

Abstract
Recent advances in localisation-based super-resolution microscopy have enabled researchers to visualise single molecular features down to individual molecular components (~5 nm), but do not yet allow manipulation of single-molecule targets in a user-prescribed, context-dependent manner. Here we report an “Action-PAINT” strategy for super-resolution labelling upon visualisation on single molecules. This approach monitors and localises DNA binding events in real-time with DNA-PAINT, and upon visualisation of binding to a desired location, photo-crosslinks the DNA to affix the molecular label. We showed the efficiency of 3-cyanovinylcarbazole nucleoside (CNVK) photo-inducible crosslinking on single molecular targets and developed a software package for real-time super-resolution imaging and crosslinking control. We then benchmarked our super-resolution labelling method on synthetic DNA nanostructures and demonstrated targeted multi-point labelling on various complex patterns with 30 nm selectivity. Finally, we performed targeted in situ labelling on fixed microtubule samples with 40 nm target size and custom-controlled, sub-diffraction spacing.
Graphical Abstract
The ability to observe and in situ modify biological systems on the molecular scale is critical for studying biology in health and in disease. Super-resolution imaging enables researchers to “see the previously invisible” by breaking the diffraction limit of light and precisely visualising biology on the molecular scale, and has broadly transformed biomedical research1–3. However, a comparable optical capability for labelling and perturbing biological systems with nanometre precision is lacking.
Although photolithography and optical masking methods have proven to be successful in high-resolution manufacturing of solid-state materials (e.g. semiconductors)4, they are less suitable for manipulating biological samples since the positions of the targets of interest are not known a priori. Optical controls in biology such as optogenetics5 typically have spatial resolution limited by the diffraction limit of light, whereas contact-based controls such as atomic force microscopes6 are often disruptive and lack the depth of sample penetration.
An optical approach that could both visualise biological structures at the molecular scale, and upon visualisation, react to the detected features to label the underlying biological system with a molecular cargo or uniquely addressable physical handle (e.g. a DNA barcode) at equal nanometre precision, would be desired (Fig. 1a). Such a capability of “labelling upon visualisation” will not only allow researchers to “see the previously invisible”, but also enable them to “touch the previously inaccessible”, presenting a possible platform of precise, single-component level interrogation of biomolecular systems, including perturbation and delivery of a functional cargo to specific protein targets, site-specific labelling and purification of macromolecular complexes, and more (see Conclusions and outlook for more details).
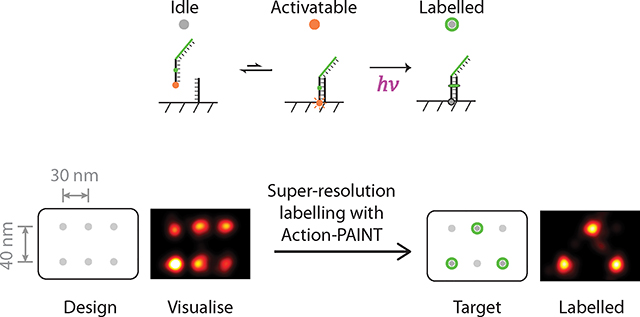
Fig. 1. Principle of super-resolution single-molecule labelling.

(a) Schematic illustration of targeted single-molecule labelling upon visualisation. Super-resolution imaging allows visualisation and selection of desired targets of interest among heterogeneous molecular complexes. Subsequent targeted single-molecule labelling allows precise delivery of molecular cargos or labels to molecular targets in various custom-defined patterns. Grey dots represent individual target molecules (notches represent defects), green and cyan halos represent successful molecular labelling (of two kinds).
(b) Strategy for implementing single-molecule labelling with DNA-PAINT combined with photo-inducible crosslinker. The crosslinker-bearing, dual-purpose imaging and labelling strand transiently hybridises to the target strand (between idle and activatable states). Exposure to the activating light pulse at the activatable state induces covalent attachment of the molecular label to the target (labelled state). The fluorophore is photobleached in the labelled state, after prolonged illumination.
(c) Experimental workflow for single-molecule labelling with Action-PAINT consists of three steps: (i) to “visualise”, a super-resolution DNA-PAINT image is captured and candidate target site locations determined, based on which the experimenter selects desired labelling target(s); (ii) to “label”, another super-resolution image stack is captured with the labelling strand, accompanied by real-time image processing and automatic crosslinking activation up detection of on-target blinking; (iii) to “confirm”, a final DNA-PAINT image is captured to assay the labelling outcome.
Recent advances in localisation-based super-resolution microscopy methods (e.g. STORM7,8, PALM9,10, PAINT11,12) have allowed researchers to optically visualise molecular features well below the diffraction limit and down to individual molecular components (~5 nm)13–21, but does not yet allow observation and manipulation or labelling of single-molecule targets in a sequential manner, which is particularly important for studying complex, spatially-varying and context-dependent biological systems. We termed this capability super-resolution labelling upon visualisation. Super-resolution microscopy methods based on the PAINT (point accumulation for imaging in nanoscale topography) principle11,12,22–24 provide a natural path towards such a capability. In these methods, a population of fluorophore-conjugated affinity probes transiently and repetitively bind to each of the imaging targets, producing apparent blinkings that can then be individually localised with high precision to synthesise the final super-resolution image. In contrast to other localisation microscopy methods, PAINT-based methods provide the extra benefit that, at any moment, typically only one affinity probe is bound to the imaging sample within a diffraction-limited area. This makes a light-induced targeting approach possible - if an activating light pulse can be introduced precisely at this moment (i.e. during the transient association between the affinity probe and a desired target), this spatiotemporal co-localisation allows selective molecular manipulation (the “action”) to be performed on the target (Fig. 1b). We termed this approach “Action-PAINT”25.
In this work, we designed and implemented such a strategy for super-resolution labelling on single molecules, based on our previous conceptual proposal25. The design combines our previously developed DNA-PAINT super-resolution microscopy method, and a fast, photo-inducible crosslinking chemistry26,27. We first assayed the efficiency of 3-cyanovinylcarbazole nucleoside (CNVK) photo-inducible crosslinking on single molecular targets, and developed a software package for real-time super-resolution image analysis and crosslinking control. Then, we benchmarked our super-resolution labelling method on synthetic DNA nanostructures and demonstrated high-efficiency (up to 65%) per-target labelling, and targeted single-molecule labelling with 70 nm selectivity (33% overall success rate). We showed that our method is capable of performing multi-point labelling on various complex 6-choose-3 patterns with 30 nm selectivity (69% success rate for at least 2 on-target labelling, 12% overall success rate, 18× higher than random labelling). Finally, we showed that our method is also compatible with in situ super-resolution imaging, and demonstrated successful targeted labelling on fixed microtubule samples with 40 nm label size and variable, diffraction-unlimited spacings (78% on-target labelling, 24% off-target, normalised 4.2× on-/off-target labelling specificity). We therefore have implemented an effective method for super-resolution labelling upon visualisation.
Results
Strategy and workflow for super-resolution labelling with Action-PAINT
We here implement our strategy for super-resolution labelling upon visualisation based on our previously developed DNA-PAINT super-resolution microscopy method12,19 (see Supplementary Methods 2.4 for more details). In brief, the DNA-PAINT method exploits transient binding between short, fluorescently labelled DNA oligonucleotides (the “imager” strands, in solution) and their complementary strands (the “docking” strands, labelled on the molecular targets), to produce apparent blinkings, when placed under z-confined illumination. With this method, we have successfully demonstrated multiplexed, 3D, cellular imaging28, quantitative super-resolution imaging29, as well as discrete molecular imaging (DMI) of molecular features down to ~5 nm in size19. Our strategy for Action-PAINT will then introduce a fast, photo-induced crosslinking reaction that will be activated at the precise moment (the “activatable” state”) when a label-bearing imager strand transiently hybridises to a selected docking strand of interest, thereby delivering the molecular label to the desired target site (Fig. 1b).
The experimental workflow for Action-PAINT consists of three steps, and is illustrated as in Fig. 1c (see Supplementary Methods 2.5 for more details). First, to “visualise”, a pre-acquisition super-resolution image of the target molecular structures is taken, with a regular, non-crosslinking DNA-PAINT imaging strand, that is complementary to the docking strands on candidate labelling targets. Based on the pre-acquisition image, one or more sub-diffraction sized region-of-interest (ROI) around the desired labelling target site(s) are manually selected. Then, to “label”, a second super-resolution image stack is recorded with a CNVK-bearing dual-purpose imaging and crosslinking strand that also carries a reporter sequence as the molecular label (the “labelling strand”). This imaging and labelling session is accompanied by real-time sub-diffraction localisation, drift correction, image analysis and laser activation control. In particular, a brief pulse of 405 nm illumination is triggered when and only when a labelling strand is detected to be hybridised within the desired target ROIs. The length of the real-time image stack is not pre-set, and is instead determined by the activation control software based on real-time analysis of the labelling progress and blinking kinetics. Finally, to “confirm”, a post-acquisition image is acquired to assay the labelling correctness and efficiency, with a third imaging strand that is complementary to the delivered reporter sequence. To assist with drift correction, DNA nanostructure drift markers are added alongside the sample, and corresponding imaging strands are supplemented to the imaging buffer for all three imaging steps.
Two technical capabilities are required for successful implementation of the above strategy. First, a fast and efficient crosslinking agent is needed, that can be optically-induced to form a covalent crosslink during the transient oligonucleotide hybridisation. The CNVK base modification was previously reported26 to provide both a fast crosslinking rate under near-UV illumination, and is compatible for incorporation into oligonucleotides, thus makes a promising candidate. Second, a real-time image analysis and crosslinking activation control software is needed for reconstructing the super-resolution image, and controlling the prompt activation of crosslinking illumination upon detection of on-target binding. Our previously developed high-accuracy single-molecule localisation and drift correction methods provides a good starting point19, and can be adapted for real-time analysis.
CNVK crosslinking test and imager design
To assay the crosslinking efficiency of CNVK-modified oligonucleotides on single molecules, we first designed and synthesised cy3b-conjugated CNVK labelling strands (Supplementary Fig. 1, see Supplementary Methods 2.3 for more details). Each CNVK labelling strand serves a dual purpose for both real-time super-resolution imaging and photo-induced crosslinking, and comprises of two parts: (i) a DNA-PAINT imaging sequence (that is complementary to the docking sequence on candidate labelling targets), which contains a CNVK base modification, and (ii) an orthogonal DNA-PAINT docking sequence, used as the reporter sequence for assaying successful crosslinking. We designed and optimised the CNVK-containing sequence to have an average binding on-time of ~1.0 s, to be compatible with the crosslinking time constant and imaging frame rate, thereby maximising the overall crosslinking efficiency (Supplementary Fig. 2, see Supplementary Methods 2.3 for more details).
After confirming the UV-induced CNVK crosslinking efficiency in bulk with a handheld 365 nm light source and a denaturing gel-shift assay (~50% for 1 s illumination at 2.5 W/cm2 intensity) (Supplementary Fig. 1, see Supplementary Methods 2.3 for more details), we performed crosslinking efficiency test at the single-molecule level, under typical super-resolution microscopy settings and with 405 nm laser line illumination (~0.5 kW/cm2) (see Supplementary Methods 2.3 and 2.4 for more details). We utilised a DNA nanostructure test platform with 12 imaging sites arranged in a rectangular grid pattern, spaced 20 nm point-to-point (the “20 nm grid”)19,30, on which we designed a test pattern with 4 corners as reference markers, and 8 internal sites as candidate labelling targets (Fig. 2a and Supplementary Fig. 3, see Supplementary Methods 2.1 for more details). We then saturated the 20 nm grid samples with CNVK labelling strands, and exposed the sample to 405 nm laser illumination under total internal reflection (TIR) configuration. After thoroughly washing the DNA grids of excess crosslinking strands, a post-illumination DNA-PAINT super-resolution image was taken, with an imager strand that is complementary to both the reference corner markers and the reporter sequence on the labelling strand. We then counted the number of labelled targets and calculated single-molecule crosslinking efficiency of ~45% after a 1 s pulse, which saturates around ~75% after 15 non-consecutive pulses (1 s each) (Fig. 2b and Supplementary Fig. 4, see Supplementary Methods 2.3 for more details), likely due to combined effects from self-assembly defects in the DNA nanostructure and undesired photo-induced crosslinking reversal27. We note that this result only reflects the obtainable crosslinking efficiency after a single (transient) oligonucleotide binding event, whereas a potentially higher labelling efficiency could be achieved during the real-time imaging and labelling session after multiple target binding events and laser crosslinking attempts (e.g. estimated efficiency >90% after up to 4 binding events, assuming 1 s average binding on-time).
Fig. 2. Crosslinking efficiency test and targeted single-molecule labelling.

(a) Test of CNVK labelling strand crosslinking efficiency on single molecules. Left, DNA nanostructure design with four corner markers and eight central crosslinking targets, spaced 20 nm point-to-point, before and after crosslinking. Right, DNA-PAINT images after crosslinking. Top, negative control, bottom, with 405 nm laser illumination.
(b) Crosslinking efficiency comparison for different illumination time. Error bars represent Poisson counting uncertainty (total number of grids counted: 103, 60, 82, respectively, each having 8 binding sites; number of labelled targets: 419, 349, 510, respectively).
(c) Schematics and representative images at different stages of targeted single-molecule labelling experiment. Top row, schematics of DNA test structure design with two candidate targets, spaced 72 nm apart; bottom row, super-resolution images. Within bottom row, left panel shows pre-acquisition image of candidate target sites together with markers, middle panel shows real-time labelling session image of CNVK labelling strand transient blinking events, and right panel shows post-acquisition image of labelled targets together with markers.
(d,e) Examples of real-time blinking and laser activation traces, comprising of (d) a single blinking event and laser pulse for the sample in (c), and (e) a series of four events for the sample in (F), left column. For each detected blinking event, XY traces (top) and time traces (bottom) are shown.
(f) Representative examples of successful labelling experiments. Top row, pre-acquisition images; middle row, real-time labelling session images; bottom row, post-acquisition images.
In (a,c,f), grey dots without and with green halo indicate idle and labelled targets, black crosses indicate fixed position markers; green and grey circles overlaid on super-resolution images indicate desired and undesired target sites, respectively.
In (d,e), XY traces are colour-coded by time (from black to orange) and overlaid with include- (green, dotted circle) and exclude- (grey, dotted circle) ROIs; time traces show detected photon count per single-molecule localisation (orange), overlaid with crosslinking laser illumination events (magenta).
See Supplementary Fig. 8–9 for additional representative examples of both successful and unsuccessful single-target labelling experiments, and Supplementary Notes 3.4 for discussion regarding statistics and reproducibility.
All scale bars, 20 nm.
Software design and crosslinking activation control
Next, we developed a software package for real-time super-resolution image analysis and crosslinking activation control (Supplementary Fig. 5, see Supplementary Notes 3.1 and Methods 2.6 for more details). We adapted our previous off-line, high-precision single-molecule localisation and drift correction algorithm for on-line processing. With moderate single-molecule point spread function (PSF) fitting precision (~3 nm by DAFL19 and <2 nm by single-molecule fitting31), a ~350 ms frame time was achieved for the complete cycle of data acquisition → super-resolution analysis → laser activation control. The resulting runtime delay for laser activation (~350 ms) is significantly less than the observed blinking on-time and crosslinking time constant (Supplementary Fig. 2). Real-time drift correction was achieved by comparing all super-resolved localisations with DNA grid positions pre-determined from the pre-acquisition image (Supplementary Fig. 5, see Supplementary Methods 2.6 for more details), a typical frame-to-frame correction of ~1 nm (r.m.s.) was observed and corrected. We benchmarked our real-time imaging quality on the 20 nm grid, and estimated ~5 nm residual drift (r.m.s.), and 12–15 nm imaging resolution (Supplementary Fig. 6, see Supplementary Notes 3.1.2 and Methods 2.6 for more details).
To maximise selective crosslinking efficiency, the crosslinking laser needs to be stringently controlled, since any off-target illumination or over-illumination may result in incorrect or unsuccessful crosslinking. After many rounds of trials and errors, we decided to implement the following laser control scheme, to optimise the targeted crosslinking performance (Supplementary Fig. 7, see Supplementary Notes 3.1.3 for more details). First, to make sure that only desired targets are being labelled, we hand-selected small target regions (circular areas with 20–40 nm diameter, depending on inter-target separation), centred around both desired (“include-ROIs”) and undesired (“exclude-ROIs”) candidate target sites. We only turned on the 405 nm laser when a single-molecule blinking event is detected within one of the include-ROIs, but not in the exclude-ROIs (Fig. 2c). To further prevent incorrect crosslinking caused by occasional mis-localised blinkings and undesired double-blinking events, we monitored each blinking event (i.e. a series of localisations from the same imager strand, imaged in consecutive frames) and excluded any blinking that either originated or has visited one of the exclude-ROIs. Second, we noticed that, although a short blinking event (and short 405 nm laser pulse) typically results in low crosslinking efficiency, a prolonged illumination could potentially reverse or damage a previously successful crosslink (see Supplementary Notes 3.2 for more details). Therefore, to maximise the crosslinking success rate, we set a maximum threshold of 3 s of UV illumination per any single blinking event. Third, each target is considered successfully crosslinked if two criteria are met: (i) the target has received at least 1 frame of 405 nm illumination, (ii) no further blinking is observed within the include-ROIs for a set monitor time window of 5–10 minutes. To avoid any unnecessary extra UV illumination, as well as to maximise the workflow efficiency, the imaging session is automatically terminated by the control software (and reported as successful) as soon as all the target sites pass the above criteria; or the session is stopped after a set time-out, and reported as unsuccessful.
Super-resolution labelling with 70 nm selectivity
We went on to test our entire Action-PAINT workflow by combining the CNVK-mediated crosslinking with our real-time imaging and labelling control software. We designed a test platform based on the 20 nm grid, by positioning two candidate target sites separated by ~70 nm, each surrounded by two reference markers to help identify their positions (Fig. 2c and S3). We note that the two candidate target sites are molecularly identical, and cannot be distinguished from each other by the imager strand, therefore we expect binding events to occur at both sites with equal frequency.
Fig. 2c–d illustrates a representative successful labelling experiment attempt. Super-resolution images of pre-acquisition, real-time labelling, and post-acquisition are shown in Fig. 2c. Analysis of the real-time blinking trace showed only one blinking event on the desired target, which triggered a pulse of 1.0 s (3 frames) of 405 nm laser illumination, and resulted in successful crosslinking onto the target (Fig. 2d). However, not all labelling attempts were as efficient, and large variations in the total number of laser pulses (or cumulative illumination dosage) were observed from molecule to molecule (Supplementary Fig. 8). Fig. 2e shows another representative real-time blinking trace comprising of four consecutive blinking events and four laser pulses (from the sample in Fig. 2f, first column). The first three transient blinking events each triggered a short pulse of 405 nm laser illumination, but did not produce a successful crosslink, as evidenced by the re-visit of the docking site by another crosslinking strand. A fourth, stable blinking event then triggered a long pulse of laser illumination (3 s, cut by the laser activation threshold) and resulted in successful crosslinking. The prolonged blinking event after the termination of laser activation also suggests permanent fixation by crosslinking. However, this effect was not always observed and could not be used as a reliable criterion for success (Supplementary Fig. 8). On the other hand, blinking events on the undesired target site did not trigger 405 nm laser activation or any crosslinking to take place.
We could only perform targeted labelling experiments on a single DNA grid in an entire field of view, due to a lack of spatial control of laser illumination in our current implementation (note that this current limitation could be overcome by implementation a highly-parallel labelling approach using a micro-mirror array). Out of a series of 33 single-molecule labelling trials attempted (Fig. 2f, see Supplementary Fig. 8–9 for more representative examples of both successful and unsuccessful single-target labelling experiments), 22 were assayed for labelling efficiencies (with post-acquisition), whereas the rest 11 were aborted half-way during the real-time imaging session due to early failing signals (such as misbehaving target site, or occasionally damaged DNA nanostructure). We estimated an on-target labelling efficiency of 59% (13 out of 22), and 11 of these trials resulted in correct on-target labelling with no off-target label, giving a 33% overall success rate, and achieving a moderate 3× increase in success rate compared with a random-labelling attempt (expected success rate 10%), assuming a stochastic labelling rate of 11% (as measured from candidate targets on non-monitored grids in the same experiment). Possible causes for these failed attempts could include: the existence of a fraction of dark fluorophores, background crosslinking by the imaging (561 nm) laser, or potential 405 illumination induced damage to the DNA nanostructure (see Supplementary Notes 3.2 for more details). These results demonstrated our ability of single-molecule discrimination and targeted labelling with sub-diffraction-limit separation (70 nm, ~1/3× diffraction-limited resolution).
Multiple-point nanoscale patterning with 30 nm selectivity
Next, we sought to demonstrate nanoscale labelling for a more complex, multi-point target, and with closer spatial separation. Such an effort would require even more stringent control of the crosslinking laser to avoid any potential off-target crosslinking. Therefore, we further modified the laser control software as below. First, to ensure high detection specificity and localisation precision, only single-molecule blinking’s with sufficiently long binding on-time (at least 2 frames) and sufficiently high photon count are considered as candidates for crosslinking. Second, due to the asynchronous nature of labelling multiple target sites and the potential of photo-induced crosslinking reversal, we extended the no-blinking monitor time limit for individual targets. For those targets which had initially passed the success criteria, but later started blinking again, likely due to crosslinking reversal27, the monitor and crosslinking workflow was restarted until the criteria was passed for all targets (Supplementary Fig. 7, see Supplementary Notes 3.1.3 for more details). We also adopted a CNVK purification procedure that was reported to remove a secondary, non-crosslinking species of CNVK27 (Supplementary Fig. 1, see Supplementary Methods 2.3 for more details).
For this test, we further designed a 6-point candidate target pattern with 30–40 nm point-to-point spacing (Fig. 3a). For each labelling experiment, we first confirmed the presence of all six docking sites during the pre-acquisition, then picked an arbitrary 3-point pattern (Fig. 3a) and manually selected the include-ROIs as well as exclude-ROIs accordingly. The real-time imaging and labelling session was then conducted under automatic software control with modified laser activation criteria as described above.
Fig. 3. Multi-point super-resolution patterning.

(a) Schematics and representative images at different stages of targeted 3-point single-molecule labelling experiment. Top row, schematics of DNA test structure design with 6 candidate targets, spaced 30–40 nm apart, bottom row, super-resolution images. Within bottom row, left panel shows pre-acquisition image of candidate target sites, middle panel shows real-time labelling session image of CNVK labelling strand transient blinking events, and right panel shows post-acquisition image of labelled targets.
(b) Example of real-time blinking and laser activation traces, comprising of three blinking events on different targets and three laser pulses for the sample in (a). For each detected blinking event, XY traces (top) and time traces (bottom) are shown.
(c) Representative examples of successful 3-point patterning experiments for each visually distinct pattern. Top row, schematics of different 6-choose-3 labelling patterns, arranged by increasing difficulty (number of 30-nm-spaced pairs necessary to be correctly distinguished for successful patterning), labelled on top, middle row, pre-acquisition images; bottom row, post-acquisition images. Last column shows the same experiment as in (a).
In (a,c), grey dots without and with green halo indicate idle and labelled targets, black crosses indicate fixed position markers; green and grey circles overlaid on super-resolution images indicate desired and undesired target sites, respectively.
In (b), XY traces are colour-coded by time (from black to orange) and overlaid with include- (green, dotted circle) and exclude- (grey, dotted circle) ROIs; time traces show detected photon count per single-molecule localisation (orange), overlaid with crosslinking laser illumination events (magenta).
See Supplementary Fig. 10–11 for more representative examples of both successful and unsuccessful 3-point labelling experiments, and Supplementary Notes 3.4 for discussion regarding statistics and reproducibility.
All scale bars, 20 nm.
Fig. 3a–b shows a successful 3-point patterning experiment. Real-time blinking trace shows three on-target blinking events detected in total, each triggered a pulse of 405 nm laser illumination (~0.6–3 s in length), and resulted in successful crosslinking. Again, not all attempts were as efficient, and typically a few attempts per target were required for successful crosslinking (Supplementary Fig. 10). Blinking events within exclude regions (Fig. 3a, middle, grey circles), however, were correctly detected and avoided for off-target crosslinking.
We demonstrated multi-point super-resolution labelling by choosing all five visually distinct 3-point patterns on the 6-point canvas in Fig. 3c, and achieved successful single-molecule patterning for all cases, including pre-acquisition and post-acquisition images for each experiment (Fig. 3d, see Supplementary Fig. 10–11 for more representative examples of both successful and unsuccessful 3-point labelling experiments). We performed a total of 84 trials of three-point patterns, out of which 62 were assayed for labelling efficiencies (with post-acquisition), and the rest 22 were aborted half-way during the real-time imaging session due to early failing signals (similar to the 2-point experiment above). Out of the 62 assayed trials, we achieved an overall per-target labelling efficiency of 65%, and a modestly low off-target labelling rate of 15% (see Supplementary Notes 3.2.1 for more details). As a result, 43 of the 62 attempts (69%) were successfully labelled with at least 2 correct sites, and 61 (98%) has at least one target correctly labelled. Overall, 10 out of 84 trials (12%) produced complete labelling (i.e. 3 on-target labels, no off-target label). This reflects not only the complexity of the 6-choose-3 pattern, but also potential crosslinking reversal caused by prolonged exposure to 405 nm illumination for already crosslinked strands (see Supplementary Notes 3.2.2 for more details). However, compared to a random labelling attempt with the same pattern complexity, we still achieved an 18× increase in success rate (12% vs 0.66%), assuming a stochastic labelling rate of 25% (as measured from candidate targets on non-monitored grids in the same experiment). We also note that, in future applications, high labelling and molecular delivery efficiency could be achieved with tandem labelling, e.g. up to 69% with two-fold and 98% with three-fold tandem labelling.
In situ super-resolution labelling on microtubules
Next, we sought to demonstrate the compatibility of Action-PAINT labelling with in situ super-resolution imaging in fixed cultured cells (Fig. 4). To present CNVK crosslinking strands on immunostained targets (microtubules), we prepared a secondary antibody that is directly conjugated to the labelling target strand. After immunostaining, the Action-PAINT workflow was conducted similarly as before (Fig. 4a), but with two important differences. First, instead of placing candidate target sites individually at regularly-spaced grid patterns as before, here we placed them continuously along microtubules, resulting in large “patches” of labelling target sites. Consequently, it was not possible to distinguish among them and select a target ROI containing a single target site; instead, each target ROI now likely contains more than one labelling target. Second, due to the unknown multiplicity of target sites, it is difficult to assay the degree of crosslinking during the real-time labelling session using the same blinking kinetics analysis as before. Therefore, we either conducted fixed-length real-time labelling sessions, or employed a different set of stopping criteria based on the total dose of crosslinking illumination received on the target.
Fig. 4. In situ super-resolution labelling on microtubules.

(a) Schematics and workflow of super-resolution labelling on immunostained and DNA-labelled microtubule samples, in fixed BSC-1 cells. Colour schemes and legends are same as in Fig. 1.
(b) Examples of successful 2-target patterning on microtubules for different inter-target spacing. Top row, schematics of desired labelling patterns; second row, pre-acquisition images; third row, real-time labelling session images; bottom row, post-acquisition images.
(c) Schematic list of the outcome of 8 experimental trials. Successfully labelled targets are shaded green, and undesired labels in exclude areas are shaded different grey levels to reflect the difference in exclude area. Arrows indicate experiments shown in (b). (*) indicates the experiment excluded from statistics, due to likely sample movement based on a visual observation of an apparent change in microtubule morphology between pre- and post-acquisition sessions.
In all panels, green and grey circles in schematics indicate desired and undesired targets; green and grey dashed rectangles indicate experimental include- and exclude-ROIs, respectively. ROIs in last row (post-acquisition images) are adjusted by 20 nm edge buffer, according to the overall localisation and labelling precision.
See Supplementary Fig. 13 for more examples of microtubule labelling experiments and Supplementary Fig. 14 for more details on labelling efficiency and specificity. See Supplementary Methods and Notes 3.3 for more details regarding crosslinking efficiency and specificity calculation, and edge-buffered ROIs, and Supplementary Notes 3.4 for discussion regarding statistics and reproducibility.
Scale bars in (b), 100 nm.
We first assayed our “batch” crosslinking efficiency and specificity on immunostained microtubules by selecting two relatively large include-ROIs (~160 nm, each containing a number of candidate labelling sites) flanked by three similar-sized exclude-ROIs, and conducting a real-time labelling session with a few different lengths (Supplementary Fig. 12). In each case, we observed multiple successful labels within the include-ROIs. However, we also observed a significant fluctuation in overall labelling efficiency, as well as several off-target labels in exclude-ROIs, likely caused by background crosslinking from the 561 nm imaging laser. We reasoned that a smaller include-ROI together with a shorter labelling session length would reduce the total dose of illumination from both the crosslinking and imaging lasers, thus allowing more uniform and specific labelling. Indeed, with reduced target ROI size (~80 nm) and a shorter labelling session, we achieved a 90% on-target labelling efficiency (9 out of 10), with a moderate 33% off-target labelling (5 out of 15) (Supplementary Fig. 12).
Next, we sought to demonstrate the controlled super-resolution targeting ability of Action-PAINT on immunostained microtubules by patterning three pairs of labels with different, diffraction-unlimited spacing (from 160 to 480 nm, Fig. 4b and S13). For these experiments, we further reduced the target ROI size (~40 nm, each including a small number of target sites only), and employed the following stopping criteria: (i) each target ROI should receive at least three separate crosslinking illuminations, (ii) ideally, at least one of the illuminations should last for two imaging frames or longer. To limit the extent of background crosslinking induced by the imaging laser, we also set a limit on the total number of frames for the real-time labelling session (see Supplementary Notes 3.1.4 for more details). Out of 7 valid labelling trials, we achieved an overall 78% on-target labelling efficiency (11 out of 14), and maintained a moderate 24% off-target labelling (5 out of 21), even with a much larger exclude-ROI area (Fig. 4c). We further quantified our labelling efficiency and specificity on a per-target basis, by comparing the number of recorded DNA-PAINT localisations in each include- or exclude-ROI, before and after Action-PAINT labelling (i.e. from pre- and post-acquisition images, ROI areas are adjusted with edge buffers, see Supplementary Methods and Notes 3.3 for more details). On average we achieved ~4.2× (median) per-target enrichment (on-target vs. off-target) in labelling efficiency (Supplementary Fig. 14), similar to our earlier observation (~4.3×) on synthetic DNA nanostructures.
Discussion
In this paper, we demonstrated super-resolution labelling and patterning with Action-PAINT, which enables targeted single-molecule “labelling upon visualisation” – i.e. successive super-resolution imaging and targeted labelling - at the single-molecule scale. Such a method allows biological researchers to not only “see the previously invisible”, but also “touch the previously inaccessible”, and could open up a broad range of new biological investigations. In our implementation, we combined real-time DNA-PAINT super-resolution microscopy and fast, photo-induced crosslinking chemistry (CNVK) to achieve this goal. We have demonstrated successful targeted single-molecule labelling with 30–70 nm spatial selectivity and high (59–65%) on-target labelling efficiency on synthetic DNA nanostructure breadboard. For single-target labelling, we have achieved an overall 33% success rate (3× higher compared to random labelling); for complex multi-point patterning (6-choose-3 targets), we have achieved a high success rate (69%) for at least 2 on-target labels, and for perfect labelling (i.e. 3 on-target labels, no off-target label) an overall 12% success rate (18× higher compared to random labelling). We also demonstrated the compatibility of our Action-PAINT method with in situ super-resolution imaging and performed successful targeted labelling of cellular targets with 40 nm label size and variable, diffraction-unlimited spacings. On microtubule samples fixed and immunostained with oligo-conjugated antibodies that present the CNVK-containing crosslinking targets, we have achieved an overall 78% on-target labelling efficiency, with a moderate 24% off-target labelling. We further quantified a normalised on-/off-target labelling specificity of ~4.2× (median) for in situ labelling on microtubules, comparable to our earlier observation (~4.3×) on synthetic DNA nanostructures. Our method can be naturally adapted for multiplexed super-resolution labelling on multiple molecular targets by exploiting programmable orthogonal binding sequences28, and for three-dimensional imaging and targeted single-molecule labelling with the application of point spread function (PSF) engineering approaches (such as astigmatism32, or double-helix PSF33).
Compared with other nanoscale patterning strategies such as electron-beam lithography or atomic force microscopy, Action-PAINT is a less disruptive, light-based labelling technique, using only standard wavelengths (405 nm, 561 nm) that can be found on common commercially available microscopes. Although light-based sub-diffraction patterning has been previously demonstrated for potential high-capacity optical storage applications (by switching off rsEGFP molecules with the RESOLFT method34), such “super-resolution writing” approach does not label or modify the targets with a physical, molecular handle. In contrast, Action-PAINT delivers a physical, addressable handle to the target of interest, which enables subsequent analysis and manipulation of the labelled targets – critical for downstream perturbation or physical manipulation to the biological system, e.g. by target-specific small molecule inhibitor delivery or handle attachment – thus making Action-PAINT a valuable tool for precision interrogation of biological systems.
Further development of this technology would likely need to address two current technical limitations. One limitation is that, due to the lack of spatial control of the crosslinking laser (currently uniformly illuminated across the entire field of view), selective single-molecule labelling could only be achieved within a small fraction of the field (up to a few diffraction-limited area). To overcome this limitation, and thus allow efficient, highly-parallel custom labelling across multiple desired regions of interest, an automated digital micro-mirror device (DMD) could be implemented to independently control the delivery of the activation laser at every diffraction-limited area across the field of view, allowing up to a million single-molecule monitoring and labelling sessions in parallel. Another current limitation is crosslinking efficiency, which ultimately constrains the overall labelling success rate and achievable patterning complexity. Possible options for improvement include using double CNVK-modified strands, or more efficient photo-inducible crosslinking chemistry. Supplementary Fig. 15 shows in silico simulation that compares the effect of different experimental conditions, including the number of include- and exclude-ROIs, characteristic binding on-time and off-time, and total imaging session length, on the overall Action-PAINT success rate.
We envision that user-defined, single-molecule labelling with Action-PAINT will provide a valuable tool for precise probing of biomolecular targets. Due to the highly heterogeneous and context-dependent nature of many biomolecular systems (such as the cellular cytoskeletal network, membrane protein clusters, cytosolic mRNAs distribution, and chromosome 3D architecture), meaningful perturbation and manipulation can only be performed based on knowledge of the particular biological system in question, which cannot be obtained a priori. Action-PAINT could provide the unique conceptual advantage which would allow targeting and perturbation decisions to be based on real-time imaging data and informed by context-relevant features such as neighbouring cytoskeletal architecture, protein distribution and interaction patterns or sequence-specific features such as topologically associated domains within chromosome 3D structure; and performed at super-resolution, single-molecule level. We outline below two future directions of targeted biological perturbation and manipulation that could be potentially enabled by Action-PAINT.
One such future direction would involve quantitative studies of the effects of cell membrane receptor and ion channel activation at the super-resolution and single-molecule level, which have thus far been limited, due to a lack of manipulation tools that allow super-resolution activation and inhibition on the single-molecule level and stoichiometric delivery of small molecule effectors. Such studies are especially important in neurons, where various studies have implied a functional significance of individual ion channels in affecting neuron generation, action potential stabilisation and spike shape; as well as the effects of ion channel clustering in affecting local ion concentration and downstream neurotransmitter36,37. Compared with current super-resolution microscopy methods that allow visualisation at the nanoscale, and optogenetic tools that allow diffraction-limited ion channel perturbation, Action-PAINT based methods could potentially allow precise activation and inhibition of individual ion channels (such as with the delivery of a single peptide toxin molecule) within crowded molecular environments. Combined with fast (sub-second) super-resolution imaging and analysis38, Action-PAINT could potentially be extended to follow single molecules in motion and perform targeted labelling on them (with simultaneous super-resolution imaging), which is suitable for targeting single ion channels during slow or cytoskeleton-confined diffusion39. Further, Action-PAINT could potentially allow studying of the effects of cooperative effects between multiple ion channels within the same cluster, and between multiple types of different ion channels.
Another such future direction would involve batch studies of proteins and protein complexes selected using imaging-based criteria, including subcellular or sub-organelle localisation, or cytoskeleton and protein interaction patterns. Action-PAINT could allow imaging-based identification followed by targeted labelling of all protein targets of interest with a universal handle (e.g. biotin, or a unique DNA handle). Following handle-based extraction and purification, a wide range of downstream analysis and manipulation could be envisioned, including extraction of labelled proteins for analysis with chromatography or single-molecule protein array40, and potential in situ proteomic analysis with mechanical force spectroscopy. One such example could be to study the different location- and direction-dependent binding partners of kinesin and dynein, using a combination of high-throughput Action-PAINT and the emerging single-molecule methods for protein identifications, thus building a protein-protein interaction map based on subcellular localisation information that are previously unachievable. Using uniquely encoded DNA handle sequences, proteins residing in multiple cellular locations, or exhibiting multiple interaction patterns could also be precisely encoded and separately extracted for analysis, potentially enabling a novel “imaging-based proteomics” approach that has not been possible with current biochemical41 and super-resolution microscopy methods.
Supplementary Material
Acknowledgments
The authors thank R. Barish, H. Soundarajan, S. Agasti, J. Woehrstein, and R. Jungmann for preliminary work on aspects of earlier versions of the project and for helpful discussions, and W. Shih, H. Sasaki, B. Beliveau, E. Boyden, and J. Paulsson for helpful discussions. This work was supported by a National Institutes of Health (NIH) Director’s New Innovator Award (1DP2OD007292), an NIH Transformative Research Award (1R01EB018659), an NIH Pioneer Award (1DP1GM133052), an NIH grant (5R21HD072481), an Office of Naval Research (ONR) Young Investigator Program Award (N000141110914), ONR grants (N000141010827, N000141310593 and N000141812549), a National Science Foundation (NSF) Faculty Early Career Development Award (CCF1054898), an NSF grant (CCF1162459) and Molecular Robotics Initiative fund at Wyss Institute for Biologically Engineering Faculty to P.Y. M.D. acknowledges support from HHMI International Predoctoral Fellowship, Department Fellowship from Systems Biology Department at the Harvard Medical School, and Technology Development Fellowship from Wyss Institute at Harvard University. S.K.S. acknowledges support from Human Frontier Science Program (HFSP) fellowship (LT000048/2016-L).
Footnotes
Competing financial interests
A U.S. patent (US App No. 15/104,570) has been filed that covers the concepts reported in this work (Inventors: Robert Barish, Peng Yin). P.Y. is cofounder of Ultivue Inc. and NuProbe Global.
Data availability
Data sets generated during the study are available from the corresponding authors upon request.
Code availability
Custom computer programs used during the study are available from the corresponding authors upon request.
References
- 1.Sahl SJ & Moerner W Super-resolution fluorescence imaging with single molecules. Curr Opin Struc Biol 23, 778–787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sydor AM, Czymmek KJ, Puchner EM & Mennella V Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol 25, 730–748 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Eggeling C, Willig KI, Sahl SJ & Hell SW Lens-based fluorescence nanoscopy. Q Rev Biophys 48, 178–243 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Sanders DP Advances in Patterning Materials for 193 nm Immersion Lithography. Chem Rev 110, 321–60 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Cohen AE Optogenetics: Turning the Microscope on Its Head. Biophysical Journal 110, 997–1003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ando T, Uchihashi T & Scheuring S Filming Biomolecular Processes by High-Speed Atomic Force Microscopy. Chem Rev 114, 3120–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rust MJ, Bates M & Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heilemann M et al. Subdiffraction-Resolution Fluorescence Imaging with Conventional Fluorescent Probes. Angewandte Chemie Int Ed 47, 6172–6176 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Betzig E et al. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Hess ST, Girirajan T & Mason MD Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys J 91, 4258–4272 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharonov A & Hochstrasser RM Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc National Acad Sci 103, 18911–18916 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jungmann R et al. Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett 10, 4756–61 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Löschberger A et al. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. Journal of cell science 125, 570–5 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Szymborska A et al. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 341, 655–658 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Pertsinidis A et al. Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc National Acad Sci 110, E2812–E2820 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang F et al. Ultra-High Resolution 3D Imaging of Whole Cells. Cell 166, 1028–1040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan C et al. Absolute Arrangement of Subunits in Cytoskeletal Septin Filaments in Cells Measured by Fluorescence Microscopy. Nano Lett 15, 3859–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikhaylova M et al. Resolving bundled microtubules using anti-tubulin nanobodies. Nature Communications 6, 7933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai M, Jungmann R & Yin P Optical imaging of individual biomolecules in densely packed clusters. Nat Nanotechnol 11, 798–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raab M, Schmied JJ, Jusuk I, Forthmann C & Tinnefeld P Fluorescence microscopy with 6 nm resolution on DNA origami. Chemphyschem : a European journal of chemical physics and physical chemistry 15, 2431–5 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Rittweger E, Han K, Irvine SE, Eggeling C & Hell SW STED microscopy reveals crystal colour centres with nanometric resolution. Nature Photonics 3, (2009). [Google Scholar]
- 22.Giannone G et al. Dynamic Superresolution Imaging of Endogenous Proteins on Living Cells at Ultra-High Density. Biophys J 99, 1303–1310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoen I, Ries J, Klotzsch E, Ewers H & Vogel V Binding-Activated Localization Microscopy of DNA Structures. Nano Lett 11, 4008–11 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Kiuchi T, Higuchi M, Takamura A, Maruoka M & Watanabe N Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat Methods 12, 743–746 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Barish R & Yin P Methods and compositions relating to optical super-resolution patterning. U.S. Patent 10,041,108 (2018).
- 26.Yoshimura Y & Fujimoto K Ultrafast Reversible Photo-Cross-Linking Reaction: Toward in Situ DNA Manipulation. Org Lett 10, 3227–30 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Vieregg JR, Nelson HM, Stoltz BM & Pierce NA Selective nucleic acid capture with shielded covalent probes. J. Am. Chem. Soc 135, 9691–9699 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jungmann R et al. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods 11, nmeth.2835 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jungmann R et al. Quantitative super-resolution imaging with qPAINT. Nature Communications 13, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothemund PW Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Smith CS, Joseph N, Rieger B & Lidke KA Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods 7, nmeth.1449 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang B, Wang W, Bates M & Zhuang X Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 319, 810–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavani et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc National Acad Sci 106, 2995–2999 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grotjohann T et al. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 478, 204–208 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Auer A, Strauss M, Schlichthaerle T & Jungmann R Fast, background-free DNA-PAINT imaging using FRET-based probes. Nano Lett 17, 6428–6434 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Dorval AD & White JA Channel Noise is Essential for Perithreshold Oscillations in Entorhinal Stellate Neurons. The Journal of Neuroscience 25, 10025–10028 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao Y-Q et al. Presynaptic Ca2+ Channels Compete for Channel Type-Preferring Slots in Altered Neurotransmission Arising from Ca2+ Channelopathy. Neuron 43, 387–400 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Huang F et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nature methods 10, 653–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albrecht D et al. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J Cell Biol 215, jcb.201603108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rondelez Y et al. Microfabricated arrays of femtoliter chambers allow single molecule enzymology. Nature Biotechnology 23, 361–365 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Rhee H-W et al. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 339, 1328–1331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.