

Abstract
In the last decades, a promising breakthrough in fluorescence imaging was represented by the advent of super-resolution microscopy (SRM). Super-resolution techniques recently became a popular method to study sub-cellular structures, providing a successful approach to observe cytoskeletal and focal adhesion proteins. Among the SR techniques, single-molecule localization microscopy plays a significant role due to its ability to unveil structures and molecular organizations in biological systems. Furthermore, since they provide information at the molecular level, these techniques are increasingly being used to study the stoichiometry and interaction between several membrane channel proteins and their accessory subunits. The aim of this review is to describe the single-molecule localization-based techniques and their applications relevant to cytoskeletal structures and membrane complexes in order to provide as future prospective an overall picture of their correlation with the mechanosensor channel expression and activity.
Keywords: Cytoskeletal structures, Membrane complexes, Mechanosensors, Super-resolution microscopy, Single molecule localization miscroscopy
Super-resolution microscopy: advances and challenges
There are a number of super-resolution techniques and these established methods, coupled together with very recently developed technological advancements in computer analysis, have led to growing usage of far-field super-resolution microscopy as a tool for studying biological processes at the sub-cellular level. The key element is represented by the ability to identify two different states (i.e., a bright and a dark one) and the possibility to induce a controlled light-driven transition from one state to the other. In particular, imaging can be based either on “stochastic” or “targeted” switching for subsequent localization of fluorescent emitters. Among the “targeted” readout family, mainly based on the RESOLFT concept (Chmyrov et al. 2013) (REversible Saturable Optical Linear Fluorescence Transitions), a prominent role is played by stimulated emission depletion (STED) (Hell and Wichmann 1994; Bierwagen et al. 2010; Vicidomini et al. 2018) and ground-state depletion microscopy (GSD) (Testa et al. 2010). A relevant role is also played by single-molecule localization (SML) microscopy, a “stochastic” approach in which single emitters can be localized with nanometric precision, thus providing direct information at the molecular scale. Several variants, corresponding to different photophysical process exploited to reach the dark state, were proposed in the last years. Among them, the most popular ones are photoactivation localization microscopy (PALM) (Betzig et al. 2006), fluorescence photoactivation localization microscopy (fPALM) (Hess et al. 2006), stochastic optical reconstruction microscopy (STORM) (Rust et al. 2006), and points accumulation for imaging in nanoscale topography (PAINT) (Schnitzbauer et al. 2017; Giannone et al. 2010). Recently, the need of extending the SR studies from sub-cellular compartments to larger samples (such as whole cells, tissues, whole organisms, or three-dimensional tissue models) leads to the development of novel SR approaches. Within this context, a further step towards volumetric imaging is represented by SR methods based on lattice microscopy (Chen et al. 2014; Aguet et al. 2016; Valm et al. 2017), light-sheet microscopy (Cella Zanacchi et al. 2011; Galland et al. 2015; Lu et al. 2019), and other techniques (Chen et al. 2018).
Despite the fact that SR techniques are currently well established, there is still room for improvement: nowadays, the current challenges are often related to their optimization to match the needs of biological samples, tuning their implementation to facilitate the study of biologically relevant mechanisms. There are several aspects that users have to face in order to properly address specific biological questions when using SR. For example, what is the best strategy for sample preparation and labeling? How should super-resolution in live cells be performed? Providing answers to such questions is becoming increasingly important for overcoming current limitations in biological applications. Indeed, additional aspects can affect the performances and they need to be considered. By way of example, when conducting live-cell imaging experiments, it is essential to minimize phototoxicity effects, thereby preserving the cellular response from light-induced impairment. Also, sample preparation procedures have a strong impact on the image quality and can affect the information gained by super-resolution. In this context, it is important to identify the optimal staining method for cell imaging, ideally able to label proteins at the endogenous expression levels with small fluorescent tags. Despite the crucial role played by these factors and the increasing possibilities available, a commonly recognized “best strategy” has still not yet been identified.
Single-molecule localization microscopy and quantitative approaches
Over the last few years, localization-based techniques, which exploit photoactivation, photoconversion, or ground-state depletion of fluorescent molecules, have become popular tools to study biological structures with unprecedented resolution. The common principle behind these techniques is the capability to collect the signal coming from a single emitter and to localize its position with nanometric precision (Deschout et al. 2014a) (Fig. 1a). All the molecules can be localized and, after a sufficient number of localization cycles, the final image can be obtained by mapping all the localized positions (Fig. 1b). Molecules are commonly detected using a widefield architecture (Fig. 1c). The localization precision typically ranges from 5 to 30 nm and depends from the number of detected photons per molecule (Mortensen et al. 2010; Deschout et al. 2014a). It can be analytically described by Eq. (1)
| 1 |
where N is the number of photons, , s is the related system point spread function (PSF), a is related to the pixel size, and b is the background (Mortensen et al. 2010, Deschout et al. 2014a).
Fig. 1.

SML concept. The localization concept behind SML microscopy (a and b). Scheme of an optical set-up for Super-Resolution Microscopy (c): OBJ, objective; DM, dichroic mirror; BP, band-pass filter; L, lens; M, mirror; A, aperture; LA1, LA2, LA3: laser lines for activation and readout
Since these techniques provide information at the molecular scale, they not only give an imaging tool the ability to overcome the diffraction barrier but they also represent a powerful tool to effectively quantify protein distributions in biological systems. Besides affording resolution far below the diffraction limit, there are several other pieces of quantitative information that can be accessed. For instance, the possibility to image single molecules at high frame rates allows also to track individual molecules and to estimate mobility and interactions of proteins (Manley et al. 2008; Balint et al. 2013; Nozaki et al. 2017). Furthermore, the advent of specific probes designed for super-resolution, such as photo-switchable dyes and photoactivatable fluorescent proteins, made super-resolution particularly attractive for quantitative studies. Single-molecule datasets can be potentially used to get precise information about the protein copy numbers at the cellular level. Advances in this field have led to the possibility for detection of single molecules and for the development of molecular counting approaches based on stepwise photobleaching (Ulbrich and Isacoff 2007). The use of genetically encoded fluorescent proteins combined with the possibility to fully characterize the photoactivation process has opened the way to stoichiometry estimation (Durisic et al. 2014). Moreover, the nature of the super-resolution stochastic approaches, thanks to the repeated localization of single fluorophores, allows direct use of the localizations number for precise protein counting. In this context, over the last few years, attention has been focused on developing quantitative approaches to discriminate between localized clusters and random distributions (Deschout et al. 2014b; Nicovich et al. 2017). In principle, even simple and intuitive methods for clustering, such as nearest-neighbor distribution, could be proposed to study whether target molecules were clustered or not. Several clustering approaches are becoming more and more popular for cluster segmentation from single-molecule localization data (Nicovich et al. 2017). Among them, approaches based on density (Ester et al. 1996; Ricci et al. 2015), mesh representation (Levet et al. 2015), and graph theory (Pavan MaP 2007; Pennacchietti et al. 2017) can be used to discriminate if points belong to the same cluster depending upon the number of molecules localized in the neighbor regions. Cluster analysis shed new light in the quantitative study of protein of interest assemblies in biological systems (Nicovich et al. 2017).
Stain proteins for single-molecule localization microscopy
Several labeling methods are available (Table 1) and the optimal staining strategy has to be identified depending upon the experiments and the sample requirements. Since the methods currently available provide different advantages and drawbacks, a balance between the needs and the performances is mandatory. Indirect immunofluorescence permits imaging at the endogenous expression level, but is not compatible with live-cell imaging. On the other side, fluorescent proteins (FPs) are suitable for live-cell staining but impose some restrictions due to overexpression and their big size. This problem can be faced using the emerging CRISPR/Cas9 technique, but the considerable size of FPs can impair the behavior of the protein of interests under investigation. For this reason, particular attention is now addressed to small chemical tags compatible with live-cell imaging.
Table 1.
Summary of staining methods for SR. Different methods suitable for live-cell imaging (upper) and for fixed cell imaging (lower)
| Fixed | Immunostaining | Immunostaining | Chemical tag |
|
Indirect (protein-specific antibody + secondary antibody) Direct (protein-specific antibody) |
Fluorescent/photoactivatable proteins antibody | HaloEnzyme-Halo-Tag-fluorophore | |
| LIVE | Fusion protein | Fusion protein (smaller peptide) | Expression System |
|
Fluorescent proteins (FPs) Photoactivatable fluorescent proteins (PA-FPs) |
Chemical tags |
Transient or stable transfection CRISP/Cas9 |
Indirect immunofluorescence is the most used labeling procedure for super-resolution imaging to identify proteins of interest (POI) in a biological sample (e.g., cell, tissue) (Vira et al. 2010; Hermanson 2013; Sednev 2015; Sednev et al. 2015). In this technique, the POI is identified by adding primary antibody specific to the target. The second step is to apply secondary antibody pre-labeled with the appropriate fluorescent dye (Vira et al. 2010; Hermanson 2013; Odell and Cook 2013; Sednev 2015; Sednev et al. 2015) (Fig.2a,b). Primary and secondary antibodies are obtained from animals (mice, rabbits, goats, donkeys, etc.) and the secondary antibody is achieved against primary antibody (again from mice, rabbits, goats, donkeys, etc.) as immunoresponse to the target antigen. The advantage of this method is that it is possible to produce secondary antibodies labeled with a wide range of fluorescent dye that is able to recognize many species-specific primary antibodies. An interesting development of typical antibodies is single-domain antibodies, called nanobodies, that are about 15 kDa that is particularly suitable in super-resolution optical microscopy application due to their small size that leads to a more correct evaluation of the dimensions of biological objects (Ries et al. 2012).
Fig. 2.
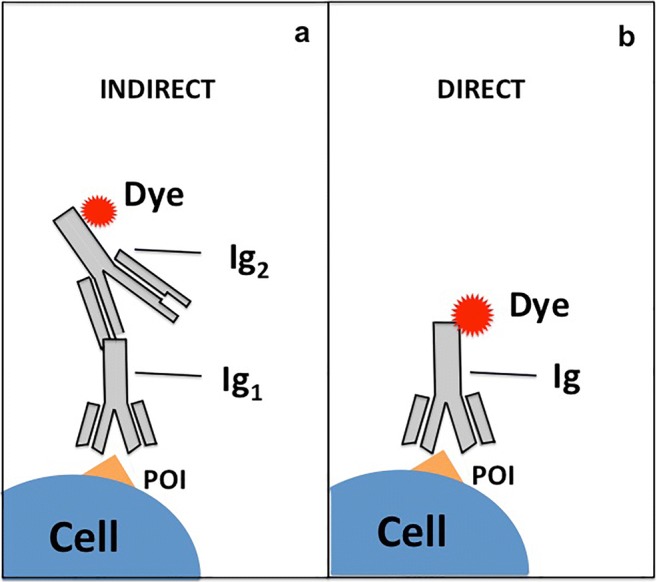
Description of immunostaining techniques. a Indirect immunostaining: protein-specific antibody (Ig1) recognizes the protein of interest (POI) expressed in cells. A second antibody (Ig2) directed against the first antibody (anti-mouse/rabbit/human, etc.) is labeled with a fluorescent dye. b Direct immunostaining: protein-specific antibody (Ig) recognizes a protein of interest (POI) and is labeled with fluorescent dye
Nevertheless, immunofluorescence microscopy is possible only by fixing and permeabilizing cells, and these processes may cause protein efflux and relocalization; therefore, it is important to combine immunofluorescence measures with in vivo cell imaging. Thus, new labeling strategies are required for implementation of the super-resolution optical microscopy in living cells (Schnell et al. 2012). Nowadays, the use of fluorescent proteins (FPs) fused with a protein of interest is a powerful and essential tool to observe cell processes and structures in vivo by using fluorescent microscopy as well as super-resolution microscopy (SRM). Fluorescent proteins are genetically encoded and they can be fused with a gene of interest and inserted into an expression vector using genetic engineering techniques (Fig. 3a). A class of fluorescent proteins, the photoactivatable fluorescent proteins (PA-FPs), have properties that make them especially suitable for single-molecule-based super-resolution imaging techniques (Siyuan Wang and Xiaowei Zhuang 2014): first of all, their high photon output means more accurate localization and leads to higher image resolution (Dempsey et al. 2011; Konstantin et al. 2005); second, the property of on–off switching rate ratio (on–off ratio), defined as the ratio between the on-switching (activation) and off-switching or photobleaching rates under the illumination of the imaging light only (Dempsey et al. 2011). The on–off ratio limits the density of dyes that can be confined, and this influences the actual image resolution based on the Nyquist sampling theorem (Shroff et al. 2008). Another PA-FPs feature is the high signaling efficiency (ratio between the number of detectable PA-FP-fusion molecules per cell): only a subset of PA-FP proteins can be photoactivated and imaged and thus, the number of fusion proteins detected could be lower than the overall expressed.
Fig. 3.
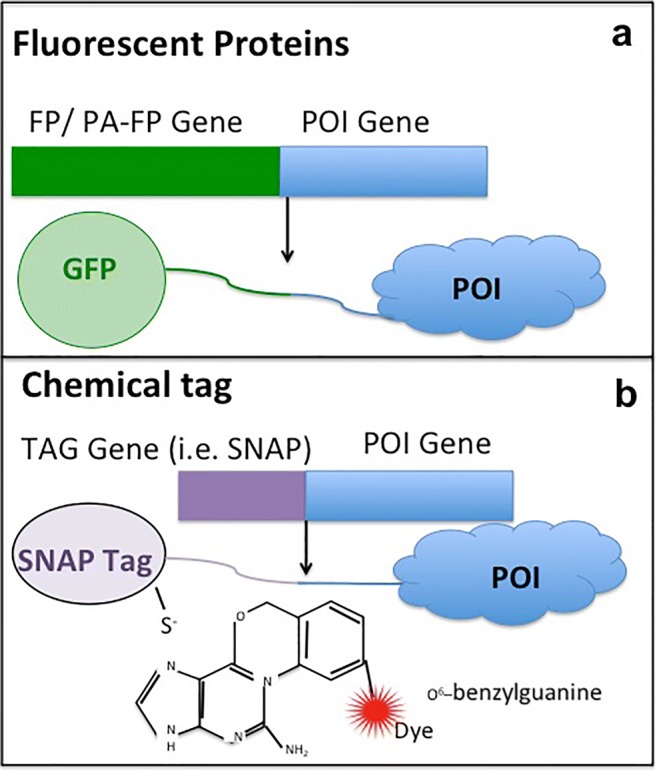
Scheme of expression of fusion proteins. a Fluorescent proteins (FPs) and photoactivatable fluorescent proteins (PA-FPs) fused with the protein of interest (POI) and b chemical tags: fluorescent ligands target SNAP (or Halo) fused with POI
Most PA-FPs are classified into two groups: EosFP-like that switches from a green fluorescent state to an orange state (Jörg Wiedenmann et al. 2004; Tsutsui et al. 2005) upon photoactivation, and PA-GFP-like that switches from a dark state to a green fluorescent state (Patterson and Lippincott-Schwartz 2002; Ryoko Ando and Miyawak 2004) upon photoactivation. The most common PA-FPs are the following: PA-GFP (Patterson and Lippincott-Schwartz 2002), Dendra2 (Gurskaya et al. 2006; Dmitriy and Chudakov 2007), mEos2 (Mingshu Zhang et al. 2012), mEos3.2 (Jörg Wiedenmann et al. 2004), tdEos (Jörg Wiedenmann et al. 2004), mKikGR (Satoshi Habuchi and Miyawaki 2008), PAmCherry (Fedor V Fedor et al. 2009), PAtagRFP (Ann et al. 2012), mMaple (Dmitriy et al. 2004), PSCFP2 (Dmitriy and Chudakov 2007; Hao Chang et al. 2012), Dronpa (Ryoko Ando and Miyawak 2004), and mGeosM (Fedor et al. 2009). Several PA-FPs (such as PAmCherry) have been especially designed for PALM/fPALM imaging (H.Patterson 2008) and the green to red photoconvertible proteins (such as Dendra 2, mEos2) have also been extensively used in SML. In case of green to red photoconvertible fluorescent proteins, UV light causes a passage from the green state to a photostable red fluorescent state due to an irreversible light-induced modification of their chromophores. Their stability and robustness make them perfect fluorescent tags to track dynamics of POI within cells (Baker et al. 2010) and to perform SML imaging (Gunewardene et al. 2011). Differently, the photoswitchable fluorescent proteins (such as Dronpa) are proteins that upon excitation at a certain wavelength can be switched on and off by light in a reversible manner. Moreover, the photoswitchable fluorescent proteins can repeat cycles of activation and deactivation, and for this, features have found a strong utility in SR time-lapse microscopy in living cells (Zhou and Lin 2013). Some chimera proteins obtained from the union of photoswitchable fluorescent proteins and POI are used in SR techniques such as reversible optically linear fluorescence transition (RESOLFT) (Hofmann et al. 2005) and stochastic optical fluctuation imaging (SOFI) (Hertel et al. 2016).
Although the use of fusion POI with PA-FPs/FPs is the main method to observe live cells by SRM, this approach has a few drawbacks mainly due to the size of the reporter proteins that could perturb the POI itself. To overcome this issue, a recent technique was developed: the chemical tag (Wombacher and Cornish 2011). This method consists of joining the gene of POI with a short sequence coding for a peptide sequence, necessary to create a protein fusion tag (Fig. 3b). Compared with FPs, chemical tags are smaller and can be combined with a large range of fluorescent dyes. Different peptides have been developed to study proteins in living system: Halo-tag (Los et al. 2008), Snap-tag (Keppler et al. 2003), TMP-tag (Gallagher et al. 2009), beta-lactamase-tag (Watanabe et al. 2010), ACP-tag (George et al. 2004), tetracysteine-tag (Hoffmann et al. 2010), and LpIA acceptor peptide (Uttamapinant et al. 2010). The most commonly used fusion peptides in optical probe development in fluorescent microscopy are the Snap-tag and the Halo-tag proteins. Snap-tag covalently reacts with O6–benzylguanine (BG) derivatives that carry fluorescent dyes (Fig. 3b) (Keppler et al. 2003; Kindermann et al. 2004). The Halo-tag protein is an engineered mutant of a bacterial dehalogenase that allows it to form an irreversible covalent compound with chloroalkane ligand (Los et al. 2008). The reaction between the Snap-tag and Halo-tag with their relevant substrates generates stable compounds for which the stoichiometry and kinetics of the reaction are rapid and predictable (Uttamapinant et al. 2010). In in vivo fluorescent microscopy, a mandatory feature of fluorescent dyes carried on substrates for tag proteins is to be permeable to the cell membranes, in order to reach intracellular fusion tag proteins. This propriety is exclusive for few chemical dyes that show cell permeability, such as the neutral fluorophores based on coumarin and rhodamine scaffolds (Keppler et al. 2003; Encell et al. 2012; Gautier et al. 2008; Lukinavicius et al. 2013).
A leitmotiv of in vivo and in vitro SRM is the attempt to create smaller and smaller fluorescent markers that allow for specific labeling in regions that, due to the dimensions, are inaccessible to traditional tags, in order to improve the imaging performance. Furthermore, smaller fluorescent tags improve the resolution and facilitate the correct localization of the POI. From this point of view, the SR performances strongly benefit with the use of chemical tags; the little DNA sequences fused together with the POI gene (to reduce perturbations in the biological systems) are marked by small tags.
Within this scenario, the use of nanobodies in indirect immunofluorescence permits more detailed imaging due to the little size of single-domain antibodies compared with a whole antibody (Ries et al. 2012).
One last observation is about how the POI gene fused with FP, PA-FPs, or Tag sequences enters inside eukaryotic living cells in order to be expressed and studied. In the majority of live cells SRM works, the method that was used is the transient or constitutive transfection. Transfection can be obtained via two possible methods: the physical ones that include electroporation, microinjection and ballistic delivery with a gene gun, and the chemical ones, based on chemical agents that create a layer around the negatively charged nucleic acids to neutralize their charge and pass through the plasma membrane (for example, calcium phosphate co-precipitation, diethylaminoethyl-dextran, or cationic lipid-based transfection reagent). Furthermore, the transfection can be transient when the cells express the foreign gene for a few days but do not integrate it into their genome. A stably transfected cell line is realized when the foreign gene becomes instead part of the genome and the transfected cells and their descendants stably express the chimera proteins. However, the forced expression of host fusion proteins causes many artifacts such as overexpression, protein aggregation (Mark et al. 2009), abnormal shape of sub-cellular organelles (Harder et al. 2004), and other unwanted effect (Voeltz et al. 2006; Birchler and Veitia 2012; Gibson et al. 2013). To overcome these artifacts affecting SRM studies, the emerging CRISPR/Cas9 technique could be employed since it offers the possibility to induce targeted deletions, insertions, and precise sequence changes in the genomic DNA of different organisms and cell types (Sander and Joung 2014). This method is based on nuclease Cas9 that cut DNA double-strand sequences at any genomic site defined by a 20-bp-long guide RNA (gRNA), complementary to the target site that contains an upstream protospacer adjacent motif (PAM) (Ratz et al. 2015). The cell endogenous homology-directed repair systems (HDR) can be used to introduce specific point mutations or to insert desired sequences (i.e., a fluorescent protein) through recombination of the target locus with exogenously supplied DNA donor templates (Sander and Joung 2014). Since insertion of the new gene occurs inside the native sequence without altering the regulatory sequence, the expression of the fusion proteins is close to that of the endogenous one.
Biological applications of SML (examples)
Live-cell super-resolution imaging
The initial use of PA-FP fusion proteins to obtain nanometric positional resolution is described in the paper of Hess et al. 2006(Betzig et al. 2006). This work introduced the PALM technique for analysis of cells transfected with specific target proteins such as dEos-fused vinculin in focal adhesion, dEosFP-fused actin visualized in cytoskeletal stress fibers and within lamellipodium, and dEosFP-fused Gag protein of human immunodeficiency virus1 retroviral protein distribution in the whole plasma membrane. Through the use of PALM tracking techniques, Frost et al. (2010) found that spines of living neurons contain localized foci of actin polymerization not exclusively at the postsynaptic site (Frost et al. 2010). This technique focused on photoactivation followed by photobleaching of the whole spine of the neuron transfected with PA-GFP actin. As the spine refills with new monomers of actin, that can be polymerized, the spine was again photoactivated. The pattern of fluorescence intensity following subsequent activation shows where new actin monomer was absorbed into the filament between the two laser pulses. With this procedure, the authors demonstrated that actin was incorporated (for the most part) into the center of the spine as well as to a remarkable amount into the tip. Many other studies have characterized the function of the actin as a key cytoskeletal protein in membrane organization by proposing different mechanisms (Sheetz 1983; Chichili and Rodgers 2007).
The work of Grimm et al. (2015) is focused on the creation of a class of tag dyes that improve brightness and photostability, in order to perform SRM experiments. In their 2015 study, the authors compared the performances of commercial Snap-tag ligand with a novel azetidinyl derivative in H2B-Snap-tag expressing HeLa cells and performed STORM experiments on fixed U20S cells expressing Halo-tag–H2B labeled with new dye JF646 Halo-tag ligand. These tags opened new possibilities for potential live imaging.
Jakobs et al. in 2014 compared the endogenous level expression of native fluorescent proteins tagged by using CRISPR/Cas9 technique with the expression of the same fused proteins in transiently transfected cells (Ratz et al. 2015). The authors studied three proteins using the CRISP/Cas9 technique by fusing the rsEGFP2 gene at HMGA1, VIM, and ZYX loci, obtaining three corresponding fusion proteins: the DNA-binding non-histone mobility protein, HMG-I-rsEGFP2, the class-III intermediate filament protein, Vimentin-rsEGFP2, and the focal adhesion plaque protein, Zyxin-rsEGP2. The expression level in knock-in cells and, upon transient expression, was compared quantitatively with fluorescence-activated cell sorting (FACS) and with the super-resolution technique RESOLFT. The authors concluded that “FACS analysis and fluorescence microscopy revealed that the expression levels of the fusion proteins were largely constant within each clonal knock-in cell line, whereas they varied by orders of magnitude in transfected cells, which likely accounts for many of the observed artefacts”. Using these gene-edited cell lines and RESOLFT nanoscopy, they demonstrated an optical resolution of about 40 nm while allowing multiple recordings to visualize sub-cellular dynamics of HMG-I-, Vimentin-, or Zyxin-rsEGFP2 fusion proteins expressed at endogenous levels in living cells.
Quantitative SML to study ion channels
The development of specific probes suitable for SR, such as photoactivatable markers, provides an attractive tool for quantitative studies. Within this scenario, the advances in the SR field lead to the possibility to count molecules using stepwise photobleaching approaches (Ulbrich and Isacoff 2007; Durisic et al. 2014; Fricke et al. 2015). This opens the way to the study of the stoichiometry of different membrane-bound proteins and to resolve the assembling of the molecular complexes. Indeed, the structure of heterodimeric complexes, such as ion channels and their subunits, can be identified. Key examples are the studies of acid-sensing ion channel (Bartoi et al. 2014), NMDA receptors (Ulbrich and Isacoff 2007), and the heteromeric glycine-gated channels (Durisic et al. 2012). One of the most biologically relevant complexes, whose stoichiometry is still controversial, is the group of the potassium channel complex that generates the repolarizing cardiac current. The union between a tetrameric voltage-gated K+ channel and type I trans-membrane regulatory subunit is known. However, the determination of the exact number of regulatory subunits is still a matter of much debate (Nakajo et al. 2010; Kobertz 2014; Plant et al. 2014). The quantitative capabilities provided by SML made this technique an extremely promising tool to study unknown heteromeric complexes formed by ion channels such as mechanosensitive channels.
SLM to study cytoskeletal and associated proteins
The cytoskeletal filaments, along with a large number of regulatory proteins and organelles, carry out multiple cellular functions contributing to mechanical, morphological, and signaling properties of the cell (Fletcher and Mullins 2010). In the last years, the advances in light microscopy allowed a better understanding of the organization, composition, and functionality of the cytoskeletal system. Both PALM and STORM provided an insight into the protein components of the cytoskeleton and their relationship with other organelles at a nanometer-scale resolution, impossible to achieve before (Pollard and Cooper 2009). Since microtubules and actin networks are well-characterized structures having dimensions comparable with the achievable spatial resolution with SMLM, they were initially used to test the resolving power of SMLM (Patterson et al. 2010; Sigal et al. 2018) and the localization efficiency (Endesfelder et al. 2014). 3D STORM imaging of microtubules in HeLa cells shows the improvement in spatial resolution that allows for the determination of each filament in the three dimensions (Fig. 4a, b).
Fig. 4.

SML imaging of microtubules. a 3D STORM image of microtubules labeled with Alexa Fluor 647 (localization precision: lateral 20 nm, axial 65 nm). b Corresponding widefield image. Scale bar 5 μm. Authors acknowledge the Nikon imaging center at IIT for STORM imaging
Several publications attest to these advances in the study of the cytoskeleton achieved through SMLM during these years. With the aid of super-resolution, for the first time, it was possible to image individual actin filaments despite their thin diameter and highly dense network. The tridimensional structure of actin polymer was resolved in sheet-like cell protrusions that support cell motility. 3D STORM imaging revealed two vertically separated actin layers forming separate and structurally different networks interacting with focal adhesions (Xu et al. 2012).
An exciting discovery concerning cytoskeleton has been the membrane-associated periodic skeleton (MPS), a quasi-1D periodic structure observed in neuronal axons by STORM imaging. MPS consists of actin filaments capped by ring-like structures (adducin) that wraps around the axon circumference. These rings are connected to each other by means of spectrin tetramers along the major axis of the axon. Sodium channels are distributed in a periodic pattern in coordination with the actin-spectrin-based cytoskeleton (Xu et al. 2013).
In recent work, Pan et al. resolved the ultra-structure of cytoskeleton in native human erythrocytes by means of STORM super-resolution microscopy. They observed spectrin tetramers connected by junctional complexes containing short-actin filaments and capping proteins. In detail, they measured an 80-nm junction-to-junction distance that means that spectrin tetramers are in a relaxed state rather than in a stretched one. This configuration may be functional for erythroid cells to allow them to undergo expansion or compression as they usually do while traversing the circulatory system (Pan et al. 2018). In addition to the investigation on the cytoskeletal polymeric network, other associated structures, such as focal adhesions, have been the focus of research. The function of focal adhesions, macromolecular assemblies connecting the intracellular actin network with the extracellular matrix, is significant and ranging from cell adhesion, migration, anchoring, mechanosensing, and signaling. Focal adhesions consist of large complexes of trans-membrane integrins and cytoplasmic proteins, such as talin, α-actinin, and vinculin, which form plaques (Sydor et al. 2015). Figure 5 shows a super-resolution image of vinculin in the focal adhesions of a HeLa cell, acquired with STORM microscopy. Single-molecule imaging and cluster analysis have shed new light on the nanoscale protein organization of focal adhesion.
Fig. 5.

SML imaging of focal adhesions. a SML imaging of vinculin labeled with Alexa Fluor 647. b Corresponding cluster analysis map. Scale bar 5 μm
Two-color PALM has been used to determine colocalization of vinculin and paxillin, showing that they actually form nano-aggregates (Shroff et al. 2007) of focal adhesions, where talin plays a central role in organizing the focal adhesion strata (Kanchanawong et al. 2010). There are several processes that are mediated by the cytoskeleton and connected with microtubule-associated proteins. One of the most fascinating aspects is the intracellular transport, in which motor proteins’ dynamics plays an important role. Motor proteins (i.e., dyneins, myosins, and kinesins), also called molecular motors, bind to the cytoskeleton and move along microtubules and actin filaments bringing cargos. The role of these nanometric motors mainly consists of the intracellular trafficking of proteins and vesicles (Kolomeisky and Fisher 2007; Kolomeisky 2013). In 2013, Bàlint et al. (Balint et al. 2013) used an all-optical correlative imaging method based on single-particle tracking and stochastic optical reconstruction microscopy to investigate how the intersection points among microtubules or between microtubules and actin filaments affect the motion of motor proteins and, as a consequence, the cargo transportation inside the cell. They discovered that, after pausing, motors can pass through intersection when the gap is smaller than ~ 100 nm without changing the direction of their movement (anterograde or retrograde) (Balint et al. 2013). In more recent work, they have further demonstrated the 3D motion of vesicles carried by motor proteins, clarifying the mechanism that permits them to circumvent obstacles, like intersections (Verdeny-Vilanova et al. 2017). Quantitative SML can be used to gain a quantitative insight, elucidating the minimum number of motors responsible for cargo transportation. Quantitative tools can be used (Cella Zanacchi et al. 2017), to show that dynein form multimers organized in nanoclusters along microtubules and to reveal the number of dyneins in each assembly (Cella Zanacchi et al. 2019).
Application to mechanosensitive channels and future perspectives
Mechanosensitive channels are proteins expressed in mechanosensitive cells that react to mechanical forces and contain a pore-forming subunit for ion conduction (Syeda et al. 2016). Emerging studies are focused on the role between cellular components that are required for gating MS channels like auxiliary subunit and cytoskeleton factors (Martinac 2014; Syeda et al. 2016). For example, in Drosophila, with regard to the mechanically activated channel NOMPC (no mechanoreceptor potential C), it was found that a cytoskeleton connection is required in order to observe mechanosensitivity properties (Zhang et al. 2015). Furthermore, it was detected that the sensitivity of the mechanosensor protein PIEZO1 to mechanical indentation is modified if the cytoskeleton is chemically disrupted (Gottlieb and Sachs 2012). In this state of the art research, a key question is the identification of which cytoskeleton structures co-works with MS channels and if they act as accessory proteins.
The proven ability of SML to investigate cytoskeletal structures suggests this technique as an optimal strategy to study their role in the MS channel activity. First, SML can help by localizing both cytoskeletal proteins and adhesion complexes involved in the regulation of MS channel activities. On the other hand, the possibility to study the stoichiometries of molecular complexes by quantitative SML will provide novel insights into the structure of MS channels and their potential accessory proteins. In this direction, a first example is given by the work of Van den Berg and colleagues (van den Berg et al. 2016), in which the channel abundance of MscS, MscL, and MscK in the E. coli plasma membrane was studied by quantitative PALM (Fig. 6a–c).
Fig. 6.
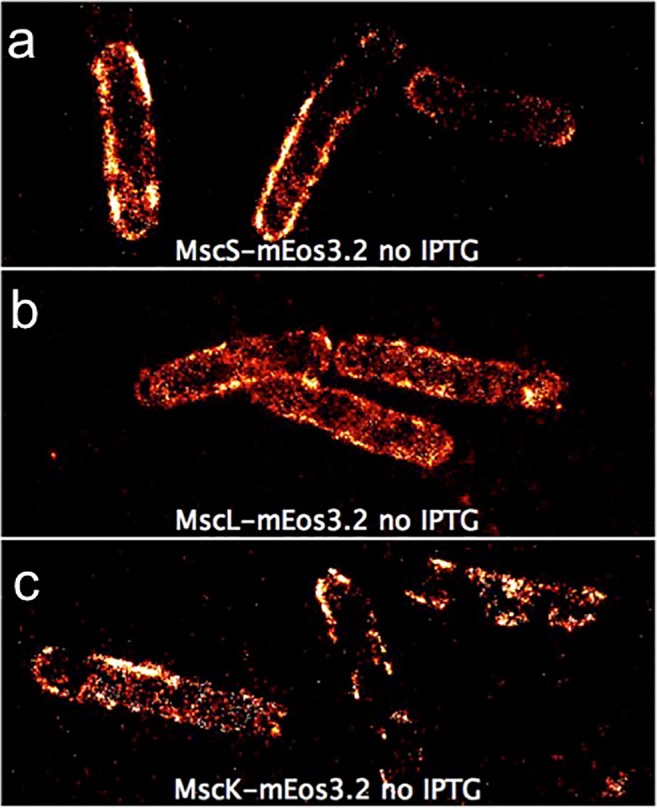
SML imaging of mechanosensitive channels. Distribution of fluorescently labeled mechanosensitive channels observed by PALM Membrane localization of MscS-mEos3.2 (a), MscL-mEos3.2 (b), and MscK-mEos3.2 (c) in E. coli MG1655. Modified from Van den Berg et al. Scientific Reports 6 (2016)
As a future perspective, the combination of these approaches will provide an overall picture of the correlation of the focal adhesion proteins, actin, and tubulin filaments with the MS channel expression and activity.
Acknowledgments
The authors acknowledge the Nanoscopy group and the Nikon imaging center at the Istituto Italiano di Tecnologia.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain studies with human participants or animals.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
R. Magrassi, Email: raffaella.magrassi@ge.ibf.cnr.it
F. Cella Zanacchi, Email: francesca.cella@iit.it
References
- Aguet F, Upadhyayula S, Gaudin R, Chou YY, Cocucci E, He K, Chen BC, Mosaliganti K, Pasham M, Skillern W, Legant WR, Liu TL, Findlay G, Marino E, Danuser G, Megason S, Betzig E, Kirchhausen T. Membrane dynamics of dividing cells imaged by lattice light-sheet microscopy. Mol Biol Cell. 2016;27:3418–3435. doi: 10.1091/mbc.E16-03-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ann L. McEvoy HH, Mark Bates, Evgenia Platonova, Paula J. Cranfill, Michelle A. Baird, Michael W. Davidson, Helge Ewers, Jan Liphardt, Robert E. Campbell (2012) A photoconvertible fluorescent protein for use in multiple imaging modalities. 12:1–15 [DOI] [PMC free article] [PubMed]
- Baker SM, Buckheit RW, 3rd, Falk MM. Green-to-red photoconvertible fluorescent proteins: tracking cell and protein dynamics on standard wide-field mercury arc-based microscopes. BMC Cell Biol. 2010;11:15. doi: 10.1186/1471-2121-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balint S, Verdeny Vilanova I, Sandoval Alvarez A, Lakadamyali M. Correlative live-cell and superresolution microscopy reveals cargo transport dynamics at microtubule intersections. Proc Natl Acad Sci U S A. 2013;110:3375–3380. doi: 10.1073/pnas.1219206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoi T, Augustinowski K, Polleichtner G, Grunder S, Ulbrich MH. Acid-sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci U S A. 2014;111:8281–8286. doi: 10.1073/pnas.1324060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Bierwagen J, Testa I, Fölling J, Wenzel D, Jakobs S, Eggeling C, Hell SW. Far-field autofluorescence nanoscopy. Nano Lett. 2010;10:4249–4252. doi: 10.1021/nl1027638. [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A. 2012;109:14746–14753. doi: 10.1073/pnas.1207726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella Zanacchi F, Lavagnino Z, Perrone Donnorso M, Del Bue A, Furia L, Faretta M, Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods. 2011;8:1047–1049. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]
- Cella Zanacchi F, Manzo C, Alvarez AS, Derr ND, Garcia-Parajo MF, Lakadamyali M. A DNA origami platform for quantifying protein copy number in super-resolution. Nat Methods. 2017;14:789–792. doi: 10.1038/nmeth.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella Zanacchi F, Manzo C, Magrassi R, Derr ND, Lakadamyali M. Quantifying protein copy number in super resolution using an imaging-invariant calibration. Biophys J. 2019;116:2195–2203. doi: 10.1016/j.bpj.2019.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, 3rd, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Bohme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science. 2014;346:1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu W, Zhang Z, Zheng C, Huang Y, Cao R, Zhu D, Xu L, Zhang M, Zhang YH, Fan J, Jin L, Xu Y, Kuang C, Liu X. Multi-color live-cell super-resolution volume imaging with multi-angle interference microscopy. Nat Commun. 2018;9:4818. doi: 10.1038/s41467-018-07244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichili GR, Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. J Biol Chem. 2007;282:36682–36691. doi: 10.1074/jbc.M702959200. [DOI] [PubMed] [Google Scholar]
- Chmyrov A, Keller J, Grotjohann T, Ratz M, d’Este E, Jakobs S, Eggeling C, Hell SW. Nanoscopy with more than 100,000 ‘doughnuts. Nat Methods. 2013;10:737. doi: 10.1038/nmeth.2556. [DOI] [PubMed] [Google Scholar]
- Dempsey GTVJ, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschout H, Cella Zanacchi F, Mlodzianoski M, Diaspro A, Bewersdorf J, Hess ST, Braeckmans K. Precisely and accurately localizing single emitters in fluorescence microscopy. Nat Methods. 2014;11:253–266. doi: 10.1038/nmeth.2843. [DOI] [PubMed] [Google Scholar]
- Deschout H, Shivanandan A, Annibale P, Scarselli M, Radenovic A. Progress in quantitative single-molecule localization microscopy. Histochem Cell Biol. 2014;142:5–17. doi: 10.1007/s00418-014-1217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriy M, Chudakov SLKAL. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc. 2007;2:2024–2032. doi: 10.1038/nprot.2007.291. [DOI] [PubMed] [Google Scholar]
- Dmitriy M, Chudakov VVV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol. 2004;22:1453–1439. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]
- Durisic N, Godin AG, Wever CM, Heyes CD, Lakadamyali M, Dent JA. Stoichiometry of the human glycine receptor revealed by direct subunit counting. J Neurosci. 2012;32:12915–12920. doi: 10.1523/JNEUROSCI.2050-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N, Laparra-Cuervo L, Sandoval-Alvarez A, Borbely JS, Lakadamyali M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat Methods. 2014;11:156–162. doi: 10.1038/nmeth.2784. [DOI] [PubMed] [Google Scholar]
- Encell LP, Friedman Ohana R, Zimmerman K, Otto P, Vidugiris G, Wood MG, Los GV, McDougall MG, Zimprich C, Karassina N, Learish RD, Hurst R, Hartnett J, Wheeler S, Stecha P, English J, Zhao K, Mendez J, Benink HA, Murphy N, Daniels DL, Slater MR, Urh M, Darzins A, Klaubert DH, Bulleit RF, Wood KV. Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr Chem Genomics. 2012;6:55–71. doi: 10.2174/1875397301206010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder U, Malkusch S, Fricke F, Heilemann M. A simple method to estimate the average localization precision of a single-molecule localization microscopy experiment. Histochem Cell Biol. 2014;141:629–638. doi: 10.1007/s00418-014-1192-3. [DOI] [PubMed] [Google Scholar]
- Ester MK, H.P Kriegel; Sander, J.; Xu, X.; (1996) A density based algorithm for discovering clusters in large spatial database with noise. Proceed of 2nd international conference on knowledge discovery and data mining 34:226-231
- Fedor V, Subach GHP, Manley S, Gillette JM, Lippincott-Schwartz J, Verkhusha VV. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke F, Beaudouin J, Eils R, Heilemann M. One, two or three? Probing the stoichiometry of membrane proteins by single-molecule localization microscopy. Sci Rep. 2015;5:14072. doi: 10.1038/srep14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost NA, Shroff H, Kong H, Betzig E, Blanpied TA. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67:86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SS, Sable JE, Sheetz MP, Cornish VW. An in vivo covalent TMP-tag based on proximity-induced reactivity. ACS Chem Biol. 2009;4:547–556. doi: 10.1021/cb900062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galland R, Grenci G, Aravind A, Viasnoff V, Studer V, Sibarita JB. 3D high- and super-resolution imaging using single-objective SPIM. Nat Methods. 2015;12:641–644. doi: 10.1038/nmeth.3402. [DOI] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- George N, Pick H, Vogel H, Johnsson N, Johnsson K. Specific labeling of cell surface proteins with chemically diverse compounds. J Am Chem Soc. 2004;126:8896–8897. doi: 10.1021/ja048396s. [DOI] [PubMed] [Google Scholar]
- Giannone G, Hosy E, Levet F, Constals A, Schulze K, Sobolevsky AI, Rosconi MP, Gouaux E, Tampe R, Choquet D, Cognet L. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys J. 2010;99:1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson TJ, Seiler M, Veitia RA. The transience of transient overexpression. Nat Methods. 2013;10:715–721. doi: 10.1038/nmeth.2534. [DOI] [PubMed] [Google Scholar]
- Gottlieb PA, Sachs F. Piezo1: properties of a cation selective mechanical channel. Channels. 2012;6:214–219. doi: 10.4161/chan.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, Lionnet T, Lavis LD. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods. 2015;12:244–250. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunewardene MS, Subach FV, Gould TJ, Penoncello GP, Gudheti MV, Verkhusha VV, Hess ST. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NGVV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Hao Chang MZ, Ji W, Chen J, Zhang Y, Liu B, Lu J, Zhang J, Xu P, Xu T. A unique series of reversibly switchable fluorescent proteins with beneficial properties for various applications. PNAS. 2012;109:4455–4460. doi: 10.1073/pnas.1113770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology : CB. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Hermanson G (2013) Bioconjugate techniques: Elsevier
- Hertel F, Mo GC, Duwe S, Dedecker P, Zhang J. RefSOFI for mapping nanoscale organization of protein-protein interactions in living cells. Cell Rep. 2016;14:390–400. doi: 10.1016/j.celrep.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Zurn A, Adams SR, Terrillon S, Ellisman MH, Tsien RY, Lohse MJ. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat Protoc. 2010;5:1666–1677. doi: 10.1038/nprot.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction<p>barrier in fluorescence microscopy at low light intensities by using reversibly<p>photoswitchable proteins. Proc Natl Acad Sci U S A. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörg Wiedenmann SI, Oswald F, Schmitt F, Röcker C, Salih A, Spindler K-D, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. PNAS. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Kindermann M, Sielaff I, Johnsson K. Synthesis and characterization of bifunctional probes for the specific labeling of fusion proteins. Bioorg Med Chem Lett. 2004;14:2725–2728. doi: 10.1016/j.bmcl.2004.03.078. [DOI] [PubMed] [Google Scholar]
- Kobertz WR. Stoichiometry of the cardiac IKs complex. Proc Natl Acad Sci U S A. 2014;111:5065–5066. doi: 10.1073/pnas.1403171111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomeisky AB. Motor proteins and molecular motors: how to operate machines at the nanoscale. J Phys Condensed matter : Institut Phys J. 2013;25:463101. doi: 10.1088/0953-8984/25/46/463101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomeisky AB, Fisher ME. Molecular motors: a theorist’s perspective. Annu Rev Phys Chem. 2007;58:675–695. doi: 10.1146/annurev.physchem.58.032806.104532. [DOI] [PubMed] [Google Scholar]
- Lukyanov Konstantin A., Chudakov Dmitry M., Lukyanov Sergey, Verkhusha Vladislav V. Photoactivatable fluorescent proteins. Nature Reviews Molecular Cell Biology. 2005;6(11):885–890. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- Levet F, Hosy E, Kechkar A, Butler C, Beghin A, Choquet D, Sibarita JB. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods. 2015;12:1065–1071. doi: 10.1038/nmeth.3579. [DOI] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Lu CH, Tang WC, Liu YT, Chang SW, Wu FCM, Chen CY, Tsai YC, Yang SM, Kuo CW, Okada Y, Hwu YK, Chen P, Chen BC. Lightsheet localization microscopy enables fast, large-scale, and three-dimensional super-resolution imaging. Commun Biol. 2019;2:177. doi: 10.1038/s42003-019-0403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Correa IR, Jr, Luo ZG, Schultz C, Lemke EA, Heppenstall P, Eggeling C, Manley S, Johnsson K. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Mark A. Rizzo MWD, and David W. Piston (2009) Fluorescent protein tracking and detection: applications using fluorescent proteins in living cells [DOI] [PubMed]
- Martinac B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim Biophys Acta. 2014;1838:682–691. doi: 10.1016/j.bbamem.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Mingshu Zhang HC, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, Zhang J, Xu PXT. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
- Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 2010;7:377–381. doi: 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY. Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc Natl Acad Sci U S A. 2010;107:18862–18867. doi: 10.1073/pnas.1010354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicovich PR, Owen DM, Gaus K. Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat Protoc. 2017;12:453–460. doi: 10.1038/nprot.2016.166. [DOI] [PubMed] [Google Scholar]
- Nozaki T, Imai R, Tanbo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, Wendt KS, Okada Y, Nagai T, Maeshima K. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol Cell. 2017;67:282. doi: 10.1016/j.molcel.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Odell ID, Cook D. Immunofluorescence techniques. J Investig Dermatol. 2013;133:e4. doi: 10.1038/jid.2012.455. [DOI] [PubMed] [Google Scholar]
- Pan L, Yan R, Li W, Xu K. Super-resolution microscopy reveals the native ultrastructure of the erythrocyte cytoskeleton. Cell Rep. 2018;22:1151–1158. doi: 10.1016/j.celrep.2017.12.107. [DOI] [PubMed] [Google Scholar]
- Patterson JS. Fluorescent proteins for photoactivation experiments. Methods Cell Biol. 2008;85:45–61. doi: 10.1016/S0091-679X(08)85003-0. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Patterson G, Davidson M, Manley S, Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavan MaP M. Dominant sets and pairwise clustering. IEEE Trans Pattern Anal Mach Intell. 2007;29:167–172. doi: 10.1109/tpami.2007.250608. [DOI] [PubMed] [Google Scholar]
- Pennacchietti F, Vascon S, Nieus T, Rosillo C, Das S, Tyagarajan SK, Diaspro A, Del Bue A, Petrini EM, Barberis A, Zanacchi FC. Nanoscale molecular reorganization of the inhibitory postsynaptic density is a determinant of GABAergic synaptic potentiation. J Neurosci. 2017;37:1747–1756. doi: 10.1523/JNEUROSCI.0514-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H, Goldstein SA. Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci U S A. 2014;111:E1438–E1446. doi: 10.1073/pnas.1323548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep. 2015;5:9592. doi: 10.1038/srep09592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods. 2012;9:582–584. doi: 10.1038/nmeth.1991. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoko Ando HM, Miyawak A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi Satoshi, Tsutsui Hidekazu, Kochaniak Anna B., Miyawaki Atsushi, van Oijen Antoine M. mKikGR, a Monomeric Photoswitchable Fluorescent Protein. PLoS ONE. 2008;3(12):e3944. doi: 10.1371/journal.pone.0003944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell U, Dijk F, Sjollema KA, Giepmans BN. Immunolabeling artifacts and the need for live-cell imaging. Nat Methods. 2012;9:152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F, Jungmann R. Super-resolution microscopy with DNA-PAINT. Nat Protoc. 2017;12:1198–1228. doi: 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]
- Sednev M (2015) A practical guide to dSTORM: super-resolution imaging with standard fluorescent probes
- Sednev MV, Belov VN, Hell SW. Fluorescent dyes with large stokes shifts for super-resolution optical microscopy of biological objects: a review. Methods Appl Fluoresc. 2015;3:042004. doi: 10.1088/2050-6120/3/4/042004. [DOI] [PubMed] [Google Scholar]
- Sheetz MP. Membrane skeletal dynamics: role in modulation of red cell deformability, mobility of transmembrane proteins, and shape. Semin Hematol. 1983;20:175–188. [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff HGC, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal YM, Zhou R, Zhuang X. Visualizing and discovering cellular structures with super-resolution microscopy. Science. 2018;361:880–887. doi: 10.1126/science.aau1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Moffitt J. R., Dempsey G. T., Xie X. S., Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proceedings of the National Academy of Sciences. 2014;111(23):8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydor AM, Czymmek KJ, Puchner EM, Mennella V. Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol. 2015;25:730–748. doi: 10.1016/j.tcb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, Patapoutian A. Piezo1 channels are inherently mechanosensitive. Cell Rep. 2016;17:1739–1746. doi: 10.1016/j.celrep.2016.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa I, Wurm CA, Medda R, Rothermel E, von Middendorf C, Folling J, Jakobs S, Schonle A, Hell SW, Eggeling C. Multicolor fluorescence nanoscopy in fixed and living cells by exciting conventional fluorophores with a single wavelength. Biophys J. 2010;99:2686–2694. doi: 10.1016/j.bpj.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 2005;6:233–238. doi: 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci U S A. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature. 2017;546:162–167. doi: 10.1038/nature22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg J, Galbiati H, Rasmussen A, Miller S, Poolman B. On the mobility, membrane location and functionality of mechanosensitive channels in Escherichia coli. Sci Rep. 2016;6:32709. doi: 10.1038/srep32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdeny-Vilanova I, Wehnekamp F, Mohan N, Sandoval Alvarez A, Borbely JS, Otterstrom JJ, Lamb DC, Lakadamyali M. 3D motion of vesicles along microtubules helps them to circumvent obstacles in cells. J Cell Sci. 2017;130:1904–1916. doi: 10.1242/jcs.201178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicidomini G, Bianchini P, Diaspro A. STED super-resolved microscopy. Nat Methods. 2018;15:173–182. doi: 10.1038/nmeth.4593. [DOI] [PubMed] [Google Scholar]
- Vira S, Mekhedov E, Humphrey G, Blank PS. Fluorescent-labeled antibodies: balancing functionality and degree of labeling. Anal Biochem. 2010;402:146–150. doi: 10.1016/j.ab.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Mizukami S, Hori Y, Kikuchi K. Multicolor protein labeling in living cells using mutant beta-lactamase-tag technology. Bioconjug Chem. 2010;21:2320–2326. doi: 10.1021/bc100333k. [DOI] [PubMed] [Google Scholar]
- Wombacher R, Cornish VW. Chemical tags: applications in live cell fluorescence imaging. J Biophotonics. 2011;4:391–402. doi: 10.1002/jbio.201100018. [DOI] [PubMed] [Google Scholar]
- Xu K, Babcock HP, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods. 2012;9:185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Cheng LE, Kittelmann M, Li J, Petkovic M, Cheng T, Jin P, Guo Z, Gopfert MC, Jan LY, Jan YN. Ankyrin Repeats Convey Force to Gate the NOMPC Mechanotransduction Channel. Cell. 2015;162:1391–1403. doi: 10.1016/j.cell.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XX, Lin MZ. Photoswitchable fluorescent proteins: ten years of colorful chemistry and exciting applications. Curr Opin Chem Biol. 2013;17:682–690. doi: 10.1016/j.cbpa.2013.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]