

Abstract
E74‐like factor 5 (ELF5) and ETS‐homologous factor (EHF) are epithelial selective ETS family transcription factors (TFs) encoded by genes at chr11p13, a region associated with cystic fibrosis (CF) lung disease severity. EHF controls many key processes in lung epithelial function so its regulatory mechanisms are important. Using CRISPR/Cas9 technology, we removed three key cis‐regulatory elements (CREs) from the chr11p13 region and also activated multiple open chromatin sites with CRISPRa in airway epithelial cells. Deletion of the CREs caused subtle changes in chromatin architecture and site‐specific increases in EHF and ELF5. CRISPRa had most effect on ELF5 transcription. ELF5 levels are low in airway cells but higher in LNCaP (prostate) and T47D (breast) cancer cells. ATAC‐seq in these lines revealed novel peaks of open chromatin at the 5’ end of chr11p13 associated with an expressed ELF5 gene. Furthermore, 4C‐seq assays identified direct interactions between the active ELF5 promoter and sites within the EHF locus, suggesting coordinate regulation between these TFs. ChIP‐seq for ELF5 in T47D cells revealed ELF5 occupancy within EHF introns 1 and 6, and siRNA‐mediated depletion of ELF5 enhanced EHF expression. These results define a new role for ELF5 in lung epithelial biology.
Keywords: airway epithelial biology, chromatin architecture, cis‐regulatory elements, E74‐like factor 5, enhancers, ETS family transcription factors, ETS‐homologous factor, lung diseases, lung epithelium, regulation of gene expression
1. INTRODUCTION
Many monogenic disorders have significant phenotypic variability that may be attributed to genetic elements outside the causative locus. These sites (modifier loci) are often identified by genome‐wide‐association studies (GWAS).1 However, defining the mechanisms whereby modifier loci contribute to phenotype is often challenging.2 One example is cystic fibrosis (CF) a life‐shortening, recessive genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Of note, mutations in CFTR do not correlate with lung disease severity, the major cause of morbidity in CF.3, 4, 5
CF impacts about 70,000 people worldwide 6 though phenotype does not correlate well with mutations in CFTR. The high frequency of the disorder facilitated a replicated GWAS, which identified single nucleotide polymorphisms (SNPs) in an intergenic region of chromosome 11p13 that significantly associated with CF lung disease severity.3, 4 The genes mapping closest to these SNPs are two epithelial–selective E twenty‐six (ETS) transcription factors (TFs), ETS homologous factor (EHF) and E74‐like factor 5 (ELF5) on the 5’ side and Apaf‐1‐interacting protein (APIP) on the 3’ side.
EHF is abundantly expressed in differentiated epithelial tissues, particularly in the prostate, pancreas, salivary gland and airway.7, 8 EHF can be a transcriptional activator or repressor, depending on cellular context, and its expression is often reduced or lost in some breast, prostate and lung carcinomas.7, 8, 9, 10 In the airway, EHF is intimately involved in many cellular processes integral to CF lung pathology such as epithelial cell differentiation and development, cell locomotion, response to wounding, barrier function, goblet cell differentiation, tissue remodelling and inflammation.11, 12 Also, EHF has a direct role in CFTR regulation, as it binds to an airway selective enhancer element of CFTR and represses its expression.13 For these reasons, EHF is a strong candidate CF modifier gene and also a potential CF therapeutic target since elevated CFTR substrate may enhance the efficacy of correctors and potentiators.14
In contrast, the role of ELF5 has been studied in breast cancer, where it drives lung metastasis by recruiting myeloid‐derived suppressor cells.15 ELF5 is also necessary for proper lung branching and epithelial differentiation in the mouse.16 It has not been well studied in the human lung, although it is a candidate gene for conferring susceptibility to asthma.17 Furthermore, ELF5 was predicted to regulate EHF expression, since ChIP‐seq with an antibody specific for ELF5 showed its occupancy at the EHF promoter and first intron in the breast cancer cell line T47D.18
Although to date, the highest p‐value SNPs identified in the replicated GWAS do not correlate with the transcription of genes at 11p13, this may reflect a paucity of data on gene expression at the single‐cell level. However, revealing the regulatory interactions of the EHF and ELF5 TFs is critical to understanding their role in lung epithelial biology. Here we investigate the cis‐regulatory elements (CREs) at chr11p13 that may control the expression of these two epithelial selective ETS transcription factors. Using CRISPR/Cas9 technology, three key CREs were deleted from the region in airway epithelial cells, and subsequent changes in gene expression and chromatin architecture were examined. We also took an unbiased screening approach to identify additional CREs at chr11p13 by activating multiple DNase I hypersensitive sites (DHS) across the region using CRISPRa,19 followed by measuring nearby gene expression. In parallel, we pursued the predicted role of ELF5 in regulating EHF by investigating open chromatin and 3D architecture in cell types with different abundance of ELF5. These genomic context studies revealed a critical role for ELF5 in coordinating the complex regulatory environment at chr11p13. Our results are relevant to the involvement of both ELF5 and EHF in lung disease, since both genes are expressed in human bronchial epithelium.
2. MATERIALS AND METHODS
2.1. Cell culture
A549,20 A549 c9 (subclone of A549 that endogenously expresses EHF protein at levels higher than WT),11 Calu3 21 and 16HBE14o‐ 22 cells were grown in Dulbecco's modified Eagle's medium (low glucose) with 10% foetal bovine serum (FBS). LNCaP and T47D cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS 23 and for T47D only, 0.2 Units/mL insulin.24
2.2. Luciferase‐based reporter assays
Luciferase promoter:enhancer constructs were generated as described previously.25 Constructs were transfected with Lipofectamine 2000 (Life Technologies) into A549 c9 and T47D cells. A modified pRL Renilla luciferase vector (Promega) was used as a transfection control. Cells were assayed for Renilla and firefly luciferase activity after 48 hours using the Dual‐Luciferase Reporter Assay System (Promega).26
2.3. CRISPR guide design, CRISPR/Cas9 transfection and screening
Two pairs of gRNAs flanking EHF intron 6, 11.2521 and 11.2516 were identified using Benchling 27 (Table S1). Both gRNAs flanking each region were sequentially cloned into a single pBlueScript (pBS) with a modified multiple cloning site. A549 c9 cells were seeded onto 6‐well plates and transfected after 24 hours with 0.1 pmol of pMJ920 (wild‐type Cas9 plasmid tagged with GFP) (Addgene, plasmid #42234) and 0.2 pmol of pBS containing the 5’ and 3’ gRNAs using Lipofectamine 2000 (Life Technologies); 48 hours after transfection, cells were prepared for fluorescence‐activated cell sorting of GFP‐positive cells and single cells were plated onto 96‐well plate. Clones were expanded and screened for homozygous deletion of each site as described previously using primers shown in Table S2.28
2.4. Reverse transcription quantitative polymerase chain reaction (RT‐qPCR)
RNA was extracted from cells using TRIzol (In Vitrogen) according to manufacturer's instructions. Reverse transcription used TaqMan Reverse Transcription Reagents (Life Technologies) by the standard protocol. qPCR assays were performed using SYBR Green reagents (Life Technologies) and primers listed in Table S3.
2.5. Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP)‐seq was performed as previously described 11 with an antibody specific for ELF5 (Santa Cruz, sc‐376737). ChIP‐qPCR was performed as described earlier.29, 30 Antibodies were specific for CTCF (Millipore 07‐729) or normal rabbit IgG (Millipore 12‐370). qPCR was performed using SYBR Green reagents with primers listed in Table S4.
2.6. Circular chromosome conformation capture and deep sequencing (4C‐seq)
4C‐seq was performed as previously described 28 in A549 c9 and its derivative CRE deletion clones, Calu3, LNCaP and T47D cells. The primers used to generate libraries for each viewpoint are listed in Table S5. Two independent 4C libraries were generated for each viewpoint for each cell type. The sequencing data were processed using the 4Cseqpipe pipeline.31 All 4C‐seq domainograms were generated using the default parameters of the pipeline.
2.7. CRISPRa/VPR‐mediated activation experiments
Single guide RNAs (sgRNAs) targeting ELF5 and EHF promoters were designed within 1 kb 5’ to the transcriptional start sites (TSS). Two to four sgRNAs were designed to target DHS cores at chr11p13 using Benchling 27 (Table S6) and were chosen based on the highest on‐target and lowest off‐target scores. sgRNAs were cloned into the pSPgRNA plasmid (Addgene #47108) and nucleofected with the SP‐dCas9‐VPR plasmid (Addgene #63798) into either 16HBE14o‐ or A549 cells with Lonza 4D‐Nucleofector kits (P3 Primary Cell V4XP‐3032 for 16HBE14o‐ and SE Cell Line V4XC‐1032 for A549). For each experiment, pSPgRNA alone was nucleofected with SP‐dCas9‐VPR and used as a negative control. Using protocols described above, RNA was extracted 48 hours post‐nucleofection and reverse transcription quantitative polymerase chain reaction (RT‐qPCR) assays were performed. Data were normalized individually to the empty vector control.
2.8. Omni Assay for transposase accessible chromatin and deep sequencing
Omni‐ATAC‐seq was performed on 50,000 Calu3, LNCaP and T47D cells as described previously32 with minor modifications. ATAC‐seq libraries were purified with Agencourt AMPure XP magnetic beads (Beckman Coulter) with a sample to bead ratio of 1:1.2 and eluted in Buffer EB (Qiagen). Data were processed by the ENCODE‐DCC/atac‐seq‐pipeline.
2.9. Transient ELF5 siRNA depletion experiments
ELF5 depletion in T47D cells was performed using Silencer select siRNA (Ambion, AM16708) as previously described.11 Using protocols described above, RNA was extracted 48 hours post‐transfection and RT‐qPCR assays were performed. For protein assays, lysates were analysed by standard western blot methods.11, 12 Antibodies were specific for EHF (Clone 5A.5, kindly donated by Dr A Tugores) and β‐tubulin (T4026, Sigma‐Aldrich).
2.10. Statistical analysis
Error bars denote standard deviation (SD) or standard error of the mean (SEM) as noted in figure legends, which also define the statistical analysis used. This was either two‐way ANOVA plus multiple comparisons tests or Student's unpaired t tests. All statistical analysis was performed in Prism software (GraphPad).
3. RESULTS
3.1. Deletion of a CRE in EHF intron 6 enhances EHF expression while impairing recruitment of a strong enhancer to the gene promoter
The EHF locus coincides with an extended track of the H3K27ac active histone mark, indicative of a stretch enhancer in airway epithelial cells.25, 33 EHF intron 6 lies at the 3’ end of this feature and coincides with a region of high occupancy by multiple transcription factors (ENCODE data 34, 35, 36). To determine the role of this element in the genomic context within airway epithelial cells, CRISPR/Cas9 was used to generate A549 c9 clones lacking an ~3.1 kb fragment coinciding with the extent of TF occupancy. Three homozygous deletion clones were evaluated and compared both to non‐targeted clones from the same experiment and wild‐type (WT) A549 c9 cells. Models of the 11p13 region (left) and the impact of removal of EHF intron 6 (right) are shown in Figure 1A. First, expression of the ELF5, EHF and APIP genes was assayed by RT‐qPCR (Figure 1B). Removal of the EHF intron 6 element did not change ELF5 or APIP transcript levels; however, it was associated with an increase in EHF (~8‐fold, P < .0001) expression.
Figure 1.
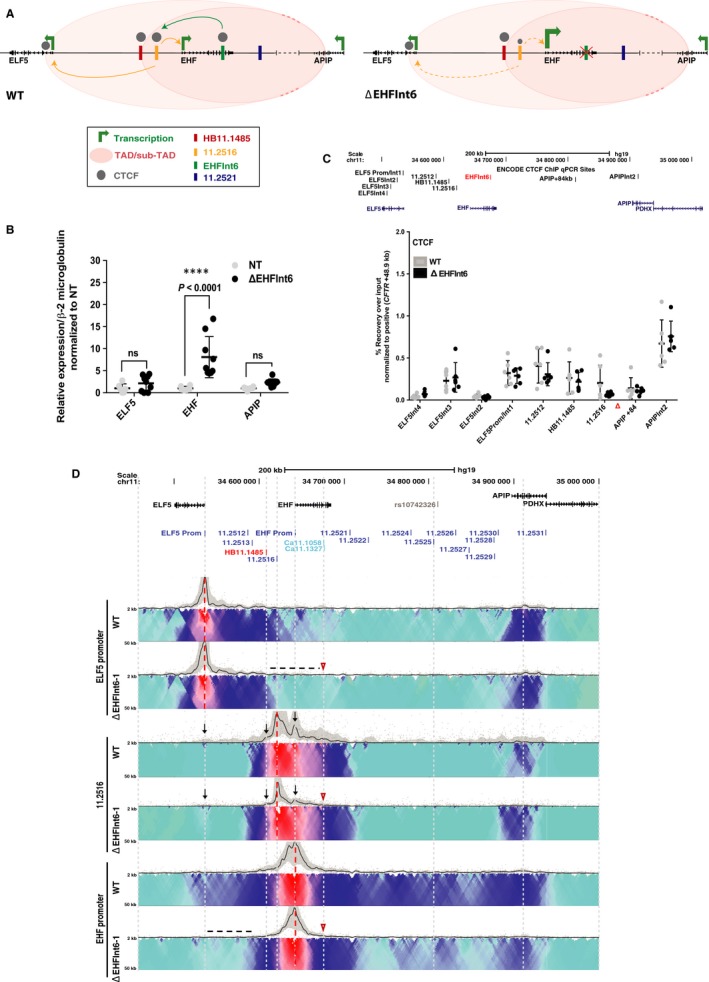
Impact of deletion of cis‐element EHF intron 6 in A549 c9 cells. (A) EHF intron 6 maintains a structural role at 11p13 and helps to facilitate looping of the 11.2516 enhancer (green arrow) to the ELF5 and EHF promoters (yellow arrows). Upon deletion of EHF intron 6 (red X), EHF expression is enhanced. Additionally, CTCF occupancy at 11.2516 is reduced (smaller grey circle) and its interactions with the ELF5 and EHF promoters are weakened (yellow dashed arrows). (B) RT‐qPCR for ELF5, EHF and APIP expression in non‐targeted WT (NT) and deletion clones. Data are normalized to β2M and relative to clonal WT. Error bars are standard deviation (SD), n ≥ 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by a two‐way ANOVA test with multiple comparisons. (C) ChIP‐qPCR for CTCF enrichment at interacting sites across the 11p13 region in WT and deletion clones. ChIP results are shown as percent recovery over input and are normalized to a positive control (CFTR + 48.9 kb (28). Error bars are SD, n ≥ 2. (D) Circular chromosome conformation capture (4C‐seq) data in WT and one EHF intron 6 deletion clone. This deletion removes the Ca11.1058 and Ca11.1327 DHS. Viewpoints at the ELF5 promoter, 11.2516 and EHF promoters are shown (red dotted lines). Open red arrowheads indicate the deletion site. The upper panel (black line) shows the main trend of interaction, and the colour‐coded domainogram indicates relative interactions with a window size ranging from 2‐50kb. Red denotes the strongest interactions; dark blue, to turquoise, to grey represent decreasing interaction frequencies. Informative interactions or loss thereof are shown as black dotted lines or black arrows
These results suggest that either transcriptional repressors of EHF are recruited to the intron 6 element or that loss of this site alters chromatin architecture at the locus, thus facilitating occupancy of activating factors at the EHF promoter or its enhancers. To investigate the latter hypothesis, we first assayed CTCF occupancy at specific sites across the chr11p13 region by ChIP‐qPCR. CCCTC‐binding factor (CTCF) is an architectural protein involved in higher order chromatin organization. It is recruited to invariant sites in the genome (topologically associated domain (TAD) boundaries) in addition to variant structural elements that may be cell‐type specific.37, 38, 39 The EHF intron 6 element binds CTCF with high affinity in several airway cell types,25 so its removal was predicted to impact CTCF recruitment at adjacent sites. Consistent with this suggestion, deletion of the EHF intron 6 element was associated with a substantial (~3‐fold) decrease in CTCF occupancy at the adjacent site DHS11.2516 (Figure 1C), though this did not reach statistical significance.
To examine the impact of loss of the EHF intron 6 element on 3D chromatin structure more broadly, we performed 4C‐seq on the deletion clones and compared them to WT A549 c9 cells (Figure 1D). A minimum of 2 deletion clones were analysed for each viewpoint, and each clone was evaluated in replica experiments. One experiment is shown in each 4C‐seq panel and is consistent with its replicates. For each 4C‐seq panel, the black line shows the main trend of interactions across the locus while below it is the domainogram,40 which uses colour‐coded intensity values to show relative interactions with window sizes varying from 2 to 50 kb.31 Red denotes the strongest interactions and dark blue, through turquoise, to grey represent gradually decreasing frequencies. Black arrows or dotted lines denote important data features, and the location of the EHF intron 6 element deletion is shown by an open red arrowhead. Here, we focus on the gain or loss of interactions at sites we showed previously to have functional importance,25 specifically those at the 5’ end of the 11p13 modifier region. Using a viewpoint at the ELF5 promoter, the deletion is seen to cause a general loss of interactions 3’ to the HB11.1485 CTCF site (black dotted line), most notably close to 11.2516 and within the first intron of EHF. With a viewpoint located 20kb 5’ to the EHF promoter (11.2516), interactions with the ELF5 and EHF promoters were notably reduced and those with HB11.1485 element were slightly diminished (black arrows). These results are consistent with our earlier data from WT cells showing that DHS11.2516 and the EHF intron 6 element interact directly.25 The EHF promoter viewpoint shows a modest loss of interactions across the whole region, most prominently between HB11.1485 and the ELF5 promoter (black dotted line). In summary, these data suggest that looping of the CRE at DHS11.2516 to the EHF and ELF5 promoters is somewhat dependent on the EHF intron 6 element and moreover that this element contributes to the maintenance of locus architecture.
3.2. Deletion of the 11.2521 CRE enhances ELF5 expression and increases interactions of the ELF5 promoter with novel sites
The 11.2521 CRE was shown previously to be a strong enhancer of both the ELF5 and EHF promoters (though not APIP), by luciferase assays in 16HBE14o‐ bronchial epithelial cells.25 These data were confirmed here in A549 c9 cells (Figure S1A‐B). To examine the function of this element in the genomic context, we used CRISPR/Cas9 to remove ~2.5 kb encompassing the site in A549 c9 cells. Two independent homozygous deletion clones were evaluated and compared both to non‐targeted clones and WT A549 c9 cells. Models of the 11p13 region (left) and the impact of removal of the 11.2521 CRE (right) are shown in Figure 2A. First, ELF5, EHF and APIP expressions were measured using RT‐qPCR (Figure 2B). Removal of this CRE did not alter EHF or APIP expression; however, it enhanced transcript abundance of ELF5 (~11‐fold, P < .0001).
Figure 2.
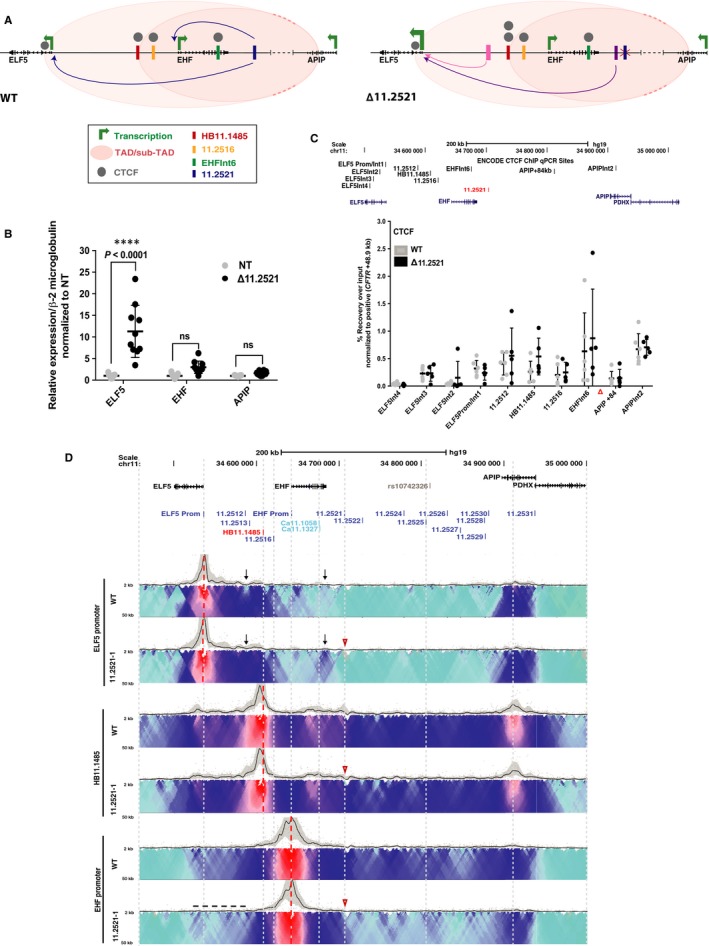
Impact of deletion of cis‐element 11.2521 in A549 c9 cells. (A) 11.2521 interacts with both the ELF5 and EHF promoters (blue arrows) and enhances their expression. Upon deletion of 11.2521 (red X), ELF5 expression is enhanced. Additionally, interactions between the ELF5 promoter and DHS 11.2512/11.2513 (pink arrow) and an unknown element 3’ to the EHF locus (purple arrow) are evident. CTCF occupancy at 5’ sub‐TAD boundary HB11.1485 is slightly increased (two grey circles). (B) RT‐qPCR for ELF5, EHF and APIP expression in non‐targeted WT (NT) and deletion clones. Data are normalized to β2M and relative to clonal WT. Error bars are SD, n ≥ 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by a two‐way ANOVA test with multiple comparisons. (C) ChIP‐qPCR for CTCF enrichment at interacting sites across the 11p13 region in WT and deletion clones. ChIP results are shown as percent recovery over input and are normalized to a positive control (CFTR + 48.9 kb). Error bars are SD, n ≥ 3. (D) 4C‐seq data as in Figure 1 in WT and one 11.2521 deletion clone. Viewpoints are at ELF5 promoter, HB11.1485 and EHF promoter (red dotted lines). Open red arrowheads indicate the deletion site. Informative interactions or loss thereof are shown as black dotted line or black arrows
Since the 11.2521 CRE is an enhancer of the ELF5 and EHF promoters in transient assays,25 the increased expression of ELF5 upon deletion of this CRE suggests that features of the genomic architecture of the region may have a dominant role in regulating gene expression. To address this hypothesis, we measured CTCF occupancy at known binding sites across the region using ChIP‐qPCR. The 11.2521 CRE itself binds CTCF with low affinity in 16HBE14o‐ cells though not in other airway cell types.25 However, this element was shown earlier by 4C‐seq to interact with many other sites of CTCF occupancy across the region,25 so we predicted that its removal could alter occupancy at some other sites. Consistent with this prediction, removal of the 11.2521 CRE was associated with moderately elevated CTCF occupancy at HB11.1485 (~2‐fold) (Figure 2C), though this alteration did not reach statistical significance.
Next, to investigate the impact of removal of the 11.2521 CRE on chromatin architecture across chr11p13, we performed 4C‐seq on the deletion clones and compared them to WT A549 c9 cells (Figure 2D). Again, two deletion clones were analysed for each viewpoint and each clone was assayed in replica experiments. One experiment is shown in each 4C‐seq panel and is consistent with its replicates. Using a viewpoint at the ELF5 promoter, loss of the 11.2521 CRE increased its interactions with DHS11.2512/11.2513 and an uncharacterized element immediately 3’ to EHF (black arrows). However, the deletion had no impact on interactions with the HB11.1485 viewpoint, while associations with the EHF promoter were modestly reduced (black dotted line). These data suggest that the 11.2521 CRE may inhibit ELF5 promoter interactions with previously uncharacterized enhancer elements, and that its functions depend upon specific chromatin architecture at chr11p13.
3.3. Deletion of the 11.2516 CRE increases ELF5 but not EHF expression and alters chromatin architecture
The 11.2516 CRE was also shown previously to be a strong enhancer of both the ELF5 and EHF promoters (though not APIP), by luciferase assays in 16HBE14o‐ cells.25 These data were confirmed here in A549 c9 cells (Figure S1A‐B). To examine the function of this element in the genomic context, we again used CRISPR/Cas9 to remove ~2.6 kb fragment encompassing the site in A549 c9 cells. Three homozygous deletion clones were evaluated and compared to both non‐targeted clones and WT A549 c9 cells. Models of the 11p13 region (left) and the impact of removal of the 11.2516 CRE (right) are shown in Figure 3A. First, ELF5, EHF and APIP expressions were measured using RT‐qPCR (Figure 3B). Removal of this CRE did not alter abundance of the EHF or APIP transcripts; however, it was associated with an increase in ELF5 expression (~7‐fold, P = .0004).
Figure 3.
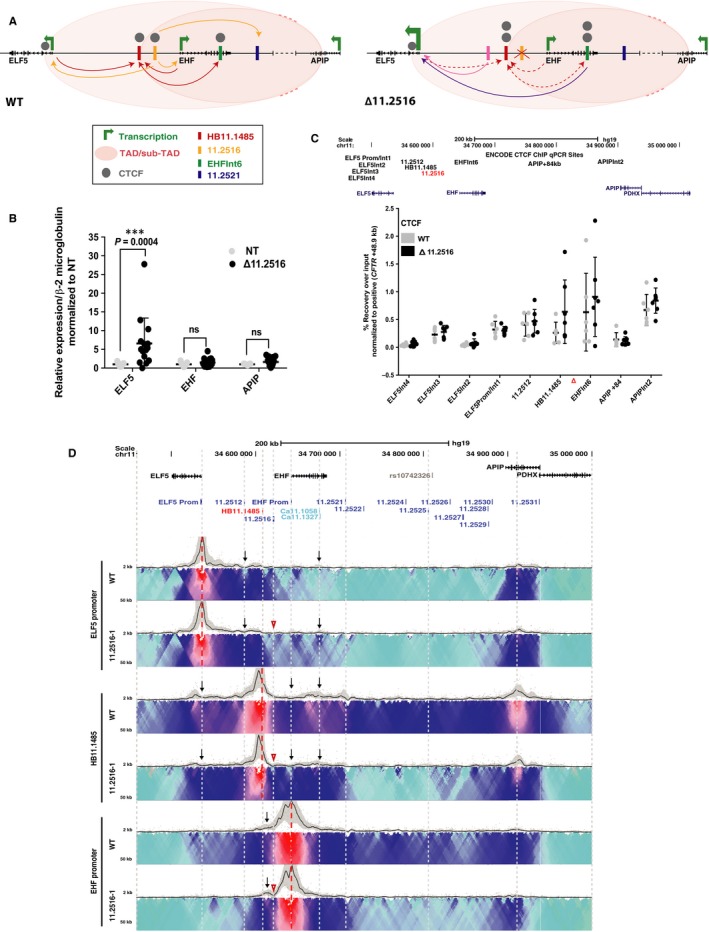
Impact of deletion of cis‐element 11.2516 in A549 c9 cells. (A) 11.2516 interacts with both the ELF5 and EHF promoters and enhances their expression (yellow arrows). 11.2516 also interacts with the 11.2521 enhancer element (yellow arrow) and helps to recruit this DHS to the gene promoters. Furthermore, 11.2516 interacts strongly with 5’ sub‐TAD boundary HB11.1485 and helps to loop this element to the ELF5 promoter, EHF promoter and EHF intron 6 (red arrows). Upon deletion of 11.2516 (red X), ELF5 expression is enhanced. Additionally, interactions between HB11.1485 and the ELF5 promoter, EHF promoter and EHF intron 6 are reduced (red dashed arrows), which allows the ELF5 promoter to gain interactions with CREs, particularly DHS 11.2512/11.2513 (pink arrow) and EHF intron 6 (purple arrow). Furthermore, CTCF occupancy increases at adjacent sites HB11.1485 and EHF intron 6 (two grey circles). (B) RT‐qPCR for ELF5, EHF and APIP expression in non‐targeted WT (NT) and deletion clones. Data are normalized to β2M and relative to clonal WT. Error bars are SD, n ≥ 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by a two‐way ANOVA test with multiple comparisons. (C) ChIP‐qPCR for CTCF enrichment at interacting sites across the 11p13 region in WT and deletion clones. ChIP results are shown as per cent recovery over input and are normalized to a positive control (CFTR + 48.9 kb). Error bars are SD, n ≥ 4. (D) 4C‐seq data as in Figure 1 in WT and one 11.2516 deletion clone. Viewpoints at the ELF5 promoter, HB11.1485 and EHF promoter (red dotted lines). Open red arrowheads indicate deletion site. Informative interactions are shown as black arrows
As for the 11.2521 CRE, it seemed probable that the in vitro enhancer activity of the 11.2516 CRE might be dominated by features of the chromatin architecture in the genomic context. To test this prediction, we measured CTCF occupancy at known sites across chr11p13 using ChIP‐qPCR. The 11.2516 CRE was shown previously to bind CTCF in multiple airway cell types.25 Hence, it was not unexpected to observe that removal of this element reproducibly enhanced CTCF occupancy at adjacent sites HB11.1485 (~2.4‐fold increase) and EHF intron 6 (~1.4‐fold increase), though these alterations did not reach statistical significance (Figure 3C). This enhancement in occupancy at nearby sites is a common feature of removal of key sites of CTCF occupancy from a genomic region.28, 41
Next, to examine the impact of removal of the 11.2516 CRE on chromatin architecture, we performed 4C‐seq on the deletion clones and compared them to WT A549 c9 cells (Figure 3D). Again, two deletion clones were analysed for each viewpoint and each clone was evaluated in replica experiments. One experiment is shown in each 4C‐seq panel and is consistent with its replicates. Removal of the 11.2516 CRE enhanced interactions between the ELF5 promoter viewpoint and the 11.2512/11.2513 and EHF intron 6 elements (black arrows). These data are consistent with our previous results showing direct interaction of 11.2516 with these sites.25 In contrast, reduced associations were evident between a viewpoint at HB11.1485 and multiple sites across the region, including the ELF5 promoter, EHF promoter and EHF intron 6 (black arrows). Loss of the 11.2516 CRE also coincided with a modest reduction of interactions between the EHF promoter and the rest of the region, with the exception of an unidentified element located between HB11.1485 and 11.2516 (black arrow), which showed an enhanced association. These data suggest that 11.2516 has an important role in the maintenance of chromatin architecture at the chr11p13 region.
3.4. Expression of ELF5 is associated with altered chromatin architecture
None of the airway cell lines used in this work and our earlier analysis of the chr11p13 region 11, 25 express detectable levels of ELF5 protein, and ELF5 transcript abundance is very low (Figure S2). However, primary cultures of human bronchial and tracheal epithelial cells have low and variable expression of ELF5,42 which may reflect the relative contribution of different cell types. Moreover, the CRISPR deletion studies described above consistently had most impact on the ELF5 gene. Hence, it was important to examine the chr11p13 region in cell types expressing both ELF5 and EHF. The T47D (breast cancer epithelial)24 and LNCaP (prostate cancer epithelial) 23 cell lines met this requirement and were chosen for further study. The abundance ratio of ELF5 to EHF transcripts is highest in T47D cells, and the reverse is seen in the LNCaP line (Figure 4A). To examine the correlation between ELF5 and EHF expression levels and chromatin architecture at chr11p13, we performed Assay for Transposase Accessible Chromatin (ATAC)‐seq 43, 44 to map open chromatin (Figure 4B) and 4C‐seq to show 3D interactions (Figure 4C) in T47D, LNCaP and Calu3 cells (which lack ELF5).
Figure 4.
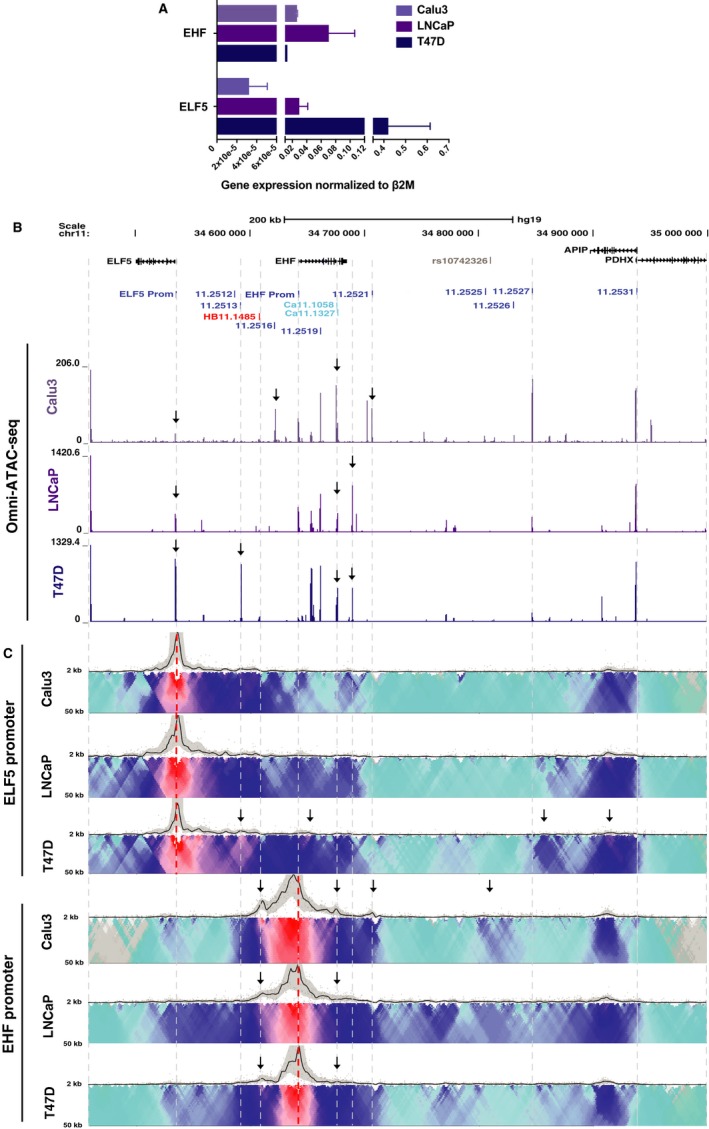
Changes in 3D organization and chromatin accessibility of 11p13 in ELF5‐expressing cells. (A) RT‐qPCR of ELF5 and EHF expression in Calu3, LNCaP and T47D cells. Data are normalized to β2M. Error bars are SEM, n = 3. (B) ATAC‐seq showing regions of open chromatin in Calu3, LNCaP and T47D cells. Key peaks are shown by black arrows. (C) 4C‐seq data as in Figure 1 in Calu3, LNCaP and T47D cells. Viewpoints at the ELF5 promoter and EHF promoter (red dotted lines). Informative interactions are shown as black arrows
Most notable are the substantially different profiles of open chromatin peaks around the ELF5 and EHF loci in the different cell lines (Figure 4B). First, the relative height of the peak at the ELF5 and EHF promoters correlated well with gene expression levels. A very prominent ELF5 promoter peak is seen in T47D cells, a smaller peak in LNCaP cells and a near background peak in Calu3 cells. In contrast, both Calu3 and LNCaP cells have a similar peak of open chromatin at the EHF promoter, which is not strongly evident in T47D cells. ATAC‐seq data also revealed large peaks of open chromatin at the two airway enhancer elements, 11.2516 and 11.2521, which were absent in both LNCaP and T47D cells, consistent with our earlier work showing these enhancers to be airway‐selective.25 In transient luciferase assays in T47D cells (Figure S1C, D), the 11.2516 fragment had only modest enhancer activity on the ELF5 and EHF promoters compared to airway cells. The 11.2521 element had no enhancer activity on the ELF5 promoter in T47D cells, consistently giving lower luciferase values than the promoter alone, and again had modest enhancer effect on the EHF promoter. Also of note is the ATAC‐seq peak at EHF intron 6 in all three cell types irrespective of EHF abundance, suggesting a ubiquitous role. A novel open chromatin peak is seen close to the 3’ end of the EHF locus in both LNCaP and T47D cells, but not in Calu3 cells suggesting a correlation of this site with ELF5 expression. Another new site of open chromatin is only seen in T47D cells at 11.2513, a region that corresponds to a DHS in tracheal epithelial cells45 but has not yet been studied.
Since expression of ELF5 is associated with novel peaks of open chromatin at chr11p13, it seemed probable that altered chromatin architecture might also be seen in ELF5 expressing cells. To investigate this hypothesis, we performed 4C‐seq on Calu3, LNCaP and T47D cells using viewpoints at the ELF5 and EHF promoters (Figure 4C). The ELF5 promoter viewpoint showed a similar pattern of interactions in Calu3 and LNCaP cells, with interactions coinciding with several peaks of open chromatin, both adjacent to ELF5 and across the EHF locus and with the 3’ TAD boundary in APIP intron 3. In contrast, high ELF5 expression in T47D cells coincided with strong interactions (black arrows) between the ELF5 promoter viewpoint, the novel peak of open chromatin at 11.2513 and a site in EHF intron 1, also associated with an ATAC‐seq peak. Interactions with the 3’ TAD boundary and an element 3’ of 11.2527 were also seen.
Using the EHF promoter viewpoint, the major specific difference seen in Calu3, LNCaP and T47D cells was a great reduction in interactions with the HB11.1485 5’ sub‐TAD boundary when the ELF5 gene is highly expressed (T47D) (black arrows). A minor reduction of interactions with the EHF intron 6 CRE was also observed in LNCaP and T47D cells (black arrows). In addition, in Calu3 cells only, interactions were evident with the airway enhancers (11.2516 and 11.2521), EHF intron 6 and the 11.2525/11.2526 sites closest to the highest p‐value SNP (rs10742326) from the GWAS4 (black arrows). In summary, these data suggest that alterations in chromatin architecture at chr11p13 and the loss of a 5’ sub‐TAD boundary enable expression of ELF5.
3.5. Activation of CREs across chr11p13 enhances ELF5 expression, but has little impact on EHF or APIP
The data shown in the previous sections suggest that in cells that express ELF5 this locus may have an important role in the functional genomics of the chr11p13 region. Our earlier in vitro studies suggested that the 11.2516 and 11.2521 CREs enhanced activity of both the ELF5 and EHF promoters 25; however, results presented here suggest that other features of the chromatin may have a dominant effect in vivo. To determine whether activation of any CREs across chr11p13 could elevate endogenous gene expression, we used CRISPRa. Briefly, nuclease‐null dCas9 was used in the dCas9‐VPR fusion, which recruits three transcriptional activators, VP64 (a transcriptional activator made from 4 tandem copies of the Herpes Simplex Viral Protein 16), NF‐kappa‐B p65 subunit [p65], and Replication and transcriptional activator [Rta] (SP‐dCas9‐VPR) to target sites. A minimum of two guide RNAs were designed to target the ELF5 and EHF promoters and multiple airway DHS across chr11p13 in 16HBE14o‐ and A549 cells (Figure 5 and Figure S3). Targeting the ELF5 promoter increased its expression by ~2600‐fold in 16HBE14o‐ and ~40‐fold in A549 cells. Targeting the EHF promoter increased its expression by ~4‐fold in 16HBE14o‐ and ~2‐fold in A549 cells. Among targeted DHS, 11.2512, which we had not studied previously, resulted in the most robust activation of ELF5 (~18‐fold in 16HBE14o‐ cells and ~7‐fold in A549). Expression of ELF5 in 16HBE14o‐ cells was also increased significantly by CRISPRa at the EHF intron 6 (~3.5‐fold), 11.2521 (~7.5‐fold), 11.2522 (~3‐fold), 11.2523 (~5‐fold), 11.2529 (~4.5‐fold) and 11.2530 (~4‐fold) DHS (Figure 5). Activation of most of the same sites together with 11.2516 also significantly increased ELF5 expression in A549 cells (Figure S3). APIP expression was not altered by activation of any of the DHS in 16HBE14o‐ cells, while only targeting the 11.2529 and 11.2530 sites slightly, but significantly, increased EHF expression (~1.3‐fold). The modest response to CRISPRa at chr11p13 suggests that either additional sequences, which are not associated with regions of open chromatin in airway cells, are key in transcriptional activation of ELF5 and EHF, or that a more complex regulatory mechanism exists for the genes in this region. The latter explanation would be consistent with our 4C‐seq observations suggesting close interactions between the ELF5 and EHF genes and their CREs.
Figure 5.
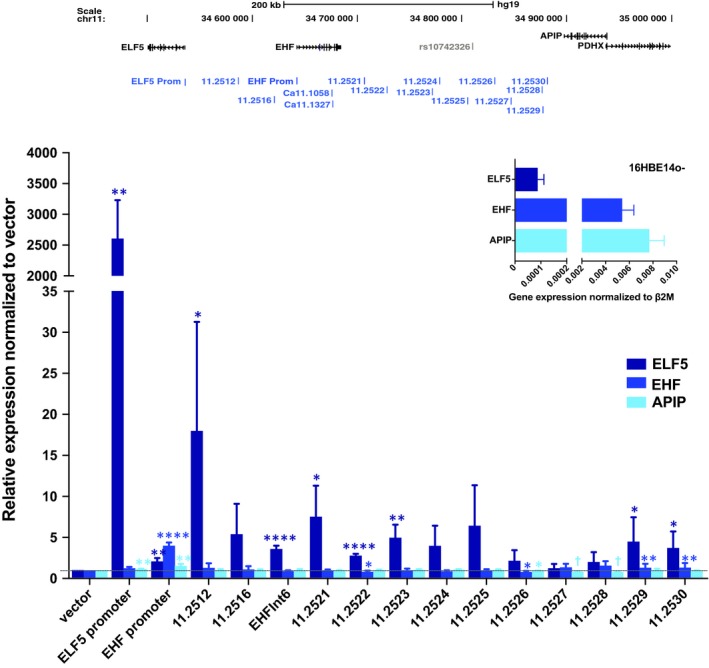
Changes in 11p13 gene expression with VPR‐mediated activation of promoters and cis‐elements in 16HBE14o‐ cells. VPR‐mediated activation of ELF5 promoter, EHF promoter and DHS at 11p13. EHF intron 6 encompasses Ca11.1058 and Ca11.1327. Error bars are SEM, n = 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by an unpaired Student's t test. † P < .0001 – statistically significant, but not biologically significant due to low expression levels. Inset panel shows ELF5, EHF and APIP gene expression for 16HBE14o‐ cells. Error bars are SEM, n = 3
3.6. ELF5 occupies regions of open chromatin at EHF and represses its expression
We next tested the hypothesis that the close physical interactions between the active ELF5 and EHF loci and their CREs might underlie the complex regulatory environment at chr11p13. We first performed siRNA‐mediated depletion of ELF5 in T47D cells, which express ELF5 and EHF, and assayed expression of both genes by RT‐qPCR. Primers located in exons 5 and 6 for ELF5 and exons 3 and 4 for EHF (Table S3) enabled detection of all major isoforms of both transcripts. Our results show that ELF5 depletion significantly increases EHF mRNA expression (Figure 6A). Correspondingly, ELF5 depletion also significantly increased EHF protein expression, suggesting that EHF transcription is repressed by ELF5 (Figure 6B).
Figure 6.
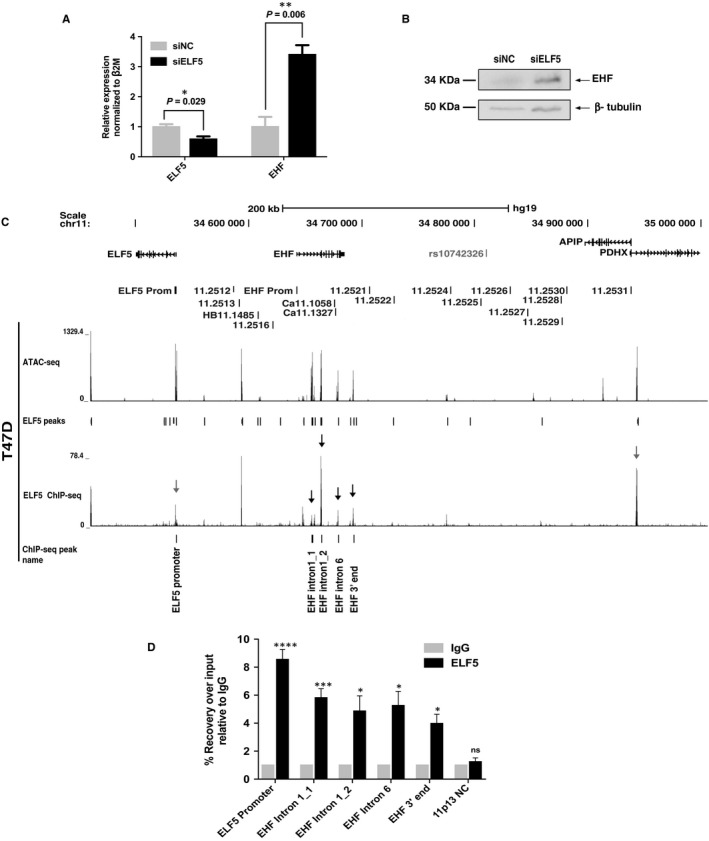
ELF5 binds to EHF locus and represses gene expression. (A) mRNA expression analysis of EHF and ELF5 during siRNA‐mediated depletion of ELF5. Error bars are SEM, n = 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by an unpaired Student's t test. (B) Western blot showing increase in EHF protein levels in response to transfection of T47D cells with siRNA against ELF5. (C) Open chromatin and ELF5 occupancy across 11p13 in T47D cells; ATAC‐seq analysis, upper track and sites of ELF5 occupancy identified by ELF5 ChIP‐seq, lower track. (D) ChIP followed by qPCR analysis confirming enrichment of ELF5 at multiple sites (marked) within the EHF locus. Error bars are SEM, n = 3. ****P < .0001, ***P < .001, **P < .01, *P < .05, ns = not significant by an unpaired Student's t test
A previous study18 suggested a direct repression of EHF by ELF5, though as ELF5 was overexpressed, there were concerns of potential off‐target effects of the TF. To determine whether the repression of EHF by ELF5 observed in our experiments was dependent on direct occupancy of CREs for the EHF locus by ELF5, we performed ChIP‐seq using an antibody specific for endogenous ELF5 in T47D cells. The Irreproducible Discovery Rate (IDR) analysis of two replica experiments is shown in Figure 6C and reveals multiple sites of ELF5 occupancy across the EHF locus, most notably in introns 1 and 6 and close to its 3’ end (black arrows). Each of the peaks of ELF5 binding also coincides with a peak of open chromatin in T47D cells, mapped by ATAC‐seq (Figure 6C upper track). The sites of ELF5 occupancy at the EHF locus were confirmed by ChIP‐qPCR analysis (Figure 6D). In addition, ELF5 occupancy coinciding with sites of open chromatin is seen at the APIP/PDHX and ELF5 gene promoters (grey arrows), suggesting that ELF5 may have a major impact across 11p13, both through controlling other genes and by autoregulation. These results are highly relevant to primary bronchial and tracheal epithelial cells, which also express both ELF5 and EHF, albeit with much lower ELF5 abundance than in T47D cells.
4. DISCUSSION
Intense interest in the chr11p13 region arose through its association with lung disease severity in CF; however, the genetic mechanisms underlying the functions of this region are not well understood. Our data reveal a complex regulatory network at chr11p13 that coordinates the expression of the ETS transcription factors ELF5 and EHF. We previously identified several putative airway epithelial cell‐selective enhancer elements of ELF5 and EHF within the region and defined important aspects of the chromatin architecture relevant to the regulation of genes at chr11p13.25 Here, we performed functional genomics experiments to elucidate the endogenous role of two predicted enhancers and a CRE that lies within a stretch enhancer at the EHF locus.
Deletion of the CRE in EHF intron 6 and the two airway epithelial cell‐selective enhancers at 11.2516 and 11.2521 using CRISPR/Cas9 protocols had unexpected effects on gene expression, and were associated with modest changes in CTCF occupancy and local higher order chromatin structure. Firstly, removal of the two enhancers that were shown to activate the ELF5 and EHF gene promoters in transient luciferase assays25 did not reduce endogenous ELF5 or EHF expression. Removal of either element enhanced ELF5 transcript abundance and had no impact on EHF levels. Though these data do not support a simple enhancer role for the 11.2521 and 11.2516 CREs, these elements are both marked with active histones (H3K27Ac) and furthermore lack repressive marks in several airway cell types, supporting their role as enhancers in the genomic context.25 This suggests additional mechanisms contribute to ELF5 and EHF gene expression levels in their endogenous location, perhaps providing functional redundancy. These factors may include features of the higher order chromatin structure and organization. This interpretation would be consistent with the luciferase assays showing enhancer activity, since within the reporter gene constructs potential CREs are sited next to a gene promoter without consideration for architectural barriers such as CTCF sites. It is also possible that eRNA transcription arising at these CREs may repress nearby gene expression.46 Global run‐on sequencing (GRO‐seq) in HCT116 cells shows eRNA transcription at the three of CREs studied here, notably from the antisense strand at EHF intron 6 (Figure S4).47 This CRE may repress EHF transcription, as intragenic eRNA expression is known to interfere with Pol II‐mediated transcription,46 in which case removal of the element could enhance EHF expression. The CRISPR/Cas9 mediated deletion experiments also uncovered a novel and important function for the CRE at EHF intron 6. This element appears to facilitate looping of the nearby enhancer, 11.2516, to both the ELF5 and EHF promoters. The observed reduction in CTCF occupancy at 11.2516 (Figure 1C), accompanied by loss of the 11.2516 interactions with the promoters as shown by 4C‐seq (Figure 1D), upon deletion of intron 6 is supportive of this role. These data are consistent with the reported mechanisms of looping of enhancers to target promoters at other loci.28, 48, 49, 50 In summary, the removal of key CREs from chr11p13 did not add clarity to our understanding of its functional genomics. However, importantly it focused our interest on the potential role of ELF5 in regulatory mechanisms in the region, since this gene was the most responsive to CRE manipulations.
ELF5 is absent from commonly used human airway epithelial cell lines, though it is expressed at low levels in primary HBE and HTE cells.42 Hence, in order to pursue our interest in ELF5 and the 5’ end of the chr11p13 region we used other epithelial cell lines (T47D and LNCaP) that express both ELF5 and EHF. Analysis of these lines revealed both novel peaks of open chromatin (probable CREs) and an altered chromatin architecture associated with expression of ELF5. Two sites are of particular interest: 11.2513, is a peak of open chromatin in primary HTE cells45 but lacks H3K27ac in the airway cell types25 so was not investigated further; also of interest is a novel element immediately 3’ to the EHF locus, which is not evident in primary HTE cells, but is clearly seen in both ELF5‐expressing cell types studied here. Upon removal of the 11.2521 enhancer from airway cells, the ELF5 promoter gains interactions with this element (Figure 2D) suggesting that this site is at least in part, responsible for the large increase in ELF5 transcript levels in these deletion clones. It seems probable the 11.2513 and novel CREs either harbour enhancers of the ELF5 promoter and/or are sites of occupancy for this transcription factor.
In addition to the novel CREs observed in T47D and LNCaP cells, 4C‐seq data showed a loss of interactions between the EHF promoter and the HB11.1485 CRE in comparison to the airway cell lines (Figure 4C). In this context, our earlier work25 where we noted that though HB11.1485 is a CTCF binding site that may demarcate a sub‐TAD boundary in airway cells, it might also have a structural role in the cell type‐specific regulation of ELF5, was prescient. The loss of interaction between the EHF promoter and HB11.1485 in ELF5‐expressing cells suggests this site is a key sub‐TAD boundary between the EHF and ELF5 promoters restricting their recruitment of nearby cell‐type specific enhancers. Upon expression of ELF5, this sub‐TAD boundary may disappear to enable interactions between the ELF5 promoter and the CREs at the 5’ end of chr11p13.
Our new data on the role of CREs 5’ to the EHF locus in ELF5 expressing cells, and the 4C‐seq results showing direct interactions of the ELF5 promoter with elements within EHF, suggested that a potential regulatory relationship between these TFs warranted further study. Our results showed that ELF5 directly regulates the EHF gene. Using ChIP‐seq for ELF5 in T47D cells, with validation of ELF5 occupancy by ChIP‐qPCR we identify multiple binding sites for ELF5 within the EHF locus, particularly within introns 1 and 6. These sites of ELF5 occupancy coincide with peaks of open chromatin, which are particularly evident in ELF5‐expressing cells. Furthermore, siRNA‐mediated depletion of the ELF5 transcript enhanced EHF mRNA and protein levels. These data suggest that ELF5 is a direct repressor of EHF expression. Our results are consistent with observations on other ETS factors, which are known to regulate each other (reviewed in51); however, the identification of an ETS family member regulating the important EHF TF is novel.
Returning to the role of chr11p13 as a modifier of lung disease severity, we do not address here the mechanisms underlying the association of high p‐value SNP close to the chr11.2525 site. However, our observations on cell‐specific recruitment of CREs across the modifier region and most importantly on the direct regulation of EHF by ELF5 are significant advances in understanding the region. EHF is intimately involved in regulating the wound response, inflammation and tissue remodelling, all of which are integral components of CF lung pathology.11, 12 For example, EHF enhances expression of S100 Calcium Binding Proteins A8 and A9 (S100A8 and S100A9), which are essential for cell motility52, 53, 54 and thus the wound response. Proper wound response is crucial for injury repair, and this process is delayed in the CF lung.12 Also, EHF enhances the transcription of genes encoding the pro‐inflammatory cytokines interleukin 8 (IL‐8) and C‐X‐C motif chemokine ligand 6 (CXCL6).11, 12 These secreted cytokines are necessary for the recruitment of neutrophils to a site of infection or injury.55, 56 Increasing the neutrophil burden in the CF lung can lead to chronic inflammation and eventual tissue remodelling,57 both of which significantly contribute to patient mortality. Furthermore, EHF promotes expression of Sam Pointed‐Domain Containing ETS Transcription Factor (SPDEF),12 which is involved in interleukin 13 (IL‐13)‐mediated goblet cell differentiation.58 Enhanced goblet cell differentiation increases expression of the mucin 5AC (MUC5AC) gene,59, 60 which encodes one of the major secreted mucins in the lung (reviewed in61) and is a hallmark of CF lung disease. Since our data show that ELF5 occupies open chromatin sites at the EHF locus and represses its expression, it is likely that this ETS factor also plays an important role in lung biology. Modulation of ELF5 expression would be predicted to impact many of the EHF‐mediated processes discussed above. Although EHF remains the primary candidate for influencing lung phenotype differences observed in CF patients, ELF5 likely also plays an important role through its regulation of EHF expression. These observations are not only relevant to CF but also to other airway diseases associated with inflammation, defective wound response and lung tissue remodelling, including asthma, chronic obstructive pulmonary disease (COPD) and bronchiectasis.62, 63
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
HS, JSE, KML and AH conceived and designed the project. HS, JSE, KML, SY, JLK, MN, CC, EMM and S‐HL designed and performed experiments. HS and AH wrote the manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health [R01 HL117843, T32 GM008056 (HS) and F31 HL146010 (HS)] and the Cystic Fibrosis Foundation [Harris 16G0, 15/17XX0, 18P0, Ebron 18F0]. We thank Dr P. Faber and staff at the University of Chicago Genomics Core for 4C‐seq sequencing and Dr A. Hoffman for generating A549 c9 11.2521 deletion clones and also Drs MR Knowles, WK O'Neal and H. Dang for helpful discussions.
Swahn H, Sabith Ebron J, Lamar K‐M, et al. Coordinate regulation of ELF5 and EHF at the chr11p13 CF modifier region. J Cell Mol Med. 2019;23:7726–7740. 10.1111/jcmm.14646
Data Availability Statement: Genome‐wide data are deposited at GEO: GSE131084.
DATA AVAILABILITY STATEMENT
Genome‐wide data are deposited at GEO: GSE131084.
REFERENCES
- 1. Genin E, Feingold J, Clerget‐Darpoux F. Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet. 2008;124:357‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Norrgard K. Genetic variation and disease: GWAS. Nature Education. 2008;1:87. [Google Scholar]
- 3. Wright FA, Strug LJ, Doshi VK, et al. Genome‐wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 2011;43:539‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corvol H, Blackman SM, Boelle PY, et al. Genome‐wide association meta‐analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun. 2015;6:8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dang H, Gallins PJ, Pace RG, et al. Novel variation at chr11p13 associated with cystic fibrosis lung disease severity. Hum Genome Var. 2016;3:16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. CF Foundation . https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/. Accessed January 2019.
- 7. Tugores A, Le J, Sorokina I, et al. The epithelium‐specific ETS protein EHF/ESE‐3 is a context‐dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem. 2001;276:20397‐20406. [DOI] [PubMed] [Google Scholar]
- 8. Kleinbaum LA, Duggan C, Ferreira E, Coffey GP, Buttice G, Burton FH. Human chromosomal localization, tissue/tumor expression, and regulatory function of the ets family gene EHF. Biochem Biophys Res Commun. 1999;264:119‐126. [DOI] [PubMed] [Google Scholar]
- 9. Taniue K, Oda T, Hayashi T, Okuno M, Akiyama T. A member of the ETS family, EHF, and the ATPase RUVBL1 inhibit p53‐mediated apoptosis. EMBO Rep. 2011;12:682‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cangemi R, Mensah A, Albertini V, et al. Reduced expression and tumor suppressor function of the ETS transcription factor ESE‐3 in prostate cancer. Oncogene. 2008;27:2877‐2885. [DOI] [PubMed] [Google Scholar]
- 11. Fossum SL, Mutolo MJ, Yang R, et al. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014;42:13588‐13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fossum SL, Mutolo MJ, Tugores A, et al. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J Biol Chem. 2017;292:10938‐10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mutolo MJ, Leir SH, Fossum SL, Browne JA, Harris A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem J. 2018;475:1323‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Solomon GM, Marshall SG, Ramsey BW, Rowe SM. Breakthrough therapies: Cystic fibrosis (CF) potentiators and correctors. Pediatr Pulmonol. 2015;50(Suppl 40):S3‐S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gallego‐Ortega D, Ledger A, Roden DL, et al. ELF5 Drives Lung Metastasis in Luminal Breast Cancer through Recruitment of Gr1+ CD11b+ Myeloid‐Derived Suppressor Cells. PLoS Biol. 2015;13:e1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Metzger DE, Stahlman MT, Shannon JM. Misexpression of ELF5 disrupts lung branching and inhibits epithelial differentiation. Dev Biol. 2008;320:149‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang JY, Shyur SD, Lam FW, Wu LS. Polymorphisms of EHF‐ELF5 genomic region and its association with pediatric asthma in the Taiwanese population. J Microbiol Immunol Infect. 2016;49:879‐884. [DOI] [PubMed] [Google Scholar]
- 18. Kalyuga M, Gallego‐Ortega D, Lee HJ, et al. ELF5 suppresses estrogen sensitivity and underpins the acquisition of antiestrogen resistance in luminal breast cancer. PLoS Biol. 2012;10:e1001461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chavez A, Scheiman J, Vora S, et al. Highly efficient Cas9‐mediated transcriptional programming. Nat Methods. 2015;12:326‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lieber M, Smith B, Szakal A, Nelson‐Rees W, Todaro G. A continuous tumor‐cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer. 1976;17:62‐70. [DOI] [PubMed] [Google Scholar]
- 21. Shen BQ, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu‐3: a human airway epithelial cell line that shows cAMP‐dependent Cl‐ secretion. Am J Physiol. 1994;266:L493‐501. [DOI] [PubMed] [Google Scholar]
- 22. Cozens AL, Yezzi MJ, Kunzelmann K, et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38‐47. [DOI] [PubMed] [Google Scholar]
- 23. Horoszewicz JS, Leong SS, Kawinski E, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809‐1818. [PubMed] [Google Scholar]
- 24. Keydar I, Chen L, Karby S, et al. Establishment and characterization of a cell line of human breast carcinoma origin. Eur J Cancer. 1979;15:659‐670. [DOI] [PubMed] [Google Scholar]
- 25. Stolzenburg LR, Yang R, Kerschner JL, et al. Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res. 2017;45:8773‐8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phylactides M, Rowntree R, Nuthall H, Ussery D, Wheeler A, Harris A. Evaluation of potential regulatory elements identified as DNase I hypersensitive sites in the CFTR gene. Eur J Biochem. 2002;269:553‐559. [DOI] [PubMed] [Google Scholar]
- 27. Benchling [Biology Software] . https://benchling.com. Accessed May 2018.
- 28. Yang R, Kerschner JL, Gosalia N, et al. Differential contribution of cis‐regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 2016;44:3082‐3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ott CJ, Blackledge NP, Kerschner JL, et al. Intronic enhancers coordinate epithelial‐specific looping of the active CFTR locus. Proc Natl Acad Sci U S A. 2009;106:19934‐19939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerschner JL, Harris A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem J. 2012;446:203‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van de Werken HJ, Landan G, Holwerda SJ, et al. Robust 4C‐seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012;9:969‐972. [DOI] [PubMed] [Google Scholar]
- 32. Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parker SC, Stitzel ML, Taylor DL, et al. National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program A, Authors NCSP. Chromatin stretch enhancer states drive cell‐specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013; 110: 17921‐17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Birney E, Stamatoyannopoulos JA, Dutta A, et al. Encode Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007; 447: 799‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489: 57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kellis M, Wold B, Snyder MP, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111:6131‐6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661‐678. [DOI] [PubMed] [Google Scholar]
- 38. Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghirlando R, Felsenfeld G. CTCF: making the right connections. Genes Dev. 2016;30:881‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Wit E, Braunschweig U, Greil F, Bussemaker HJ, van Steensel B. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008;4:e1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barutcu AR, Maass PG, Lewandowski JP, Weiner CL, Rinn JL. A TAD boundary is preserved upon deletion of the CTCF‐rich Firre locus. Nat Commun. 2018;9:1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gillen AE, Yang R, Cotton CU, et al. Molecular characterization of gene regulatory networks in primary human tracheal and bronchial epithelial cells. J Cyst Fibros. 2018;17:444‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC‐seq: A method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol. 2015; 109: 1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods. 2013;10:1213‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bischof JM, Ott CJ, Leir SH, et al. A genome‐wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 2012;67:385‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cinghu S, Yang P, Kosak JP, et al. Intragenic enhancers attenuate host gene expression. Mol Cell. 2017; 68(1):104‐117.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. GRO‐seq . https://nascent.colorado.edu/. Accessed July 2019.
- 48. Holwerda SJ, de Laat W. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mora A, Sandve GK, Gabrielsen OS, Eskeland R. In the loop: promoter‐enhancer interactions and bioinformatics. Brief Bioinform. 2016;17:980‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ren G, Jin W, Cui K, et al. CTCF‐Mediated Enhancer‐Promoter Interaction Is a Critical Regulator of Cell‐to‐Cell Variation of Gene Expression. Mol Cell. 2017; 67(6):1049‐1058.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharrocks AD. The ETS‐domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827‐837. [DOI] [PubMed] [Google Scholar]
- 52. Kwon CH, Moon HJ, Park HJ, Choi JH, Park DY. S100A8 and S100A9 promotes invasion and migration through p38 mitogen‐activated protein kinase‐dependent NF‐kappaB activation in gastric cancer cells. Mol Cells. 2013;35:226‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lim SY, Yuzhalin AE, Gordon‐Weeks AN, Muschel RJ. Tumor‐infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene. 2016;35:5735‐5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Y, Zhang Z, Zhang L, et al. S100A8 promotes migration and infiltration of inflammatory cells in acute anterior uveitis. Sci Rep. 2016;6:36140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171‐197. [DOI] [PubMed] [Google Scholar]
- 56. Jovic S, Linge HM, Shikhagaie MM, et al. The neutrophil‐recruiting chemokine GCP‐2/CXCL6 is expressed in cystic fibrosis airways and retains its functional properties after binding to extracellular DNA. Mucosal Immunol. 2016;9:112‐123. [DOI] [PubMed] [Google Scholar]
- 57. Castellani S, Conese M. Not All Is CFTR ‐ Neutrophils and Cholesterol in Cystic Fibrosis. EBioMedicine. 2017;24:28‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park KS, Korfhagen TR, Bruno MD, et al. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest. 2007;117:978‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kanoh S, Tanabe T, Rubin BK. IL‐13‐induced MUC5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin Exp Allergy. 2011;41:1747‐1756. [DOI] [PubMed] [Google Scholar]
- 60. Kondo M, Tamaoki J, Takeyama K, Nakata J, Nagai A. Interleukin‐13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am J Respir Cell Mol Biol. 2002;27:536‐541. [DOI] [PubMed] [Google Scholar]
- 61. Ridley C, Thornton DJ. Mucins: the frontline defence of the lung. Biochem Soc Trans. 2018;46:1099‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Athanazio R. Airway disease: similarities and differences between asthma, COPD and bronchiectasis. Clinics. 2012;67:1335‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gernez Y, Tirouvanziam R, Chanez P. Neutrophils in chronic inflammatory airway diseases: can we target them and how? Eur Respir J. 2010;35:467‐469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome‐wide data are deposited at GEO: GSE131084.