

Abstract
Background & Aims
Epithelial tight junctions are compromised in patients with gastrointestinal disease. Occludin is a protein component of tight junctions; its removal from tight junctions or reduced expression results in barrier loss. Knockout (KO) of occludin in mice, however, does not appear to affect intestinal in tight junction structure or function. We investigated whether mucosal injury or repair are compromised in occludin-KO mice.
Methods
We performed studies with occludin-KO mice, mice with intestinal epithelial cell-specific KO of occludin, and B6 (control) mice, as well as mice that overexpress occludin from a transgene in the intestinal epithelium. Colitis was induced by administration of dextran sulfate sodium or 2,4,6-trinitrobenzene sulphonic acid. Intestinal tissues were collected from mice, patients with Crohn’s disease or ulcerative colitis, or subject without these diseases (controls) and analyzed by histology, immunohistochemistry, quantitative real-time PCR, and immunoblots. Occludin was knocked down in Caco-2BBe cells.
Results
Mice with intestinal epithelial cell-specific KO of occludin developed less-severe colitis than control mice. This protection was due to reduced activation of intrinsic and extrinsic apoptotic pathways. Promoter analysis revealed that occludin increased transcription of the caspase 3 gene (CASP3). Mucosal biopsies from patients with Crohn’s disease or ulcerative colitis had lower levels of occludin than controls; reduced occludin correlated with lower levels of CASP3. Incubation of Caco-2BBe monolayers with tumor necrosis factor caused occludin downregulation, which reduced CASP3 expression and prevented induction of apoptosis via the intrinsic pathway (stimulated by 5-fluorouracil) or extrinsic pathway (stimulated by tumor necrosis factor).
Conclusions
In intestinal epithelial cells, the tight junction protein occludin increases expression of CASP3. Occludin loss reduces expression of CASP3 and susceptibility of these cells to apoptosis. Reduced levels of occludin and CASP3 in intestinal epithelial cells of patients with inflammatory bowel diseases might promote restoration of mucosal homeostasis in response to inflammatory conditions.
Keywords: survival, IBD, gene regulation, cell death
Lay Summary
Intestinal tissues from patients with inflammatory bowel diseases have reduced expression of the barrier protein occludin. Occludin loss protects cells from programmed cell death. Inflammation-induced downregulation of occludin might prevent damage in intestinal tissues.
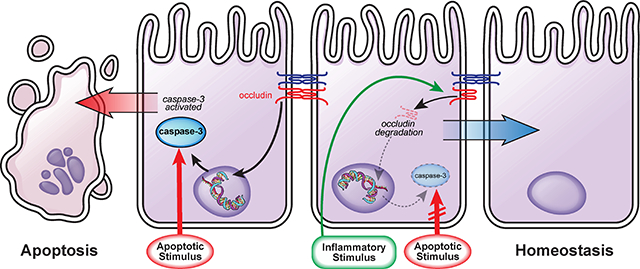
Graphical Abstract
Introduction
The first transmembrane tight junction protein discovered, occludin, is present within tight junction strands1 and, when overexpressed, induces intracellular, multi-lamellar bodies with fused, tight junction-like membranes.2 The occludin C-terminal coiled-coil domain binds directly to ZO-13 via interactions regulated by phosphorylation within and adjacent to that region.4–7 A range of in vitro studies, including analyses of peptides corresponding to occludin domains,8 occludin knockdown,9–11 or expression of occludin mutants, e.g., those lacking the C- terminal tail, 12 suggest that occludin limits paracellular permeability to macromolecular probes. The observation that occludin removal from the tight junction, either in response to F-actin depolymerization or cytokine stimulation10, 13, 14 increases leak pathway permeability is consistent with this function. Conversely, cytokine stimulation of occludin-deficient monolayers does not reduce barrier function beyond the increased permeability already present as a consequence of occludin deletion.10, 11 These data indicate that this form of cytokine-induced barrier loss is largely, and perhaps entirely, caused by occludin removal from the tight junction.
Despite extensive in vitro data, the viability of and absence of obvious structural or functional tight junction defects within the small intestine, colon, and bladder of occludin knockout (KO) mice15, 16 has led many to conclude that occludin does not contribute to mucosal homeostasis. This conclusion fails to account for the multiple abnormalities of occludin KO mice including hearing loss, growth retardation, chronic gastritis, cerebellar and basal ganglia calcification, male sterility, and inability of females to suckle their pups.15, 17 In addition, transgenic EGFP-occludin expression within the intestinal epithelium, i.e., overexpression, limited TNF-induced depletion of tight junction-associated occludin, leak pathway permeability increases, and diarrhea.18 Nevertheless, the lack of baseline intestinal dysfunction in occludin KO mice suggests that compensatory changes might be masking relevant deficits. Experience with other KO mice has shown that underlying phenotypes can often be exposed by stress.19–21
We hypothesized that occludin KO mice might be hypersensitive to colitogenic stimuli. In contrast, mice lacking intestinal epithelial occludin were markedly protected from disease. This unexpected result reflected insensitivity of occludin-deficient intestinal epithelial cells to a variety of intrinsic and extrinsic apoptotic pathway agonists. This protection was due to loss of occludin-dependent CASP3 transcription that led to reduced caspase-3 expression. Moreover, TNF-induced occludin downregulation protected epithelia from apoptosis by reducing caspase-3 expression. Finally, we confirmed previous reports of intestinal epithelial occludin downregulation in Crohn’s disease and ulcerative colitis and found that this was strongly correlated with reduced caspase-3 expression. These data indicate that the adaptive process initiated by occludin downregulation is active and may enhance epithelial survival in human disease. We conclude that occludin serves as a critical regulator of epithelial apoptosis and survival by modulating CASP3 transcription and caspase-3 expression.
Results
Epithelial occludin expression exacerbates DSS colitis
Extensive in vitro studies indicate that the tight junction protein occludin is involved in epithelial barrier regulation.8–12 We initially asked if occludin expression contributes to epithelial homeostasis by comparing mucosal architecture, epithelial proliferation, and epithelial migration in wildtype (WT) and occludin KO mice; no differences were detected (Suppl. Fig. 1). Nevertheless, reduced occludin expression in a range of intestinal disorders22 suggests that occludin may be involved in disease. To test this hypothesis, we assessed the sensitivity of occludin KO mice to epithelial damage. Exposure to DSS induced expected weight loss (Fig. 1A, Suppl. Fig. 2A), clinical disease activity (Fig. 1B, Suppl. Fig. 2B), colon shortening (Fig. 1C), histologic damage (Fig. 1D), and neutrophil infiltration (Fig. 1E) in WT mice. However, universal occludin KO mice, in which all tissues lacked occludin, were remarkably resistant to DSS-induced colitis by all of these measures (Fig. 1A–E). Complementation by transgenic intestinal epithelia-specific EGFP-occludin expression23 restored sensitivity to DSS colitis (Fig. 1A–E). These data thereby link the observed phenotype specifically to intestinal epithelia and demonstrate that intestinal epithelial occludin expression promotes pathogenesis of DSS colitis.
Figure 1. Intestinal epithelial occludin expression promotes DSS-induced intestinal disease.

A. DSS-induced weight loss was attenuated in occludin KO mice. Transgenic intestinal epithelial EGFP-occludin expression (Tg/KO) restored DSS sensitivity to that of WT mice. Analysis at day 7 is shown (n ≥ 3 for water groups and n ≥ 4 for DSS-treated groups). The complete time course is shown in Suppl. Fig. 2A.
B. Disease activity clinical scores were attenuated by occludin KO and restored by in Tg/KO mice (Day 7 data, n ≥ 6 per group). The complete time course is shown in Suppl. Fig. 2B.
C. Colonic shortening occurred in DSS-treated WT and Tg/KO mice but markedly reduced in occludin KO mice (n ≥ 6 per group).
D. Histopathology shows preservation surface epithelium and some crypts in DSS-treated occludin KO, relative to WT or KO mice with transgenic occludin expression. Bar = 50μm.
E. DSS-induced increases in myeloperoxidase activity were attenuated in occludin KO mice but restored to WT levels in Tg/KO mice (n ≥ 3 for water groups and n ≥ 4 for DSS-treated groups).
F. DSS-induced increases in intestinal permeability to 4 kD dextran were markedly attenuated in occludin KOiec mice (n ≥ 5 per group).
G. DSS enhanced Il6, Il1b, Il17, and Cxcll (KC, IL-8) expression in WT, but not occludin KOIEC, mice (n ≥ 5 per group).
H. DSS increased colonic epithelial proliferation, as assessed by Ki-67 staining, similarly in WT and occludin KOIEC mice (n ≥ 5 per group).
I. DSS increased numbers of TUNEL-positive cells in WT, but not occludin KOIEC, mice (n = 4 per group).
J. DSS increased numbers of ISOL-positive cells (red) in WT, but not occludin KOIEC, mice. Nuclei shown in blue. Bar = 20μm (n = 4–6 per group).
K. DSS increased numbers of cleaved caspase-3-positive cells (green) in WT, but not occludin KOiec, mice. Occludin (cyan) and E-cadherin (red) are shown for reference. Bar = 10μm (n ≥ 4 per group).
Ocln+/+ littermates were used as WT controls for Ocln−/− and Ocln−/− x vil-EGFP-occludin (Tg/KO) mice and Oclnf/f littermates were used as WT controls for Oclnf/f x vil-Cre (occludin KOIEC) mice in this and all other figures. Statistical analyses by one-way ANOVA with Bonferroni post-test. *, P<0.05; **, P<0.01.
Although complementation with transgenic occludin demonstrates the essential role of intestinal epithelial occludin in DSS sensitivity, tissue-specific occludin KO is more definitive. We therefore generated intestinal epithelial-specific occludin KO (occludin KOIEC) mice. As with universal occludin KO mice, intestinal mucosal architecture of occludin KOIEC (Oclnf/fx Vil-Cre) mice was indistinguishable from WT (Oclnf/f) mice (Suppl. Fig. 3A). Occludin KOIEC did not, however, demonstrate male infertility or any of the other phenotypic abnormalities reported in universal occludin KO mice.15, 17 Similar to universal KO mice, DSS colitis severity was reduced in Occludin KOiec mice. The protection observed included attenuation of DSS-induced increases in intestinal permeability (Fig. 1F) and inflammatory cytokine production (Fig. 1G) relative to occludin-expressing controls.
Intestinal epithelial occludin promotes DSS-induced apoptosis
The damage that characterizes DSS colitis represents an imbalance between chemical injury and mucosal repair. Occludin could potentially affect either or both of these processes. Epithelial proliferative responses to DSS-induced injury were, however, indistinguishable in occludin KOiec and WT mice (Fig. 1H). We therefore turned our attention to damage. Epithelial cell death, assessed as DNA fragmentation using terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL), was similar in unstressed WT and occludin KOIEC mice. DSS markedly increased the number of TUNEL-positive epithelial cells in WT mice, but this increase was largely suppressed in occludin KOIEC mice (Fig. 1I). To better assess the mechanism of cell death, sections were labeled by in situ oligonucleotide ligation (ISOL), which specifically detects double-strand DNA breaks that are blunt-ended or have a one-base 3’-overhang. This is more specific for apoptosis than TUNEL, which detects DNA fragmentation induced by multiple processes. As with TUNEL, DSS-induced increases in ISOL labeling were significantly attenuated in treated occludin KOIEC mice (Fig. 1J). Finally, cleaved caspase-3 staining, a hallmark of apoptosis, was increased by DSS treatment of WT, but not occludin KOIEC, mice (Fig. 1K). Collectively, these data indicate that intestinal epithelial occludin expression sensitizes cells to DSS-induced apoptosis.
Intestinal epithelial occludin deletion limits TNBS-induced colitis and tissue damage
DSS induces colitis via direct chemical injury to intestinal epithelial cells.24 We therefore considered the possibility that the protection afforded by occludin deletion was specific to direct chemical injury. To determine whether the effects of intestinal epithelial occludin deletion extended to other forms of injury, we assessed the sensitivity of occludin KOIEC and control mice to TNBS colitis. TNBS haptenates autologous and luminal proteins to trigger immune-mediated damage. Despite this distinct pathogenic mechanism, resistance of occludin KOIEC mice to TNBS was similar to that after DSS challenge. This protection was obvious on physical examination (Fig. 2A) and could be validated quantitatively by reduced disease activity scores (Fig. 2B). Histologic damage (Fig. 2C), intestinal barrier loss (Fig. 2D), and cytokine production (Fig. 2E) were also limited in TNBS-treated occludin KOIEC mice relative to occludin-sufficient WT controls.
Figure 2. Intestinal epithelial-specific occludin KO limits epithelial apoptosis and overall severity of TNBS colitis.

A. TNBS induced wasting and loss of normal fur texture in WT (Oclnf/f) mice. At day 4 after rectal TNBS instillation these features were almost undetectable in occludin KOIEC (Oclnf/f x vil-Cre) mice. Bar = 1cm.
B. TNBS-induced increases in disease activity scores were attenuated in occludin KOIEC mice (Day 4 data, n = 5 WT, 10 KOIEC).
C. Histopathology shows severe damage in TNBS-treated WT, but not occludin KOIEC, mice. Bar = 50μm.
D. TNBS-induced increases in intestinal permeability to 4 kD dextran were markedly attenuated in occludin KOIEC, relative to WT, mice (n ≥ 5 per group).
E. TNBS-induced Il6, Il1b, Cxcll, and Tnf upregulation in WT, but not occludin KOIEC, mice (n = 5 per group).
F. TNBS increased colonic epithelial proliferation similarly in WT and occludin KOIEC mice (n = 3 per group). Representative photomicrographs are shown in Suppl. Fig 3B.
G. TNBS increased numbers of TUNEL-positive cells in WT, but not occludin KOIEC, mice (n = 3 per group). Representative photomicrographs are shown in Suppl. Fig 3B.
H. TNBS increased numbers of ISOL-positive cells in WT, but not occludin KOIEC, mice (n = 3 per group).
I. TNBS increased numbers of cleaved caspase-3-positive cells in WT, but not occludin KOIEC, mice (n = 3 per group). Representative photomicrographs are shown in Suppl. Fig 3B.
Statistical analyses by Student’s f-test or one-way ANOVA with Bonferroni post-test. *, P<0.05; **, P<0.01.
TNBS-induced injury caused similar epithelial proliferative responses in occludin KOIEC and WT mice (Fig. 2F, Suppl. Fig. 3B). As with DSS, epithelial occludin deletion attenuated TNBS-induced apoptosis. Moreover, TUNEL (Fig. 2G, Suppl. Fig. 3B), ISOL (Fig. 2H, Suppl. Fig. 3B), and cleaved caspase-3 (Fig. 2I, Suppl. Fig. 3B) staining were markedly reduced in TNBS-treated occludin KOiec mice relative to WT controls. These data indicate that the protection from apoptotic cell death afforded by intestinal epithelial occludin deletion extends to multiple forms of epithelial injury.
Occludin KO limits activation of both intrinsic and extrinsic apoptotic pathways
Thus far, the results demonstrate that occludin-deficient epithelia are resistant to colitis-associated apoptosis. Given the immune-mediated mechanism of TNBS colitis, it is likely that this resistance to apoptosis includes insensitivity to extrinsic pathway activation by pro-apoptotic mediators, such as tumor necrosis factor-α (TNF). In contrast, the relative contributions of extrinsic, versus intrinsic, i.e., cellular stress, pathways to DSS-induced epithelial apoptosis have not been defined. To determine whether occludin expression facilitates apoptosis via the intrinsic pathway (Fig. 3A), mice were treated with the chemotherapeutic nucleoside analogue 5-fluorouracil (5-FU), which increased epithelial apoptosis, as demonstrated morphologically (Fig. 3B), by TUNEL staining (Fig. 3C), and by cleaved caspase-3 staining (Fig. 3D). These increases were largely absent in occludin KO mice. Occludin therefore facilitates, and occludin deletion prevents, apoptosis via the intrinsic pathway.
Figure 3. Occludin increases intrinsic and extrinsic pathway apoptosis in vivo.

A. Schematic highlighting the intrinsic and extrinsic apoptotic signaling pathways, which are activated by 5-FU and TNF, respectively.
B. 5-FU-induced crypt epithelial apoptosis (arrowheads) was reduced in occludin KO mice, particularly within the lower and middle crypt (positions 1–6). Bar = 50μm (n = 4 per group).
C. TUNEL staining (red) shows fewer 5-FU-induced DNA breaks in occludin KO crypts. Nuclei are shown in blue. Bar = 20mm (n = 4 per group).
D. Fewer cleaved caspase-3 positive cells (red) were present in WT, relative to occludin KO, mice after 5-FU treatment. Nuclei are shown in blue. Bar = 20μm (n = 4 per group).
E. Acute TNF treatment-induced caspase-3 cleavage (red) within villus epithelia was markedly reduced in occludin KO mice. Nuclei are shown in blue. Bar = 50μm (n = 3 per group).
F. Systemic T cell activation (anti-CD3 IgG) resulted in increased numbers of TUNEL-positive small intestinal epithelia in WT mice. This was markedly attenuated in occludin KOIEC mice (n = 4 per group).
G. Systemic T cell activation increased ISOL-positive (red) cell numbers in WT, but not occludin KOiec, mice. Nuclei are shown in blue. Bar = 50μm (n = 4–6 per group).
H. Western blot of intestinal epithelia isolated from vehicle- and TNF-treated WT and occludin KO mice (as in panel E). With the exception of cleaved caspase-3, markers of TNF signaling and caspase activation were not affected by occludin KO. Quantitative analyses are shown in Suppl. Fig. 4.
Statistical analyses by Student’s t-test or one-way ANOVA with Bonferroni post-test. *, P<0.05; **, P<0.01.
To assess the effect of occludin loss on extrinsic pathway apoptosis (Fig. 3A), mice were treated with recombinant TNF, which increased small intestinal epithelial apoptosis 30-fold (Fig. 3E). As with DSS- and 5-FU-induced epithelial apoptosis, TNF-induced apoptosis was almost entirely blocked by occludin deletion. We also assessed small intestinal epithelial apoptosis in response to a more complex stimulus, cytokine storm induced by systemic T cell activation.25, 26 Small intestinal epithelial apoptosis was readily detected 2 days after anti-CD3 administration, as indicated by TUNEL (Fig. 3F) and ISOL (Fig. 3G) labeling, in WT mice. Occludin KOIEC mice were, however, protected. Occludin deletion therefore prevents apoptosis of intestinal epithelia in response to intrinsic and extrinsic pathway stimuli.
Caspase-3 activation is defective in occludin-deficient epithelia in vivo and in vitro
To identify the events downstream of TNF receptor activation affected by occludin, small intestinal epithelial cells from WT and occludin KO mice that received vehicle or recombinant TNF were isolated and probed for ERK and p38 MAP kinase activation, IκB degradation, and caspase cleavage (Fig. 3H). ERK and p38 MAP kinase activation as well as IκB and caspase-8 degradation were similar in epithelia from TNF-treated WT and occludin KO mice (Fig. 3H, Suppl. Fig. 4A–D). Caspase-9 was not degraded, consistent with direct TNF-induced caspase-8 activation (Fig. 3E, Suppl. Fig. 4E). In contrast to WT mice, where substantial caspase-3 cleavage was present, almost no cleaved caspase-3 was detected in intestinal epithelial cells from TNF-treated occludin KO mice (Fig. 3H; Suppl. Fig. 4F). These data suggest that insufficient caspase-3 activation may be responsible for the apoptotic resistance of occludin KO epithelia. Because caspase-3 is a terminal, executioner caspase, this single defect induced by occludin deletion can explain resistance to both intrinsic and extrinsic pathway activation.
While the in vivo data are striking, they do not address whether the effects of occludin KO on intestinal epithelial apoptosis reflect interactions with other cell types, such as immune cells, or are cell autonomous. To assess this, occludin knockdown (KD) intestinal epithelial monolayers were tested for sensitivity to intrinsic and extrinsic pathway-induced apoptosis induced by staurosporine (STS) or TNF and cycloheximide (CHX), respectively (Suppl. Fig. 5). Both epithelial loss, as demonstrated by reduced nuclear density, and caspase-3 activation were limited in occludin KD monolayers (Suppl. Fig. 5). ERK and p38 MAP kinase activation were similar in occludin-sufficient and KD monolayers following STS or TNF treatment. Caspase-3 activity, indicated by cleavage of poly (ADP-ribose) polymerase (PARP), a caspase-3 substrate and key intermediate in apoptotic progression, was also significantly reduced in both STS- and TNF-treated occludin KD, relative to WT, monolayers. These data demonstrate that in vitro occludin KD accurately recapitulates the effects of occludin KO in vivo and suggest that this is due to reduced capase-3 activity. The results further indicate that occludin deficiency limits epithelial apoptosis in a cell autonomous manner.
To measure caspase activation directly, cytosolic extracts from occludin-sufficient and occludin-deficient epithelia were studied. In the absence of activating stimuli, caspases-8, −9, and −3 all displayed low protease activity that was quantitatively similar in WT and occludin KD extracts (Suppl. Fig. 6). Cytochrome C and dATP activated caspases-8 and −9 in all cells. In contrast, caspase-3 activity increased 12.0±1.6-fold in occludin-sufficient extracts but only 4.6±0.4-fold in cytosol of occludin-deficient epithelia. These data indicate that occludin depletion reduces caspase-3 activity and further support the hypothesis that occludin deficiency prevents apoptosis by suppressing caspase-3.
Occludin enhances transcription from the CASP3 promoter
Because the results point to a defect at the level of caspase-3 activation, we tested expression of genes that could affect caspase-3 activity downstream of caspase-8 and −9 activation. Intestinal epithelial caspase-3 protein and Casp3 mRNA expression were both markedly reduced in intestinal epithelia from occludin KO mice (Fig. 4A–B). This was specific for caspase-3, as caspase-8 and −9 expression were not affected by occludin KO (Fig. 3H, Suppl. Fig. 4D–F). Moreover, western blot analyses comparing expression of a panel of apoptosis-related proteins, including the inhibitor of apoptosis (IAP) family, pro- and anti-apoptotic bcl-2 proteins, and apoptosis-related mitochondrial proteins, failed to identify differences between WT and occludin KO epithelia (Fig. 4C). We also considered the possibility that occludin promotes caspase-3 expression by interacting with ZO-1, as ZO-1 is known to regulate gene expression via the ZO-1-associated nucleic acid binding protein (ZONAB)/DbpA.27 However, Casp3 transcription was indistinguishable in WT and intestinal-epithelial specific ZO-1 KO (ZO-1 KOIEC) mice28 (Suppl. Fig. 4G). These data indicate that occludin specifically promotes caspase-3 expression via a mechanism that is unrelated to ZO-1 and further suggests that reduced caspase-3 expression is the primary means by which occludin-deficiency limits apoptosis.
Figure 4. Occludin promotes CASP3 transcription.

A. Caspase-3 expression was reduced in jejunal epithelial cells isolated from occludin KO mice. Caspase-8, caspase-9, and E-cadherin expression were not affected (n = 3 per group).
B. qRT-PCR of jejunal epithelial cells from WT or occludin KO mice shows reduced Casp3 mRNA abundance in occludin KO mice (n = 5 per group).
C. Expression of apoptosis-related proteins was similar in jejunal epithelial cells from WT and occludin KO mice. Data shown are representative of n ≥ 3 per group.
D. Caspase-3 protein expression was reduced in occludin KD Caco-2BBe monolayers. Caspase-8, caspase-9, and E-cadherin expression were not affected (n = 3 per group).
E. qRT-PCR shows reduced CASP3 mRNA abundance in occludin KD, relative to WT, Caco-2BBe (n = 3 per group).
F. Luciferase reporter assay shows reduced activity of the CASP3 promoter in occludin KD compared to WT monolayers (n = 6 per group).
G. Induction of EGFP-occludin, but not EGFP, expression, increased CASP3 promoter activity in occludin KD Caco-2BBe (n = 5 per group).
H. Caco-2BBe were transfected with PGK promoter-driven luciferase expression plasmid containing the CASP3 3’ UTR. Luciferase activity was similar in WT and occludin KD monolayers (n ≥ 5 per group).
Statistical analyses by Student’s t-test or one-way ANOVA with Bonferroni post-test. *, P<0.05; **, P<0.01.
To determine whether occludin regulates CASP3 mRNA content via transcriptional activation we took advantage of apoptosis resistance in occludin KD Caco-2BBe intestinal epithelial cells (Suppl. Fig. 5). Western blot analyses confirmed that, like native epithelia, occludin loss in vitro reduced caspase-3, but not caspase-8 or −9, protein expression (Fig. 4D), indicating that, in this context, the in vitro model accurately recapitulates in vivo biology. CASP3 mRNA was markedly reduced in occludin KD relative to WT Caco-2BBe (Fig. 4E). To further analyze CASP3 transcriptional regulation, the human CASP3 promoter was used to drive luciferase expression in transfected Caco-2BBe cells. Luciferase activity in occludin KD cells was only 31 ±3% of that in occludin-sufficient cells (Fig. 4F). The specificity of this effect was confirmed by analyzing the activity of the same reporter after induction of EGFP or EGFP-occludin expression in occludin KD cells. Luciferase activity was significantly increased by induction of EGFP-occludin, but not by EGFP, expression, confirming occludin-dependent CASP3 promoter regulation (Fig. 4G). In contrast, a reporter construct containing the CASP3 3’UTR generated similar luciferase activity in occludin-deficient and occludin-sufficient epithelial monolayers (Fig. 4H). These data show that occludin expression results in increased CASP3 promoter activation and mRNA transcription.
Reduced caspase-3 expression is sufficient to confer apoptosis resistance
To determine if reduced caspase-3 expression was sufficient to explain the apoptotic resistance of occludin KO mice, we analyzed mice in which one Casp3 allele was deleted (Casp3+/−). Intestinal epithelial caspase-3 expression in Casp3+/− mice was reduced by 40% (Fig. 5A), similar to the reductions in occludin KO epithelia. Casp3+/− mice were also protected from DSS-induced apoptosis of colonic epithelia (Fig. 5B) and TNF-induced apoptosis of small intestinal epithelia (Fig. 5C). The effects of deleting one Casp3 allele are therefore similar to the effect of occludin KO in terms of intestinal caspase-3 protein expression and apoptotic sensitivity. While these data cannot be interpreted as indicating that reduced caspase-3 expression is the sole means by which occludin modulates apoptosis, they do show that the partial caspase-3 downregulation induced by occludin loss is sufficient to explain apoptotic resistance.
Figure 5. Partial loss of caspase-3 expression is sufficient to limit DSS-induced intestinal epithelial apoptosis.

A. Caspase-3 expression was reduced in colonic epithelia from Casp3+/− (+/−), relative to WT (+/+), mice (n = 5 per group). Intestinal epithelial caspase-3 expression was similar in in Casp3+/− and occludin KO mice (n = 3 per group).
B. TUNEL staining (red) shows DSS-induced epithelial apoptosis in colonic mucosa from Casp3+/−, relative to WT, mice. Bar = 100 μm (n = 4 per group).
C. Cleaved caspase-3 staining shows attenuated TNF-induced apoptosis of small intestinal epithelium of Casp3+/−, relative to WT, mice. Bar = 100 μm (n = 3 per group).
Littermates from Casp3+/− × Casp3+/− matings were used in these experiments Statistical analysis by one-way ANOVA with Bonferroni post test; **, P<0.01.
Caspase-3 expression is reduced in parallel with occludin loss in Crohn’s disease and ulcerative colitis
Intestinal epithelial occludin expression is reduced in Crohn’s disease and ulcerative colitis.22, 29, 30 If the model suggested by in vivo studies of mice and in vitro studies of human cell lines are correct, this should be associated with reduced caspase-3 expression. Quantitative morphometry of occludin and caspase-3 expression confirmed reduced epithelial occludin expression and demonstrated that this was associated with reduced caspase-3 expression in ileal biopsies from Crohn’s disease patients, relative to healthy control subjects (Fig. 6A–C). Similarly, epithelial occludin and caspase-3 expression were both reduced in colonic biopsies from ulcerative colitis patients (Fig. 6D, Suppl. Fig. 7A, B). Caspase-3 downregulation correlated directly with occludin downregulation in both forms of inflammatory bowel disease. These data suggest that occludin downregulation is the mechanism of reduced caspase-3 expression in human disease.
Figure 6. Ileal epithelia from Crohn’s disease patients demonstrate reduced expression of occludin and caspase-3.

A, B. H&E and multiplex immunohistochemical staining of ileal biopsies from normal subjects and Crohn’s disease patients. Expression of occludin and caspase-3 (green, as indicated) were reduced in Crohn’s disease. E-cadherin (red) and nuclei (blue) are shown for reference. Bars = 100 μm (A), 50 μm (B).
C. Quantitative morphometry of occludin, caspase-3, and E-cadherin staining intensity within the intestinal epithelium (n = 8 to 11).
D. Ileal epithelial caspase-3 expression correlated with occludin expression in biopsies from Crohn’s disease patients (r2 = 0.76).
Statistical analysis by Student’s t-test. *, P<0.05; **, P<0.01.
Proinflammatory cytokine treatment decreases occludin and caspase-3 expression to cause apoptosis-resistance
Previous studies have shown that prolonged, i.e., >20 h, TNF treatment reduces occludin transcription in HT29 intestinal epithelial cells.31 Consistent with this, treatment of WT Caco-2BBe monolayers with low-dose TNF (0.5 ng/ml) for 24 h reduced occludin expression by 32% (Fig. 7A, B) and was accompanied by a 41% reduction in caspase-3 expression. As a result, caspase-3 expression in TNF-treated WT monolayers was similar to that of untreated occludin KD epithelia (Fig. 7A, B). In contrast, low-dose TNF treatment did not affect caspase-3 expression in occludin-deficient monolayers (Fig. 7A, B). Cytokine-induced occludin downregulation is, therefore, sufficient to reduce caspase-3 expression in vitro in a manner similar to that observed in inflammatory bowel disease.
Figure 7. TNF-induced occludin downregulation prevents apoptosis of occludin-sufficient cells.

A, B. Low-dose (0.5 ng/ml) TNF treatment decreased occludin and caspase-3 expression in occludin-sufficient, but not occludin-deficient, cells.
C, D. TNF pre-treatment diminished apoptosis and cell loss induced by STS (C) or TNF and CHX. Cleaved caspase-3 (red) and nuclei (blue) are shown. Low-dose TNF pre-treatment did not affect occludin KD monolayers. Bar = 100 μm (n = 3 per group).
Statistical analysis by one-way ANOVA with Bonferroni post test; *, P<0.05; *, P<0.01.
We next assessed the effect of cytokine-induced caspase-3 downregulation on activation of intrinsic and extrinsic apoptotic pathways. After STS or high-dose TNF and CHX challenge, 23% and 25% of occludin-sufficient cells stained positively for cleaved caspase-3, respectively, whereas only 9% and 6% were positive after a protective low-dose TNF pre-treatment (Fig. 7C, D). This low level of apoptosis was similar to untreated occludin KD epithelia, which were not affected by low-dose TNF pretreatment (Fig. 7C, D). The data demonstrate that mild inflammatory stimuli that trigger occludin downregulation also downregulate caspase-3 and, in turn, confer resistance to intrinsic and extrinsic pathway apoptosis. The failure of low-dose TNF to affect caspase-3 expression or apoptotic sensitivity of occludin-deficient epithelia confirms that occludin is required for this mechanism of caspase-3 downregulation and apoptotic resistance. Cytokine-induced occludin downregulation may therefore be an adaptive process that enhances epithelial survival in the inflammatory environments.
Discussion
In vivo and in vitro studies have demonstrated that occludin loss is critical to epithelial tight junction leak pathway permeability increases induced by inflammatory stimuli. 10, 11, 14, 18, 25, 32 Nevertheless, the observation that occludin KO mice have essentially normal tight junction structure and barrier function15, 16 has caused many to question the importance of this protein. A possible explanation for the lack of intestinal defects in occludin KO mice could be compensation by tricellulin or MARVELD3, which together with occludin comprise the tight junction associated MARVEL protein (TAMP) family.33 We therefore asked if occludin KO mice would display an intestinal epithelial phenotype in response to stress.
We hypothesized that mild barrier defects might render occludin KO mice hypersensitive to injury, as has been shown in other mice lacking tight junction proteins.34, 35 Instead, occludin KO mice were markedly protected from DSS-induced chemical injury. We initially considered that this unexpected result might be explained by occludin loss in non-epithelial cells, such as intraepithelial lymphocytes.36 Transgenic occludin expression within intestinal epithelia was, however, able to complement universal occludin KO. Moreover, we developed intestinal epithelial-specific occludin KO (KOIEC) mice and found that they too were protected from both DSS-induced chemical damage and immune-mediated damage triggered by TNBS. Finally, in vitro studies using occludin KD intestinal epithelial cell lines confirmed that apoptotic resistance was a cell autonomous consequence of occludin loss.
To better characterize the mechanism by which occludin deletion confers apoptotic resistance, we challenged mice with prototypic activators of intrinsic and extrinsic apoptotic pathways. Occludin-deficient epithelia were protected from both, in vivo and in vitro. Events upstream of caspase-3 cleavage were excluded as potential causes of this resistance to apoptosis. Occludin depletion did, however, reduce CASP3 transcription, protein expression, and enzymatic activity. Consistent with this, cleavage of PARP, a proteolytic target of activated caspase-3, was reduced in occludin-deficient epithelia. Although these data suggested a mechanism for apoptotic resistance of occludin-deficient epithelium, we asked whether an ~50% reduction in caspase-3 expression was sufficient to limit apoptosis using Casp3+/− mice. Remarkably, these mice, in which caspase-3 expression was reduced by ~50%, were protected from apoptotic stimuli to the same extent as in occludin KO mice. These data demonstrate that the downregulation of capase-3 expression associated with occludin loss is sufficient to limit epithelial apoptosis. Occludin downregulation therefore limits apoptosis as a result of reduced CASP3 transcription.
Our studies are not the first to link occludin to transcriptional regulation. Previous work demonstrated that transformation of a salivary gland epithelial cell line, by transfection of oncogenic Raf-1, downregulated occludin and claudin-1 expression while inducing epithelial-mesenchymal transformation and disrupting monolayer growth in vitro.37, 38 Remarkably, transfection-induced occludin overexpression restored claudin-1 expression, reversed epithelial-mesenchymal transformation, and prevented in vivo growth of the transformed line.37, 38 The mechanisms by which occludin reversed neoplastic transformation were not defined, but the extent to which occludin protein expression was restored did correlate directly with claudin-1 mRNA content.37 Thus, in addition to our discovery of occludin-dependent caspase-3 transcription, occludin may also be a transcriptional activator of claudin-1 and, possibly, other genes that regulate epithelial-mesenchymal transformation.
In hepatocellular carcinoma cell lines, occludin overexpression has been reported to enhance caspase-3 expression and apoptotic sensitivity,39 i.e. the inverse of occludin deletion. Those studies linked occludin effects to the C-terminal coiled-coil domain, as an occludin splice variant lacking exon 9, which includes that domain, did not affect apoptosis.39 Interestingly, this region also includes the occludin-ELL (OCEL) domain.40, 41 ELL encodes an elongation factor that increases the catalytic rate of RNA polymerase II transcription; chromosomal translocations that create a bifunctional protein by fusing ELL to an RNA polymerase II, MLL, are common in acute myeloid leukemia.42 As a whole, these studies, our results, and data indicating that a C-terminal occludin fragment enhances epithelial apoptosis43 suggest that the occludin OCEL domain may regulate caspase-3 transcription.
We have previously reported that the occludin OCEL domain is essential for to TNF-induced reductions in occludin anchoring at the tight junction and subsequent endocytosis.10 While not fully-characterized, it is thought that this endocytosis accelerates degradation and contributes to reduced occludin expression in colitis.22 Our data indicate this occludin downregulation promotes mucosal homeostasis. How this effect interacts with the occludin loss-induced increases in leak pathway permeability, which has been hypothesized to exacerbate disease, remains to be defined.
Overall, our data indicate that inflammation-associated occludin downregulation leads to reduced caspase-3 expression and generalized apoptotic resistance. This may therefore represent an adaptive mechanism that limits epithelial damage in the context of disease. We further speculate that, in chronic inflammation, e.g., inflammatory bowel disease, this apoptotic insensitivity might also allow accumulation of cells with genetic defects and, in turn, facilitate neoplastic transformation. This hypothesis will need to be tested, but the results presented here demonstrate that occludin downregulation has beneficial effects. It will also be important for future studies to better define this new relationship between tight junction molecular structure and epithelial survival.
Materials and Methods
Mice
Occludin KO,15 intestinal epithelial specific EGFP-occludin transgenic,18 caspase-3 KO,44 and ZO-1 KOIEC28 mice have been described. Occludin KOIEC mice were generated using OClntm1a(EUCOMM)wtsi embryonic stem cells to create C57BL/6J-Oclnf/f mice, which were crossed with villin-Cre45 mice. Littermate Oclnf/f lacking Cre were used as wild type controls in experiments with occludin KOIEC mice.
Further experimental details are available in the Supplemental Methods.
Supplementary Material
What you need to know.
BACKGROUND AND CONTEXT
Loss of the intestinal barrier in patients with inflammatory bowel diseases (IBD) is associated with reduced expression of the tight junction protein occludin. We investigated the function of occludin in cell lines, mice, and patient biopsies.
NEW FINDINGS
Occludin increases expression of caspase 3, which induces apoptosis. Mice that lack occludin in the intestinal epithelium are protected from colitis via reduced caspase 3 and apoptosis. Biopsies from patients with IBD have reduced occludin, which correlates with lower levels of caspase 3.
LIMITATIONS
These studies were performed in mice, cell lines, and human tissue samples. Studies are needed to determine mechanisms by which occludin regulates transcription.
IMPACT
Occludin downregulation by inflammatory stimuli might be a mechanism of epithelial preservation.
Acknowledgements
We would like to thank the Brigham and Women’s Hospital Pathology Core and University of Chicago Human Tissue Resource Center for their expert assistance, Linda Degenstein and the University of Chicago Transgenic Core for assistance with development of occludin KOIEC mice, Margaret Neville (University of Colorado) and Sachiko Tsukita (Osaka University, Japan) for providing occludin KO mice, and Mingtao Li (Sun Yat-sen University, China) for the human CASP3 promoter-containing vector. This work was supported by NIH grants R01DK61931 (JRT), R01DK68271 (JRT), R24DK099803 (JRT), K01DK092381 (LS), and the Harvard Digestive Disease Center (P30DK034854) and by National Science Foundation of China grant 81800464 (LZ).
Footnotes
The authors have declared that no conflicts of interest exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993; 123:1777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furuse M, Fujimoto K, Sato N, et al. Overexpression of occludin, a tight junction-associated integral membrane protein, induces the formation of intracellular multilamellar bodies bearing tight junction-like structures. J Cell Sci 1996; 109:429–35. [DOI] [PubMed] [Google Scholar]
- 3.Fanning AS, Jameson BJ, Jesaitis LA, et al. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 1998; 273:29745–53. [DOI] [PubMed] [Google Scholar]
- 4.Bolinger MT, Ramshekar A, Waldschmidt HV, et al. Occludin S471 Phosphorylation Contributes to Epithelial Monolayer Maturation. Mol Cell Biol 2016; 36:2051–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elias BC, Suzuki T, Seth A, et al. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J Biol Chem 2009; 284:1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raleigh DR, Boe DM, Yu D, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol 2011; 193:565–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong V Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am J Physiol 1997; 273:C1859–67. [DOI] [PubMed] [Google Scholar]
- 8.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol 1997; 136:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu AS, McCarthy KM, Francis SA, et al. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol 2005; 288:C1231–41. [DOI] [PubMed] [Google Scholar]
- 10.Buschmann MM, Shen L, Rajapakse H, et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol Biol Cell 2013; 24:3056–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Itallie CM, Fanning AS, Holmes J, et al. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci 2010; 123:2844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balda MS, Whitney JA, Flores C, et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol 1996; 134:1031–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell 2005; 16:3919–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz BT, Wang F, Shen L, et al. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 2007; 132:2383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitou M, Furuse M, Sasaki H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000; 11:4131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulzke JD, Gitter AH, Mankertz J, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta 2005; 1669:34–42. [DOI] [PubMed] [Google Scholar]
- 17.Kitajiri SI, Katsuno T, Sasaki H, et al. Deafness in occludin-deficient mice with dislocation of tricellulin and progressive apoptosis of the hair cells. Biol Open 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchiando AM, Shen L, Graham WV, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol 2010; 189:111–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrary E, Cohen-Tannoudji M, Pehau-Arnaudet G, et al. In vivo, villin is required for Ca(2+)-dependent F-actin disruption in intestinal brush borders. J Cell Biol 1999; 146:819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinkai Y, Rathbun G, Lam KP, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992; 68:855–67. [DOI] [PubMed] [Google Scholar]
- 21.Ma A, Datta M, Margosian E, et al. T cells, but not B cells, are required for bowel inflammation in interleukin 2-deficient mice. J Exp Med 1995; 182:1567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005; 129:550–64. [DOI] [PubMed] [Google Scholar]
- 23.Pinto D, Robine S, Jaisser F, et al. Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J Biol Chem 1999; 274:6476–82. [DOI] [PubMed] [Google Scholar]
- 24.Wirtz S, Neufert C, Weigmann B, et al. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2007; 2:541–6. [DOI] [PubMed] [Google Scholar]
- 25.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest 2005; 115:2702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vyas D, Robertson CM, Stromberg PE, et al. Epithelial apoptosis in mechanistically distinct methods of injury in the murine small intestine. Histol Histopathol 2007; 22:623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nie M, Balda MS, Matter K. Stress- and Rho-activated ZO-1-associated nucleic acid binding protein binding to p21 mRNA mediates stabilization, translation, and cell survival. Proc Natl Acad Sci U S A 2012; 109:10897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Odenwald MA, Choi W, Kuo WT, et al. The scaffolding protein ZO-1 coordinates actomyosin and epithelial apical specializations in vitro and in vivo. J Biol Chem 2018; 293:17317–17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007; 56:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kucharzik T, Walsh SV, Chen J, et al. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol 2001; 159:2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mankertz J, Tavalali S, Schmitz H, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci 2000; 113:2085–90. [DOI] [PubMed] [Google Scholar]
- 32.Mir H, Meena AS, Chaudhry KK, et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim Biophys Acta 2016; 1860:765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raleigh DR, Marchiando AM, Zhang Y, et al. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell 2010; 21:1200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 2007; 204:3067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vetrano S, Rescigno M, Cera MR, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 2008; 135:173–84. [DOI] [PubMed] [Google Scholar]
- 36.Edelblum KL, Shen L, Weber CR, et al. Dynamic migration of gammadelta intraepithelial lymphocytes requires occludin. Proc Natl Acad Sci U S A 2012; 109:7097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Mrsny RJ. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J Cell Biol 2000; 148:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Mandell KJ, Parkos CA, et al. The second loop of occludin is required for suppression of Raf1-induced tumor growth. Oncogene 2005; 24:4412–20. [DOI] [PubMed] [Google Scholar]
- 39.Gu JM, Lim SO, Park YM, et al. A novel splice variant of occludin deleted in exon 9 and its role in cell apoptosis and invasion. FEBS J 2008; 275:3145–56. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai K, Michiue T, Kikuchi A, et al. Inhibition of the canonical Wnt signaling pathway in cytoplasm: a novel property of the carboxyl terminal domains of two Xenopus ELL genes. Zoolog Sci 2004; 21:407–16. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Fanning AS, Anderson JM, et al. Structure of the conserved cytoplasmic C-terminal domain of occludin: identification of the ZO-1 binding surface. J Mol Biol 2005; 352:151–64. [DOI] [PubMed] [Google Scholar]
- 42.Shilatifard A, Lane WS, Jackson KW, et al. An RNA polymerase II elongation factor encoded by the human ELL gene. Science 1996; 271:1873–6. [DOI] [PubMed] [Google Scholar]
- 43.Beeman NE, Baumgartner HK, Webb PG, et al. Disruption of occludin function in polarized epithelial cells activates the extrinsic pathway of apoptosis leading to cell extrusion without loss of transepithelial resistance. BMC Cell Biol 2009; 10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuida K, Zheng TS, Na S, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996; 384:368. [DOI] [PubMed] [Google Scholar]
- 45.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 2004; 39:186–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.