

Abstract
Background:
Finding a therapy for Alzheimer's disease (AD) is perhaps the greatest challenge for modern medicine. The chemical scaffolds of many drugs in the clinic today are based upon natural products from plants, yet Cannabis has not been extensively examined as a source of potential AD drug candidates. Here we determine if a number of non-psychoactive cannabinoids are neuroprotective in a novel preclinical AD and neurodegeneration drug screening platform that is based upon toxicities associated with the aging brain. This drug discovery paradigm has yielded several compounds in or approaching clinical trials for AD.
Methods:
Eleven cannabinoids were assayed for neuroprotection in assays that recapitulate proteotoxicity, loss of trophic support, oxidative stress, energy loss and inflammation. These compounds were also assayed for their ability to remove intraneuronal amyloid and subjected to a structure-activity relationship analysis. Pairwise combinations were assayed for their ability to synergize to produce neuroprotective effects that were greater than additive.
Results:
Nine of the eleven cannabinoids have the ability to protect cells in four distinct phenotypic neurodegeneration screening assays, including those using neurons that lack CB1 and CB2 receptors. They are able to remove intraneuronal Aβ, reduce oxidative damage, and protect from the loss of energy or trophic support. Structure-activity relationship (SAR) data show that functional antioxidant groups such as aromatic hydroxyls are necessary but not sufficient for neuroprotection. Therefore, there is a need to focus upon CB1 agonists that have these functionalities if neuroprotection is the goal. Pairwise combinations of THC and CBN lead to a synergistic neuroprotective interaction.
Conclusion:
Together these results significantly extend the published data by showing that non-psychoactive cannabinoids are potential lead drug candidates for AD and other neurodegenerative diseases.
Keywords: Cannabinoids, Alzheimer’s, neuroprotection, drug discovery, phenotypic screening
Background
Alzheimer's disease (AD) is the most common form of dementia, affecting over 50 million people worldwide. This number is expected to exceed 130 million by 2050 [1]. There are, however, no therapies that reduce the rate of progression of AD or any other old age-associated neurodegenerative disease. There are likely many reasons for this failure, including the complexity of the diseases and their association with their greatest risk factor, old age. Another contributory factor is the insistence of drug developers to focus on pre-selected molecular targets, usually associated with very rare, genetic forms of the disease. With AD, most drug candidates have been directed against components of the amyloid cascade that are genetically linked to familial Alzheimer's disease (FAD). To date, all of the drug candidates involving this pathway have failed in the clinic [2].
In contrast to this targeted approach, the chemical scaffolds for the majority of drugs in the clinic today were derived from plants and were initially discovered by phenotypic screening [3]. This process is defined as drug screening in cellular or animal disease models that mimic aspects of the human disease with the goal of preventing the disease phenotype. It does not require prior knowledge of the drug target.
Cannabis once held a prominent position in medicine, but due to political and social factors it is currently understudied in the context of AD drug discovery. Cannabis contains nearly 500 compounds, many unique to the plant, including over 100 cannabinoids [4]. These compounds are particularly interesting for medicine because the activities of some cannabinoids very likely overlap with endocannabinoids, an incompletely understood group of endogenous compounds with receptors involved in multiple aspects of cellular physiology [5-7]. Therefore, many potential therapeutic uses for the compounds in Cannabis remain undiscovered.
Proteotoxicity is associated with aging in all organisms from bacteria to humans [8]. Because neurons do not divide, they have no mechanism to dilute aggregated proteins and are therefore more vulnerable to proteotoxicity than dividing cells. We have shown that when aggregated proteins are removed from the fly brain by stimulating autophagy, the flies live longer, while decreasing autophagy in the brain reduces lifespan [9]. Therefore, proteotoxicity in the brain is not only central to disease, but also to lifespan.
MC65 is a human neuron-like cell line that is an excellent model for proteotoxicity. It has an inducible plasmid that generates the C99 fragment of the amyloid precursor protein (APP) upon the removal of tetracycline from the culture medium. Aβ is produced by endogenous γ secretase, and the cells die within a few days [10]. The parent cell line is SK-N-MC from a human tumor and has an electrical membrane typical of neurons [11]. This assay therefore recapitulates an event, proteotoxicity, which occurs in all aging cells and specifically in AD. We have shown that when MC65 cells are treated with THC during the induction of Aβ, the cells do not accumulate the toxic peptide and do not die [12]. It has also been shown that THC directly inhibits Aβ aggregation [13, 14], while cannabinoids reduce the amount of Aβ that accumulates in the brains of transgenic hFAD mice [15, 16].
Although THC is effective at removing intraneuronal Aβ, it has the social burden of being psychoactive. We therefore asked if a group of pure, non-psychoactive cannabinoids have neuroprotective activity in a screening platform used for AD drug discovery. We screened these compounds in assays based upon toxicities associated with the old brain, including proteotoxicity, loss of trophic support, loss of energy and oxidative stress [3]. We also examined their effect on microglial inflammation. It is shown here that over half of the cannabinoids that were studied were very neuroprotective in these assays, and therefore have the potential to be translated into the clinic for the treatment of neurodegenerative diseases.
Methods
Reagents
All cannabinoids were purchased from Cayman Chemicals under a Schedule 1 License from the DEA. The other chemicals were from Sigma.
Cell Culture Assays
Assay Protocol:
The methodology for all of the cell culture assays was the same. Initial compound screening was done at least twice in triplicate between 10 μM and 250 nM using a 2-fold serial dilution. Lower dilutions were used starting at 1 μM if required. The dose response curves were plotted and the EC50s determined as the concentration that inhibited the death of 50% of the cells. The data in the tables is presented as the average of triplicate assays. In most cases, the survival ranged from 0-10% for control cells (no compound) and 90-100% for compound-treated cells. Concentrations that were either toxic or had no effect at 10 μM are noted in the tables and these compounds were not studied further.
Intraneuronal Amyloid Toxicity:
The culture and induction of C99 in MC65 cells was performed as previously described [10, 17]. Briefly, cells were grown in the presence of tetracycline. When tetracycline is removed, the cells are induced to make the C99 fragment of APP that is then cleaved by γ secretase to generate Aβ, which aggregates and its toxicity leads to cell death within 4 days.
Oxytosis Assay:
HT22 cells were plated at 2 × 103 cells per well in 96-well tissue culture dishes in Dulbecco's modified Eagle medium (DMEM) plus 10% fetal calf serum (FCS). The following day the test compounds were added in triplicate at appropriate concentrations. Thirty minutes after compound addition, 5 mM glutamate was added to initiate the cell death cascade [3]. Twenty hr later, the MTT cell viability assay was performed [3]. The results are presented as the percentage of the control with vehicle alone that remain alive.
Energy Loss Assay:
HT22 cells were seeded onto 96-well tissue culture dishses at a density of 5 × 103 cells per well in DMEM plus 10% FCS. The next day, the medium was replaced with DMEM supplemented with 7.5% FCS and the cells were treated with 20 μM iodoacetic acid (IAA) and the compounds [18]. After 2 hr, the medium in each well was aspirated and replaced with fresh medium without IAA but containing the test compounds. 20 hr later, viability was measured by the MTT assay.
Trophic Factor Withdrawal Assay:
Primary cortical neurons were prepared from 18-day-old rat embryos according to published procedures and cultured at a low cell density of 1 × 106/35 mm dish in DMEM/F12 (2 ml) containing N2 supplements (Invitrogen) and the compounds to be assayed [3]. Viability was assayed 2 days later using a fluorescent live-dead assay (Molecular Probes, Eugene, OR, USA) and the data is presented as the percentage of input cells surviving.
Inflammation Assay:
Inflammation was induced by bacterial lipopolysaccharide (LPS) in the microglial cell line BV2 using production of NO as a primary read-out. NO production was measured in the presence or absence of the test compound 24 hr after LPS addition using the Griess assay.
SDS-PAGE and Immunoblotting
Cultured cells were washed twice in cold phosphate-buffered saline, scraped into lysis buffer and proteins separated on 12% SDS-polyacrylamide gels [17]. The following primary antibodies were used: Beta Amyloid (6E10; Wako); actin (Enzo Life Sciences).
RNA Analysis
RNA was isolated from HT22 and MC65 cells using Qiagen RNeasy Mini Kit (Qiagen). RNA-Seq libraries were prepared using the Illumina TruSeq Stranded mRNA Sample Prep Kit according to the manufacturer’s instructions. The libraries were pooled and sequenced (single-end 50 bp) using the HiSeq 2500 platform (Illumina).
Methylation of Cannabinol (CBN)
The chemical methylation was carried out as described. A solution of CBN (10 mg, 0.032 mmol) was added to a mixture of toluene (3 mL) and methanol (2 mL) with an excess of trimethylsilyldiazomethane (TMSCHN2, 2 M in hexane, 0.5 mL, 1 mmol). The reaction solution was stirred at room temperature for 16 h and the solvent was evaporated with N2 gas. The residue was washed sequentially with methanol and chloroform to yield cannabinol methyl ether (MCBN) with over 95% purity as determined by ESI-Quadrupole-Orbitrap-MS analysis and 1H and 13C NMR spectroscopic data.
Results
We have used a unique drug screening platform to identify natural products that are neuroprotective and then to do the medicinal chemistry required to make the natural product into a pharmacologically viable drug candidate when necessary. As an initial attempt to identify neuroprotective compounds in Cannabis, we screened eleven commercially available cannabinoids and related compounds in the five major assays we use for drug discovery [3]. These assays are outlined below. The results are presented in Table 1 and the structures of the compounds shown in Figure 1. Representative dose-response curves for two of the cannabinoids are shown in Figure 2.
Table 1.
Biological Activity of Cannabinoids
| No | Compound | Oxytosis (EC50) |
Energy Loss (EC50) |
AβToxicity (EC50) |
Trophic Withdrawal (EC50) |
Inflammation (EC50) |
|---|---|---|---|---|---|---|
| 1 | 9ΔTHCA | 8 μM | 20 μM | - | - | >10 μM |
| 2 | CBDA | >10 μM | 500 nM | 70 nM | 120 nM | >10 μM |
| 3 | CBGA | - | - | - | - | 9 μM |
| 4 | 9ΔTHC | 550 nM | 500 nM | 100 nM | 430 nM | 7 μM |
| 5 | DMCBD | - | - | >1 μM | >5 μM | 9 μM |
| 6 | CBDV | 1.1 μM | 90 nM | 100 nM | 350 nM | >10 μM |
| 7 | 8ΔTHC | 400 nM | 60 nM | 85 nM | 275 nM | >10 μM |
| 8 | CBG | 1.9 μM | 2 μM | 80 nM | >1.5 μM | >10 μM |
| 9 | CBC | 750 nM | 750 nM | 100 nM | 200 nM | >10 μM |
| 10 | CBN | 700 nM | 300 nM | 90 nM | 120 nM | >10 μM |
| 11 | CBD | 610 nM | 200 nM | 30 nM | 75 nM | 10 μM |
EC50 = 50% of maximal protection; Oxytosis and Energy Loss use HT22 cells; Aβ Toxicity uses MC65 cells; Trophic Withdrawal uses primary neurons; Inflammation uses BV2 microglia. THCA, tetrahydrocannabinol acid; CBDA, cannabidiolic acid; CBGA, cannabigerolic acid; Δ9THC; CBD, cannabidiol; CBDV, cannabidivarin, Δ8THC; CBG, cannabigerol; CBC, cannabichromene; CBN, cannabinol; DMCBD, dimethyl cannabidiol. Toxicity at 10 μM is indicated by -. EC50s below 1 μM are shaded in grey.
Figure 1:

Structures of compounds used in Table 1.
Figure 2:

Biological activities of CBG and CBC. Each compound was tested in triplicate at the indicated concentrations as described in the text. The average cell viability is presented as a function of the concentration. (A) Oxytosis (HT22 cells); (B) Energy loss (HT22 cells); (C) Aβ toxicity (MC65 cells); (D) Loss of trophic factor support (primary rat cortical neurons).
Oxidative stress is a major component of aging and is elevated in AD [19]. Oxytosis is a programmed cell death pathway in which glutamate inhibits the uptake of cystine into neurons, thereby reducing the synthesis of glutathione, stimulating mitochondrial ROS production, oxidative stress, the influx of calcium and ultimately cell death [20]. The standard model for this screening assay is the HT22 mouse hippocampal nerve cell line. The details of this cell death pathway have been very well studied [for recent reviews see [20,21]]. Oxytosis is distinct from excitotoxicity, but identical to the recently named ferroptosis pathway [22]. Oxytosis can be a downstream result of excitotoxicity and morphological features of oxytosis are found in the aging brain [23, 24]. Cannabidivarin (CBDV), cannabigerol (CBG), cannabichromene (CBC), cannabinol (CBN), 8ΔTHC, the acid (THCA), and cannabidiol (CBD) all prevented oxytosis (Table 1, Fig. 2). Only cannabigerolic acid (CBGA), which is universally toxic in all of our assays at 10 μM, cannabidiolic acid (CBDA) and cannabidiol 2’,6’ dimethyl ether (DMCBD), a synthetic CBD derivative and 15-lipoxygenase inhibitor [25], were inactive.
To mimic the energy loss that occurs with aging and the acute loss of ATP in stroke we developed an assay in which neurons are treated with iodoacetic acid, inhibiting glyceraldehyde 3-phosphate dehydrogenase, resulting in the loss of ATP [18]. Neuroprotective compounds maintain ATP levels and cell viability under these conditions [26]. All cannabinoids except CBGA and DMCBD were able to sustain energy metabolism in the absence of glycolysis (Table 1, Fig. 2).
The loss of trophic factors is a common feature of aging and AD [27]. When embryonic neurons are removed from their brain environment and put into tissue culture at low density, they lose their trophic support and die within 24 hr. Growth factors or compounds that activate growth factor pathways lead to cell survival. Thus, when E18 embryonic cortical neurons are put into culture in the presence of increasing amounts of a drug candidate, those that mimic growth factor activity are neuroprotective [3]. All of the cannabinoids tested except THCA, DMCBD and CBGA are active in this assay (Table 1, Fig. 2).
Compounds were also screened for their ability to suppress the pro-inflammatory response of microglial cells to LPS. Cannabinoids have anti-inflammatory activity in a variety of conditions [28] and microglial cells express both CB1 and CB2 receptors [29]. However, only CBD, DMCBD, CBGA, and 9ΔTHC had EC50 values ≤ 10 μM in this assay (Table 1).
Finally, proteotoxicity was assessed by inducing MC65 cells to generate the C99 fragment of the amyloid precursor protein (APP) upon removal of tetracycline from the culture medium. As previously shown, 9ΔTHC was very effective at preventing intracellular Aβ toxicity, as was another psychoactive cannabinoid, 8ΔTHC, with EC50 values of 100 and 85 nM, respectively (Table 1). The biosynthetic precursor acid form of THC, THCA, was toxic to the MC65 cells at one μM. Seven non-psychoactive cannabinoids were also tested, and several of these were also very potent inhibitors of amyloid toxicity. CBDV, CBC, CBN, CBDA and CBD all prevented amyloid toxicity at 100 nM or less (Table 1, Fig. 2).
Several of the cannabinoids prevent the death of human MC65 neurons when applied at the same time as the cells are induced to make C99 and Aβ. Cell death is caused by the accumulation of amyloid inside the cells; none is secreted [10]. To determine if the cannabinoids protect the cells by preventing the accumulation of Aβ, the cells were induced to make Aβ in the presence or absence of the cannabinoids and after 2 days all extracts were analyzed with an Aβ antibody that also recognizes the amyloid precursor protein (APP). Figure 3 shows that THC, CBDV, CBG, CBC, CBN and CBD all block cell death and prevent the cells from accumulating the highly aggregated Aβ seen at the top of the gel. The 100 kD doublet in Figure 3 is the amyloid precursor protein APP and the 37 kD protein is a non-specific band recognized by the 6E10 antibody used to detect Aβ and APP.
Figure 3:
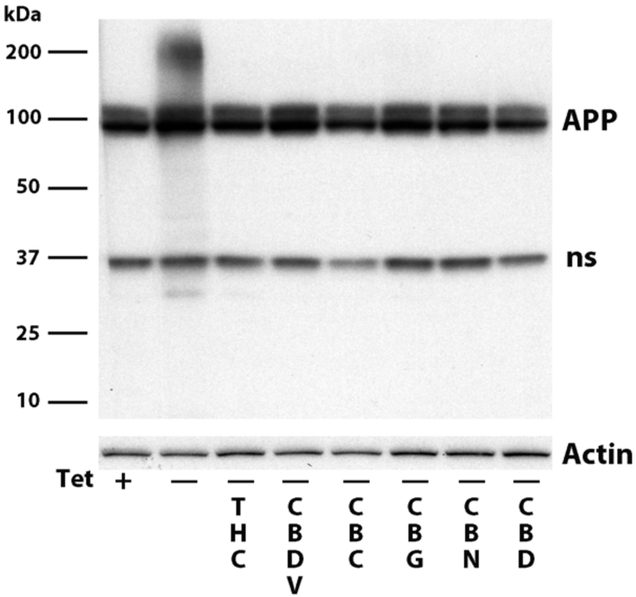
MC65 cells were induced (- tet) to make Aβ in the presence or absence of 100 nM cannabinoid. 48 hrs later the cells were blotted with the 6E10 antibody that recognizes aggregated Aβ, as well as APP and a nonspecific band at ~37 kd.
To determine if the cannabinoids can also speed up the degradation of preexisting amyloid, MC65 cells were induced to make Aβ for 24 hrs. At this time (Fig. 4, lane I 24), one set of cells was harvested, and both tetracycline, that stops C99 production, and a γ secretase inhibitor that inhibits Aβ cleavage from C99 (SI), were added to the remaining cultures. At the same time, half of the cultures were exposed to the different neuroprotective cannabinoids, and the other half vehicle alone. After another 24 hr, all the cells were harvested. Figures 4A and B show by Western blotting that there were significantly lower levels of Aβ aggregates in the cannabinoid-treated cultures as compared to the cultures where only Aβ production was halted by the γ secretase inhibitor which represents spontaneous degradation. Thus, CBDV, CBG, CBC, CBN and CBD all have the ability to stimulate the degradation and removal of preformed, aggregated Aβ from neurons.
Figure 4:

The nonpsychoactive cannabinoids CBDV, CBG, CBC, CBN and CBD all expedite the rate of removal of aggregated Aβ. MC65 cells were induced to make Aβ for 24 hrs (I24) or non-induced (NI). At this point, the cells were exposed to 100 nM cannabinoid, with or without tet and a γ secretase inhibitor (SI) (1 μM). Cells were harvested 24 hr later. Six lanes were with tet and SI and 5 lanes with the 5 cannabinoids. For statistics, all lanes were scanned for band density and the six control lanes were compared to the 5 treated lanes. (A) Western blot with the 6E10 antibody. (B) Bar graph of the quantified Western blot data. CAN is the sum of the cannabinoids.
It is generally assumed that THC functions via its interaction with the receptors CB1 or CB2, and that the non-psychoactive cannabinoids interact with these same receptors to a lesser extent, but also with other receptors [7]. However, neither HT22 cells (oxytosis and energy depletion assays) nor MC65 cells (proteotoxicity assay) express CB1 or CB2 receptors, as determined by RNAseq (cutoff: average FPKM <0.1) (not shown).
To gain insight into the structure-activity relationship (SAR) of cannabinoids in neuroprotection, we screened a number of agonists and antagonists of CB1 and CB2 in MC65 and HT22 cells. Table 2 shows that some synthetic CB1 agonists as well as the endocannabinoid AEA and its analog AM404 are protective in MC65 cells, while two CB1 antagonists, AM281 and AM251, potentiate toxicity. The structures of these compounds are shown in Figures 5 and 6. However, other potent CB1 and CB2 agonists like RCS-8 and MDA19 are completely inactive in MC65 cells. These data support the observation that MC65 do not respond to synthetic cannabinoids in a manner predicted for the involvement of CB1 or CB2.
Table 2.
Pharmacology
| MC65 | HT22 | ||||
|---|---|---|---|---|---|
| Compounds | MOA | Activity | EC50 | Activity | EC50 |
| R-1 methanandamide | CB1 agonist | protects | 3 μM | protects | 4 μM |
| 2 AG | CB1/2 agonist | protects | 0.05 μM | potentiates | 0.35 μM |
| Arvanil | CB1 agonist | protects | 0.4 μM | potentiates | 0.1 μM |
| AEA | Endocannabinoid | protects | 0.3 μM | protects | 0.35 μM |
| AM 404 | AEA analogue | protects | 0.4 μM | protects | 10 μM |
| SLV319 | CB1 antagonist | no effect | — | no effect | — |
| AM 281 | CB1 antagonist | potentiates | 0.9 μM | potentiates | 1.5 μM |
| AM 251 | CB1 antagonist | potentiates | 3.5 μM | potentiates | 1 μM |
| CP-55, 940 | CB1 and 2 agonist | protects | 5 μM | protects | 6 μM |
| NADA | CB1 and CB2 agonist | protects | 0.08 μM | protects | 1.1 μM |
| WIN-55, 212 | CB2 agonist | protects | 1.5 μM | no effect | — |
| JWH 133 | CB2 agonist | no effect | >10 μM | no effect | — |
| Q-3-Carboxamide | CB2 agonist | protects | 0.09 μM | no effect | — |
| AM 1241 | CB2 agonist | protects | 0.3 μM | protects | 0.45 μM |
| AM 630 | CB2 antagonist | potentiates | 0.01 μM | potentiates | 0.3 μM |
| UBR 597 | Inhibits AEA degradation | protects | 1.1 μM | protects | 0.5 μM |
| RCS-8 | CB1 agonist | no effect | — | no effect | — |
| MDA-19 | CB2 agonist | no effect | — | no effect | — |
| MCBN | Methyl ether of CBN | no effect | — | no effect | — |
| Hu210 | CB1 agonist | protects | 0.95 μM | protects | 0.6 μM |
| Hu211 | Inactive enantomer of Hu210 | protects | 1.5 μM | protects | 0.72 μM |
MC65 cells were induced to make Aβ or HT22 cells were exposed to a toxic glutamate concentration in the presence of three-fold serial dilutions between 10 nM and 20 μM of the indicated compounds. Cell viability was monitored and the compounds either had no effect, potentiated, or inhibited toxicity. There was no direct toxicity to cells at the concentrations indicated. The EC50S are given for either protection or potentiation. NADA is N-arachidonoyl dopamine, Q-3-carboxamide is 4-quinolone-3 carboxamide, and AEA is arachidonyl ethanolamide, 2 AG is arachidonoylglycerol, and MCBN is methyl CBN. (—) means no value determined.
Figure 5:

Structures of some of the compounds in Table 2.
Figure 6:

Structures of endocannabinoids and synthetic derivatives.
In contrast to the MC65 cells, the responses to both CB1 and CB2 receptor agonists and antagonists are somewhat different in HT22 cells. For example, the CB2 agonists, WIN-55,212 and Q-3-carboxamide that are protective in MC65 cells are ineffective in HT22 cells, and the CB1/2 agonists 2AG and arvanil that are also protective with MC65 cells, potentiate glutamate toxicity in HT22 cells. However, AEA and an inhibitor of its breakdown, UBR597, are protective in both assays, along with the AEA analog AM404. These data further support the idea that cannabinoids engage neuroprotective pathways that are independent of CB1 and CB2, and that the mechanisms responsible for the initiation of the protective response are different between MC65 and HT22 cells.
An examination of the structure-activity relationship among the compounds listed in Table 2 suggests that either an aromatic hydroxyl group or a keto group is necessary but not sufficient for the compound to be neuroprotective. Most of the neuroprotective compounds in Table 2 have one of these groups. Since JWH133, a Δ8-THC analogue without the aromatic hydroxyl group at the 1-position is completely inactive in both assays, additional experiments were done to test the hypothesis that aromatic hydroxyl groups are required for activity. The aromatic hydroxyl group at the 1-position of CBN was converted to a 1-methoxy and the resulting methylated CBN (MCBN) was examined. Table 2 shows that MCBN is inactive in both assays. Two other potent CB1 agonists, RCS-8 and MDA19 that lack aromatic hydroxyl groups are also inactive. Moreover, Hu210 is a potent CB1 agonist, while its enantiomer, Hu211, does not bind CB1 [30]; both are active in our assays (Table 2). Hu211 is neuroprotective in stroke and is a weak NMDA receptor antagonist [31]. These data further support the idea that the ability of a compound to act as an agonist for CB1 or CB2 receptors is not required for neuroprotection in the two assays, and that the aromatic hydroxyl groups play a critical role in the neuroprotective activity. The neuroprotective compounds that have keto groups, such as AM1241 and UBR597 all have hydrophobic domains distal to the ketone. These hydrophobic components may function as a membrane anchor for specific interactions of the compounds with protein receptors/enzymes in cells and assist their neuroprotective actions.
Endogenous cannabinoids such as 2AG and AEA have a polyunsaturated 20-carbon chain derived from arachidonic acid (ARA) (Fig. 6). This ARA chain is known to help maintain neuronal cell membrane fluidity [32], stimulate cell membrane expansion [33], and protect from cellular oxidative stress [34]. Impairment of ARA content in the brain may contribute to neurodegenerative disorders such as AD [35]. We have previously shown that AEA exerts neuroprotection against Aβ proteotoxicity and inflammation [12]. Table 2 shows that other endocannabinoids and their analogues like 2AG, R-1 methanandamide, arvanil, AM404, and NADA effectively protect from Aβ toxicity in MC65 cells (Table 2). Interestingly, some of these endocannabinoid analogues also contain aromatic hydroxyl groups. However, since 2AG and arvanil potentiate toxicity in HT22 cells, it is likely that there is a specificity to the mode of action in addition to a general antioxidative effect.
The entourage effect is a concept arguing that a synergistic interaction between compounds in Cannabis modulates the human biological responses to the plant [36]. This concept is primarily applied to the psychoactive effects of THC in conjunction with additional compounds in the plant. While not recapitulating the entourage effect, the heterogeneity of the response to cannabinoids in neurons that lack CB1 and CB2 suggests that there may be an interaction between the different cannabinoids in neuroprotection assays. This can be easily tested by constructing a screening matrix on a 96-well tissue culture dish in which the concentration of the first compound is increased in one direction and the concentration of the second is increased perpendicular to the first (Fig. 7). In the oxytosis assay, HT22 cells are treated with glutamate and nerve cell survival monitored 20 hr later. The viability data are then plotted in a 3-dimensional diagram, and it is asked if there is a synergistic or additive effect between two compounds. Controls for additive effects are each compound vs itself. With THC vs CBD there is a clear additive effect, but no synergistic effect (Fig. 7A). In contrast, there is a large synergistic (greater than additive) effect of THC vs. CBN (Fig. 7B). These data show that combinations of cannabinoids can positively interact in the absence of CB1 and CB2 receptors in the context of neuroprotection.
Figure 7:

Synergistic interaction between CBN and THC, but not CBD and THC. Increasing concentrations of CBD or CBN and THC were added to 96 well tissue culture dishes perpendicular to each other. Sufficient glutamate was added to kill 90% of the cells in the absence of cannabinoids. The percent survival in each well is then plotted against the concentrations of the two compounds. The colors are consistent for each concentration of THC (0, blue; 0.1 μM, red; 0.2 μM, green). Higher concentrations of CBD were used than CBN. The arrows indicate conditions of maximum synergy in the case of CBN vs THC. There was only an additive effect of CBD with THC.
Discussion
The plant genus Cannabis consists of one species, sativa. It is generally divided into three major varieties, those with high 9ΔTHC and low CBD (drug type), those with high CBD and very little 9ΔTHC (fiber type) and an intermediate type with both THC and CBD at moderate levels [37]. However, all groups contain about 500 distinct compounds and at least 100 cannabinoids [38]. Of these, only a few have been studied in detail. 9ΔTHC is a partial agonist of CB1 and CB2 receptors, and suppresses glutamate and GABA neurotransmission [39, 40]. CBD lacks psychoactivity, only very weakly interacts with CB1 and CB2 [41], but is an antioxidant and excellent anti-inflammatory compound in animal models [42, 43]. It is also a weak agonist for the G protein-coupled receptors GPR55 and GPR18, the vanilloid receptor, and several neurotransmitter receptors [43-46]. DMCBD is a synthetic derivative of CBD and is a potent 15-lipoxygenase inhibitor [25]. There is evidence that CBN, CBG, and THCA are also able to interact with several neurotransmitter receptors as well as acting as CB1 and CB2 antagonists [47-49]. Finally, a very large number of synthetic CB1 and CB2 receptor agonists and antagonists have been made, but their development was largely based upon receptor screening with minimum associated biology [50]. Several of these potent CB1 agonists, such as RCS-8 and MOA-19, have no activity in our screening assays.
CBD is the best studied cannabinoid in the context of AD [51,52]. CBD is neuroprotective in several cell lines and primary cortical neurons [14, 53]. Importantly, there have been a few in vivo rodent studies demonstrating that CBD reduces the inflammatory response associated with AD [54, 55]. In transgenic AD mice, CBD is able to prevent long-term memory defects, but has no effect on Aβ levels or oxidative stress [56, 57]. CBD in combination with THC has also been examined in AD mice. In some studies, the combination of CBD and THC as well as THC alone had therapeutic efficacy [15, 58-60]. We have shown that THC reduces the accumulation of intracellular Aβ, a toxic peptide that many believe is involved in AD pathogenesis [12]. Together, these results along with our data presented here suggest that cannabinoids have significant potential for the treatment of AD.
It is generally assumed that the biological activities of THC are mediated by CB1 and CB2 receptors. Both are G protein-coupled and cause a decrease in cAMP. CB1 is most highly expressed in the brain while CB2 is frequently associated with immune cells [29]. To determine if these receptors mediated the neuroprotective effects of the cannabinoids in our neuroprotection screening assays, we assessed the effects of CB1 and CB2 agonists and antagonists in two nerve-like cell lines that lack CB1 and CB2, the mouse hippocampal nerve cell line, HT22 and the human nerve cell line, MC65. In MC65, the majority of the CB1 agonists were protective, while the antagonists potentiated toxicity. CB2 agonists were mostly protective. In HT22 cells, CB2 agonists were mostly ineffective and the CB2 antagonist, AM630, potentiated toxicity. Results with CB1 agonists and antagonist were mixed. Since neither HT22 nor MC65 cells express CB1 or CB2 receptors, these data suggest that the neuroprotective aspects of cannabinoids are mediated by alternative mechanisms. A similar conclusion was made from a study using oxidatively stressed HT22, PC12 and SH-SYSY cells [42, 53].
It has been argued that the neuroprotective effects of the natural cannabinoids are due to their antioxidant activities [42]. They all contain an aromatic hydroxyl group that can reduce the rates of oxidation of radicals by transferring a proton (hydrogen) and one electron between the two oxygen atoms. However, the magnitude of this transfer, the stabilization of the oxidized compound, and therefore their antioxidant effect, is very dependent upon the environment in which the reaction occurs [61]. Because cannabinoids are amphiphilic, with a hydrophobic aliphatic tail and an ionizable polar head group, they could also insert into membranes and act as chain breakers of lipid peroxidation. However, because of the very low EC50s of these compounds and the pharmacology of synthetic CB1 and CB2 agonists and antagonists, it is very unlikely that the antioxidant properties of cannabinoids are solely responsible for their neuroprotective properties. Nevertheless, it does appear that either aromatic hydroxyl or keto groups are required for neuroprotection and that synthetic CB1 agonists that lack these groups will lack neuroprotective activities.
The entourage effect with Cannabis has been widely debated, but with little science to back up its existence [36]. Essentially all drugs interact with each other in some manner. These can lead to therapeutic strategies for cancer [62] or, more frequently, toxic drug interactions [63]. While not duplicating the interaction of the multiple compounds in Cannabis, it is easy to examine pairwise interactions of cannabinoids in cell culture. When this was done with THC, CBD and CBN in an assay based upon the oxytosis cell death pathway, a significant synergistic enhancement of neuroprotection between THC and CBN was seen, while the same assay using THC and CBD only produced an additive effect. Since HT22 cells do not have cannabinoid receptors, the therapeutic relevance of this interaction is unknown, but it does suggest that CBN and CBD are targeting distinct molecular pathways. This conclusion is also supported by the limited pharmacology shown in Table 2 indicating that the responses to CB1 and CB2 agonists in the two cell lines are distinct.
Conclusion
The above results demonstrate that some non-psychoactive cannabinoids are effective at nanomolar levels in a drug-screening platform that has been used to identify bona fide AD drug candidates [3]. Moreover, these cannabinoids are also able to stimulate the clearance of intraneuronal Aβ and protect nerve cells from three additional old age-associated neurotoxic insults. Importantly, they are functional in the absence of CB1 and CB2 receptors and, in some cases, show a synergistic neuroprotective effect with each other. We conclude that the cannabinoids are engaging currently undefined targets and that one or more of these cannabinoids could provide a lead compound for AD drug therapy.
Acknowledgments
Funding: This study was supported by grants from the Paul F. Glenn Center for Aging at the Salk Institute (JG), NIH (RO1 AG046153 and RF1 AG054714 to PM and DS, and R41AI104034 to PM), the Edward N. & Della Thome Memorial Foundation (PM), and the Salk Core Facility (NIH-NCI:P30 014195, the Chapman Foundation and the Helmsley Trust).
Abbreviations
- AD
Alzheimer's Disease
- AEA
Arachidonoyl ethanolamide
- APP
Amyloid precursor protein
- ARA
Arachidonic acid
- ATP
Adenosine triphosphate
- cAMP
Cyclic adenosine
- CBC
Cannabichromene
- CBD
Cannabidiol
- CBD
Cannabidiolic acid
- CBDV
Cannabidivarin
- CBG
Cannabigerol
- CBGA
Cannabigerolic acid
- CBN
Cannabinol
- DEA
Drug Enforcement Agency
- DMCBD
cannabidiol 2',6' dimethyl ether
- DMEM
Dulbecco's modified Eagle medium
- FAD
Familial Alzheimer's Disease
- FCS
Fetal calf serum
- LPS
Lipopolysaccharide
- MCBN
methylated cannabinol
- MTT
Tetrazolium-based colorimetric assay
- NADA
N-arachidonoly dopamine
- NMR
Nuclear magnetic resonance
- ROS
Reactive oxygen species
- SAR
Structure activity relationship
- THC
Tetrahydrocannabinol
- THCA
Tetrahydrocannabinol acid
- TMSCHN2
Trimethylsilyldiazomethane.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Availability of data and materials: All of the cell lines used in the screening assays are available. All data generated or analyzed during this study are included in the manuscript or available upon request.
Competing interests: The authors declare that they have no competing interests.
References
- 1.Alzheimer's Disease International (2015, www.alz.co.uk/research/statistics) Dementia statistics. Vol. 2018, n.p. [Google Scholar]
- 2.Cummings J Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci. 2018; 11:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prior M, Chiruta C, Currais A, Goldberg J, Dargusch R, Maher P, and Schubert D Back to the future with phenotypic screening. ACS Chem Neurosci. 2014;5:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenneisen R (2007) Chemistry and analysis of phytocannabinoids and other cannabis constituents In Marijuana and th Cannabinoids (322 pgs) (Eisohly MA, ed) pp. 17–49, Humana Press, Totowa, New Jersey, [Google Scholar]
- 5.Mastinu A, Premoli M, Ferrari-Toninelli G, Tambaro S, Maccarinelli G, Memo M, and Bonini SA Cannabinoids in health and disease: pharmacological potential in metabolic syndrome and neuroinflammation. Horm Mol Biol Clin Investig. 2018. March 30. pii:/j/hmbci.ahead-of-print/hmbci-2018-0013/hmbci-2018-0013.xml. doi: 10.1515/hmbci-2018-0013. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6.Paez JA, and Campillo NE Innovative Therapeutic Potential of Cannabinoid Receptors as Targets in Alzheimer's disease and Less Well-Known Diseases. Curr Med Chem 2018. February 25 [E-pub ahead of print] doi 102174/0929867325666180226095132. [DOI] [PubMed] [Google Scholar]
- 7.Mechoulam R, and Parker LA The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. [DOI] [PubMed] [Google Scholar]
- 8.Currais A, Fischer W, Maher P, and Schubert D Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 2017;31:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, and Finley KD Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184 [DOI] [PubMed] [Google Scholar]
- 10.Sopher BL, Fukuchi K, Smith AC, Leppig KA, Furlong CE, and Martin GM Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the beta-amyloid precursor protein. Brain Res Mol Brain Res. 1994;26:207–217. [DOI] [PubMed] [Google Scholar]
- 11.Elinder F, Akanda N, Tofighi R, Shimizu S, Tsujimoto Y, Orrenius S, and Ceccatelli S Opening of plasma membrane voltage-dependent anion channels (VDAC) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death Differ. 2005;12:1134–1140. [DOI] [PubMed] [Google Scholar]
- 12.Currais A, Quehenberger O, A MA, Daugherty D, Maher P, and Schubert D Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. NPJ Aging Mech Dis. 2016;2:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao C, Li Y, Liu H, Bai G, Mayl J, Lin X, Sutherland K, Nabar N, and Cai J The potential therapeutic effects of THC on Alzheimer's disease. J Alzheimers Dis. 2014;42:973–984. [DOI] [PubMed] [Google Scholar]
- 14.Janefjord E, Maag JL, Harvey BS, and Smid SD Cannabinoid effects on beta amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell Mol Neurobiol. 2014;34:31–42. [DOI] [PubMed] [Google Scholar]
- 15.Aso E, Andres-Benito P, and Ferrer I Delineating the Efficacy of a Cannabis-Based Medicine at Advanced Stages of Dementia in a Murine Model. J Alzheimers Dis. 2016;54:903–912. [DOI] [PubMed] [Google Scholar]
- 16.Aso E, Juves S, Maldonado R, and Ferrer I CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AbetaPP/PS1 mice. J Alzheimers Dis. 2013;35:847–858. [DOI] [PubMed] [Google Scholar]
- 17.Valera E, Dargusch R, Maher PA, and Schubert D Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer's disease. J Neurosci. 2013;33:10512–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maher P, Salgado KF, Zivin JA, and Lapchak PA A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 2007;1173:117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beal MF Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. [DOI] [PubMed] [Google Scholar]
- 20.Lewerenz J, and Maher P Control of Redox State and Redox Signaling by Neural Antioxidant Systems. Antioxid Redox Signal. 2011; 14: 1449–1465 [DOI] [PubMed] [Google Scholar]
- 21.Maher P How fisetin reduces the impact of age and disease on CNS function. Front Biosci (Schol Ed). 2015;7:58–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewerenz J, Ates G, Methner A, Conrad M, and Maher P Oxytosis/Ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Frontiers Neurosci 2018. April:20; 12:214 doi. 2018; 12:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schubert D, and Piasecki D Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewerenz J, and Maher P Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front Neurosci. 2015;9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda S, Hirayama A, Urata S, Mano N, Fukagawa K, Imamura M, Irii A, Kitajima S, Masuyama T, Nomiyama M, Tatei S, Tomita S, Kudo T, Noguchi M, Yamaguchi Y, Okamoto Y, Amamoto T, Fukunishi Y, Watanabe K, Omiecinski CJ, and Aramaki H Cannabidiol-2',6'-dimethyl ether as an effective protector of 15-lipoxygenase-mediated low-density lipoprotein oxidation in vitro. Biol Pharm Bull. 2011;34:1252–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldberg J, Currais A, Prior M, Fischer W, Ratiff E, Daugherty D, Dargusch R, Finley K, Esparza-Molto PB, Cuezva JM, Maher P, Petrascheck M, and Schubert D The mitochondrial ATP synthase is a shared drug target among aging and dementia. Aging Cell 2018:e12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen SJ, Watson JJ, Shoemark DK, Barua NU, and Patel GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138:155–175. [DOI] [PubMed] [Google Scholar]
- 28.McCoy KL Interaction between Cannabinoid System and Toll-Like Receptors Controls Inflammation. Mediators Inflamm. 2016;2016:5831315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabral GA, and Marciano-Cabral F Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78:1192–1197. [DOI] [PubMed] [Google Scholar]
- 30.Mechoulam R, Feigenbaum JJ, Lander N, Segal M, Jarbe TU, Hiltunen AJ, and Consroe P Enantiomeric cannabinoids: stereospecificity of psychotropic activity. Experientia. 1988;44:762–764. [DOI] [PubMed] [Google Scholar]
- 31.Darlington CL Dexanabinol: a novel cannabinoid with neuroprotective properties. IDrugs. 2003;6:976–979. [PubMed] [Google Scholar]
- 32.Fukaya T, Gondaira T, Kashiyae Y, Kotani S, Ishikura Y, Fujikawa S, Kiso Y, and Sakakibara M Arachidonic acid preserves hippocampal neuron membrane fluidity in senescent rats. Neurobiol Aging. 2007;28:1179–1186. [DOI] [PubMed] [Google Scholar]
- 33.Darios F, and Davletov B Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature. 2006;440:813–817. [DOI] [PubMed] [Google Scholar]
- 34.Wang ZJ, Liang CL, Li GM, Yu CY, and Yin M Neuroprotective effects of arachidonic acid against oxidative stress on rat hippocampal slices. Chem Biol Interact. 2006;163:207–217. [DOI] [PubMed] [Google Scholar]
- 35.Amtul Z, Uhrig M, Wang L, Rozmahel RF, and Beyreuther K Detrimental effects of arachidonic acid and its metabolites in cellular and mouse models of Alzheimer's disease: structural insight. Neurobiol Aging. 2012;33:831 e821–831. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Ramos J The entourage effect of the phytocannabinoids. Ann Neurol. 2015;77:1083. [DOI] [PubMed] [Google Scholar]
- 37.ElSohly MA, Radwan MM, Gul W, Chandra S, and Galal A Phytochemistry of Cannabis sativa L. Prog Chem Org Nat Prod. 2017;103:1–36. [DOI] [PubMed] [Google Scholar]
- 38.Elsohly MA, and Slade D Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci. 2005;78:539–548. [DOI] [PubMed] [Google Scholar]
- 39.Pertwee RG Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem. 2010;17:1360–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajos N, Ledent C, and Freund TF Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience. 2001;106:1–4. [DOI] [PubMed] [Google Scholar]
- 41.Petitet F, Jeantaud B, Reibaud M, Imperato A, and Dubroeucq MC Complex pharmacology of natural cannabinoids: evidence for partial agonist activity of delta9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci. 1998;63:PL1–6. [DOI] [PubMed] [Google Scholar]
- 42.Marsicano G, Moosmann B, Hermann H, Lutz B, and Behl C Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem. 2002;80:448–456. [DOI] [PubMed] [Google Scholar]
- 43.Izzo AA, Borrelli F, Capasso R, Di Marzo V, and Mechoulam R Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci. 2009;30:515–527. [DOI] [PubMed] [Google Scholar]
- 44.Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, Moriello AS, Davis JB, Mechoulam R, and Di Marzo V Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russo EB, Burnett A, Hall B, and Parker KK Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res. 2005;30:1037–1043. [DOI] [PubMed] [Google Scholar]
- 46.McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, and Bradshaw HB N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010; 11:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cascio MG, Gauson LA, Stevenson LA, Ross RA, and Pertwee RG Evidence that the plant cannabinoid cannabigerol is a highly potent alpha2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br J Pharmacol. 2010;159:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill TD, Cascio MG, Romano B, Duncan N, Pertwee RG, Williams CM, Whalley BJ, and Hill AJ Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br J Pharmacol. 2013;170:679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, and Pertwee RG Evidence that the plant cannabinoid Delta-9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor agonist. Br J Pharmacol. 2005;146:917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tai S, and Fantegrossi WE Pharmacological and Toxicological Effects of Synthetic Cannabinoids and Their Metabolites. Curr Top Behav Neurosci. 2017;32:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watt G, and Karl T In vivo Evidence for Therapeutic Properties of Cannabidiol (CBD) for Alzheimer's Disease. Front Pharmacol. 2017;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fonseca BM, Teixeira NA, and Correia-da-Silva G Cannabinoids as Modulators of Cell Death: Clinical Applications and Future Directions. Rev Physiol Biochem Pharmacol. 2017;173:63–88. [DOI] [PubMed] [Google Scholar]
- 53.Harvey BS, Ohlsson KS, Maag JL, Musgrave IF, and Smid SD Contrasting protective effects of cannabinoids against oxidative stress and amyloid-beta evoked neurotoxicity in vitro. Neurotoxicology. 2012;33:138–146. [DOI] [PubMed] [Google Scholar]
- 54.Esposito G, Scuderi C, Savani C, Steardo L Jr., De Filippis D, Cottone P, Iuvone T, Cuomo V, and Steardo L Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol. 2007;151:1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Esposito G, Scuderi C, Valenza M, Togna GI, Latina V, De Filippis D, Cipriano M, Carratu MR, Iuvone T, and Steardo L Cannabidiol reduces Abeta-induced neuroinflammation and promotes hippocampal neurogenesis through PPARgamma involvement. PLoS One. 2011;6:e28668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng D, Low JK, Logge W, Garner B, and Karl T Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1E9 mice. Psychopharmacology (Berl). 2014;231:3009–3017. [DOI] [PubMed] [Google Scholar]
- 57.Cheng D, Spiro AS, Jenner AM, Garner B, and Karl T Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer's disease transgenic mice. J Alzheimers Dis. 2014;42:1383–1396. [DOI] [PubMed] [Google Scholar]
- 58.Casarejos MJ, Perucho J, Gomez A, Munoz MP, Fernandez-Estevez M, Sagredo O, Fernandez Ruiz J, Guzman M, de Yebenes JG, and Mena MA Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J Alzheimers Dis. 2013;35:525–539. [DOI] [PubMed] [Google Scholar]
- 59.Aso E, and Ferrer I Cannabinoids for treatment of Alzheimer's disease: moving toward the clinic. Front Pharmacol. 2014;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aso E, Sanchez-Pla A, Vegas-Lozano E, Maldonado R, and Ferrer I Cannabis-based medicine reduces multiple pathological processes in AbetaPP/PS1 mice. J Alzheimers Dis. 2015;43:977–991. [DOI] [PubMed] [Google Scholar]
- 61.Foti MC Antioxidant properties of phenols. J Pharm Pharmacol. 2007;59:1673–1685. [DOI] [PubMed] [Google Scholar]
- 62.Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, and Yeger H Combination therapy in combating cancer. Oncotarget. 2017;8:38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tannenbaum C, and Sheehan NL Understanding and preventing drug-drug and drug-gene interactions. Expert Rev Clin Pharmacol. 2014;7:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]