prenylated Trp-Pro substrate 11 in vitro (Supplementary Fig. 19). The 2.4-Å crystal structure of PhqE in complex with cofactor (NADP+/NAD+) and premalbrancheamide (Supplementary Fig. 20) is highly similar to the MalC structure (1.0 Å Cα R.M.S.D.; Fig. 4b).
Figure 4. Structures of MalC and PhqE.

a. MalC tetramer colored by subunit. b. Superposition of MalC and PhqE product complex (gray); NADP+ (black C) and premalbrancheamide (green C) are shown as spheres. c. Active site of PhqE·(+)-1·NADP+ complex showing close arrangement of the product and the cofactor. d. Omit electron density (Fo-Fc; contoured at 2.2 σ) for the substrate 11 (cyan) in the PhqE/D166N·11·NADP+ complex structure. e. Omit electron density (Fo-Fc; contoured at 2.2 σ) for the product 1 (green) in the PhqE·(+)-1·NADP+ complex structure. f. Pre-organization for cycloaddition. Substrate 11 binds with the prenyl group poised for the IMDA (dashed lines) in the substrate complex g. Overlay of 11 and premalbrancheamide (+)-1 from the product complex. h. Surface representation of the product complex showing high shape complementarity between premalbrancheamide and PhqE.
Consistent with our kinetic data, PhqE crystals grown with NADP+ showed strong electron density for the cofactor bound in a manner conserved with bacterial SDR homologs, while NAD+ showed weak electron density (Supplementary Fig. 21). Lys50 accounts for preferential binding of NADP+ through a salt bridge with the cofactor 2’-phosphate. Premalbrancheamide binds in a surface groove and is surrounded by hydrophobic residues. The bicyclo[2.2.2]diazaoctane ring system is buried against the nicotinamide and several amino acids including Arg131 (Fig. 4c). The indole lies in a pocket formed by Ala, Leu and Val side chains. The gem-dimethyl contacts Asp166 and Trp169, which are part of a “PDPGW” motif unique to these bifunctional enzymes and not present in other SDRs (Supplementary Fig. 24–25). Given the dual functions (reductase and Diels-Alderase) of MalC/PhqE, the product complex reveals PhqE to be well-adapted in its capacity as a stereo- and enantioselective biocatalyst due to the shape complementarity between the active site pocket and the product (Fig. 4f). Additionally, the short distance (~4 Å) between nicotinamide C4 and the deoxy C5 of premalbrancheamide suggests that reduction to the reactive azadiene also occurs in the same location of the active site and indicates that reduction and cycloaddition are highly coordinated.
We sought a non-reactive substrate complex with NADP+ and 11, and obtained strong electron density for 11 with indication of a flexible orientation in PhqE/D166N (Fig. 4d), whereas the wild-type PhqE yielded ambiguous density for 11. The indole of 11 binds in the same pocket as premalbrancheamide while the pyrazinone is pushed towards the nicotinamide (Fig. 4e). Deoxy C5 of 11 lies 3.6 Å from the nicotinamide C4 consistent with hydride delivery to this position. In addition to hydride transfer, protonation of the pyrazinone alkoxide is required to form the reactive azadiene 12. Curiously the corresponding oxygen atom is part of a hydrogen-bonding network involving the NADP+ 2’-hydroxyl and Arg131, suggesting that the cofactor may play a role in proton transfer during reduction. Superposition of the substrate and product complexes revealed a high degree of pre-organization of 11 towards the Diels-Alder reaction and also affirmed that the reduction and IMDA reactions are spatially confined.
We used molecular dynamics (MD) simulations to explore the concepts of coordinated hydride delivery, proton transfer and pre-organization in the active site. First, we monitored the distance between the putative hydride acceptor (C5 of 11) and nicotinamide C4 over a 1.2 μs simulation. The average distance between these carbon atoms was 4.2 Å, consistent with the crystal structures (Supplementary Fig. 22). We next explored alkoxide protonation. In the crystal structure, the NADP+ ribose 2’-hydroxyl is hydrogen-bonded to the alkoxide oxygen and Arg131. Within the first few ns of the simulation Arg131 displaces the ribose hydroxyl and interacts with the alkoxide for the remainder of the simulation, lending significance to the role of Arg131 in protonation of 11. Notably, Asp131 also interacts with D109 during the simulation, supporting the proton donor role for Arg131 by lowering its pKa and allowing access to bulk solvent (Supplementary Fig. 22). To assess facial selectivity in the cycloaddition reaction that forms the (+)- and (−)-premalbrancheamide enantiomers, we monitored the dihedral angle along N15-C5a-C12a-C13 (Supplementary Fig. 23). Comparison of this dihedral angle in the constrained premalbrancheamide and unconstrained 11 revealed that the untethered diene explores only a single face of the pyrazinone ring corresponding to the natural (+)-enantiomer (Supplementary Fig. 23). Interestingly, this pre-organization was lost when NADP+ was omitted and the simulation was performed with substrate 12, consistent with our observation that the cofactor is required for enantio-controlled cycloaddition. Together these results further support the conclusion that MalC/PhqE-catalyzed reduction and cycloaddition are coordinated and take place in the same active site pocket where the enzyme-cofactor complex provides stereocontrol by positioning the diene for [4+2] cycloaddition as the reactive azadiene is generated by reduction of 11.
Based on this information, we probed the reaction mechanism by site-directed mutagenesis. MalC was chosen for this analysis to directly compare results with the reconstitution assay. All of the targeted amino acids are conserved in MalC and PhqE. With the in vitro reconstitution assay, MalC variants were assayed in the presence of the MalG NRPS and MalE prenyltransferase (Fig. 5a, b). Reductase activity was assessed by the levels of oxidized intermediate 11: higher levels indicate less reductase activity. The effect on the IMDA reaction was determined by measuring levels of unnatural (−)-1 as a percent of all premalbrancheamide: ~50% (−)-1 formation indicates loss of enzymatic IMDA function. We identified five MalC substitutions that greatly diminished reductase activity (D108A: 17% of wild type levels, R130A: 19%, D165A: 0%, D165N: 6%, W168L: 16%) (Supplementary Fig. 24), and found that loss of function is highly correlated with loss of stereocontrol in the IMDA reaction. A single exception is MalC D165A, which produced mainly (+)-1, suggesting that Asp165 is required for reduction but not the IMDA reaction. The activity of MalC variants was also measured in assays with synthetic 11 (Fig. 5c). In agreement with the reconstitution assay, Asp108, Arg130 and Asp165 were required for reduction (D108A: 8% of wild type levels, R130A: 8%, D165A: 3%, D165N: 1%). Based on these data, we propose a mechanism for MalC in which Arg130 serves as a proton donor potentially in conjunction with 2’-OH of NADPH ribose (Fig. 5d). NADPH is the hydride donor, and Asp165 may stabilize the positive charge of the zwitterionic substrate 11 and facilitate formation of the reactive azadiene intermediate 12. Asp108 interacts with Arg130, allowing access to bulk solvent. Stereocontrol of the IMDA reaction is primarily driven by shape complementarity, with contacts between substrate and Trp168 and the cofactor playing a critical role in the IMDA process.
Figure 5. Catalytic mechanism of the MalC/PhqE-catalyzed Diels-Alder reaction.

a – b. Profiles of MalC substrate 11 (blue), MalC product (+)-1 (dark green), and (−)-1 (light green) in the “MalG+MalE+MalC” reconstitution assay. c. MalC product formation assessed by conversion of synthetic 11 to (+)-1. The results agree with those of the reconstitution assay in panel b. Product levels due to non-enzymatic conversion by NADPH were subtracted in all cases. All data represent the average of triplicate independent experiments (center values, mean; error bars, SD; n = 3). d. Proposed catalytic mechanism for MalC/PhqE, with residue numbers for MalC (PhqE residue number = MalC residue number + 1). Arg130 is the indirect proton donor, possibly mediated via the 2’-OH of NADPH ribose. Arg130 forms a salt bridge with Asp108, which is accessible to bulk solvent. Asp165 stabilizes the positive charge of 11, and hydride transfer from NADPH completes the first reduction step, forming an unstable azadiene intermediate. The subsequent IMDA reaction is accelerated primarily via entropy trapping, with diastereo- and enantioselectivity achieved via close packing of the NADP+ nicotinamide, the azadiene and MalC Trp168, which together restrain the conformations of both the diene ring and the dienophile to ensure a single cycloaddition mode.
The MalC/PhqE Diels-Alderases clearly evolved from an ancestral SDR but belong to a remote fungal sub-network of extant SDRs (Supplementary Fig. 24–26). Interestingly, genes encoding these IMDAs are generally linked to genes for Pro-Trp NRPS with reductive offloading domains. The SDR catalytic Tyr and Lys were replaced by shorter, non-polar residues (Ile and Cys) providing space to accommodate the substrate. The “PDPGW” motif positions the essential Asp165 3.0 Å closer to the substrate compared to the corresponding amino acid in canonical SDRs (Supplementary Fig. 24). The SDR hydrogen bonding network is partially maintained since the catalytic Arg side chain of MalC/PhqE occupies the position of the SDR catalytic Lys, providing a compelling example of protein evolution in molecular detail.
Conclusions
Our comprehensive approach to studying the Diels-Alder mediated construction of bicyclo[2.2.2]diazaoctane indole alkaloids represents a culmination of conceptual, experimental and computational studies initiated almost a half-century ago by Birch.37 The divergent biogenesis to create the monoketo- and diketopiperazine-type molecules employed by diverse fungi was revealed through characterization of the respective biosynthetic gene clusters, which suggested a differential release mechanism from the functionally related bimodular NPRS systems (Supplementary Fig. 1). This information was leveraged to design a biomimetic total synthesis of premalbrancheamide, providing a direct validation of the prenylated dipeptide azadiene intermediate and IMDA construction of the target natural product in racemic form. The basis for creating the (+)-antipodal form of premalbrancheamide via a presumed stereoselective Diels-Alderase motivated our search for the corresponding enzyme from the Mal and Phq pathways, resulting in identification of a novel Diels-Alderase and a mechanistic understanding of enantio-induction. We have demonstrated that during biosynthetic assembly, the key step to produce the polycyclic core is catalyzed by a bifunctional reductase and intramolecular [4+2] Diels-Alderase, MalC/PhqE, providing exquisite diastereo- and enantiocontrol in cooperation with reductive offloading of a dipeptide aldehyde from the NRPS. Derived from SDR ancestors, the active site of MalC/PhqE evolved to accommodate an aromatic zwitterion substrate, and both the reduction and the IMDA steps are NADP(H)-dependent. In contradistinction to all other known putative Diels-Alderases2 which are either redox-neutral cyclases or oxidases, we have discovered the first reductase-dependent Diels-Alderase. Our work reveals a distinct class of Diels-Alder enzymes and provides insights into the nature of IMDA catalysis as well as providing a bold evolutionary thesis. The availability of key intermediates provided from biomimetic synthesis enabled us to probe the molecular mechanism of this transformation in unprecedented detail. The MalC/PhqE-catalyzed reaction includes the remarkable step of “rescuing” an aromatic zwitterionic substrate 11 to create the bicyclic product 1 and avoid premature pathway termination. This brilliant evolutionary solution to protect the structural and stereochemical integrity of this architecturally unique family of alkaloids is, to the best of our knowledge, unprecedented and underscores the expanding plasticity and adaptability of secondary metabolite genes and enzymes. The Mal/Phq biosynthetic pathway represents a novel “toolkit” for chemoenzymatic diversification of indole alkaloids with opportunities for facile access to improved calmodulin inhibitors, anthelmintics and other therapeutics to treat human and animal diseases.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health R01 CA070375 (to R.M.W. and D.H.S.), R35 GM118101, the Hans W. Vahlteich Professorship (to D.H.S.), and R01 DK042303 and the Margaret J. Hunter Professorship (to J.L.S.). J.N.S. and K.N.H. acknowledge the support of the National Institute of General Medical Sciences of the National Institutes of Health under Award Numbers F32GM122218 (to J.N.S.) and R01GM124480 (to K.N.H.). Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). Anton 2 computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research. GM/CA@APS is supported by the National Institutes of Health, National Institute of General Medical Sciences (AGM-12006) and National Cancer Institute (ACB-12002). We thank Prof. Stephen Ragsdale for his assistance with anaerobic enzyme assays and Prof. Pavel Nagorny for his assistance with polarimetry measurements.
Footnotes
Data Availability
Coordinates and associated structure factors have been deposited with the PDB under accession codes 6NKH (MalC), 6NKI (PhqB R·NADPH), 6NKK (PhqE·1·NADP+) and 6NKM (PhqE D166N·11·NADP+).
Author Information The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
REFERENCES
- 1.Finefield JM, Frisvad JC, Sherman DH & Williams RM Fungal origins of the bicyclo[2.2.2]diazaoctane ring system of prenylated indole alkaloids. J Nat Prod 75, 812–833 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klas K, Tsukamoto S, Sherman DH & Williams RM Natural Diels-Alderases: elusive and irresistable. J Org Chem 80, 11672–11685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klas KR et al. Structural and stereochemical diversity in prenylated indole alkaloids containing the bicyclo[2.2.2]diazaoctane ring system from marine and terrestrial fungi. Nat Prod Rep 35, 532–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson AP et al. Paraherquamide and 2-deoxy-paraherquamide distinguish cholinergic receptor subtypes in ascaris muscle. J Pharmacol Exp Ther 303, 853–860 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Little PR et al. Efficacy of a combined oral formulation of derquantel-abamectin against the adult and larval stages of nematodes in sheep, including anthelmintic-resistant strains. Vet Parasitol 181, 180–193 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Buxton SK et al. Investigation of acetylcholine receptor diversity in a nematode parasite leads to characterization of tribendimidine-and derquantel-sensitive nAChRs. Plos Pathog 10, e1003870 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mugishima T et al. Absolute stereochemistry of citrinadins A and B from marine-derived fungus. J Org Chem 70, 9430–9435 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Mercado-Marin EV et al. Total synthesis and isolation of citrinalin and cyclopiamine congeners. Nature 509, 318–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter AEA & Sammes PG A Diels-Alder reaction of possible biosynthetic importance. J Chem Soc Chem Comm, 1103 (1970). [Google Scholar]
- 10.Stocking EM & Williams RM Chemistry and biology of biosynthetic Diels-Alder reactions. Angew Chem Int Ed Engl 42, 3078–3115 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Li S et al. Comparative analysis of the biosynthetic systems for fungal bicyclo[2.2.2]diazaoctane indole alkaloids: the (+)/(−)-notoamide, paraherquamide and malbrancheamide pathways. MedChemComm 3, 987–996 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stocking EM, Sanz-Cervera JF & Williams RM Studies on the biosynthesis of paraherquamide: synthesis and incorporation of a hexacyclic indole derivative as an advanced metabolite. Angew Chem Int Ed Engl 40, 1296–1298 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Ding YS et al. Detection of VM55599 and preparaherquamide from Aspergillus japonicus and Penicillium fellutanum: biosynthetic implications. J Nat Prod 71, 1574–1578 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding YS, Greshock TJ, Miller KA, Sherman DH & Williams RM Premalbrancheamide: synthesis, isotopic labeling, biosynthetic incorporation, and detection in cultures of Malbranchea aurantiaca. Org Lett 10, 4863–4866 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding Y et al. Genome-based characterization of two prenylation steps in the assembly of the stephacidin and notoamide anticancer agents in a marine-derived Aspergillus sp. J Am Chem Soc 132, 12733–12740 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu CJ, Li CW, Gao H, Huang XJ & Cui CB Penicimutamides D–E: two new prenylated indole alkaloids from a mutant of the marine-derived Penicillium purpurogenum G59. RSC Advances 7, 24718–24722 (2017). [Google Scholar]
- 17.Martinez-Luis S et al. Malbrancheamide, a new calmodulin inhibitor from the fungus Malbranchea aurantiaca. Tetrahedron 62, 1817–1822 (2006). [Google Scholar]
- 18.Kim HJ, Ruszczycky MW, Choi SH, Liu YN & Liu HW Enzyme-catalysed [4+2] cycloaddition is a key step in the biosynthesis of spinosyn A. Nature 473, 109–112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudson GA, Zhang ZG, Tietz JI, Mitchell DA & van der Donk WA In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J Am Chem Soc 137, 16012–16015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wever WJ et al. Chemoenzymatic synthesis of thiazolyl peptide natural products featuring an enzyme-catalyzed formal [4+2] cycloaddition. J Am Chem Soc 137, 3494–3497 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian ZH et al. An enzymatic [4+2] cyclization cascade creates the pentacyclic core of pyrroindomycins. Nat Chem Biol 11, 259–265 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Ohashi M et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L et al. Genome mining and assembly-line biosynthesis of the UCS1025A pyrrolizidinone family of fungal alkaloids. J Am Chem Soc 140, 2067–2071 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato N et al. Control of the stereochemical course of [4+2] cycloaddition during transdecalin formation by Fsa2-family enzymes. Angew Chem Int Ed Engl 57, 9754–9758 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Fage CD et al. The structure of SpnF, a standalone enzyme that catalyzes [4 + 2] cycloaddition. Nat Chem Biol 11, 256–258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Q et al. Enzyme-dependent [4 + 2] cycloaddition depends on lid-like interaction of the N-terminal sequence with the catalytic core in PyrI4. Cell Chem Biol 23, 352–360 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Byrne MJ et al. The catalytic mechanism of a natural Diels-Alderase revealed in molecular detail. J Am Chem Soc 138, 6095–6098 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Zheng QF et al. Structural insights into a flavin-dependent [4+2] cyclase that catalyzes trans-decalin formation in pyrroindomycin biosynthesis. Cell Chem Biol 25, 718–728 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Cai Y et al. Structural basis for stereoselective dehydration and hydrogen-bonding catalysis by the SAM-dependent pericyclase LepI. bioRxiv, doi: 10.1101/491761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domingo LR, Zaragoza RJ & Williams RM Studies on the biosynthesis of paraherquamide A and VM99955. A theoretical study of intramolecular Diels-Alder cycloaddition. J Org Chem 68, 2895–2902 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Quadri LEN et al. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37, 1585–1595 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Nodvig CS, Nielsen JB, Kogle ME & Mortensen UH A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10, e0133085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraley AE et al. Function and structure of MalA/MalA’, iterative halogenases for late-stage C-H functionalization of indole alkaloids. J Am Chem Soc 139, 12060–12068 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filling C et al. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J Biol Chem 277, 25677–25684 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Oppermann U et al. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact 143–144, 247–253 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Man H et al. Structures of alcohol dehydrogenases from Ralstonia and Sphingobium spp. reveal the molecular basis for their recognition of ‘bulky-bulky’ ketones. Top Catal 57, 356–365 (2014). [Google Scholar]
- 37.Birch AJ & Wright JJ Studies in relation to biosynthesis. XLII. The structural elucidation and some aspects of the biosynthesis of the brevianamides-A and -E. Tetrahedron 26, 2329–2344 (1970). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.