

Abstract
The last few decades of longevity research have been very exciting. We now know that longevity and healthspan can be manipulated across species, from unicellular eukaryotes to nonhuman primates, and that while aging itself is inevitable, how we age is malleable. Numerous dietary, genetic, and pharmacological studies now point to links between metabolism and growth regulation as a central aspect in determining longevity and, perhaps more importantly, health with advancing age. Here, we focus on a relatively new player in aging studies GSK3, glycogen synthase kinase, a key factor in growth and metabolism whose name fails to convey the extensive breadth of its role in cellular adaptation. First, we provide a brief overview of GSK3, touching on those aspects that are likely relevant to aging. Then, we outline the role of GSK3 in cellular functions including growth signaling, cell fate, and metabolism. Next, we describe evidence demonstrating a direct role for GSK3 in a range of age-related diseases, despite the fact that they differ considerably in their etiology and pathology. Finally, we discuss the role that GSK3 may play in normative aging and how GSK3 might be a suitable target to oppose age-related disease vulnerability.
Keywords: GSK3, Glycogen synthase kinase 3, Aging, Metabolism, Age-related disease
Introduction
Aging is the greatest risk factor for a range of chronic diseases and disorders including cancer, diabetes, and neurodegenerative disease, and significant effort is being invested to identify casual aspects in morbidity and loss of resilience as a function of age (Kennedy et al. 2014). Dietary excess and a sedentary lifestyle appear to increase vulnerability to diseases and disorders traditionally viewed as age-related (Bonomini et al. 2015), potentially linking the pace of aging to metabolic dysfunction. Caloric restriction (CR) without malnutrition prolongs lifespan and healthspan in a wide range of species including non-human primates, and although the mechanisms yet to be established, a growing literature points to a central role for metabolism in the beneficial effects of CR (Balasubramanian et al. 2017). A prevailing theme in genetic studies of aging is that repression of growth and growth signaling is also strongly linked to longevity (Bartke 2017). Evidence from yeast, worms, flies, and rodents links lifespan extension to insulin and IGF-1 signaling pathway components (Fontana and Partridge 2015). Pathways regulating growth signaling and metabolism are known to be highly interconnected, raising the possibility that this integrated network is what is intrinsically linked to the increase in disease vulnerability that accompanies age. Factors that coordinate growth signaling and metabolism are strong candidates as targets in the treatment of age-related diseases and in development of preventative interventions to prolong good health with advancing age. One such effector of growth signaling is glycogen synthase-kinase 3 (GSK3), a broad specificity serine-threonine kinase that has been linked to insulin resistance, systemic inflammation, and several aspects of Alzheimer’s disease (AD) pathology (Beurel et al. 2015).
There are two isoforms of GSK3 enzyme, GSK3a and GSK3b, collectively referred to as GSK3. The genes encoding these two isoforms reside on separate chromosomes and are ubiquitously expressed (Woodgett 1990). Loss of function mutants have revealed that they have partially overlapping functions; GSK3a knockout (KO) mice are viable due to compensatory activity from GSK3b, but GSK3b KO is embryonically lethal (Hoeflich et al. 2000; Kaidanovich-Beilin et al. 2009; MacAulay et al. 2007). Genetic studies have place GSK3 as a critical regulator of growth and development that also impinges on metabolic homeostatic mechanisms (Table 1). GSK3 is constitutively active and can be inhibited through phosphorylation or by sequestration in a cytosolic complex (Cross et al. 1995; Dominguez et al. 1995). Signaling through insulin and WNT pathways appears to regulate distinct pools of GSK3: AKT activation leads to GSK3 phosphorylation and inhibition but does not affect beta-catenin, while WNT causes dislocation of GSK3 from its beta-catenin targeting destruction complex, leading to stabilized and active beta-catenin (Ding et al. 2000). Despite these differences in mechanistic detail, both WNT and insulin pathways share GSK3b as an effector in signaling and both converge on cell growth and metabolism. Over 70 GSK3 substrates have been validated, representing diverse roles in cellular function. Many GSK3b targets have established relevance to aging, including PI3K, mTOR complex 1 (mTORC1), AMP kinase (AMPK), and peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1a), among others. This breadth of influence implies that GSK3 may be a central coordinator of the cellular response to growth stimulus or repression (Sutherland 2011). Genetic studies have revealed general details of GSK3 function at the cellular level, including signaling downstream of growth and inflammation, and modulation of cell cycle (Jope and Johnson 2004; Jope et al. 2017). GSK3b in particular is enriched in the brain where it has brain-specific roles and is required for neurogenesis, regulation of synaptic plasticity, and neurotransmission (Beurel et al. 2015). These aspects of GSK3b have been particularly well studied in the context of psychiatric disorders, where the GSK3b inhibitor lithium has been used for over a century as a mood-stabilizing agent (Klein and Melton 1996). Intriguingly, GSK3b has also been identified as a major tau kinase implicated in the formation of neurofibrillary tangles, making it a potential target for AD therapeutics (Hanger et al. 1992). As outlined below, recent evidence implicates GSK3b in models of delayed and accelerated aging and interesting new roles for GSK3b in cellular function have been discovered. The goal of this review is to shed light on GSK3b as a factor that links metabolism and growth signaling, and to discuss how it might play a role in aging as a driver of age-related pathology.
Table 1.
Transgenic mouse models reveal the biology of GSK3
| PMID | Year | Isoform | Expression system | Functional outcomes |
|---|---|---|---|---|
| GSK3 gain of function | ||||
|
11007782 12182887 16943560 |
2000 2002 2006 |
GSK3b (S9A) | Thy1-Cre |
Tau hyperphosphorylation Reduced brain size Hypophagia, increased locomotor activity |
| 11226152 | 2001 | GSK3b | CamkIIa-Cre (Tet-off) | Astrogliosis, apoptosis, and neurodegeneration |
| 12472906 | 2002 | Impaired spatial memory | ||
| 17241269 | 2007 | Impaired long-term potentiation | ||
| 15375789 | 2004 | GSK3b | a-actin-Cre | Impaired glucoregulation, hyperlipidemia, and adiposity |
| 15791206 | 2005 | GSK3a (S21A) | Whole body | Normal glucoregulation |
| 27140617 | 2016 | GSK3b (S9A) | Resistance to HFD-induced adiposity and glucoregulatory dysfunction, increased adiponectin | |
| 18219478 | 2008 | GSK3b (S9A) | Insulin2-Cre | Reduced beta-cell mass, impaired glucoregulation |
| GSK3 loss of function | ||||
| 10894547 | 2000 | GSK3b | Whole body | Embryonic lethality mediated by TNFa |
|
17908561 19925672 23549082 |
2007 2009 2013 |
GSK3a | Whole body |
Enhanced glucoregulation and IRS1 expression Reduced aggression, impaired motor coordination Reduced lifespan, sarcopenia, increased cellular senescence |
| 18694957 | 2008 | GSK3b |
Albumin-Cre MLC1f-Cre |
Normal glucoregulation Enhanced glucoregulation and skeletal muscle glycogen |
| 19801986 | 2009 |
GSK3a GSK3b |
Whole body Nestin-Cre |
Progenitor pool expansion, impaired neurogenesis |
| 20821187 | 2010 | GSK3b | Insulin2-Cre | Enhanced glucoregulation, resistance to HFD-induced diabetes |
GSK3 and growth signaling
Upstream mechanisms controlling GSK3 activity, including protein complex formation, sequestration, and autoregulation, have been thoroughly reviewed elsewhere (Beurel et al. 2015). Briefly, inhibitory phosphorylation of GSK3b occurs at the serine-9 position in response to growth stimulus, subsequently impacting GSK3 target proteins involved in glycogen synthesis, translation, and cell survival (Stambolic and Woodgett 1994; Frame and Cohen 2001). Many GSK3 substrates require priming phosphorylation four residues C-terminal to the GSK3 consensus site (S/TXXXS/T) and serine-9 phosphorylation of GSK3 only prevents the binding of the kinase to primed substrates (Frame and Cohen 2001). Both AKT and S6K have been shown to phosphorylate GSK3b at the serine 9 site (serine X in SK3a) in response to growth signaling, while phosphatases including PP2A result in dephosphorylation at this site (Stambolic and Woodgett 1994; Cross et al. 1995; Eldar-Finkelman et al. 1995). Interestingly PI3K and mTOR complex 1 are key nodes that regulate longevity, and significant cross-talk between these pathways has been heavily implicated in upstream regulation of GSK3 activity (Manning and Toker 2017). More recent evidence also places both mTOR complex 1 and PI3K signaling downstream of GSK3, revealing a complex network of signals that impacts cellular function and health (Hermida et al. 2017).
GSK3 phosphorylates and activates TSC2 resulting in inhibition of the GTPase Rheb with subsequent inhibition of the mTOR complex 1 pathway. Interestingly, expression of TSC2 lacking the GSK3b phosphorylation sites resulted in persistent activation of mTOR complex 1 and apoptosis in response to nutrient deprivation (Inoki et al. 2006). This potentially places GSK3b upstream of other mTORC1 functions, including lipid and protein biosynthesis, nucleotide metabolism, and glycolysis; however, the role of GSK3 in controlling cell activity at the functional level has not been very well defined. Glucose uptake and coincident GLUT1 expression is increased in response to GSK3 inhibition in vascular smooth muscle (Buller et al. 2008). Translation is reportedly blocked by GSK3b activity, but it is unclear whether this effect is mediated by mTOR complex 1 inhibition or by direct phosphorylation of eIF2B, another GSK3b substrate (Welsh et al. 1998). More recently, GSK3 has been implicated as a negative regulator of mTOR complex 2 function, where the complex 2 binding partner Rictor was identified as a GSK3 substrate that is subsequently targeted for proteasomal degradation (Koo et al. 2015). GSK3 itself has also been implicated as a target downstream of mTOR complex 1. TSC2 deficiency in cells results in persistent S6K-dependent inhibition of GSK3b, although this regulatory mechanism may only be relevant in the context of insulin resistance, since in these cells signaling through insulin pathways was blunted and AKT, the usual dominant kinase for GSK3, was inhibited (Zhang et al. 2006). Altogether, these complex interactions with insulin and mTOR demonstrate that GSK3b is important in redirecting cellular growth and synthetic pathways in response to changes in nutrient availability (Fig.1).
Fig. 1.
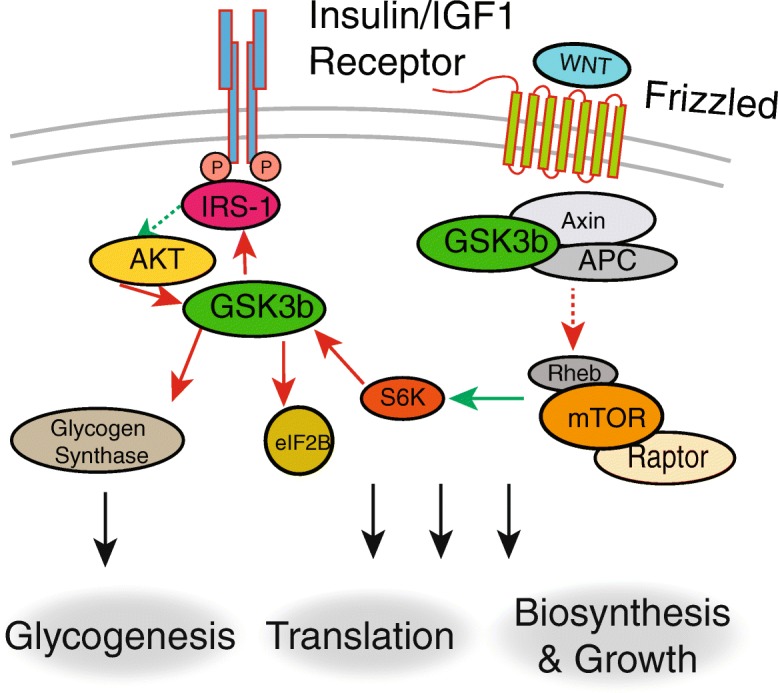
GSK3b and growth regulation. GSK3b activity is suppressed by the insulin/IGF1 and WNT signaling pathways. In the resting state, GSK3b is constitutively active and suppresses growth signaling, translation, and glycogenesis by phosphorylating multiple targets. Activation of Akt by the insulin signaling pathway leads to phosphorylation and suppression of GSK3b activity. GSK3b is also active in the absence of WNT signaling leading to inhibition of mTORC1 and cell growth. Conversely, active mTORC1 inhibits GSK3b activity in a negative feedback loop through S6K
In addition to controlling aspects of mTOR signaling, GSK3 has multiple interactions within the insulin/IGF1 signaling pathway. Although GSK3 is canonically inhibited through insulin signaling, it also exerts feedback control on this pathway. GSK3 was shown to phosphorylate IRS-1 and suppress insulin signaling activity in cells (Liberman and Eldar-Finkelman 2005). Inhibitory phosphorylation by GSK3 has also been extended to IRS-2 in the stress response (Sharfi and Eldar-Finkelman 2008). In another study, GSK3 was found to target IRS-1 for proteasomal degradation under conditions of insulin resistance, demonstrating that GSK3 controls both the stability and activity of this protein (Leng et al. 2010) (Fig.2). The stability of the insulin-signaling antagonist PTEN is also regulated by GSK3 phosphorylation, although the functional consequences of this interaction are unclear (Maccario et al. 2007). These data collectively implicate GSK3 as a regulator of insulin sensitivity that may be mechanistically important in conditions such as type 2 diabetes. This notion is supported by data showing that GSK3 can impact key aspects of glucoregulatory health and is discussed in more detail below.
Fig. 2.
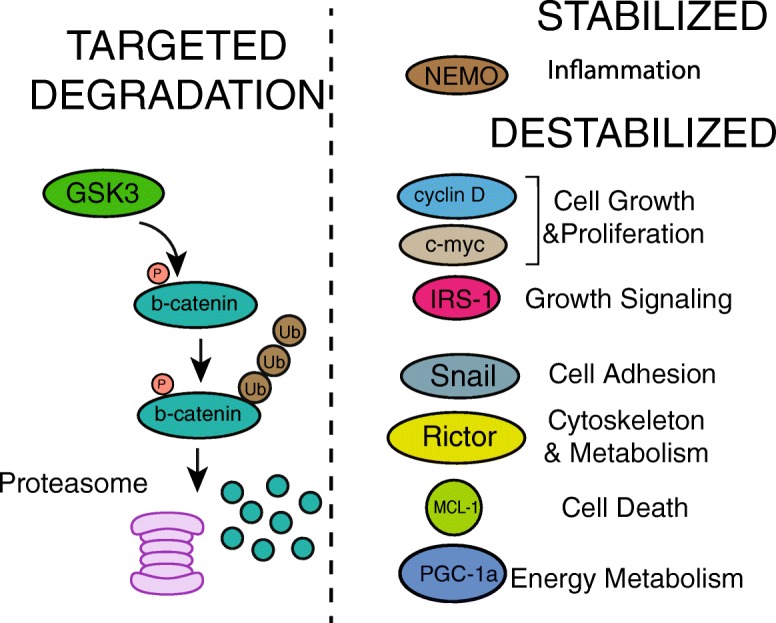
GSK3 phosphorylation regulates the stability of diverse protein targets. Many proteins phosphorylated by GSK3b are targeted for ubiquitin-mediated degradation, most notably beta-catenin in the absence of WNT signaling. These proteins are involved in diverse pathways that impact aging and disease vulnerability including inflammation, metabolism, growth, and different aspects of cell fate
GSK3 and cell fate
Downstream of its involvement in growth and nutrient signaling, GSK3b directly impacts cell survival and proliferation (Fig.3). Early work demonstrated that GSK3b activation was necessary for growth factor withdrawal-induced apoptosis, and has since been extended to a wide variety of toxicities, including oxidative stress, ER stress, and neuronal excitotoxicity (Maurer et al. 2014). MCL-1 (myeloid leukemia cell differentiation protein) is targeted for degradation by GSK3b in response to both inflammatory stimuli and growth factor withdrawal, resulting in activation of the intrinsic apoptotic pathway (Maurer et al. 2006). GSK3 additionally phosphorylates VDAC1, which blocks its interaction with hexokinase II. The loss of interaction disrupts mitochondrial localization and anti-apoptotic function of hexokinase II and increases the vulnerability of cells to apoptosis (Pastorino et al. 2005). It was subsequently shown that GSK3 is a positive regulator of the mitochondrial permeability transition pore (mPTP). GSK3b association with VDAC2 is necessary for maximal permeability of the mPTP under conditions of oxidative stress (Tanno et al. 2014). Intriguingly, GSK3b also upregulates activity of the tumor suppressor TP53, resulting in cell cycle arrest and apoptosis (Watcharasit et al. 2003). In contrast to the pro-apoptotic effects of GSK3b activity, GSK3b inhibition through WNT and growth signaling promotes cell survival and proliferation. The effect of GSK3b on cell proliferation has been demonstrated in stem cells, where inhibition of GSK3b promotes self-renewal and blocks differentiation (Kim et al. 2009). Deletion of both GSK3a and GSK3b in neural progenitor cells resulted in a massive expansion of progenitor cells; however, these progenitors showed compromised differentiation. GSK3-directed deregulation of stem cells was linked to beta-catenin, although activation of c-myc and c-jun that are also direct substrates of GSK3 could reasonably play a role too (Sutherland 2011). Expression of constitutively active GSK3 mutants resistant to N-terminal phosphorylation (mutated at S21A and S9A for GSK3a and GSK3b respectively) results in blockage of neural precursor cell proliferation (Eom and Jope 2009). The ability of GSK3 control cell fate has significant implications for the aging field: stem cell and progenitor cell renewal declines with age resulting in a diminished capacity of tissues to regenerate (Lopez-Otin et al. 2013). Taken together, these multiple interactions ensure that GSK3b can rapidly induce changes in cell survival in response to extracellular stimuli or when conditions are appropriate can stimulate renewal through stem cell recruitment. Therapeutic inhibition of GSK3 has the potential to counteract age-related decline in cellular renewal and stem cell recruitment; however, given the role of GSK3 in the balance of growth and cell survival, the risks of such a therapy would need to be thoroughly evaluated.
Fig. 3.
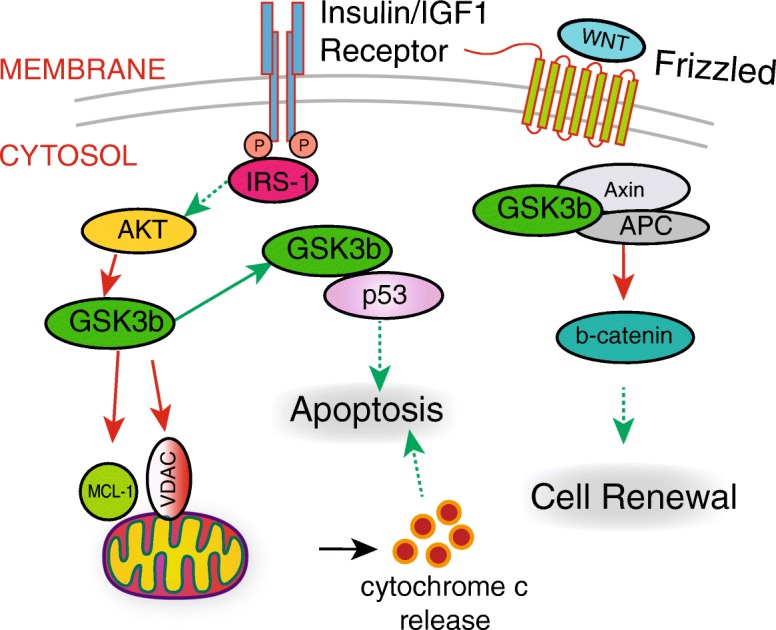
GSK3 regulates cell death, renewal, and differentiation. Active GSK3b increases mitochondrial permeability by phosphorylating proteins on the mitochondrial outer membrane. Subsequent release of cytochrome c triggers the intrinsic pathway leading to apoptosis. GSK3b also interacts with p53 and promotes cell cycle arrest and apoptosis. Inhibitory phosphorylation of GSK3b in response to insulin signaling promotes cell survival by blocking these pathways. Stem cell function is regulated in part through the activity of the WNT signaling pathway. GSK3b phosphorylates and targets beta-catenin for degradation in the absence of WNT signaling, blocking stem cell renewal
GSK3 and inflammation
Chronic inflammation increases with age and growth signaling pathways intersect with inflammation through GSK3. The regulation of inflammation by GSK3 is best characterized through its differential control of CREB and NF-kB transcriptional activity (Fig.4). GSK3 is required for the integrity of Toll-like receptor (TLR) signaling (Martin et al. 2005). Downstream of TLR activation, the status of GSK3 tips the balance between pro- and anti-inflammatory cytokine production. GSK3 activity is required for production of pro-inflammatory IL-6, IL-1B, and IFNy following TLR stimulation, and inhibition of GSK3 during TLR activation favors IL-10 production. Mechanistically, these outcomes are linked to increased nuclear CREB activity and lower NFkB activity in GSK3-inhibited TLR-activated monocytes, where CREB competes and sequesters CPB, a binding partner it shares with NFkB. Inhibition of GSK3 with lithium has also been shown to elevate CREB activity in neuronal cells (Grimes and Jope 2001), supporting the notion that GSK3 could regulate neuroinflammation. This is supported by in vivo evidence showing that adult onset neuron-specific overexpression of GSK3b (CamKII Tet) induces astrogliosis; however, this effect appears to be cell non-autonomous as glia do not express the transgene (Lucas et al. 2001)(Table 1). Both CREB and NFkB have been identified as direct substrates of GSK3b. Phosphorylation of CREB occurs after priming phosphorylation by PKA, preventing CREB DNA binding and activity thus ensuring that CREB activity is temporally controlled (Bullock and Habener 1998; Grimes and Jope 2001). NEMO is an indirect NFkB regulator that along with IKK targets NFkB inhibitor IkB for proteasomal degradation. NEMO interacts with and is phosphorylated by GSK3b at multiple sites resulting in its stabilization (Medunjanin et al. 2016). Importantly, neuroinflammation has been linked to peripheral metabolic dysfunction in mice (Zhang et al. 2008), and it is not unreasonable to think it could also contribute to pathological aging through this same IKK/NFkB axis. Cross-talk between growth and inflammation has been established in Th17 cells through GSK3a. Here, IKK is activated in response to IL-1 signaling resulting in GSK3a inhibition and stimulation of AKT-mTORC1 (Gulen et al. 2012), linking GSK3 to regulation of adaptive immunity. The importance of GSK3 in mediating the inflammatory response is underscored by the effectiveness of GSK3 inhibitors in treating inflammation. Administration of a GSK3 inhibitor prevented the death of mice resulting from a lethal dose (LD100) of lipopolysaccharide (Martin et al. 2005). Elsewhere, GSK3b plays a pro-inflammatory role in mouse models of arthritis and peritonitis (Hu et al. 2006). Here too GSK3 resides at the intersection of pro-and anti-inflammatory cytokine production and mediates cross-talk between Interferon gamma (IFNg) and TLR signaling. These studies suggest that GSK3 inhibitors could be therapeutically useful in the treatment of inflammatory diseases. In mouse models of accelerated aging, the observed increased inflammation and is associated with activation of NEMO (Osorio et al. 2012). Indeed, aging is associated with low-grade sterile inflammation (Franceschi and Campisi 2014) that includes elevated pro-inflammatory cytokines in the serum, activation of NF-kB signaling, and increased expression of immune and inflammation-related genes (Salminen et al. 2012). Aging studies investigating the impact of long-term GSK3 inhibition are needed to clarify whether targeting GSK3 could be useful in attenuating age-related increases in inflammation.
Fig. 4.
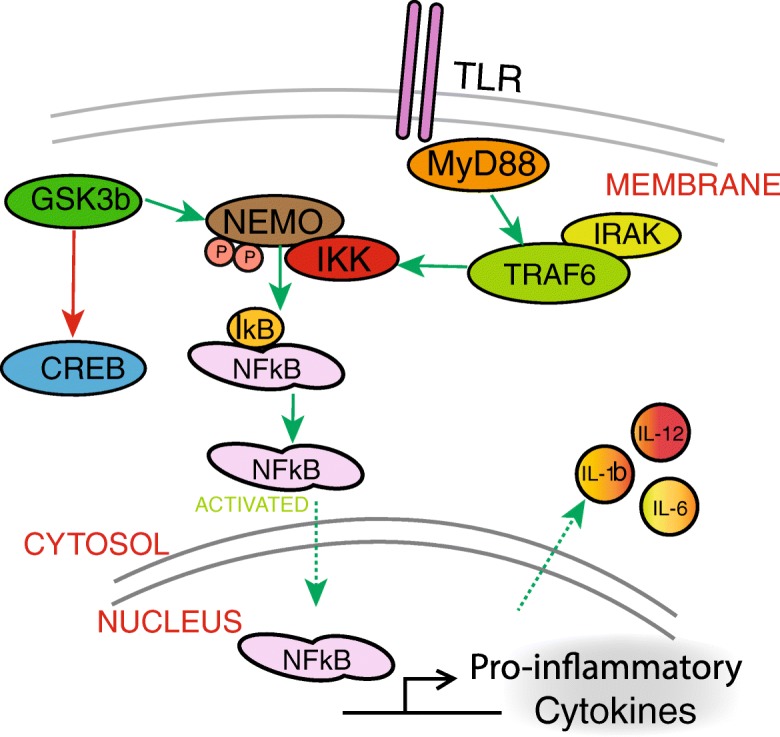
GSK3b regulates inflammation. Active GSK3b promotes NFkB stability and nuclear localization downstream of toll-like receptor (TLR) signaling, leading to the production of pro-inflammatory cytokines. Conversely, GSK3b blocks nuclear accumulation of CREB by promoting its degradation. GSK3b also promotes the integrity of TLR signaling by stabilizing NEMO, a factor that promotes the DNA binding activity of NFkB
GSK3 and cellular senescence
Another age-related phenomenon linked to GSK3 is cellular senescence, a state of permanent cell cycle arrest caused by diverse adverse signals, including contact inhibition, genotoxic injury, and mitochondrial stress (Campisi and d’Adda di Fagagna 2007; Wiley et al. 2016). Transplantation of senescent cells into mice resulted in aging pathology, while removal of these cells pharmacologically alleviated symptoms suggesting that cell senescence directly contributes to the aging process (Xu et al. 2018). N-terminal phosphorylation of both GSK3 isoforms is reported to accompany hepatocyte senescence and treatment with lithium was sufficient to induce a senescent phenotype that is coincident with augmented anabolism including protein synthesis and glycogenesis (Seo et al. 2008). GSK3b was shown to accumulate in the nucleus of human fibroblasts that had undergone replicative senescence, where it formed a stable complex with p53 (Zmijewski and Jope 2004). Intriguingly, treatment of these cells with lithium blocked the interaction of GSK3b with p53 and caused cells to enter a reversible quiescent state. Thus, it appears GSK3 could both promote and oppose cellular senescence under different circumstances. The role of mitochondrial metabolism in cellular senescence has yet to be fully resolved. Mitochondrial dysfunction has been associated with senescence and is detected in senescent cells; however, mitochondrial dysfunction has also been identified as a causal agent in senescence (Kwon et al. 2019). Growing evidence shows a role for cellular senescence and the senescence-associated secretory phenotype in promoting aging phenotypes (Kirkland and Tchkonia 2017). GSK3, residing as it does at the intersection of growth and metabolism, is likely to be important in senescence and may even contribute senescent cell accumulation as a function of age.
GSK3 and energy metabolism
Growth signaling was one of the first pathways to be associated with longevity and the impact of growth inhibition been consistently observed among species (Fontana and Partridge 2015). Extended lifespan has been shown in several genetic mouse models of somatotropic axis deficiency (Bartke 2017), and blockage of growth by CR is thought to partially mediate its benefits (Anderson and Weindruch 2010). The reduction of growth with CR is accompanied by a coordinated upregulation of mitochondrial energy metabolism pathways. Intrigingly, this effect is highly conserved in multiple tissues in mice and among species on CR, suggesting that it is fundamentally important how CR works (Barger et al. 2015). An important question is how cross-talk between growth and metabolic pathways might mediate the effects of CR. As a key effector of growth signaling, GSK3 is perfectly positioned as a regulator of this cross-talk and recent studies have explored the role of both GSK3 isoforms in controlling key metabolic proteins, intermediary metabolism, and mitochondrial function.
AMPK is a major player in regulating cellular energetics (Steinberg and Carling 2019). GSK3b phosphorylation inhibits AMPK activity by making the activation loop site more vulnerable to inhibitory phosphatases (Suzuki et al. 2013). Ablation of the GSK3b site on AMPK in cells resulted in constitutive autophagy and inability to respond to anabolic conditions. Interestingly, GSK3b phosphorylation of AMPK requires a priming phosphorylation by AKT indicating that GSK3b may play a role in fine-tuning the balance of anabolism and catabolism to energetic status. Control over AMPK raises the possibility that GSK3 could be a negative regulator of mitochondrial energy production. GSK3 has previously been shown to inhibit the activity of pyruvate dehydrogenase, which may attenuate mitochondrial activity (Hoshi et al. 1996). A more direct relationship between GSK3 and mitochondria has been established through Drp1, which promotes mitochondrial fission (Chou et al. 2012). Phosphorylation of Drp1 by GSK3 promoted elongated mitochondrial morphology, while inhibition with lithium resulted in more fragmented mitochondria. The ability of GSK3 to regulate mitochondria through PGC-1a, a transcriptional co-activator and master regulator of mitochondrial function, has also been reported (Anderson et al. 2008), where GSK3b was shown to target PGC-1a for nuclear proteasomal degradation (Fig.5). Another study in primary neurons demonstrated that GSK3b phosphorylation of PGC-1a at T-295 was required for recognition by an E3 ubiquitin ligase (Olson et al. 2008). Several additional questions surrounding this regulation remain, including the identity of the kinase that primes PGC-1a for GSK3b targeting. In liver, GSK3b phosphorylates PPARa at serine 73, resulting in ubiquitination and proteasomal degradation. GSK3 inhibition in this study was associated with attenuated hepatic steatosis in high-fat-diet-fed mice, highlighting an important connection between GSK3b and metabolic disease (Hinds et al. 2016). The functional consequences of GSK3b manipulation have recently been investigated in the context of the brain. GSK3b is enriched in neurons, which are highly dependent on mitochondrial function. Brain aging is associated with changes in neuronal energy metabolism (Martin et al. 2016), and treatment of mice with GSK3 inhibitor lithium carbonate resulted in upregulation of PGC-1a and elevation of cytochrome c oxidase activity in the hippocampus (Martin et al. 2018). Inhibition of GSK3 stabilized PGC-1a protein and enhanced mitochondrial activity in neuroglioma cells, with concomitant changes in redox metabolism. Collectively, these studies support the idea that active GSK3 could regulate energy metabolism through multiple mechanisms. Identifying metabolic pathways downstream of both GSK3 isoforms will open the door to therapeutics that target age-related metabolic impairment.
Fig. 5.
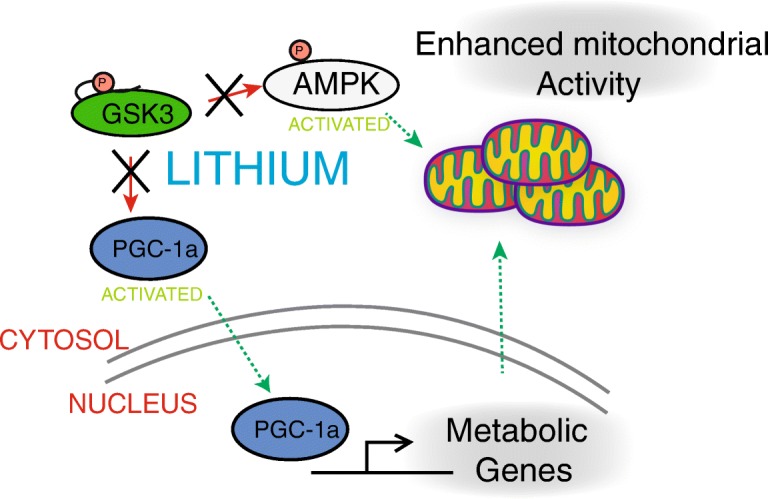
GSK3b regulates mitochondrial energy metabolism. Inhibition of GSK3b by lithium stabilizes PGC1a and increases mitochondrial respiration. Activated PGC1a increases expression of target genes coincident with increased respiration. GSK3 negatively regulates AMPK and activating phosphorylation of AMPK is increased with lithium. These nutrient sensing pathways converge on mitochondrial function, suggesting that GSK3 is a key upstream regulator of metabolism
GSK3 and age-related disease
Changes in GSK3 expression and function are strongly related to several diseases and disorders that share an increase in risk of incidence as a function of age. Among these are type 2 diabetes, cancer, inflammatory conditions, and Alzheimer’s disease (Beurel et al. 2015). There is a substantial volume of research published on each of these chronic conditions and the role of GSK3 in each is rather scattered about the literature. Here we will briefly describe some of the studies that favor a role for GSK3 in progression of age-related diseases and suggest that GSK3 could be an agent in creating age-related disease vulnerability in the first place.
Diabetes
GSK3b expression levels have been found to correlate positively with glucoregulatory dysfunction in diabetes patients (Nikoulina et al. 2000). The significance of this association is unclear; however, genetic studies have revealed that GSK3 is a powerful mediator of systemic glucose and lipid homeostasis and that these actions have a high degree of tissue specificity (Table 1). The whole-body GSK3b KO is embryonic lethal but the whole-body GSK3a KO mice display improved glucoregulatory function early in life (MacAulay et al. 2007). Skeletal muscle-specific GSK3b overexpression resulted in hyperlipidemia, elevated fasting insulin, and glucose intolerance in male mice (Pearce et al. 2004). Conversely, mice lacking GSK3b in skeletal muscle exhibit improved glucose tolerance and greater insulin-stimulated glycogen synthase regulation (Patel et al. 2008). Liver-specific deletion of GSK3b did not exhibit an overt phenotype, indicating that the glucoregulatory role of GSK3 appears to be primarily extra-hepatic (Patel et al. 2008). It seems likely that at least some of the phenotypes GSK3 overexpression are linked to its role downstream of insulin and mTOR signaling. Double knock-in mice expressing AKT insensitive GSK3a and GSK3b were resistant to high-fat-diet-induced obesity, dyslipidemia, and glucoregulatory impairment (Chen et al. 2016). These mice also showed greater circulating levels of adiponectin, the adipose tissue-derived endocrine factor that signals through AMPK. Likely linked to the increase in adiponectin, the mice also showed an increase in energy expenditure. Further evidence suggests that GSK3 could be important for pancreatic endocrine function. Beta cell-specific expression of a constitutively active GSK3b transgene (GSK3b S9A) resulted in a reduction in beta cell mass and function (Liu et al. 2008). Conversely, GSK3b deletion conferred resistance to high-fat-diet-induced diabetes and resulted in beta cell expansion in mice (Liu et al. 2010). Small-molecule inhibitors of GSK3 increased the replication of beta cells from isolated rat islets, suggesting that targeting GSK3 may have therapeutic value (Mussmann et al. 2007).These data show that GSK3 can impact multiple aspects of the metabolic syndrome, including insulin sensitivity, glucose clearance and storage, and lipid homeostasis. These studies highlight the distinction that needs to be made in terms of GSK3 protein levels and phosphorylation status, tissue specificity in GSK3 actions, and how its sensitivity to different signaling inputs not only allows for distinct outcomes of its activation but also means that context must be considered if GSK3 is to be a druggable target for metabolic dysfunction.
Alzheimer’s disease
The role of GSK3 as a mediator of Alzheimer’s disease pathology has been studied on and off for a few decades. GSK3a and GSK3b isoforms can each phosphorylate residues on tau in vitro (Hanger et al. 1992); however, GSK3b but not GSK3a has been shown to co-localize with neurofibrillary tangles (NFTs) and active but not inactive (phosphorylated at S9) GSK3b was found in neurons in the early stages of tangle formation, suggesting that kinase active GSK3b is involved in the pathogenesis of NFTs (Yamaguchi et al. 1996; Pei et al. 1999). This is supported by genetic evidence showing that neuron-specific overexpression of GSK3b in mice resulted in hyperphosphorylation of tau on residues that lead to paired helical filament formation (Lucas et al. 2001). GSK3 has also been connected to amyloid plaque pathology. Lithium treatment blunted processing and secretion of amyloid precursor protein (APP) peptide fragments from cells and lowered levels of A-beta in the brains of a genetic model of Alzheimer’s disease (Phiel et al. 2003). In cells, the effect of lithium to promote amyloid secretion was attributed to GSK3a, but not GSK3b. This finding is supported by in vivo evidence showing that GSK3a, but not GSK3b, knock-down ameliorates amyloid plaque load in a mouse model of Alzheimer’s disease (Hurtado et al. 2012). This same study demonstrated that knock-down of both GSK3 isoforms reduced tau phosphorylation, misfolding, and memory deficit. GSK3 has also been implicated as a mediator of neuroinflammation, neuritic damage, and learning and memory deficits (Lucas et al. 2001; Hernandez et al. 2002). Mice with induced overexpression of GSK3b exhibit phosphorylation and mislocalization of tau, along with increased nuclear beta-catenin. These cellular phenotypes were associated with cognitive impairments and gliosis even in the absence of NFT deposition. GSK3b associated neuronal apoptosis, decreased brain volume, and impairment in learning in memory were completely reversible after 6 weeks of transgene silencing, pointing to a direct role of GSK3 in producing Alzheimer’s disease-related traits (Engel et al. 2006). Additional studies have established that GSK3b promotes long-term depression and inhibits long-term potentiation, further connecting GSK3 to memory impairment (Hooper et al. 2007). More recently, GSK3 was shown to be activated in mouse models of Alzheimer’s disease and inhibition was shown to attenuate dendritic spine loss (DaRocha-Souto et al. 2012). Mechanistically, the protective effect of GSK3 inhibition was linked to preserved CREB activity and increased expression of the CREB target gene BDNF. A recent clinical trial in patients who were already being treated for Alzheimer’s disease suggests that disease progression and extent of GSK3 inhibition will both be important factors to consider in using GSK3 as a treatment target (Lovestone et al. 2015). Peptides that mimic GSK3-primed substrates are currently being developed and may provide advantages over ATP-competitive inhibitors, including higher selectivity and weaker GSK3 inhibition (Eldar-Finkelman and Martinez 2011). These studies and others convincingly show GSK3 as a player in Alzheimer’s disease (Llorens-Marítin et al. 2014), but raise questions about the sequence of events in the etiology and progression of spontaneous age-related Alzheimer’s disease. It will be of profound interest to understand which of the roles identified for GSK3 in Alzheimer disease models, as a factor promoting amyloid and NFT pathology or as a factor responding to the burden of amyloid and NFT pathology, is the more physiologically important one during human disease development and how it might be effectively targeted as a clinical intervention.
Cancer
As a key regulator of cell fate, GSK3 has been implicated in the biology of many different cancers, both as an oncogene and as a tumor suppressor. Elevated GSK3b expression has been observed in pancreatic cancer cell lines, where it was responsible for promoting NF-kB activity and was necessary for NF-kB-mediated proliferation and survival (Ougolkov et al. 2005). High levels of GSK3 expression have been observed in colon, liver, and ovarian cancers; however, the physiological significance of this is not known (McCubrey et al. 2014). The role of GSK3 as a tumor suppressor is perhaps better understood. GSK3 is an effector of the WNT signaling pathway that targets beta-catenin for degradation, thus blocking the transcription of oncogenic targets. Expression of a kinase-inactive form of GSK3b in mice acts as a dominant negative and promotes the formation of mammary tumors that express high levels of beta-catenin and cyclin D1 (Farago et al. 2005). GSK3 may also play a role in the enhanced survival of cancer cells by controlling the localization and activation of bcl-2 family protein Bax, a key regulator of apoptosis (Maurer et al. 2014). Mcl-1 is another bcl-2 family protein linked to GSK3. Ablation of the GSK3b phosphorylation site on stabilizes Mcl-1, blocks apoptosis, and desensitizes cells to chemotherapeutics, again supporting a role for GSK3 in cancer development and as a target for enhancing cancer therapies (Ding et al. 2007). Another aspect of cancer biology linked to GSK3 is the epithelial-mesenchymal transition (EMT), a process that renders cancer cells more motile and metastatic. GSK3 targets the transcriptional repressor Snail for proteasomal degradation resulting in maintenance of E cadherin expression (Doble and Woodgett 2007). E cadherin expression is required for normal adhesion of epithelial cells, so loss of this protein is a key step in the EMT. Taken together, these studies reveal a complex and often contradictory role for GSK3 in the biology of cancer. Importantly, the stabilization of beta-catenin remains an unwanted consequence of GSK3 inhibitors. Development of isoform-selective inhibitors for GSK3 has promise for the treatment of acute myeloid leukemia (Wagner et al. 2018). In this study, selective inhibition of GSK3a was shown to block AML colony formation without causing beta-catenin stabilization, and impaired leukemia initiation and prolonged survival in vivo. Many GSK3 inhibitors fail clinical trials because of off-target toxicities. A better understanding of the biology of GSK3 will be imperative if treatments focusing on this key growth responsive kinase are to be selective and effective.
Normative aging
The role of GSK3 in normative aging has only recently been investigated. GSK3b protein levels increase in multiple regions of the rat brain with age (Lee et al. 2006). Conversely, the neuroprotective intervention of CR is associated with lower protein levels of GSK3b in both mouse and rhesus hippocampus and relatively greater levels of inhibitory serine-9 phosphorylation (Martin et al. 2016). Further evidence for a role of GSK3 in aging comes from shorter lived species. In worms, GSK3 inhibition resulted in dose-dependent increases in lifespan that was accompanied by chromatin remodeling (McColl et al. 2008). Another study found that lithium increased mitochondrial energetics in worms and postulated that lithium resulted in selective autophagy of dysfunctional mitochondria (Tam et al. 2014). In flies, lithium resulted in dose-dependent reduction in triglycerides and increased xenobiotic resistance, and the lifespan promoting effect of lithium was dependent upon activation of NRF-1 (Castillo-Quan et al. 2016). Conversely, overexpression of Shaggy, the GSK3 homolog in flies, resulted in shortened lifespan that was partially rescued by lithium. These results show that GSK3 is relevant to longevity and suggest that its ability regulate metabolism is central to its role in aging. Although GSK3b KO is embryonically lethal, GSK3a KO mice exhibit shortened lifespans and increased age-related pathology (Zhou et al. 2013). This includes cardiac dysfunction, early onset of sarcopenia, and increases in cellular senescence, some of which are thought to occur partially through activation of mTOR complex 1. Therefore, it appears that both GSK3 loss of function and gain of function can negatively impact lifespan under different circumstances. Given the established role of GSK3 in development, studies that use conditional modulation of GSK3 would be required to dissect its role specifically in the aging process.
Conclusion
From its initial identification as a regulator of glycogen metabolism, the known functions of GSK3 have expanded to encompass numerous fundamental pathways. GSK3 plays multifactorial roles in growth signaling, inflammation, senescence, cell fate, and energy metabolism. Each of these pathways has been implicated in age-related dysfunction, suggesting that GSK3 may play a central role in the increased disease vulnerability that accompanies aging. Many of the disease and disorders of age have been studied in isolation and many transgenic mouse studies employ young animals. Nonetheless, across the literature, there is an interesting case to be made for GSK3 as a driver in aging and even perhaps as a potential target for intervention. Evidence to date suggests that GSK3 actions and responsivity to perturbation are highly context-dependent: differences in interactions and flux through GSK3-regulated pathways occur as a function of cell type, tissue type, and background metabolic status. High granularity studies focusing on GSK3 biology, in particular the impact of status and combinations of signaling inputs on cellular outcome, should be highly informative. If dysregulation of GSK3 is indeed causal age-related disease vulnerability, targeted therapeutic strategies could prove effective across a spectrum of age-related diseases.
Acknowledgments
The study was conducted with the use of resources and facilities at the William S. Middleton Memorial Veterans Hospital, Madison WI. The authors declare no conflict of interest.
Funding information
This work was supported by NIH AG057408, The Glenn Foundation for Medical Research, the American Federation for Aging Research, and NIH training fellowship AG000213 (DSC).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Anderson RM, Weindruch R. Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab. 2010;21:134–141. doi: 10.1016/j.tem.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian P, Howell PR, Anderson RM. Aging and caloric restriction research: a biological perspective with translational potential. EBioMedicine. 2017;21:37–44. doi: 10.1016/j.ebiom.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger JL, Anderson RM, Newton MA, da Silva C, Vann JA, Pugh TD, Someya S, Prolla TA, Weindruch R. A conserved transcriptional signature of delayed aging and reduced disease vulnerability is partially mediated by SIRT3. PLoS One. 2015;10:e0120738. doi: 10.1371/journal.pone.0120738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A. Somatic growth, aging, and longevity. NPJ Aging Mech Dis. 2017;3:14. doi: 10.1038/s41514-017-0014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomini Francesca, Rodella Luigi Fabrizio, Rezzani Rita. Metabolic Syndrome, Aging and Involvement of Oxidative Stress. Aging and Disease. 2015;6(2):109. doi: 10.14336/AD.2014.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL, Brosius FC., 3rd A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Phys Cell Phys. 2008;295:C836–C843. doi: 10.1152/ajpcell.00554.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase a and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Castillo-Quan JI, Li L, Kinghorn KJ, Ivanov DK, Tain LS, Slack C, Kerr F, Nespital T, Thornton J, Hardy J, Bjedov I, Partridge L. Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 2016;15:638–650. doi: 10.1016/j.celrep.2016.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fajol A, Hoene M, Zhang B, Schleicher ED, Lin Y, Calaminus C, Pichler BJ, Weigert C, Häring HU, Lang F, Föller M. PI3K-resistant GSK3 controls adiponectin formation and protects from metabolic syndrome. Proc Natl Acad Sci U S A. 2016;113:5754–5759. doi: 10.1073/pnas.1601355113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, Tu WY, Kao TC, Hsu CM, Cheng JT, Chou AK, Lee CI, Loh JK, Howng SL, Hong YR. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One. 2012;7:e49112. doi: 10.1371/journal.pone.0049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- DaRocha-Souto B, Coma M, Perez-Nievas BG, Scotton TC, Siao M, Sanchez-Ferrer P, Hashimoto T, Fan Z, Hudry E, Barroeta I, Sereno L, Rodriguez M, Sanchez MB, Hyman BT, Gomez-Isla T. Activation of glycogen synthase kinase-3 beta mediates beta-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol Dis. 2012;45:425–437. doi: 10.1016/j.nbd.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–32481. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185:73–84. doi: 10.1159/000101306. [DOI] [PubMed] [Google Scholar]
- Dominguez I., Itoh K., Sokol S. Y. Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proceedings of the National Academy of Sciences. 1995;92(18):8498–8502. doi: 10.1073/pnas.92.18.8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J Biol Chem. 1995;270:987–990. doi: 10.1074/jbc.270.3.987. [DOI] [PubMed] [Google Scholar]
- Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom TY, Jope RS. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol Psychiatry. 2009;66:494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD, Seldin DC. Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65:5792–5801. doi: 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106–118. doi: 10.1016/j.cell.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, Beurel E, Kaidanovich-Beilin O, Fox PL, DiCorleto PE, Wang JA, Qin J, Wald DN, Woodgett JR, Jope RS, Carman J, Dongre A, Li X. Inactivation of the enzyme GSK3alpha by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity. 2012;37:800–812. doi: 10.1016/j.immuni.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul. 2017;65:5–15. doi: 10.1016/j.jbior.2017.06.003. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem. 2002;83:1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- Hinds TD, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin reductase a attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α*. J Biol Chem. 2016;291:25179–25191. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25:81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci U S A. 1996;93(7):2719–2723. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Carroll JC, Macdonald C, Aboagye AK, Trojanowski JQ, Lee VM. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS, Cheng Y, Lowell J, Worthen RJ, Sitbon YH, Beurel E. Stressed and inflamed, can GSK3 be blamed? Trends Biochem Sci. 2017;42:180–192. doi: 10.1016/j.tibs.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, MacAulay K, Doble BW, Henkelman M, Miyakawa T, Roder J, Woodgett JR. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2:35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F. Aging: a common driver of chronic diseases and a target for novel interventions. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W-Y, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–28. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo J, Wu X, Mao Z, Khuri FR, Sun SY. Rictor undergoes glycogen synthase kinase 3 (GSK3)-dependent, FBXW7-mediated ubiquitination and proteasomal degradation. J Biol Chem. 2015;290:14120–14129. doi: 10.1074/jbc.M114.633057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SM, Hong SM, Lee YK, Min S, Yoon G. Metabolic features and regulation in cell senescence. BMB Rep. 2019;52:5–12. doi: 10.5483/BMBRep.2019.52.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Chung YH, Joo KM, Lim HC, Jeon GS, Kim D, Lee WB, Kim YS, Cha CI. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci Lett. 2006;409:134–139. doi: 10.1016/j.neulet.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Leng S, Zhang W, Zheng Y, Liberman Z, Rhodes CJ, Eldar-Finkelman H, Sun XJ (2010) Glycogen synthase kinase 3 beta mediates highglucose-induced ubiquitination and proteasome degradation of insulin receptor substrate 1. J Endocrinol 206:171–181 [DOI] [PMC free article] [PubMed]
- Liberman Ziva, Eldar-Finkelman Hagit. Serine 332 Phosphorylation of Insulin Receptor Substrate-1 by Glycogen Synthase Kinase-3 Attenuates Insulin Signaling. Journal of Biological Chemistry. 2004;280(6):4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia. 2008;51:623–631. doi: 10.1007/s00125-007-0914-7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tanabe K, Baronnier D, Patel S, Woodgett J, Cras-Méneur C, Permutt MA. Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53:2600–2610. doi: 10.1007/s00125-010-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Marítin M, Jurado J, Hernández F, Ávila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovestone S, Boada M, Dubois B, Hull M, Rinne JO, Huppertz HJ, Calero M, Andres MV, Gomez-Carrillo B, Leon T, del Ser T. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis. 2015;45:75–88. doi: 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phosphorylation on Thr366. Biochem J. 2007;405:439–444. doi: 10.1042/BJ20061837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor – mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Imunnol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SA, DeMuth TM, Miller KN, Pugh TD, Polewski MA, Colman RJ, Eliceiri KW, Beasley TM, Johnson SC, Anderson RM. Regional metabolic heterogeneity of the hippocampus is nonuniformly impacted by age and caloric restriction. Aging Cell. 2016;15:100–110. doi: 10.1111/acel.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SA, Souder DC, Miller KN, Clark JP, Sagar AK, Eliceiri KW, Puglielli L, Beasley TM, Anderson RM. GSK3beta regulates brain energy metabolism. Cell Rep. 2018;23:1922–1931.e4. doi: 10.1016/j.celrep.2018.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. GSK-3 - at the crossroads of cell death and survival. J Cell Sci. 2014;127:1369–1378. doi: 10.1242/jcs.138057. [DOI] [PubMed] [Google Scholar]
- McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350–357. doi: 10.1074/jbc.M705028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko Z, Cocco L, Martelli AM, Cervello M. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881–2911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medunjanin S, Schleithoff L, Fiegehenn C, Weinert S, Zuschratter W, Braun-Dullaeus RC. GSK-3beta controls NF-kappaB activity via IKKgamma/NEMO. Sci Rep. 2016;6:38553. doi: 10.1038/srep38553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussmann R, Geese M, Harder F, Kegel S, Andag U, Lomow A, Burk U, Onichtchouk D, Dohrmann C, Austen M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J Biol Chem. 2007;282:12030–12037. doi: 10.1074/jbc.M609637200. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- Olson B. L., Hock M. B., Ekholm-Reed S., Wohlschlegel J. A., Dev K. K., Kralli A., Reed S. I. SCFCdc4 acts antagonistically to the PGC-1 transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes & Development. 2008;22(2):252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, Lopez-Otin C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA, Llano A, Moore GB, Murphy GJ, Smith SA, Taylor CM, Yates JW, Morrison AD, Harper AJ, Roxbee-Cox L, Abuin A, Wargent E, Holder JC. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3beta on a muscle-specific promoter. Metabolism. 2004;53:1322–1330. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012;4:166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YH, Jung HJ, Shin HT, Kim YM, Yim H, Chung HY, Lim IK, Yoon G. Enhanced glycogenesis is involved in cellular senescence via GSK3/GS modulation. Aging Cell. 2008;7:894–907. doi: 10.1111/j.1474-9726.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Sharfi H, Eldar-Finkelman H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am J Physiol Endocrinol Metab. 2008;294:E307–E315. doi: 10.1152/ajpendo.00534.2007. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303(Pt 3):701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18:527–551. doi: 10.1038/s41573-019-0019-2. [DOI] [PubMed] [Google Scholar]
- Sutherland C. What are the bona fide GSK3 substrates? Int J Alzheimers Dis. 2011;2011:505607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin J, Saltiel AR, Inoki K. Inhibition of AMPK catabolic action by GSK3. Mol Cell. 2013;50:407–419. doi: 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam ZY, Gruber J, Ng LF, Halliwell B, Gunawan R. Effects of lithium on age-related decline in mitochondrial turnover and function in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 2014;69:810–820. doi: 10.1093/gerona/glt210. [DOI] [PubMed] [Google Scholar]
- Tanno M, Kuno A, Ishikawa S, Miki T, Kouzu H, Yano T, Murase H, Tobisawa T, Ogasawara M, Horio Y, Miura T. Translocation of glycogen synthase kinase-3beta (GSK-3beta), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2) J Biol Chem. 2014;289:29285–29296. doi: 10.1074/jbc.M114.563924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner FF, Benajiba L, Campbell AJ, Weiwer M, Sacher JR, Gale JP, Ross L, Puissant A, Alexe G, Conway A, Back M, Pikman Y, Galinsky I, DeAngelo DJ, Stone RM, Kaya T, Shi X, Robers MB, Machleidt T, Wilkinson J, Hermine O, Kung A, Stein AJ, Lakshminarasimhan D, Hemann MT, Scolnick E, Zhang YL, Pan JQ, Stegmaier K, Holson EB. Exploiting an asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci Transl Med. 2018;10:eaam8460. doi: 10.1126/scitranslmed.aam8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 1998;421:125–130. doi: 10.1016/s0014-5793(97)01548-2. [DOI] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor a. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedernhofer LJ, Khosla S, Tchkonia T, Kirkland JL. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24:1246–1256. doi: 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol Cell. 2006;24:185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, Vagnozzi RJ, Lal H, Force T. GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest. 2013;123:1821–1832. doi: 10.1172/JCI64398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski JW, Jope RS. Nuclear accumulation of glycogen synthase kinase-3 during replicative senescence of human fibroblasts. Aging Cell. 2004;3:309–317. doi: 10.1111/j.1474-9728.2004.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]