

Abstract
The protozoan Trypanosoma cruzi is the causative agent of Chagas disease, endemic in Latin America but present worldwide. Research efforts have focused on the examination of immune mechanisms that mediate host-protection as well as immunopathology during this parasitic infection. The study of CD8+ T cell immunity emerges as a key aspect given the critical importance of parasite-specific CD8+ T cells for host resistance throughout the infection. In the last years, new research has shed light about novel pathways that modulate the induction, maintenance and regulation of CD8+ T cell responses to T. cruzi. This new knowledge is setting the ground for future vaccines and/or immunotherapies. Herein, we critically review and analyze the latest results published in the field.
Keywords: Trypanosoma cruzi, CD8+ T cells, Chagas disease
CD8+ T cell immunity against Chagas disease
Infection with the protozoan T. cruzi causes Chagas disease whose progression, from symptomless to severe, is linked to parasite heterogeneity and a variable host immune response (Box 1). In particular, pathogen-specific effector CD8+ T cells are critical for resistance to protozoa as well as to infections with most intracellular microbes [1]. Accordingly, initial studies about the immune response triggered by T. cruzi demonstrated that depletion of CD8+ T cells [2] or deficiency in the β2-microglobulin [3] favors parasite replication and increases host susceptibility during the acute phase of the experimental infection. Also, early evidences indicated that expansion and effector function of CD8+ T cells are required along the entire infection to control parasite load and prevent excessive inflammation in hearts of chronically infected mice [4]. These grounding reports established the concept that CD8+ T cell immunity is critical for survival during acute T. cruzi infection and kicked-off several research lines aimed to understand the dynamics of that response. In the following sections, we review the latest results about CD8+ T cell immunity to T. cruzi together with fundamental knowledge in the field to integrate the available information into a comprehensive picture.
Box1. Chagas disease - epidemiology, transmission and pathology.
Chagas disease (American Trypanosomiasis) is a life-threatening illness caused by the protozoan parasite Trypanosoma cruzi [96]. Last estimates calculated an infected population of about 6 million in Latin America, with more than 70 million people living at risk of infection and 40000 new cases diagnosed per year [97]. Modern migration has led to Chagas disease spreading beyond endemic areas, becoming a global public health concern [98].
In areas where Chagas disease is common, the main way of transmission is vector-borne, through blood-sucking insects of the triatomine family. Other routes of T. cruzi transmission include blood transfusion, transplantation, consumption of contaminated food or vertical transmission (from mother to fetus). When vectorially acquired, Chagas disease has two major phases: acute and chronic. Severe acute disease occurs in less than 5% of patients and around 30-40% of the chronically infected people can develop cardiac, digestive, neurological or mixed alterations. Chronic chagasic cardiomyopathy (CCC) is the most serious manifestation of the chronic form of Chagas disease and constitutes the most common type of infectious myocarditis in the world [99]. In addition to CCC, skeletal muscle alterations such as myositis, vasculitis, atrophy and necrosis of myofibrils may be responsible for the physical dysfunction of patients with severe chronic Chagas disease [100]. Although much less studied, adipose tissue is also an important target tissue of T. cruzi and its infection is associated with a profound impact on systemic metabolism, increasing the risk of metabolic syndrome [101].
It is generally accepted that parasite persistence and chronic inflammation play an important role in host tissue damage [102]. In the setting of a chronic infection, a balance exists between immune activation that controls parasite replication, and immune suppression, which prevents immunopathology. Despite many decades of research on the subject, the infection remains incurable, and the factors that steer chronic Chagas disease from an asymptomatic state to clinical onset are still unclear.
General features of CD8+ T cell responses during experimental T. cruzi infection
As described for model CD8+ T cell responses (Box 2), a robust parasite-specific CD8+ T cell immunity emerges upon natural T. cruzi infection but it shows a delayed kinetics when compared to other microbial infections [5, 6]. Of note, this response is extremely focused on a few immunodominant peptides derived from surface parasite proteins such as trans-sialidase (TS) and amastigote surface protein 2 (ASP2) that exhibit considerable intra and inter-strain variability in sequence and expression pattern. Indeed, the immunodominance (see Glossary) pattern is particular to each parasite strain [7, 8]. Immunodominance has been suggested as detrimental to the host by restricting the breadth, and therefore the effectiveness, of the anti-parasitic CD8+ T cell response. However, experimental manipulations to eliminate CD8+ T cells specific for immunodominant epitopes highlighted the plasticity of the T. cruzi-specific CD8+ T cell repertoire and demonstrated that immunodominance neither contribute to, nor detract from, the ability to control T. cruzi infection [9, 10].
Box 2. Development of CD8+ T cell responses during acute versus chronic infections.
Adaptive immune responses consist of distinct phases: antigen recognition and activation of lymphocytes (the induction phase) followed by elimination of the pathogen (the effector phase). Afterwards, the immune response contracts as antigen-stimulated lymphocytes die by apoptosis, restoring homeostasis. Few antigen-specific cells survive and become long-lived cells responsible of the immunological memory. The duration of each phase may vary in immune responses triggered by different challenges.
During acute infection or following vaccination, antigen-specific naïve CD8+ T cells undergo robust proliferation and clonal expansion to differentiate into an effector population that includes KLRG1hi CD127lo short-lived effector cells and KLRG1lo CD127hi memory precursor cells. Effector T cell differentiation is accompanied by transcriptional, epigenetic, and metabolic reprogramming, with the acquisition of hallmark effector features such as the ability to produce cytokines and cytotoxic molecules. Following antigen clearance and resolution of inflammation, the CD8+ T cell response suffers a contraction in which the majority of activated effector T cells die. A small subset, however, persists and differentiates into memory T (Tmem) cells. Tmem cells downregulate their effector program and acquire a stem cell–like ability to survive in an antigen-independent fashion as long-lived cells that undergo slow homeostatic self-renewal driven by IL-7 and IL-15 [103]. CD8+ Tmem cells retain the ability to re-expand upon secondary antigen encounter, resulting in an anamnestic response that controls the infection more rapidly than during the primary response [104]. In the chronic infection, antigen-specific naïve CD8+ T cells differentiate into an effector T cell population similar to that observed following acutely resolved antigen encounter. However, antigen persistence and the consequently sustained inflammatory microenvironment drive cell exhaustion, a phenomenon in which pathogen-specific T cells gradually loose effector function [23]. Exhausted T cells arise from the KLRG110 CD127hi subset, and therefore share certain features with memory T cells [105]. To date, exhausted T cells have been described in the context of chronic infections (and other chronic pathologies) in mice and humans [106-110].
Once effector immunity clears the circulating parasites and reduces parasite loads in tissues, T. cruzi specific CD8+ T cells acquire memory traits, exhibiting proliferative responses after stimulation with both parasite antigens and homeostatic cytokines [11, 12]. Remarkably, specific CD8+ T cells isolated during the chronic phase of experimental T. cruzi infection exhibit effector competence and critically contribute to the persistent control of parasite outgrowth [13]. Altogether, the data obtained using experimental infection models in immunocompetent hosts (summarized in Supplementary Table S1) demonstrated that CD8+ T cell immunity against T. cruzi is robust and critical for parasite control during the acute phase. Furthermore, it generates immunological memory and remains functional to restrain parasite replication even in the context of chronic T. cruzi persistence.
The efficiency of the natural CD8+ T cell response may be interpreted as discouraging for strategies aimed to enhance or manipulate CD8+ T cell immunity as a rational approach to further improve resistance to T. cruzi. However, global analysis of results obtained using mice that bear defects in immune pathways that results in altered CD8+ T cell immunity, together with data obtained with samples of Chagas disease patients, support alternative interpretations discussed along this review.
CD8+ T cell immunity in human Chagas Disease
Although restricted by the inherent limitations of studies in humans, the investigation of the CD8+ T cell immunity in patients with Chagas disease underscored some similarities with the response described in experimental infection settings (Table 1). In this regard, the relevance of CD8+ T cell immunity in human T. cruzi infection is well illustrated by a recent transcriptomics study [14]. It describes that patients with whole blood transcriptional signatures enriched in genes related to NK/CD8+ T cell cytotoxicity exhibit reduced parasitism as well as less severe chagasic chronic cardiomyopathy. Furthermore, though immunodominance of particular epitopes was not as obvious as in mice, CD8+ T cells from T. cruzi infected individuals also recognize TS derived peptides [7, 15] and few other parasite epitopes [16-18]. On the other hand, different from responses in mice that remain functional in chronicity, CD8+ T cell responses in patients with chronic Chagas disease show several evidences of dysfunction that were associated with the clinical severity of the disease (Table 1). Early studies reported that an important proportion of memory CD8+ T cells from T. cruzi infected patients exhibit a phenotype of terminal differentiation, likely associated with chronic activation [19]. These cells are characterized by downregulation of CD28 and CD27, increased susceptibility to apoptosis and reduced effector function upon stimulation with parasite antigens. Furthermore, the proportion of this senescent effector CD8+ T cell subset is increased while CD8+ T cells with features of stem cell memory are diminished in patients with more severe chronic disease [20]. The differential distribution of “bulk” CD8+ T cell subsets in patients with different grades of cardiomyopathy was further confirmed at clonal level. Thus, CD8+ T cells able to recognize four parasite epitopes restricted to the HLA-A*02:01 molecule display naive traits in non-symptomatic patients but a terminal effector and senescent phenotype in patients with cardiac symptoms [21]. Concomitant with terminal differentiation, CD8+ T cells from patients with severe disease have a higher frequency of cells co-expressing inhibitory receptors such as PD-1, CTLA-4, 2B4, CD160, and TIM-3, and a lower frequency of polyfunctional parasite-specific CD8+ T cells compared with patients without symptoms or with mild disease [22]. These features resemble those of dysfunctional or exhausted T cells that arise in the context of chronic viral infections [23] (Box 2). Indeed, chronic parasite persistence arises as a possible cause of CD8+ T cell dysfunction given that antiparasitic treatment in asymptomatic patients improves the quality of antigen-specific CD8+ T cell responses associated with a decrease in inhibitory receptor co-expression [24]. In addition to sustained antigenic stimulation, chronic exposure to inflammatory mediators such as nitric oxide may also contribute to the dysfunctional state of CD8+ T cells from infected patients. The mechanism underlying this effect seems to be the nitration of surface T cell proteins that leads to T cell unresponsiveness and apoptosis [25]. In a similar direction, perturbed signaling downstream the IL-7 receptor, which is critical for T cell survival, has been suggested as one cell-intrinsic mechanism of CD8+ (and CD4+) T cell exhaustion during chronic Chagas disease [26].
Table 1.
Reported similarities and differences of CD8+ T cell immunity in mouse versus humans
| Mouse | Human | ||
|---|---|---|---|
| Similarities | Relevance | CD8+ T cell depletion during acute [2] and chronic stages [13] as well as b2-microglobulin deficiency [3] favor parasite replication and increase host susceptibility. | Patients with whole blood transcriptional signatures enriched in genes related to NK/CD8+ T cell cytotoxicity exhibit reduced parasitism as well as less severe chagasic chronic cardiomyopathy [14]. |
| Immunodominance | Parasite-specific CD8+ T cell response is focused on few immunodominant peptides derived from trans-sialidase and amastigote surface protein 2 [7, 8]. The immunodominance pattern is particular to each parasite strain [7] | Immunodominance not as obvious as in mice but CD8+ T cells from T. cruzi infected individuals recognize TS derived peptides [7, 15] and few other parasite epitopes [16-18]. | |
| Differences | Differentiation status | After initial control of parasite replication, T. cruzi specific CD8+ T cells acquire memory traits and proliferate after stimulation with parasite antigens and homeostatic cytokines [11, 12]. | A proportion of memory CD8+ T cells from T. cruzi infected patients exhibit an immunosenescent phenotype with CD28 and CD27 downregulation and increased susceptibility to apoptosis [19]. The frequency of this cell subset is increased while CD8+T cells with stem cell memory phenotype are diminished in patients with more severe chronic disease [20, 21]. |
| Functionality | Specific CD8+ T cells isolated from chronically infected WT mice exhibit little expression of inhibitory receptors such as PD-1, Lag3 and Tim3, are functional competent and critically contribute to the persistent control of parasite outgrowth [13] | Compared with patients without symptoms or with mild disease, patients with severe disease have CD8+ T cells characterized by co-expression of inhibitory receptors such as PD-1, CTLA-4, 2B4, CD160, and TIM-3, a perturbed IL-7R signaling [26] and reduced poly-functional effector response [22, 26]. Antigen-specific CD8+ T cell functionality is partially restored by conventional antiparasitic treatment [24]. |
The reasons of the difference between experimental and human T. cruzi infection concerning the differentiation status and functionality of CD8+ T cells remain unclear. One possible explanation is that the longer life span of humans may allow longer chronic infectious processes, promoting cell exhaustion. Alternatively, given the heterogeneity of host responses in humans compared to laboratory animals, it is possible that particular immune effector pathways and/or different levels of parasite loads may promote CD8+ T cell dysfunction in certain patients but not others. In this regard, it is critical to continue our efforts to completely understand the cellular and molecular mechanisms underlying the induction and maintenance of protective CD8+ T cell immunity against T. cruzi. Taking appropriate note of their inherent limitations, studies exploiting models of experimental infections in genetically modified mice could certainly be instrumental in this direction.
Pathways that participate in the induction of parasite specific CD8+ T cells
Initial activation and priming are the less studied phases of the CD8+ T cell response to T. cruzi. Our current picture is based only on few studies that focus on particular aspects of priming and induction during experimental infections (Figure 1, Key Figure). Therefore, a systematic evaluation of these initial and critical steps is still missing.
Figure 1, Key Figure. Priming and maintenance of T. cruzi CD8+ T cell response.
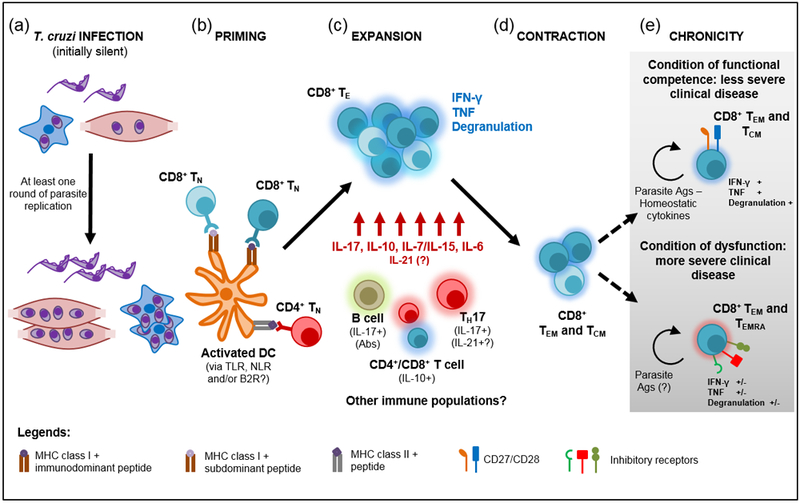
a) T. cruzi infects different cell types within the host but at least one round of parasite replication is necessary to allow the accumulation of sufficient amounts of parasite antigens (Ags) and ligands able to activate antigen presenting cells with T cell priming ability. Dendritic cells (DC) recognize T. cruzi through ligation of toll-like receptors (TLR) that are nevertheless not essential for CD8+ T cell priming. Alternatively, Nod-like receptors (NLR) or bradykinin 2 receptors (B2R) may be involved in this process. b) Activated DCs prime naïve CD8+ T cells (TN) that recognizes immunodominant as well as subdominant parasite epitopes. CD4+ T cell help seems to be mainly required in the induction of CD8+ T cells specific for immunodominant peptides. c) Upon priming, there is a robust expansion of parasite-specific effector CD8+ T cells (TE) that show polyfunctional effector response and are critical for the control of parasite replication. TE CD8+ T cell survival and effector function is sustained by particular cytokines and soluble mediators produced by different cell subsets. d) After the limitation of parasite burden, the CD8+ T cell response contracts and gives rise to effector memory (TEM) and central memory (TCM) CD8+ T cells that persist during the chronic phase. e) Although memory CD8+ T cells remains functional to limit parasite outgrowth, at least two scenarios have been reported in the chronic phase in relation to CD8+ T cell phenotype and functional competence.
T. cruzi recognition by the immune system relies on parasite molecules able to ligate receptors expressed by innate immune cells such as dendritic cells and macrophages, particularly toll-like receptors (TLR). Identified T. cruzi TLR ligads include mucin-like glycoproteins that bind TLR2 and 6, glycoinositolphospholipids that are recognized by TLR4, and parasite DNA (which contains abundant oligodeoxynucleotide unmethylated CpG motifs) and total RNA that potently activate TLR9 and 7, respectively [27]. Remarkably, the initial steps of T. cruzi infection seem to be relatively silent and therefore, the primary parasite inoculum does not promote the induction of parasite-specific CD8+ T cells [6, 28]. Rather, effector CD8+ T cells emerge with slow kinetics only after a round of parasite multiplication that likely results in the accumulation of T. cruzi antigens and ligands with adjuvant properties that, in turn, promote the maturation of antigen presenting cells. Accordingly, injection of irradiated (non-proliferating) parasites is not able to induce protective CD8+ T cell immunity while higher parasite inoculum or infection with a fast replicating parasite strain accelerate T. cruzi parasitemia and expansion of parasite-specific CD8+ T cells [6, 28]. In the same line, the kinetics of the CD8+ T cell response to a moderate initial T. cruzi infection dose can be speeded by the injection of different TLR ligands that would act as adjuvants [6]. Strikingly, however, specific CD8+ T cell immunity triggered by natural T. cruzi infection is preserved in mice deficient for TLR2, TLR4, TLR9 or the TLR adaptor molecule MyD88 despite the increased susceptibility of these mouse strains [29]. Thus, it is likely that TLR-dependent pathways are dispensable for adaptive CD8+ T cell immunity to T. cruzi or their contribution can be compensated by other innate recognition systems such as Nod-like receptors [30] or bradykinin B2 receptors [31] able to drive innate immune cell activation.
Besides requiring the activation of antigen presenting cells, provision of MHC class I-restricted epitopes for T cell priming critically depends on the cytosolic degradation of mature proteins by a specialized type of proteasome called immunoproteasome [32]. In fact, CD8+ T cell responses to many viral infections show diminished magnitude or quality (immunodominance pattern) in the absence of immunoproteasome expression [32]. Similarly, T. cruzi infected dendritic cells derived from bone marrow of mice lacking the expression of the three immunoproteasome subunits exhibit reduced antigen presentation of MHC class I-restricted parasite epitopes and are less efficient to activate IFN-γ production by CD8+ T cells purified from infected mice [33]. Furthermore, immunoproteasome deficient-mice present a drastically diminished response of CD8+ T cells specific for immunodominant and subdominant epitopes after T. cruzi infection together with a conserved CD4+ T cell response. Accordingly, these infected mice showed higher tissue parasitism and increased susceptibility to this parasitic infection.
CD4+ T cells are essential for the development of primary and, specially, memory CD8+ T cell responses in many settings including infection and cancer [34]. However, the role of CD4+ T cell help in the development of T. cruzi specific CD8+ T cell responses has been scarcely investigated. An initial report described that mice lacking CD4+ T cells due to MHC class II deficiency generate functional (IFN-γ producing and cytotoxic) parasite-specific CD8+ T cells after T. cruzi infection [35]. However, these infected CD4+ T cell-deficient mice exhibit a significant decrease in the frequency of CD8+ T cells recognizing dominant but not subdominant parasite epitopes [35]. In contrast, a second study reported that MHC clas II and CD4 deficient mice developed marginal levels of specific cytotoxic activity in vivo after infection with a high dose of a different T. cruzi strain [28]. Although apparently contradictory, these findings could be reconciled by the idea that CD4+ T cell helps to modulate strain-specific immunodominance patterns during primary CD8+ T cell response against T. cruzi, licensing dendritic cells to prime maximal response mainly to dominant epitopes, as previously reported for viral infections [36, 37].
Once primed, T. cruzi-specific CD8+ T cells will continue with the next steps of a conventional T cell response (Box 2), which includes expansion concomitantly with effector cell differentiation, followed by contraction and memory generation (Figure 1). Several signals provided by soluble mediators and cell surface molecules will influence the development of these steps in the CD8+ T cell fate, as discussed below.
Mediators that sustain the maintenance of CD8+ T cell immunity to T. cruzi
The signals that regulate the expansion and survival of effector CD8+ T cells and promote the generation of memory CD8+ T cells have not been completely elucidated. Nevertheless, some cytokines and immune cell populations have been identified as involved in the maintenance of sustained CD8+ T cell response during T. cruzi infection (Figure 1).
Cytokines: IL-10, IL-17, and others
Recently, we demonstrated that mice deficient in IL-17RA show an abortive CD8+ T cell response during T. cruzi infection [38]. This phenotype is not a consequence of a reduced induction or expansion of parasite-specific CD8+ T cells but rather evidenced a premature contraction. Remarkably, IL-17 signaling is required once the parasite-specific CD8+ T cell response is established to promote survival of effector cells. Accordingly, IL-17A, but not IL-17F, IL-17C or IL-17E, is able to stimulate in vitro activated CD8+ T in cells in a direct fashion, downregulating the pro-apoptotic protein BAD and promoting cell survival. Furthermore, effector CD8+ T cells elicited by T. cruzi infection in absence of IL-17RA exhibit a phenotypic, functional and transcriptomic profile compatible with cell exhaustion. In agreement with their deficient CD8+ T cell response, infected IL-17RA knockout mice show poor control of the parasite in target tissues such as spleen, liver and heart that can be partially reverted by inhibiting the PD-1/PD-L1 inhibitory pathway [38]. Altogether, our results underscore that during T. cruzi infection, cell populations that produce IL-17, which include Th17, Tc17, NK cells and B cells [39, 40]. may sustain and potentiate parasite-specific CD8+ T cell responses.
Additionally, IL-10 has been recently shown to modulate CD8+ T cell responses during T. cruzi infection [41]. Initial evidences in this regard came from the observation that infected C57BL/6J mice exhibit an exceptional high IL-10 expression that was associated with increased frequency of cytokine-producing CD8+ T cells in infected hearts [42]. Likewise, recent data show that IL-10 deficient mice exhibit an impaired expansion of CD8+ T cells following acute T. cruzi infection, confirming the participation of IL-10 in the sustenance of CD8+ T cell immunity in this inflammatory context [41]. CD8+ T cells from infected IL-10 deficient mice show diminished proliferation, cytotoxic potential and IFN-γ production in comparison to their WT counterparts. Furthermore, IL-10 absence selectively affects survival and increases the expression of the inhibitory receptor PD-1 on CD8+ T cells without altering these parameters on CD4+ T cells. The effects of IL-10 on CD8+ T cells from T. cruzi infected mice are not achieved through direct signaling as recombinant IL-10 failed to up-regulate CD8+ T cell function in vitro.
Besides IL-17 and IL-10, few other cytokines have been investigated in their role to sustain CD8+ T cell responses in the context of T. cruzi infection. In this regard, human CD8+ T cell lines derived from inflammatory heart infiltrates show enhanced survival and expansion in the presence of IL-7 and IL-15 [43]. Indeed, increased local production of both cytokines is associated with the predominant presence of CD8+ T cells in heart biopsies of patients with Chagas disease. In addition, IL-6, which is induced during experimental [44] and human [45] T. cruzi infection, promote the survival of human CD8+ T cells from chagasic patients [25]. As discussed above, peripheral leukocytes from chagasic patients present increased tyrosine nitration of CD8+ T cells that leads to increased apoptotic rate, loss of the TCRζ-chain, and reduced effector function. IL-6 stimulation of peripheral blood mononuclear cells obtained from healthy donors and infected in vitro with T. cruzi blunts parasite-induced nitration and increases survival of CD8+T cells. In contrast, and despite the fact that type I interferons are evident early after T. cruzi infection, infected mice lacking the receptor for type I interferon showed conserved frequencies of immunodominant TSKB20- and subdominant TSKB18-specific CD8+ T cells [46].
Cells: B lymphocytes and Th17 cells
There is scarce knowledge about the cells and signals able to sustain CD8+ T cell response in T. cruzi infection once established. However, B cells were one of the first cell populations reported to participate in the generation of effector/memory CD4+ and CD8+ T cells during T. cruzi infection [47]. Infected muMT mice, wich lack mature B cells due to absence of surface-IgM expression, exhibit low CD8+ T cell numbers. Unfortunately, the absence of a kinetics evaluation in this study does not allow to definitively conclude whether the reduced magnitude of the responses a consequence of deficient maintenance or induction of effector T cells. More recently, we and others described that B cells are dispensable for the priming but not for the maintenance of CD8+ T cell responses against T. cruzi [48, 49], although the underlying mechanisms were found to be different. In this way, JhD mice, which have a deletion in the immunoglobulin heavy chain locus that results in lack of functional B cells, show a reduction in parasite-specific CD8+ T cell numbers and effector function after mucosal vaccination followed by challenge with a virulent T. cruzi strain. This phenotype can be reversed by injection with T. cruzi immune serum obtained from WT mice, underscoring a role for antibodies in the B cell mediated modulation of CD8+ T cell immunity [49]. In a different direction, we determined that B cell depletion by anti-CD20 injection during T. cruzi infection affects the magnitude and quality of the specific CD8+ T cell response. This effect is associated with a reduction in the frequency of IL-17A producing B and non-B cell cell populations. Also, parasite-specific CD8+ T cells from B cell depleted infected mice exhibit increased apoptosis and poor effector function, a phenotype similar to that observed in infected IL-17RA deficient mice. Furthermore, B cell depletion partially arrested CD8+ T cell expansion, leading to a premature contraction of the response. Of note, treatment with rIL-17A partially restored CD8+ T cell immunity and parasite control in anti-CD20-treated T. cruzi infected mice [48]. Our results highlight important cytokine-dependent (possibly antibody-independent) mechanisms of immunomodulation exerted by B cells, reinforcing the notion that IL-17 is a key cytokine for the sustenance of CD8+ T cell immunity during T. cruzi infection.
Besides B cells, TCR-transgenic CD4+ T cells specific for an immunodominant peptide of T. cruzi and polarized in vitro into Th17 cells were shown to potentiate CD8+ T cell immunity when co-transferred with polyclonal CD8+ T cells into infected RAG KO mice. Indeed, these Th17 cells augment CD8+ T cell proliferation and cytokine production and confer a stronger protection against T. cruzi-related mortality compared to Th1 cells [50]. Remarkably, these parasite-specific Th17 cells acts through a mechanism that is independent of IL17 but IL-21-dependent [50]. The differences between these findings and our results with IL-17RA deficient mice [38] may arise from the notion that in vitro generated Th17 cells evaluated by Cai et al. [50] may have different phenotype and functional capacity than those generated in vivo during the natural infection. Despite this, Th17 cells are a poorly represented T helper subset during T. cruzi infection and thus other IL-17 producing cell populations may play a more significant role to sustain CD8+ T cell immunity [39, 40]. In the end, it is likely that robust parasite-specific CD8+ T cell responses rely on a crosstalk among several cell subsets able to secrete different mediators including IL-17 and IL-21, as well as other cytokines.
Regulation of CD8+ T cell responses
Immunoregulatory pathways are fundamental for host resistance to T. cruzi as they orchestrate balanced effector immune responses able to achieve parasite control without extensive tissue damage [51]. The characteristics of different suppressor cell populations and anti-inflammatory cytokine responses during T. cruzi infection have been recently reviewed [52, 53]. In particular, the role of Foxp3+ regulatory T (Treg) cells have been extensively studied under different T. cruzi infection models, where contradictory results have been reported. These discrepancies were attributed to the variety of parasite and mouse strains used as well as the infection dose [53], but they also might be consequence of limitations in Treg cell approaching strategies, the diversity of parameters studied, the tissues analyzed and the focus on different stages of the infection. In this way, most studies targeted Treg cells by the use of anti-CD25 depleting antibodies and few of them investigated its impact on CD8+ T cell immunity towards T. cruzi. Initial studies concluded a limited role for Treg cells during T. cruzi infection as a slight effect is observed in the frequency of antigen-specific CD8+ T cells in blood of Treg cell-depleted mice at the acute phase of infection, without affecting parasitemia levels and survival curves [54]. Additionally, the functionality of CD8+ T cells remains unaltered in the acute and/or chronic infection of Treg cell-depleted mice, and even after long periods of anti-CD25 treatment. Other groups reported no effects on CD8+ T cell responses following Treg cell depletion, however in these cases the analysis of the cytotoxic response was very limited [55-57].
The use of strategies that specifically target Foxp3+ Treg cells have recently shed light in understanding the impact of this population over CD8+ T cell immunity after T. cruzi infection. In this way, specific-Treg cell depletion immediately after T. cruzi infection of DEREG (DEpletion of REGulatory T cells) mice improves the numbers of splenic parasite-specific CD8+ T cells, as well as their cytokine production capacity, suggesting that Treg cells are able to regulate the induction of CD8+ T cell responses [58]. In line with these observations, we demonstrated that Treg cells activated in the context of T. cruzi infection are able to suppress total and parasite-specific CD8+ T cell immunity [59]. Given that we determined a significant reduction in the frequencies of Treg cells during the acute phase of T. cruzi infection, we decided to use a strategy that opposed that of the previous work, potentiating the Treg cell response through the injection of in vitro differentiated Treg cells. This Treg cell adoptive transfer results in an impaired CD8+ T cell response accompanied by increased parasite levels in the spleen and liver of acutely infected animals. More importantly, our results indicate that the natural contraction of the activated Treg cell response observed during the acute phase of T. cruzi infection may be critical to allow the emergence of protective anti-parasite CD8+ T cell immunity. Indeed, we showed that Treg cells and parasite-specific CD8+ T cells inversely correlate in the spleen of mice during the acute infection, in concordance to what has been suggested in humans with chronic Chagas disease [60, 61].
The mechanisms used by Treg cells to achieve CD8+ T cell suppression during T. cruzi infection remain to be fully elucidated. Considering the phenotypic profile acquired by Treg cells after the infection, it is plausible that both direct and indirect regulation of CD8+ T cell immunity may be involved [59]. Accordingly, TGF-β and CTLA-4 blocking suggested that these molecules may be partially involved in Treg cell suppression of CD8+ T cell priming, affecting CD8+ T cell proliferation and effector cytokine production, respectively [58]. Furthermore, at least two studies report that immunomodulatory molecules that support Treg cells concomitantly turn down the magnitude of the CD8+ T cell response after T. cruzi infection. Pharmacological inhibition of the enzyme Haeme Oxygenase-1 during acute infection results in decreased spleen Treg cell numbers and reduced cardiac Foxp3 expression in parallel with a raise in the CD8+ T cell heart infiltrate [62]. In a similar direction, mice deficient in Galectin-1, a beta-galactoside-binding protein that participates in several immunoregulatory circuits [63], exhibit increased frequencies of CD8+ T cells and decreased parasite burden in skeletal muscle during the acute phase of T. cruzi infection, accompanied by a reduced frequency of Treg cells in the spleen and lymph nodes [64]. It remains to be elucidated whether Haeme Oxygenase-1 and Galectin-1 exert a direct effect on the CD8+ T cell population and/or an indirect mechanism through suppression by regulatory cells.
Myeloid-derived suppressor cells (MDSCs) have also been implied in CD8+ T cell control during T. cruzi infection. MDSCs that produce peroxynitrites increase in the spleen and liver of acutely infected mice and associate with augmented numbers of CD8+ T cells that undergo surface tyrosine nitration. The interplay between these cell subsets was confirmed when MDSCs depletion with 5-fluorouracil decreased the frequency of tyrosine nitrated CD8+ T cells [65]. MDSC depletion also recovers the proliferative response of splenocytes and raises CD107a+ CD8+ T cell frequencies in T. cruzi-infected mice [65]. Altogether, these evidences suggest a suppressive function for MDSCs over CD8+ T cell response, although the direct impact of this suppression in parasite control could not be established given the reduced survival of 5-FU treated infected mice.It is likely that in spite of the enhanced cellular response, 5-FU mediated MDSC depletion unleashed inflammation and compromised tissue integrity, which in turn favored parasite circulation and reduced host survival. Whether MDSC suppressive role is exerted by granulocytic and/or monocytic subpopulations, and if other mediators apart from peroxynitrites are involved in MDSCs mediated CD8 T cell suppression is yet to be determined.
In addition to regulatory cells, cytokines released by different cell types in response to T. cruzi can also suppress cytotoxic responses. By the use of mice carrying a dominant negative form of the TGF-β type II receptor, Martin et al described that TGF- β can directly target total and parasite-specific CD8+ T cells to impair their expansion and therefore prevent their exacerbated accumulation, especially in the chronic phase of the infection [66]. In addition, Ebi3, likely as a part of IL-27 but not IL-35, was reported as a key modulator of CD8+ (and CD4+) T cell IFN-γ responses in the heart and spleen at the acute phase of T. cruzi infected mice [67]. IL-27 is produced in hearts by myeloid MHC class II+ CD11b+ cells and induces the expansion of CD3+ CD4+ IL-10+ Foxp3− Tr1 cells. Tr1-derived IL-10 would play a role in controlling IFN-γ-producing T cell responses in the context of this parasitic infection [67, 68]. Interestingly, the pro-inflammatory cytokine IL-18 could also act as a modulator of the CD8+ T cell effector cytokine response, as deficiency in this molecule increased frequency of memory CD8+ T cells that produce IFN-γ in the spleen of mice chronically infected with T. cruzi Colombian strain [69]. As these mice also showed decreased proportion of splenic CD4+ CD25+ T cells and Foxp3 expression during the acute phase, it is likely that IL-18 play an indirect role through Treg cells.
Altogether, the evidences discussed above underscore that multiple regulatory pathways act in concert to modulate CD8+ T cell immunity to T. cruzi (Figure 2). Currently, several groups are focused on targeting these immunoregulatory pathways as a mean to improve protective CD8+ T cell responses against T. cruzi without a deleterious exacerbation of tissue damage.
Figure 2. Pathways of CD8+ T cell suppression during T. cruzi infection.
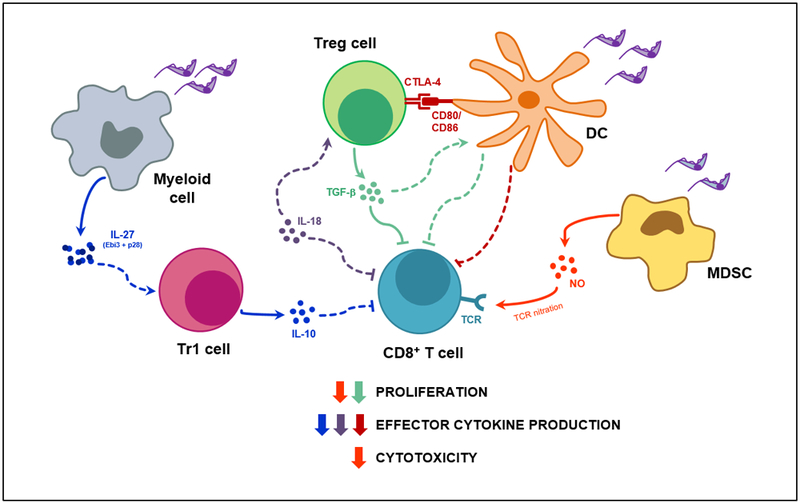
T. cruzi infection induces the secretion of soluble anti- and pro-inflammatory mediators by a wide range of immune cell populations as well as the upregulation of inhibitory receptors, such as CTLA-4, by Treg cells. In turn, each mediator may target one or several functional mechanisms of the CD8+ T cell response, either acting directly on CD8+ T cells or indirectly through other cell subtypes. Concurrently, each aspect of the CD8+ T cell effector response might be controlled by more than one inhibitory mechanism. Suppression pathways are represented by different colors that show correspondence with the color of the arrow next to the CD8+ T cell process that they suppress (i.e proliferation, effector cytokine production or cytotoxicity). Solid lines indicate a demonstrated mechanism, while dashed lines illustrate possible interactions; arrow heads stand for activation/production while blunt ends denote inhibition/suppression. Treg cell, CD4+ Foxp3+ regulatory T cell; Tr1 cell, CD4+ Foxp3- IL-10+ regulatory T cell; DC, dendritic cell; MDSC, myeloid derived suppressor cell; NO, Nitric Oxide; CTLA-4, Cytotoxic T-Lymphocyte Antigen 4; TGF-b, transforming growth factor beta; IL, interleukin; TCR, T cell receptor.
Strategies to enhance parasite-specific CD8+ T cell immunity
Given the features of CD8+ T cell responses discussed in the precedent sections, many research groups have focused on the development of different strategies aimed to prophylactically induce CD8+ T cell immunity against T. cruzi and/or potentiate it, particularly during the acute infection. Although the maximal goal during this infection stage is achieving parasite elimination and cure, a more realistic objective would be to reduce parasite levels to a minimum in order to prevent transmission and reduce the frequency of symptomatic individuals. Below we discuss different approaches aimed at boosting parasite-specific CD8+ T cell immunity before or during the acute phase.
Whether potentiating CD8+ T cell responses during the chronic phase may be beneficial for the host is still matter of debate. However, recent reports have linked increased CD8+ T cell terminal differentiation and dysfunction in chronically infected individuals with a more severe clinical disease [22] and even enhanced congenital transmission [70]. These reports raise the possibility that reprogramming and/or reinvigorating CD8+ T cells through checkpoint blockade (i.e. anti-PD-1, anti-CTLA-4) or other immunomodulatory approaches may be useful by reducing parasite loads in tissues and ameliorating clinical disease. Extensive research in this direction is needed to evaluate the potentiality of these strategies.
Vaccines
Several prophylactic and therapeutic vaccine prototypes for Chagas disease have been developed exploiting a variety of delivery systems (plasmids, adenoviruses and recombinant proteins/peptides) and adjuvants. Of note, only a fraction of these vaccines explicitly evaluated the effects on the potentiation of parasite-specific CD8+ T cell responses (Table 2). Among them, the conventional design approach is to use as immunogen those parasite antigens that dominate the response against natural infection such as TS and ASP2 [71-76]. In a similar direction, recent alternatives consist in the usage of fragments from one or more parasite proteins and even individual epitopes [77-79]. However, given the remaining questions about the potential detrimental role of immunodominant CD8+ T cell responses for protection against T. cruzi, other research groups searched for alternative candidates. This strategy consisted in the selection of antigens based on in silico analysis that followed the premises of identifying proteins that are phylogenetically conserved in diverse T. cruzi strains and expressed in the infective and intracellular mammalian stages of T. cruzi [80, 81]. All these different approaches showed prophylactic effect inducing, in a variable extent, different effector immune mechanisms that result in the reduction of parasite loads and tissue damage, in parallel with increased host survival. Furthermore, a few of these vaccines were also shown to have therapeutic potential.
Table 2.
Prophylactic and therapeutic vaccine prototypes for Chagas disease
| Delivery system + Adjuvant |
Immunization route Mouse strain |
T. cruzi Protein(s) | Effect on CD8+ T cells | Effect on other immune mechanisms |
Challenge | Outcome | Ref |
|---|---|---|---|---|---|---|---|
| Heterologous Plasmid DNA Prime/Recombinant Human Ad5 Boost Vaccine | Intramuscular – 2- week interval between prime and boost. Female C57BL/6 and A/Sn mice |
ASP2. | Induction of parasite-specific CD8+ T cells determined by multimer staining and effector function (CD107a mobilization, IFN-γ and TNF production, in vivocytotoxic activity). Long-lived response. |
Not evaluated but protection shown to be partially dependent on CD4+ T cells. | Prophylactic: Infection with Y strain 2 or 14 weeks after boost. |
Reduced parasitemia. Increased survival. |
[71-73] |
| Homologous prime/boost protocol with a combination of Ad5 encoding 2 parasite proteins | Subcutaneous – 2 doses with 4-6-week interval. Female C57BL/6 mice |
ASP2 and TS proteins of the Y T. cruzi Type II strain. | Induction of parasite-specific CD8+ T cells that exhibit cytotoxic capacity in vivo and produce IFN-γ after specific stimulation. | Induction of ASP2 and TS specific IgM and IgG. | Prophylactic: Infection with Colombiana or CL Brener strains 4 weeks after last immunization |
Reduced parasite load and electrical abnormalities in hearts. | [74] |
| Conserved frequency of parasite-specific IFN-γ producing CD8+ T cells. Reduction in polyclonal T cells proliferation and effector function. |
Reduction in plasma Nitric Oxide. Reduction in the frequency of cardiac perforin-1+ cells and mRNA levels of inducible nitric oxide synthase. Preserved numbers of cardiac IFN-γ+ cells. |
Therapeutic: Infection with Colombiana followed by 2 doses of immunization at 120 and 160 days after infection. |
Reversion of functional and histological heart abnormalities. No differences in survival or in cardiac parasitism. |
||||
| Recombinant protein + ISCOMATRIX adjuvant | Subcutaneous – 3 doses with 2-week interval. Female BALB/c mice |
Glycosylated mutant inactive TS. | Induction of CD8+ T cells that produce IFN-γ after stimulation with TS. |
Induction of strong TS specific IgG response. Increase in antigen-specific IFN-γ production by total splenocytes and CD4+ T cells. |
Prophylactic: Infection with Tulahuen strain 2 weeks after last immunization |
Marked reduction in parasitemia. Increased host survival. Reduced parasite load, inflammatory infiltrate and fibrosis in hearts. |
[75] |
| Heterologous Plasmid DNA-prime/protein-boost vaccine. Plasmids encoding IL-12 and GM-CSF were used during priming as adjuvants. |
Intramuscular: DNA-prime followed by Intradermal: protein-boost with a 21-day interval. Female C57BL/6 mice. |
TcG2 and TcG4 proteins selected by in silico analysis. | Increase in total numbers of spleen effector and central memory CD8+ T cells. Induction of parasite-specific CD8+ T cell proliferation and effector function. Long lived response. |
Increase in total numbers of spleen effector and central memory CD4+ T cells. Induction of parasite-specific CD4+ T cell proliferation and effector function. Long lived response. |
Prophylactic: Infection with Sylvio X10/4 strain after 120 days of last immunization. |
Reduced tissue parasitism in spleen, heart and skeletal muscle. | [81] |
| Recombinant protein in a poly (lactic-co-glycolic acid) nanoparticle delivery system + CpG ODN adjuvant. | Subcutaneous – 2 doses with 4-week interval. Female BALB/c mice |
Tc24 protein (24 kDa flagellar Ca2+ binding protein). | Induction of antigen-specific CD8+ proliferation | Induction of parasite- specific IFN-γ producing splenocytes. Increased concentration of IFN-γ and IL-4 in plasma with augmented IFN-γ/IL-4 ratio. Generation of high titers of antigen-specific IgG |
Therapeutic: Infection with H1 strain followed by 2 dose immunization at 7 and 17 days after infection. |
Decreased parasitemia and amastigote nests in heart. Reduced cardiac inflammatory infiltrate. |
[80] |
| DNA vaccine encoding a parasite enzymatic domain or dendritic cell-based vaccine matured with LPS and pulsed with different parasite peptides | Intramuscular with DNA vaccine - 2 doses with 2-week interval. Intravenous with cellular vaccine – 2-3 doses with 1-2-week intervals. Balb/c mice. |
Consensus TS enzymatic domain (aa 34-678). Recombinant CD8+ and CD4+ epitopes from TS. |
Induction of parasite antigen-specific IFN-γ producing CD8+ T cells | Induction of TS specific IgG | Prophylactic: Infection with Tulahuen strain one month after final immunization. |
Decreased parasitemia and increased survival. | [76] |
| Ad5 encoding parasite epitopes within the capsid protein pIX | Intramuscular – Prime, boost and reboost with 2 weeks intervals. C3H/He mice. |
Epitopes from surface glycoprotein 83 (gp83) or ASP2 (ASP2-M) | Induction of CD8+ T cells that mobilize CD107a and produce IFN-γ and TNF after stimulation with the parasite peptide. | No effect on CD4+ T cells as ASP2-M is a MHC classI restricted epitope. Induction of neutralizing antibodies only when combined with Ad5-gp83 |
Prophylactic: Infection with Tulahuen strain 2 weeks after boost |
Decreased parasitemia and increased survival. Moderate effect in Ad5-ASP-M immunized mice and maximal effect in Ad5-ASP-M+Ad5-gp2 co-immunized mice. |
[77] |
| Recombinant protein + liposomes with cage-like structures | Subcutaneous – 3 doses every 2 weeks. Female Balb/c mice. |
TS fragment (aa 338-627) | Increase in the production of IFN-γ by spleen CD8+ T cells stimulated with parasite-antigens. | Slight increase in spleen Treg cells numbers. Reduction in spleen G-MDSC numbers. |
Prophylactic: Infection with Tulahuen strain 15 days after last immunization. |
Decreased parasitemia. Increased survival. Reduced heart fibrosis. |
[78] |
| Chimeric antigen containing domains of 3 parasite proteins + 3′5′-c-di-AMP (STING agonist). | Intranasal – 3 doses every 15 days. Female C3H/HeN mice. |
Cruzipain (N-terminal domain). Inactive TS (a-helix linker). ASP2 (central region). |
Induction of parasite-specific CD8+ T cells determined by multimer staining and effector function | Induction of:
|
Prophylactic: Infection with virulent RA strain 15-30 days after last immunization. |
Increased survival. Reduced parasitemia and tissue parasitism. Reduced tissue damage determined by histology and biochemical markers. |
[79] |
AMP: adenosine 5' monophosphate; STING: Stimulator of interferon genes; TS: Trans-sialidase; ASP2: amastigote surface protein 2; Treg cells: Foxp3+ regulatory CD4+ T cells; G-MDSC: Granulocytic myeloid derived suppressor cells; Ad5: Human Adenovirus 5 (non-replicating); CpG ODN: oligonucleotides containing CpG motifs (TLR9 agonitics).
Besides evaluating the vaccine efficiency, two of the reports summarized in Table 2 provided interesting information about the characteristics of the elicited CD8+ T cell response that may be associated with their effectiveness. In this way, protection by heterologous prime-boost vaccination was linked to their ability to induce parasite-specific CD8+ T cells that exhibit phenotypic and functional attributes of superior quality in comparison to those induced by natural infection [71-73]. Vaccine-elicited CD8+ T cells show low expression of the death receptor CD95 and a phenotype compatible with an effector T cell fate. These cells, in turn, give rise to long-lived effector memory T cells that do not extensively proliferate but migrate and differentiate into effector cells upon infection challenge. These authors propose that even though the naturally-triggered immunity to T. cruzi is strong, vaccination could be exploited to induce CD8+ T cells of improved quality and fitness to cope with the infection. Moreover, a combination of genetic and cell-based immunization approaches allowed to obtain important data related to epitope immunodominance [76]. These authors showed that a DNA vaccine encoding an enzymatically active TS and an immunodominant CD8+ T cell epitope is able to enhance subdominant pathogen-specific CD8+ T cell responses as consequence of a co-stimulatory effect mediated by active TS. Remarkably, vaccines inducing both immunodominant and subdominant epitope responses are significantly less protective than those inducing only immunodominant-specific responses. Altogether, these results suggest that increasing breadth of T cell epitope responses, at least during vaccination, is not necessarily advantageous for resistance against T. cruzi.
The knowledge gained in experimental models could guide efforts to move forward towards a vaccine candidate in humans. This translational step presents a new layer of difficulty as the selected parasite epitopes need not only to be immunogenic and conserved among different parasite strains, but also to bind the extremely polymorphic human HLA molecules. So far, attempts in this direction include immunoinformatic approaches aimed to identify potential parasite epitopes that could bind HLA classI supertypes (group of HLA alleles with largely overlapping peptide binding specificities) such as A2, that are able to induce CD8+ T cell immunity with a high population coverage [82]. Moreover, HLA class II epitopes for CD4+ T cell induction as well as lineal and conformational epitopes for B cell activation, are also being explored for the design of multiepitope subunit vaccines capable of triggering different arms of the anti-parasitic immune response [83].
Immunomodulatory drugs
In the last years, it has become increasingly clear that the combination of antiparasitic agents with strategies aimed to modulate particular immunological pathways triggered by T. cruzi infection may be useful to reduce the associated pathology. Also, there is growing interest in evaluating possible immunomodulatory potential of conventional chemotherapeutics as well as novel drugs that showed promising trypanocidal effect. Accumulating evidences indicate that treatment with conventional drugs for chemotherapy of Chagas disease markedly impact on the quality of the host immune response. Benznidazole treatment initially increases and lately reduces the frequency of IFN-γ producing T cells [84], improves CD8+ T cell response [24, 85] and restores the phenotype of CD8+ (and CD4+) T cells, as evidenced by a decrease in the frequency of activated and effector cells [86]. Although it remains to be established whether these are direct effects or rather indirect consequences of parasite load reduction, the immunological changes induced by conventional drugs have been postulated to improve treatment efficacy or at least, to serve as biomarkers of treatment efficacy.
In regard to novel drug candidates, it was reported that the Tryptophan-derived catabolite 3-hydroxykynurenine (3-HK) shows a direct antiparasitic effect [87] and also induces Treg cells and suppresses Th1 and Th2 responses, reducing the incidence and severity of chronic cardiomyopathy in an experimental infection model [88]. The mechanism underlying 3-HK immunomodulatory role seems to be linked to its ability to ligate the Aryl hydrocarbon Receptor (AhR), a ligand-activated transcription factor that plays important roles in the modulation of immune responses. Of note, AhR was recently reported to have a dichotomous role in the generation of memory CD8+ T cells during T. cruzi infection [89]. Strong and/or sustained AhR activation induced by different ligands has negative effects in the development of parasite-specific memory CD8+ T cell subsets. In contrast, very weak (or lack of) AhR activation observed upon T. cruzi infection in AhRd mice, which express the hyporesponsive variant d of the Ah allele, enhances CD8+ T cell immunity and parasite control. It remains to be specifically evaluated whether drugs able to inhibit AhR pathways may be useful to improve CD8+ T cell responses during T. cruzi infection or vaccination.
In addition, promising chemotherapeutic candidates that resulted from the Drugs for Neglected Diseases Initiative (DNDi) such as K777, a vinyl sulfone cysteine protease inhibitor of cruzain, are being evaluated by their immunomodulatory properties. In particular for CD8+ T cells, in vitro K777 treatment of peripheral blood mononuclear cells from patients with Chagas disease increases the frequency of cells producing IFN-γ, TNF and IL-10, suggesting it may induce beneficial changes in the immunological profile of infected individuals [90].
Finally, unspecific therapies used to mitigate cardiovascular symptoms may also modulate immunological pathways showing an impact on disease progression [91-93]. Particularly, pentoxifylline (PTX), a methylxanthine phosphodiesterase inhibitor used as cardioprotective and as treatment for peripheral vascular diseases, show important effects on CD8+ T cells activated during T. cruzi infection. PTX administration reduces the frequency of spleen CD8+ T cells expressing activation and migration markers and more remarkably, decreases the cardiac infiltration of perforin+ CD8+ T cells preserving the presence of IFN-γ producers. Consequently, PTX hampers the progression of heart injury and reverse functional cardiac abnormalities without compromising tissue parasite control [94].
Of note, conventional chemotherapeutic drugs result in the modulation of several immune parameters including subpopulation distribution and function of CD8+ T cells that have been attributed to the success of the treatment to reduce parasite burden [24, 95]. Therefore, future work is required to establish whether the immunomodulatory effects of novel candidates are directly exerted on particular immune pathways or an indirect consequence of the trypanocidal activity.
Concluding remarks
Described more than a century ago, Chagas disease remains a major health problem in endemic areas and is becoming a global concern due to migratory movements. There is consensus in that variable disease progression results from complex host-microbe interactions. After decades of research, it is well-accepted that the magnitude and quality of the host CD8+ T cell immunity is a key element for resistance to T. cruzi. Particular features of parasite-specific CD8+ T cell responses include slow induction, immunodominance, high functional competence or cell dysfunction according to the particular infection setting, among others. However, continued efforts are required to improve our understanding of the biological pathways underlying the development and maintenance of a balanced CD8+ T cell immunity to T. cruzi (see Outstanding Questions). Furthermore, given that features of the CD8+ T cell immunity to T. cruzi have been delineated mainly based on data from experimental infection, future work should also be oriented to overcome the scarcity of detailed studies in the context of human Chagas disease. Altogether, data obtained from experimental and human infections will be critical to guide future work aimed to develop prophylactic and/or therapeutic strategies to achieve maximal parasite control (and ideally elimination), together with minimal tissue damage during the acute phase, in order to prevent clinical disease in chronicity.
Outstanding Questions.
Would the acceleration in the development of pathogen-specific CD8+ T cells be the best approach to achieve complete elimination of T. cruzi?
Is it possible to target immune pathways in order to potentiate T. cruzi-specific CD8+ T cell immunity, and thereby enhance the control of parasite replication, without unleashing uncontrolled inflammation?
Is the repertoire breadth of human CD8+ T cell responses to T. cruzi associated with the severity of clinical disease?
The dysfunction of CD8+ T cells observed in Chagas disease patients, is a cause or rather a consequence of high parasite burden and severity of the clinical disease?
Could treatments oriented to restore proliferative potential and/or effector function of dysfunctional CD8+ T cells be useful to ameliorate chronic clinical pathology in patients with Chagas disease?
Is it possible to design a unique vaccine able to confer protection for all the diverse parasite strains described?
Could the combination of selected trypanocidal drugs and immunomodulatory agents achieve superior anti-parasitic effect by limiting parasite replication and, concomitantly, potentiating CD8+ T cell immunity?
Supplementary Material
Highlights.
CD8+ T cells are critical for host resistance during T. cruzi infection given their effectiveness to control parasite outgrowth throughout all infection stages.
T. cruzi-specific CD8+ T cell immunity shows a significant magnitude but develops with a delayed kinetics, displays a relatively reduced breadth and may acquire dysfunctional features.
Proliferation, survival and cell exhaustion of T. cruzi-specific CD8+ T cells are conditioned by particular cytokines and soluble mediators secreted by different effector and regulatory immune cell populations.
Dysfunctional CD8+ T cells in patients with Chagas disease are associated with higher parasite loads and more severe clinical disease.
Several strategies including vaccines and trypanocidal drugs with immunomodulatory properties have shown diverse success to enhance CD8+ T cell immunity, reduce parasite burden and limit clinical severity.
AKNOWLEDGEMENTS
This work was supported by: Agencia Nacional de Promoción Científica y Técnica (PICT 2015-0127 to EAR; PICT 2015-0645 to AG), Consejo Nacional de Investigaciones Científicas y Técnicas, CONICET, (PIP 112-20110100378) Secretaría de Ciencia y Técnica-Universidad Nacional de Córdoba and National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers R01AI110340 to EAR and R01AI116432 to AG. EAR, CM and AG are researchers from CONICET. CLAF thank CONICET for the fellowship awarded. This article reflects only the authors’ views, and the agency is not responsible for any use that may be made of the information it contains.
GLOSSARY
- DEREG (DEpletion of REGulatory T cells) mice
These mice express a simian diphtheria toxin receptor-enhanced green fluorescent protein (DTR-eGFP) fusion protein under control of the endogenous forkhead box P3 promoter/enhancer regions. DTR-eGFP expression is observed in fully functional Foxp3+CD4+regulatory T cell populations allowing fluorescent detection or diphtheria toxin-induced ablation of Foxp3+ Treg cells.
- Exhausted T cells
Distinct CD8+ T cell lineage that arises during chronic infections and cancers. Exhausted T cells are characterized by progressive loss of effector functions, high and sustained inhibitory receptor expression, metabolic dysregulation, poor memory recall and homeostatic self-renewal, as well as distinct transcriptional and epigenetic programs.
- Foxp3+ regulatory T (Treg) cells
a population of T cells that inhibits the activation of other immune cells and is necessary to maintain peripheral tolerance to self-antigens. Treg cells are CD4+ and express the α chain of the IL-2 receptor (CD25), CTLA-4 as well as other inhibitory receptors.
- Immunodominance
The dominance of an antigen (over all others) in its ability to induce an immune response.
- Immunogen
is a specific type of antigen (typically above 20kDa) that is able to elicit an immune response.
- Immunoproteasome
highly efficient proteolytic machinery abundantly expressed in immune cells, that after exposition to inflammatory stimuli (i.e. interferons) replace the three subunits of conventional proteasomes. It plays an essential role in the immune system, degrading intracellular proteins to allow peptide presentation in an MHC class I context.
- Myeloid-derived suppressor cells (MDSCs)
a heterogeneous group of immature myeloid precursors that suppress immune responses. This cell population expresses Ly6C or Ly6G and CD11b in mice, and CD33, CD11b, and CD15 in humans.
- Priming
It is the first contact of a naive T or B cell with its specific antigen that leads to cell activation and differentiation into effector T or B cells.
- Senescent effector CD8+ T cells
Particular population of effector CD8+ T cells described in humans during aging, chronic infection and cancer characterized by cell cycle arrest, critical telomere shortening, loss of co-stimulatory molecules CD27 and CD28 expression, increased expression of CD57 and KLRG1, and increased expression of proteins involved in DNA damage responses.
- Stem cell memory
a rare subset of memory lymphocytes endowed with the stem cell–like ability to self-renew and the multipotent capacity to reconstitute the entire spectrum of memory and effector T cell subsets.
- Tyrosine nitration
is a form of post-traslational protein modification induced by peroxynitrites that may affect molecules of the TCR: CD8 complex, leading to apoptosis, unresponsiveness and dysfunction.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wong P and Pamer E (2003) CD8 T cell responses to infectious pathogens. Annu Rev Immunol 21,29–70 [DOI] [PubMed] [Google Scholar]
- 2.Tarleton RL (1990) Depletion of CD8+ T cells increases susceptibility and reverses vaccine-induced immunity in mice infected with Trypanosoma cruzi. J Immunol 144, 717–724 [PubMed] [Google Scholar]
- 3.Tarleton RL, et al. (1992) Susceptibility of beta 2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature 356, 338–340 [DOI] [PubMed] [Google Scholar]
- 4.Tarleton RL, et al. (1994) Depletion of T-cell subpopulations results in exacerbation of myocarditis and parasitism in experimental Chagas' disease. Infect Immun 62, 1820–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzelepis F, et al. (2006) Distinct kinetics of effector CD8+ cytotoxic T cells after infection with Trypanosoma cruzi in naive or vaccinated mice. Infect Immun 74, 2477–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Padilla A, et al. (2009) Insufficient TLR activation contributes to the slow development of CD8+ T cell responses in Trypanosoma cruzi infection. J Immunol 183, 1245–1252 [DOI] [PubMed] [Google Scholar]
- 7.Martin DL, et al. (2006) CD8+ T-Cell responses to Trypanosoma cruzi are highly focused on strain-variant trans-sialidase epitopes. PLoS Pathog 2, e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzelepis F, et al. (2008) Infection with Trypanosoma cruzi restricts the repertoire of parasite-specific CD8+ T cells leading to immunodominance. J Immunol 180, 1737–1748 [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg C, et al. (2010) CD8+ T cells specific for immunodominant trans-sialidase epitopes contribute to control of Trypanosoma cruzi infection but are not required for resistance. J Immunol 185, 560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg CS, et al. (2016) Long-Term Immunity to Trypanosoma cruzi in the Absence of Immunodominant trans-Sialidase-Specific CD8+ T Cells. Infect Immun 84, 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin DL and Tarleton RL (2005) Antigen-specific T cells maintain an effector memory phenotype during persistent Trypanosoma cruzi infection. J Immunol 174, 1594–1601 [DOI] [PubMed] [Google Scholar]
- 12.Bixby LM and Tarleton RL (2008) Stable CD8+ T cell memory during persistent Trypanosoma cruzi infection. J Immunol 181,2644–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pack AD, et al. (2018) Highly competent, non-exhausted CD8+ T cells continue to tightly control pathogen load throughout chronic Trypanosoma cruzi infection. PLoS pathogens 14, e1007410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laugier L, et al. (2017) Whole-Genome Cardiac DNA Methylation Fingerprint and Gene Expression Analysis Provide New Insights in the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Clin Infect Dis 65, 1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wizel B, et al. (1997) Identification of Trypanosoma cruzi trans-sialidase family members as targets of protective CD8+ TC1 responses. J Immunol 159, 6120–6130 [PubMed] [Google Scholar]
- 16.Egui A, et al. (2012) Trypanosoma cruzi paraflagellar rod proteins 2 and 3 contain immunodominant CD8(+) T-cell epitopes that are recognized by cytotoxic T cells from Chagas disease patients. Mol Immunol 52, 289–298 [DOI] [PubMed] [Google Scholar]
- 17.Diez H, et al. (2006) Evaluation of IFN-gamma production by CD8 T lymphocytes in response to the K1 peptide from KMP-11 protein in patients infected with Trypanosoma cruzi. Parasite Immunol 28, 101–105 [DOI] [PubMed] [Google Scholar]
- 18.Maranon C, et al. (2011) Identification of HLA-A *02:01-restricted CTL epitopes in Trypanosoma cruzi heat shock protein-70 recognized by Chagas disease patients. Microbes Infect 13, 1025–1032 [DOI] [PubMed] [Google Scholar]
- 19.Albareda MC, et al. (2006) Trypanosoma cruzi modulates the profile of memory CD8+ T cells in chronic Chagas' disease patients. Int Immunol 18, 465–471 [DOI] [PubMed] [Google Scholar]
- 20.Mateus J, et al. (2015) Low frequency of circulating CD8+ T stem cell memory cells in chronic chagasic patients with severe forms of the disease. PLoS Negl Trop Dis 9, e3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egui A, et al. (2015) Differential phenotypic and functional profiles of TcCA-2-specific cytotoxic CD8+ T cells in the asymptomatic versus cardiac phase in Chagasic patients. PLoS One 10, e0122115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lasso P, et al. (2015) Inhibitory Receptor Expression on CD8+ T Cells Is Linked to Functional Responses against Trypanosoma cruzi Antigens in Chronic Chagasic Patients. J Immunol 195, 3748–3758 [DOI] [PubMed] [Google Scholar]
- 23.Wherry EJ (2011) T cell exhaustion. Nat Immunol 12, 492–499 [DOI] [PubMed] [Google Scholar]
- 24.Mateus J, et al. (2017) Antiparasitic Treatment Induces an Improved CD8(+) T Cell Response in Chronic Chagasic Patients. J Immunol 198, 3170–3180 [DOI] [PubMed] [Google Scholar]
- 25.Sanmarco LM, et al. (2016) IL-6 Improves the Nitric Oxide-Induced Cytotoxic CD8+ T Cell Dysfunction in Human Chagas Disease. Front Immunol 7, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albareda MC, et al. (2015) Perturbed T cell IL-7 receptor signaling in chronic Chagas disease. J Immunol 194, 3883–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acevedo GR, et al. (2018) The Unsolved Jigsaw Puzzle of the Immune Response in Chagas Disease. Front Immunol 9, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzelepis F, et al. (2007) Modulation of CD4(+) T cell-dependent specific cytotoxic CD8(+) T cells differentiation and proliferation by the timing of increase in the pathogen load. PLoS ONE 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira A-C, et al. (2010) Impaired innate immunity in Tlr4(−/−) mice but preserved CD8+ T cell responses against Trypanosoma cruzi in Tlr4-, Tlr2-, Tlr9- or Myd88-deficient mice. PLoS Pathog 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva GK, et al. (2010) Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol 184, 1148–1152 [DOI] [PubMed] [Google Scholar]
- 31.Monteiro AC, et al. (2007) Bradykinin B2 Receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-1 responses. PLoS Pathog 3, e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy MK and Weinberg JB (2015) The immunoproteasome and viral infection: a complex regulator of inflammation. Front Microbiol 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ersching J, et al. (2016) The Combined Deficiency of Immunoproteasome Subunits Affects Both the Magnitude and Quality of Pathogen- and Genetic Vaccination-Induced CD8+ T Cell Responses to the Human Protozoan Parasite Trypanosoma cruzi. PLoS Pathog 12, e1005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laidlaw BJ, et al. (2016) The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padilla A, et al. (2007) Limited role for CD4+ T-cell help in the initial priming of Trypanosoma cruzi-specific CD8+ T cells. Infect Immun 75, 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi EY, et al. (2002) Immunodominance of H60 is caused by an abnormally high precursor T cell pool directed against its unique minor histocompatibility antigen peptide. Immunity 17, 593–603 [DOI] [PubMed] [Google Scholar]
- 37.Freeman ML, et al. (2014) Promotion of a subdominant CD8 T cell response during murine gammaherpesvirus 68 infection in the absence of CD4 T cell help. J Virol 88, 7862–7869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tosello Boari J, et al. (2018) IL-17RA-Signaling Modulates CD8+ T Cell Survival and Exhaustion During Trypanosoma cruzi Infection. Front Immunol 9, 2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bermejo DA, et al. (2013) Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORgammat and Ahr that leads to IL-17 production by activated B cells. Nat Immunol 14, 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tosello Boari J, et al. (2012) IL-17RA Signaling Reduces Inflammation and Mortality during Trypanosoma cruzi Infection by Recruiting Suppressive IL- 10-Producing Neutrophils. PLoS Pathog 8, e1002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pino-Martinez AM, et al. (2019) IL-10 participates in the expansion and functional activation of CD8(+) T cells during acute infection with Trypanosoma cruzi. J Leukoc Biol 105, 163–175 [DOI] [PubMed] [Google Scholar]
- 42.Roffe E, et al. (2012) IL-10 limits parasite burden and protects against fatal myocarditis in a mouse model of Trypanosoma cruzi infection. J Immunol 188, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fonseca SG, et al. (2007) Locally produced survival cytokines IL-15 and IL-7 may be associated to the predominance of CD8+ T cells at heart lesions of human chronic Chagas disease cardiomyopathy. Scand J Immunol 66, 362–371 [DOI] [PubMed] [Google Scholar]
- 44.Truyens C, et al. (1994) Interleukin-6 (IL-6) production in mice infected with Trypanosoma cruzi: effect of its paradoxical increase by anti-IL-6 monoclonal antibody treatment on infection and acute-phase and humoral immune responses. Infect Immun 62, 692–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volta BJ, et al. (2016) Serum Cytokines as Biomarkers of Early Trypanosoma cruzi infection by Congenital Exposure. J Immunol 196, 4596–4602 [DOI] [PubMed] [Google Scholar]
- 46.Martin D, et al. (2010) Generation of Trypanosoma cruzi-specific CD8+ T-cell immunity is unaffected by the absence of type I interferon signaling. Infect Immun 78, 3154–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardillo F, et al. (2007) B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology 122, 584–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiocca Vernengo F, et al. (2019) CD8+ T cell immunity is compromised by anti-CD20 treatment and rescued by IL-17A. bioRxiv 10.1101/642801, 642801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan NL, et al. (2015) Deficiency of antigen-specific B cells results in decreased Trypanosoma cruzi systemic but not mucosal immunity due to CD8 T cell exhaustion. J Immunol 194, 1806–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai CW, et al. (2016) Th17 Cells Are More Protective Than Th1 Cells Against the Intracellular Parasite Trypanosoma cruzi. PLoS Pathog 12, e1005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dutra WO, et al. (2014) Immunoregulatory networks in human Chagas disease. Parasite Immunol 36, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cabrera G and Marcipar I (2019) Vaccines and the regulatory arm of the immune system. An overview from the Trypanosoma cruzi infection model. Vaccine 37, 3628–3637 [DOI] [PubMed] [Google Scholar]
- 53.Fresno M and Girones N (2018) Regulatory Lymphoid and Myeloid Cells Determine the Cardiac Immunopathogenesis of Trypanosoma cruzi Infection. Front Microbiol 9, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotner J and Tarleton R (2007) Endogenous CD4(+) CD25(+) regulatory T cells have a limited role in the control of Trypanosoma cruzi infection in mice. Infect Immun 75, 861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mariano FS, et al. (2008) The involvement of CD4+CD25+ T cells in the acute phase of Trypanosoma cruzi infection. Microbes Infect 10, 825–833 [DOI] [PubMed] [Google Scholar]
- 56.Sales PA Jr., et al. (2008) The regulatory CD4+CD25+ T cells have a limited role on pathogenesis of infection with Trypanosoma cruzi. Microbes Infect 10, 680–688 [DOI] [PubMed] [Google Scholar]
- 57.Nihei J, et al. (2014) Administration of a nondepleting anti-CD25 monoclonal antibody reduces disease severity in mice infected with Trypanosoma cruzi. Eur J Microbiol Immunol (Bp) 4, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ersching J, et al. (2016) A Human Trypanosome Suppresses CD8+ T Cell Priming by Dendritic Cells through the Induction of Immune Regulatory CD4+ Foxp3+ T Cells. PLoS Pathog 12, e1005698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Araujo Furlan CL, et al. (2018) Limited Foxp3(+) Regulatory T Cells Response During Acute Trypanosoma cruzi Infection Is Required to Allow the Emergence of Robust Parasite-Specific CD8(+) T Cell Immunity. Front Immunol 9, 2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vitelli-Avelar DM, et al. (2005) Chagasic patients with indeterminate clinical form of the disease have high frequencies of circulating CD3+CD16-CD56+ natural killer T cells and CD4+CD25High regulatory T lymphocytes. Scand J Immunol 62, 297–308 [DOI] [PubMed] [Google Scholar]
- 61.de Araujo FF, et al. (2011) Regulatory T cells phenotype in different clinical forms of Chagas' disease. PLoS Negl Trop Dis 5, e992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gutierrez FR, et al. (2014) Haeme oxygenase activity protects the host against excessive cardiac inflammation during experimental Trypanosoma cruzi infection. Microbes Infect 16, 28–39 [DOI] [PubMed] [Google Scholar]
- 63.Sundblad V, et al. (2017) Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol 199, 3721–3730 [DOI] [PubMed] [Google Scholar]
- 64.Poncini CV, et al. (2015) Trypanosoma cruzi Infection Imparts a Regulatory Program in Dendritic Cells and T Cells via Galectin-1-Dependent Mechanisms. J Immunol 195, 3311–3324 [DOI] [PubMed] [Google Scholar]
- 65.Arocena AR, et al. (2014) Myeloid-derived suppressor cells are key players in the resolution of inflammation during a model of acute infection. Eur J Immunol 44, 184–194 [DOI] [PubMed] [Google Scholar]
- 66.Martin DL, et al. (2007) TGF-beta regulates pathology but not tissue CD8+ T cell dysfunction during experimental Trypanosoma cruzi infection. Eur J Immunol 37, 2764–2771 [DOI] [PubMed] [Google Scholar]
- 67.Medina TS, et al. (2017) Ebi3 Prevents Trypanosoma cruzi-Induced Myocarditis by Dampening IFN-gamma-Driven Inflammation. Front Immunol 8, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bohme J, et al. (2016) Epstein-Barr virus-induced gene 3 suppresses T helper type 1, type 17 and type 2 immune responses after Trypanosoma cruzi infection and inhibits parasite replication by interfering with alternative macrophage activation. Immunology 147, 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Esper L, et al. (2014) Regulatory effects of IL-18 on cytokine profiles and development of myocarditis during Trypanosoma cruzi infection. Microbes Infect 16, 481–490 [DOI] [PubMed] [Google Scholar]
- 70.Egui A, et al. (2017) Expression of inhibitory receptors and polyfunctional responses of T cells are linked to the risk of congenital transmission of T. cruzi. PLoS Negl Trop Dis 11, e0005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Alencar B, et al. (2009) Perforin and gamma interferon expression are required for CD4+ and CD8+ T-cell-dependent protective immunity against a human parasite, Trypanosoma cruzi, elicited by heterologous plasmid DNA prime-recombinant adenovirus 5 boost vaccination. Infect Immun 77, 4383–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rigato P, et al. (2011) Heterologous plasmid DNA prime-recombinant human adenovirus 5 boost vaccination generates a stable pool of protective long-lived CD8(+) T effector memory cells specific for a human parasite, Trypanosoma cruzi. Infect Immun 79, 2120–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vasconcelos JR, et al. (2014) Adenovirus vector-induced CD8(+) T effector memory cell differentiation and recirculation, but not proliferation, are important for protective immunity against experimental Trypanosoma cruzi Infection. Hum Gene Ther 25, 350–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pereira IR, et al. (2015) A human type 5 adenovirus-based Trypanosoma cruzi therapeutic vaccine re-programs immune response and reverses chronic cardiomyopathy. PLoS Pathog 11, e1004594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bontempi IA, et al. (2015) Efficacy of a trans-sialidase-ISCOMATRIX subunit vaccine candidate to protect against experimental Chagas disease. Vaccine 33, 1274–1283 [DOI] [PubMed] [Google Scholar]
- 76.Eickhoff CS, et al. (2016) Costimulatory Effects of an Immunodominant Parasite Antigen Paradoxically Prevent Induction of Optimal CD8 T Cell Protective Immunity. PLoS Pathog 12, e1005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matthews QL, et al. (2016) Epitope Capsid-Incorporation: New Effective Approach for Vaccine Development for Chagas Disease. Pathog Immun 1, 214–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Prochetto E, et al. (2017) Trans-sialidase-based vaccine candidate protects against Trypanosoma cruzi infection, not only inducing an effector immune response but also affecting cells with regulatory/suppressor phenotype. Oncotarget 8, 58003–58020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanchez Alberti A, et al. (2017) Engineered trivalent immunogen adjuvanted with a STING agonist confers protection against Trypanosoma cruzi infection. NPJ Vaccines 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barry MA, et al. (2016) A therapeutic nanoparticle vaccine against Trypanosoma cruzi in a BALB/c mouse model of Chagas disease. Hum Vaccin Immunother 12, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta S and Garg NJ (2015) A Two-Component DNA-Prime/Protein-Boost Vaccination Strategy for Eliciting Long-Term, Protective T Cell Immunity against Trypanosoma cruzi. PLoS Pathog 11, e1004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eickhoff CS, et al. (2015) An immunoinformatic approach for identification of Trypanosoma cruzi HLA-A2-restricted CD8(+) T cell epitopes. Hum Vaccin Immunother 11,2322–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khatoon N, et al. (2018) Examination of antigenic proteins of Trypanosoma cruzi to fabricate an epitope-based subunit vaccine by exploiting epitope mapping mechanism. Vaccine 36, 6290–6300 [DOI] [PubMed] [Google Scholar]
- 84.Laucella SA, et al. (2009) Changes in Trypanosoma cruzi-specific immune responses after treatment: surrogate markers of treatment efficacy. Clin Infect Dis 49, 1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Egui A, et al. (2019) A parasite biomarker set for evaluating benznidazole treatment efficacy in patients with chronic asymptomatic Trypanosoma cruzi infection. Antimicrob Agents Chemother [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Albareda MC, et al. (2018) Distinct Treatment Outcomes of Antiparasitic Therapy in Trypanosoma cruzi-Infected Children Is Associated With Early Changes in Cytokines, Chemokines, and T-Cell Phenotypes. Front Immunol 9, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knubel CP, et al. (2010) Indoleamine 2,3-dioxigenase (IDO) is critical for host resistance against Trypanosoma cruzi. FASEB J 24, 2689–2701 [DOI] [PubMed] [Google Scholar]
- 88.Knubel CP, et al. (2011) 3-Hydroxy kynurenine treatment controls T. cruzi replication and the inflammatory pathology preventing the clinical symptoms of chronic Chagas disease. PLoS One 6, e26550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ambrosio LF, et al. (2019) Role of Aryl Hydrocarbon Receptor (AhR) in the Regulation of Immunity and Immunopathology During Trypanosoma cruzi Infection. Front Immunol 10, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Otta DA, et al. (2018) Identification of Anti-Trypanosoma cruzi Lead Compounds with Putative Immunomodulatory Activity. Antimicrob Agents Chemother 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leite ALJ, et al. (2017) The immunomodulatory effects of the Enalapril in combination with Benznidazole during acute and chronic phases of the experimental infection with Trypanosoma cruzi. Acta Trop 174, 136–145 [DOI] [PubMed] [Google Scholar]
- 92.Horta AL, et al. (2018) The beta-blocker carvedilol and the benznidazole modulate the cardiac immune response in the acute infection induced by Colombian strain of the Trypanosoma cruzi. Mem Inst Oswaldo Cruz 113, e180271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vilar-Pereira G, et al. (2016) Combination Chemotherapy with Suboptimal Doses of Benznidazole and Pentoxifylline Sustains Partial Reversion of Experimental Chagas' Heart Disease. Antimicrob Agents Chemother 60, 4297–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pereira IR, et al. (2015) Pentoxifylline reverses chronic experimental Chagasic cardiomyopathy in association with repositioning of abnormal CD8+ T-cell response. PLoS Negl Trop Dis 9, e0003659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perez-Mazliah DE, et al. (2013) Sequential combined treatment with allopurinol and benznidazole in the chronic phase of Trypanosoma cruzi infection: a pilot study. J Antimicrob Chemother 68, 424–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perez-Molina JA and Molina I (2018) Chagas disease. Lancet 391, 82–94 [DOI] [PubMed] [Google Scholar]
- 97.PanAmerican Health Organization (2006) Estimacion Cuantitativa de la Enfermedad de Chagas en las Americas OPS/HDM/CD/425-06 [Google Scholar]
- 98.Tanowitz HB, et al. (2011) Chagas disease has now gone global. PLoS Negl Trop Dis 5, e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rassi A Jr., et al. (2017) Chronic Chagas cardiomyopathy: a review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) trial. Mem Inst Oswaldo Cruz, 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roffe E, et al. (2016) Trypanosoma cruzi Causes Paralyzing Systemic Necrotizing Vasculitis Driven by Pathogen-Specific Type I Immunity in Mice. Infect Immun 84, 1123–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanowitz HB, et al. (2017) Adipose tissue: a safe haven for parasites? Trends in parasitology 33, 276–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Machado FS, et al. (2012) Pathogenesis of Chagas disease: time to move on. Front Biosci (Elite Ed) 4, 1743–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kaech S and Cui W (2012) Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12, 749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cui W and Kaech SM (2010) Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev 236, 151–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Angelosanto JM, et al. (2012) Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol 86, 8161–8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gallimore A, et al. (1998) Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med 187, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Day CL, et al. (2006) PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 [DOI] [PubMed] [Google Scholar]
- 108.Lechner F, et al. (2000) Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med 191, 1499–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McLane LM, et al. (2019) CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495 [DOI] [PubMed] [Google Scholar]
- 110.McKinney EF, et al. (2015) T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.