

Abstract
The A‐kinase anchoring proteins (AKAPs) are a group of structurally diverse proteins identified in various species and tissues. These proteins are able to anchor protein kinase and other signalling proteins to regulate cardiac function. Acting as a scaffold protein, AKAPs ensure specificity in signal transduction by enzymes close to their appropriate effectors and substrates. Over the decades, more than 70 different AKAPs have been discovered. Accumulative evidence indicates that AKAPs play crucial roles in the functional regulation of cardiac diseases, including cardiac hypertrophy, myofibre contractility dysfunction and arrhythmias. By anchoring different partner proteins (PKA, PKC, PKD and LTCCs), AKAPs take part in different regulatory pathways to function as regulators in the heart, and a damaged structure can influence the activities of these complexes. In this review, we highlight recent advances in AKAP‐associated protein complexes, focusing on local signalling events that are perturbed in cardiac diseases and their roles in interacting with ion channels and their regulatory molecules. These new findings suggest that AKAPs might have potential therapeutic value in patients with cardiac diseases, particularly malignant rhythm.
Keywords: A‐kinase anchoring proteins, arrhythmia, calmodulin, cardiomyocytes, hypertrophy, large‐conductance Ca2+‐activated K+ channels, sudden cardiac death
1. INTRODUCTION
A‐kinase anchoring proteins (AKAPs) have been identified in a number of species and tissues and are related to the composition of a wide variety of complexes implicated in different signalling cascades. AKAPs are distinguished by their ability to bind cyclic adenosine monophosphate (cAMP)‐dependent protein kinase A (PKA) as well as other signalling enzymes at focal points within the cell to ensure the integration and processing of multiple signalling pathways.1 Additional signalling proteins including adenylyl cyclases (ACs), phosphodiesterases (PDEs), protein kinases, phosphatases, GTPases and ion channels locate on the anchoring protein. AKAPs recruit these signalling molecules and form multifunctional complexes to generate protein‐protein interactions.2, 3, 4 AKAPs have been traditionally named on the basis of their apparent molecular weight, whereas in different species, there is disparity in the naming of the same anchoring protein. Hence, the Human Genome Organisation (HUGO) gives approval for the nomenclature of the AKAPs.1
Cyclic AMP is a widespread intracellular second messenger that regulates numerous physiological and pathological processes. The concentration and signalling of cAMP are tightly controlled and co‐ordinated through the involvement of molecular machinery co‐ordinating the spatial and temporal processes of localized cAMP signalling events.5, 6 Stimulating G protein‐coupled receptors (GPCRs), such as β adrenoceptor, by interacting with α subunit of Gs protein (αs) promotes signal transduction through the cAMP pathway via specific extracellular ligands, leading to the activation of most ACs, which convert ATP into cAMP. Through the generation of cAMP following AC activation, cAMP‐dependent PKA is activated, and ligands that stimulate GPCRs coupled to Gi can inhibit AC activity. Furthermore, cAMP may be degraded by PDEs7, 8, 9, 10 (see Figure 1A).
Figure 1.
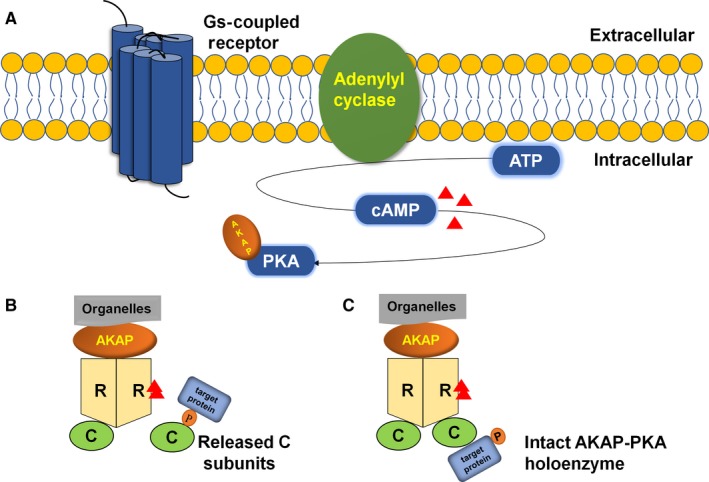
A, Stimulating GPCRs promotes signal transduction through the cAMP pathway via specific extracellular ligands leading to the activation of AC, which converts ATP into cAMP. AKAP anchors in PKA. B, cAMP binding to the R subunits of PKA increases, and the active catalytic subunits are released to phosphorylate their targets. C, This compact state may provide for the phosphorylation of associated target proteins
The PKA holoenzyme is a heterotetramer consisting of two regulatory (R) subunits that maintain two catalytic (C) subunits in an inhibited state.11 The holoenzyme can dissociate into a regulatory subunit dimer (each monomer binds two cAMP) and two free but active catalytic subunits when binding four molecules of cAMP.12 There are three genes for the C subunit gene products, including Cα, Cβ and Cγ. The R genes have been divided into four different types: RIα, RIβ, RIIα and RIIβ.13 Cα1, Cα2 and Cα3 are contained in Cα isoforms. Cα1 exists in a wide variety of human tissues; Cα2 is mainly expressed in sperm cells; and the expression of Cα3 remains to be elucidated. The Cβ isoform has been found in human tissues, and the function of the Cγ isoform, which is expressed in testis tissue, remains unclear.14, 15 The four PKA R subunit isoforms share a universal domain organization containing the N‐terminal dimerization/docking (D/D) domain, a linker including the inhibitor site, and two consecutive cAMP‐binding domains. Differs in cAMP responsiveness and subcellular localization show that RI isoforms are predominantly diffuse in the cytoplasm and are more sensitive to cAMP signalling, whereas the RII isoforms are more localized in cells and less responsive to cAMP signalling.16 The RI subunits have a pseudosubstrate binding site, and the RII subunits are not only substrates but also inhibitors of the C subunit. However, the phosphorylated RII dimer does not dissociate from the C subunits in the absence of cAMP. The R subunits are tightly bound to the C subunits, thereby preventing the C subunit from interacting with external protein substrates. The cAMP‐binding domain allows the cells to turn the second messenger cAMP signal into a biological response. Therefore, when the structure of PKA is changed, cAMP‐dependent activation is decreased.12, 17, 18, 19 AKAPs are a family of functionally related proteins that interact with the regulatory subunits of the PKA holoenzyme. Through interaction between the hydrophobic pocket of PKA and the 14‐18 amino acid amphipathic helix region of AKAPs, AKAPs anchor the R subunit dimer D/D domain, and AKAPs are responsible for anchoring the two R subunits specifically. Although some AKAPs show specificity for RI and RII subunits, most AKAPs tend to show more specificity for the RII subunit than that for the RI subunit.3, 20 The RIIα D/D domain can accommodate various side chains at numerous positions of the AKAP peptide; the flexible N terminus of the D/D domain is the crucial one. At many cellular microdomains, cAMP signalling is amplified by facilitating PKA interactions with many AKAPs, which results from this kind plasticity of the D/D domain.21 Moreover, spatially restricted activation of PKA is guaranteed by the binding of this kinase with AKAPs21, 22, 23, 24 (see Figure 1B). In contrast, a recent study has shown that even local cAMP production stimulates kinase activity, and AKAP79:2RII:2C assemblies remain intact, which means AKAP‐PKA holoenzyme assemblies remain intact (see Figure 1C). cAMP production in response to physiological effectors of GPCR signalling appears not to promote catalytic subunit release from anchored PKA holoenzymes.25 But this result has been challenged recently by Gray's group,26 which proved that catalytic subunits are released from regulatory subunits by cAMP, and during cAMP activation, tether to R subunits does not restrict C subunit activity. These views remain controversial and have yet to be explored.
Over the decades, more than 70 different AKAPs have been discovered in various cells, and accumulating evidence has indicated that several AKAPs play key roles in modulating multiple signalling pathways in the vasculature and in the heart. By co‐ordinating signalling pathways, AKAPs modulate the physiological and pathological function of cardiomyocytes and endothelial and smooth muscle cells, thereby influencing vascular and cardiac function (Table 1). AKAPs can function in the heart to influence contractility, action potential, arrhythmias, hypoxia adaptation, heart failure and hypertrophy.2, 27, 28, 29, 30, 31, 32 In this review, we will provide an overview of recent results describing the functional regulation of AKAPs in cardiac pathophysiology.
Table 1.
Characterization of AKAPs in cardiomyocytes
| Function | Gene Name | Alternative Name | Binding Partners | Intracellular Localization | References |
|---|---|---|---|---|---|
| Pro‐hypertrophic | AKAP6 | mAKAP | PKAII, PDE4D3, AC5, RyR2, CaNA, PP2A, NFATc, ERK5, MEK5, Epac1, Rap1, Siah2, PDK1, RSK3, NCX1, nesprin‐1α, | Nuclear envelope | 1, 2, 38, 39, 40, 41, 42, 43, 44, 45, 46, 96, 97, 98 |
| AKAP13 | AKAP‐Lbc, Ht31 | PKA RII, RhoA, Actin, PKC, PKD, KSR1, Raf, MEK1/2, ERK1/2, PKNα | Cytoskeleton | 1, 31, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 99, 100 | |
| Anti‐hypertrophic | AKAP7 | AKAP15, AKAP18 | PKAII, L‐type Ca2+ channel, phospholamban, PP1, inhibitor 1 | Plasma membrane, endoplasmic reticulum | 1, 4, 59, 60, 61, 62, 63, 101 |
| AKAP1 | D‐AKAP1, AKAP121, AKAP149 | PKAI and II, PKCα, Src, RSK1, PP1, PP2A, CaN, PTPD1, Lfc | Mitochondria, nuclear envelope, endoplasmic reticulum | 1, 2, 35, 36, 37, 102, 103, 104 | |
| AKAP5 | AKAP79, AKAP150 | PKAII, PKC, CaN, KCNQ2, L‐type Ca2+ channel, β‐AR, AC5 and AC‐6, SAP97, caveolin‐3 | Plasma membrane, T tubules | 1, 2, 3, 22, 28, 65, 66, 67, 84, 85, 86, 87, 105, 106 | |
| Contractility | AKAP5 | AKAP79, AKAP150 | PKAII, PKC, CaN, KCNQ2, L‐type Ca2+ channel, β‐AR, AC5 and AC‐6, SAP97, caveolin‐3 | Plasma membrane, T tubules | 1, 2, 3, 22, 28, 65, 66, 67, 84, 85, 86, 87, 105, 106 |
| AKAP12 | Gravin, AKAP250 | PKA RII, β‐AR, PKC, PDE4D, Src | Plasma membrane | 1, 2, 80, 81, 82, 83, 107, 108 | |
| Arrhythmias | AKAP9 | Yotiao, AKAP350, AKAP450 | PKAII, PP1, PP2A, PKC, PKN1, kinase 1, AC, PDE4D3, KCNQ1, CLIC | Plasma membrane, Golgi, centrosome | 1, 2, 3, 84, 85, 86, 87, 88, 109, 110 |
| AKAP5 | AKAP79, AKAP150 | PKAII, PKC, CaN, KCNQ2, L‐type Ca2+ channel, β‐AR, AC5 and AC‐6, SAP97, caveolin‐3 | Plasma membrane, T tubules | 1, 2, 3, 22, 28, 65, 66, 67, 84, 85, 86, 87, 105, 106 |
2. CARDIAC HYPERTROPHY
2.1. D‐AKAP1
D‐AKAP1, which means a dual‐specificity A‐kinase anchoring protein, binds to both the RI and RII subunits of PKA.33 Several D‐AKAP1 isoforms or homologues were identified in various species. These isoforms include mouse AKAP121, rat AKAP121 and human AKAP149.34 It has been shown that down‐regulation of D‐AKAP1 is related to oxidative stress, mitochondrial dysfunction, cardiomyocyte hypertrophy and apoptosis.35, 36, 37
Previous studies have shown that knockdown of D‐AKAP1 induces, rather than inhibits, hypertrophy. In contrast, overexpression of D‐AKAP1 has the opposite effect on cell size. On one hand, cell size is reduced by increased D‐AKAP1 expression. The effect of the hypertrophic adrenergic agonist isoproterenol is inhibited. The result of D‐AKAP1 knockdown on hypertrophy is mediated by the activation of the calcineurin (CaN)/nuclear factor of activated T cell (NFATc) pathway, as shown by alterations in intracellular NFATc3 localization. Small hairpin RNA (shRNA) experiments have been performed to show that D‐AKAP1 knockdown induces NFATc3 dephosphorylation and translocation to the nucleus, resulting in hypertrophy.35
In addition, other studies demonstrated that D‐AKAP1 is an important regulator of mitochondrial function and cell survival, and thus, D‐AKAP1 down‐regulation may represent an important event in the development of cardiac dysfunction. Displacement of D‐AKAP1 from mitochondria is closely related to increased reactive oxygen species (ROS) generation, and ROS production induces D‐AKAP1 degradation. Accumulation of ROS also promotes cardiomyocyte apoptosis. This is an important observation because it suggests that improper cAMP signalling can spill over into mitochondrial regulatory pathways, connecting cardiomyocyte survival and oxidative stress.36
2.2. mAKAP
The scaffolding protein muscle‐selective AKAP (mAKAP), also known as AKAP6,38 is a PKA‐anchoring partner that is expressed in the brain, heart and skeletal muscle. α and β are two alternatively spliced forms of mAKAP, which is required in cardiac myocytes for the induction of cardiac hypertrophy by transverse aortic constriction and isoproterenol infusion.39, 40 However, mAKAP‐β, mainly expressed in heart and skeletal muscle, plays a crucial role in myoblast differentiation, myotube formation and muscle regeneration.38, 41 The classical view is that mAKAP complex anchoring extracellular‐regulated protein kinases 5 (ERK5) can induce cardiac hypertrophy.42 During the past few years, some literature has revealed several novel signalling pathways by which mAKAP regulates cardiac hypertrophy.39, 40, 41, 42, 43, 44, 45
First, phospholipase Cε (PLCε) scaffolded to mAKAP is a multifunctional enzyme implicated in cardiovascular, pancreatic and inflammatory functions. Evidence shows that PLCε generates second messengers at the nuclear envelope that are required for hypertrophy, and phosphatidylinositol 4‐phosphate (PI4P) is a perinuclear substrate in the Golgi apparatus for mAKAP‐scaffolded PLCε.43, 44 PI4P, together with PLCε, is a substrate for mammalian PLC isoforms. Activation of mAKAP‐scaffolded PLCε is directly involved in perinuclear PI4P depletion, which means that, as a perinuclear enzyme, PLCε can hydrolyse PI4P to produce diacylglycerol (DAG). Notably, cardiac hypertrophy development is significantly reduced after the cardiac‐specific deletion of PLCε. This strongly suggests that mAKAP‐PLCε signalling in cardiac myocytes is important for hypertrophy development. Furthermore, the neonatal myocyte analysis of PLCε function is largely relevant to the function of the whole heart.44
In addition, the scaffolding protein mAKAP organizes a calcineurin/myocyte enhancer factor 2 (MEF2) signalling complex in myocytes to regulate gene transcription. In the stressed heart, MEF2 is significant for the transactivation of hypertrophic gene transcription.45, 46 A laboratory used primary neonatal rat cardiac myocytes transfected with expression plasmids for either control mCherry or mCherry‐CaNBD and then stimulated the cells for two days with norepinephrine, which is a type of adrenergic agonist that can increase the cross‐sectional area of cells. By measuring the cellular cross‐sectional area on images, it was found that there was no distinct difference in size between treated and untreated myocytes expressing mCherry‐CaNBD. Meanwhile, the expression of atrial natriuretic factor (ANF), a marker for hypertrophy encoded by the MEF2‐transactivated Nppa gene, was down‐regulated in mCherry‐CaNBD‐expressing myocytes after adrenergic stimulation. Taken together, these data suggest that calcineurin binding to mAKAP is required for the induction of cardiac hypertrophy and that this event is mediated by MEF2.46
Additionally, mAKAP‐β contributes to the orchestration of Ca2+‐dependent signalling transduction. During states of elevated sympathetic stimulation, PKA‐catalysed ryanodine receptor Ca2+ release channel (RyR2) phosphorylation could increase local Ca2+ release with the participation of mAKAP‐β. Ca2+ is released to induce sarcomeric contraction, and mAKAP‐β complexes may connect contractility to the induction of hypertrophy.38 Lee et al further studied skeletal myoblast differentiation and muscle regeneration based on mAKAP. mAKAP knockdown was shown to markedly impede the formation of myotubes and decrease myoblast differentiation and skeletal muscle regeneration.41
2.3. AKAP‐Lbc
AKAP‐Lbc (also known as AKAP13 and Ht3147) is a Rho‐specific guanine nucleotide exchange factor inside cells, and it functions as a scaffolding protein to co‐ordinate the Rho signalling pathway. AKAP‐Lbc not only anchors PKA but can also activate Rho.48 Diviani's group has undertaken a number of fundamental studies on AKAP‐Lbc, and they identified AKAP‐Lbc as the first Rho‐guanine nucleotide exchange factor (GEF) involved in signalling pathways leading to cardiomyocyte hypertrophy by activating RhoA and transducing hypertrophic signals downstream of α1‐adrenergic receptors (ARs).49 It has been demonstrated that AKAP‐Lbc is up‐regulated in human hypertrophic cardiomyopathy.50 However, AKAP‐Lbc assembles a macromolecular signalling complex to co‐ordinate the activity of transduction enzymes, which has a direct impact on compensatory hypertrophy and maintenance of cardiac function during the early‐phase of cardiac remodelling.31
AKAP‐Lbc‐ΔPKD1 is a truncated form of AKAP‐Lbc that is unable to bind PKD1. Because of the deletion of the PKD1 binding domain on AKAP‐Lbc, AKAP‐Lbc‐ΔPKD mice exhibit reduced myocyte hypertrophy with increased cardiac extracellular collagen synthesis and apoptosis in response to transaortic constriction (TAC)–induced pressure overload or angiotensin (AT‐II) and phenylephrine (PE) infusion.51 Furthermore, AKAP‐Lbc‐∆PKD1 mice display an altered cardiac transcriptional response to TAC‐induced pressure overload, which means AKAP‐Lbc‐PKD1 signalling is critical for transcriptional regulation during the development of compensatory hypertrophy.52 However, the AKAP‐Lbc/PKD1 complex has been shown to prevent mitochondrial dysfunction and cardiomyocyte death induced by doxorubicin. As a molecular platform, AKAP‐Lbc co‐ordinates protective signals preventing DOX‐induced cardiomyocyte toxicity. Stimulation of α1‐adrenergic receptors (ARs) contributes to the activation of AKAP‐Lbc‐anchored PKD1, and in DOX‐treated cardiomyocytes, two anti‐apoptotic pathways are activated to enhance the expression of Bcl2 and inhibit the mitochondrial translocation of the pro‐apoptotic protein Bax. AKAP‐Lbc/PKD1 complex functions to prevent mitochondrial dysfunction and cardiomyocyte death induced by DOX.53
The tyrosine phosphatase Shp2 is a component of the A‐kinase anchoring protein (AKAP)‐Lbc complex, and the interaction of AKAP‐Lbc and Shp2 inside cells is complicated. Shp2 is a PKA substrate; Shp2 is phosphorylated by PKA in cardiac myocytes in response to isoproterenol stimulation. At the same time, AKAP‐Lbc plays an important role in the regulation of Shp2 activity by facilitating the phosphorylation of Shp2 by PKA.54 Two key amino acids in Shp2, Thr‐73 and Ser‐189 are phosphorylated by PKA.55 In summary, chronic activation of PKA in the hypertrophic heart promotes the inhibition of Shp2 activity associated with AKAP‐Lbc54 (see Figure 2).
Figure 2.
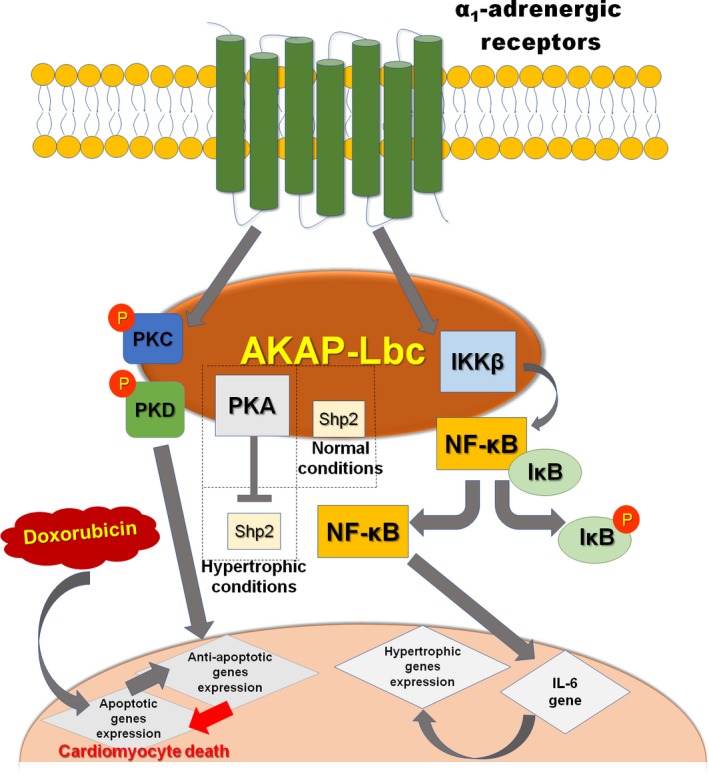
In cardiomyocytes, AKAP‐Lbc mediates IKKβ activation after stimulation of α1‐AR. Activated IKKβ leads to NF‐κB‐dependent production of IL‐6, which in turn engages signalling pathways controlling the transcription of cardiomyocyte hypertrophic genes. AKAP‐Lbc assembles a signalling complex composed of PKA and Shp2 in cardiac myocytes. Some conditions lead to PKA activation, thereby promoting inhibition of Shp2 activity, which may contribute to the induction of cardiac hypertrophy, and the AKAP‐Lbc/PKD signalling complex mediates protection against doxorubicin (DOX)‐induced cardiomyocyte death
IκB is an inhibitor of the transcription factor NF‐κB, which is a mediator of the growth responses induced by a variety of pro‐hypertrophic agonists.56 NF‐κB is recognized as a key transcription factor mediating cardiac hypertrophy.57 The inhibitor of IκB kinase (IKK) complex (IKKα, IKKβ and IKKγ) contributes to the phosphorylation of IκB under stimulation.58, 59 AKAP‐Lbc promotes the activation of anchored IKKβ, which in turn results in the phosphorylation and degradation of IκB and activation of NF‐κB. Finally, activated NF‐κB induces transcription of the IL‐6 gene and subsequent stimulation of IL‐6‐mediated pathways to control foetal gene transcription and cardiomyocyte hypertrophy60 (Figure 2).
2.4. AKAP15
AKAP15 (also known as AKAP7 and AKAP1861), which anchors PKA to calcium channels, is a family of alternatively spliced isoforms (α, β, γ and δ) that are known to play a role in cardiac L‐type calcium dynamics.62 A transgenic mouse with destructed AKAP15/L‐type Ca2+ channel (LTCCs) binding is not sensitive to cAMP stimulation, and the mice also suffer from cardiac hypertrophy.4 AKAP15 directly interacts with the distal C terminus of the cardiac CaV1.2 channel via a leucine zipper‐like motif, and AKAP15 facilitates cardiac contraction via regulation of beta‐adrenergic (β‐AR)‐stimulated L‐type Ca2+ channels.62, 63, 64 The distal C‐terminal domain (DCT) maintains a non‐covalent interaction with the truncated CaV1.2 channel, acting as an autoinhibitor of the channel and reducing the channel's sensitivity. DCT is required for normal responses to β‐AR signalling and AKAP15 localization. Deletion of the DCT induces cardiac hypertrophy, possibly as a result of impairment of regulation of the peripheral vasculature, such as increased peripheral vascular resistance. CaV1.2 channels without DCT cannot be regulated by the β‐AR/PKA signalling pathway and cannot support normal expression and localization of AKAP15. This result underscores the importance of AKAP15 in normal excitation‐contraction coupling and suggests that AKAP15 plays a role as an inhibitor of cardiac hypertrophy.65
2.5. AKAP5
Although the expression of AKAP5 (murine AKAP150, human AKAP7961) in the heart is low, it is widely expressed in the periphery and plays a major role in forming discrete signalling networks. AKAP5 is able to bind and inactivate Ca2+/calmodulin‐dependent phosphatase (CaN). On this condition, CaN‐mediated cardiac hypertrophy can be inhibited.22, 28 However, endogenous CaN activity does not directly regulate cardiac Ca2+ channel activity in mouse myocytes. AKAP5 mice with significantly diminished endogenous CaN activity can retain normal myocyte size.66 Cardiac β‐ARs are key regulators of cardiac size. AKAP5 is a key regulator of myocardial signalling by β‐ARs. Deletion of AKAP5 was associated with significant cardiac hypertrophy.28, 67 Because deletion of AKAP5 prevented the recycling of internal β1‐AR, the influence did not include the internalization of β1‐AR in mouse cardiac myocytes.68
3. MYOFIBRE CONTRACTILITY DYSFUNCTION
3.1. AKAP79/150
AKAP79/150 interacts with PKA, protein kinase C(PKC), Ca2+/calmodulin‐dependent phosphatase (CaN), calmodulin (CaM) and other signalling molecules to regulate vascular tone and blood pressure.28, 29, 30, 69, 70
During hyperglycaemia and diabetes, AKAP79/150 is reported to contribute to enhancing vascular tone through facilitating large‐conductance Ca2+‐activated K+ (BK) channel remodelling. AKAP150 anchors CaN and mediates nuclear factor of activated T cell c3 (NFATc3) activation and the transcriptional suppression of regulatory BK‐β1 subunit during diabetes induced by glucose,29 and the BK‐β1 subunit is a crucial regulatory factor of vascular tone.71 In conclusion, anchoring of calcineurin by AKAP150 is required for BK channel impairment during hyperglycaemia and diabetes, which promotes enhanced vascular tone.29
In addition, hypercontractility of arterial myocytes and enhanced vascular tone during diabetes are attributed to the effects of increased glucose on L‐type CaV1.2 channels.72 α1C is a subpopulation of the CaV1.2 channel pore‐forming subunit, and Ser1928 is a highly conserved PKA consensus phosphorylation site located within the intracellular C terminus of α1C. As a key molecular signalling event underlying the potentiation of CaV1.2 channel activity and vasoconstriction upon acute increases in extracellular D‐glucose and diabetes, the AKAP‐dependent, PKA‐meditated phosphorylation of α1C at Ser1928 plays a vital role in this progression.73
The expression of transient receptor potential vanilloid 4 (TRPV4) channels is comprehensive, and they belong to a kind of Ca2+‐permeable, non‐selective cation channel.74 Contractile function is closely tied to TRPV4 channels in that cardiomyocyte TRPV4 is a novel mediator of enhanced contractile function early in ischaemia‐reperfusion.75 Endothelial impairment can influence the regulation of vascular tone, and endothelial cells (ECs) are assumed to be an important regulator of vasodilatory function. Stimulating some receptors on ECs excites TRPV4 channels, which are localized at myoendothelial projections (MEPs). The PKC‐anchoring protein AKAP79/150 mainly localizes to MEPs, which contributes to the opening of TRPV4 and enhances local Ca2+ influx. In contrast, in hypertension, this molecular assembly is disrupted.76 However, in the sarcolemma of arterial myocytes, the PKCα‐associated, AKAP150‐dependent modulation of TRPV4 channels relies on the distance between these two proteins.77
3.2. Gravin
Beta‐adrenergic receptors (β‐ARs), and especially β2‐AR, are identified as significant regulators of cardiac contractility by activating PKA.78 Gravin, also known as AKAP12 and AKAP250, has the ability to bind β2‐AR.79 Therefore, gravin plays an indispensable role in the β‐AR‐mediated regulation of cardiac contractility.80
In one experiment, isoproterenol (ISO) was applied in wild‐type (WT) and gravin mutant (gravin‐t/t) mice to detect cardiac contractility, and it was found that, at diastole, there was no obvious difference between WT and gravin‐t/t mice. However, at systole, left ventricular internal dimensions (LVID) were decreased in the gravin‐t/t mice compared with WT mice. Moreover, cardiomyocytes isolated from gravin‐t/t mice had enhanced cardiomyocyte contractility in the presence of a proportionally lower diastolic baseline and maximum height of intracellular Ca2+ transients. These results indicated that gravin is a key factor in the desensitization/resensitization cycle of β2‐AR. The signalling mechanism resulting from disruption of gravin's scaffold is such that when the gravin gene is mutant in mice, the baseline cardiac function is increased, and contractility is enhanced in response to acute β‐AR stimulation. At the same time, the phosphorylation of β2‐AR is decreased, which in turn attenuates receptor desensitization.80
Li et al used right ventricles of gravin mutant (gravin‐t/t) mice to test the effect of acute β‐AR stimulation on cardiac contractility in vivo on the absence of gravin binding to β2‐AR, PKA and other signalling molecules. It was shown that gravin‐t/t muscles exhibited increased myofilament Ca2+ responsiveness while maintaining their ability to release Ca2+ from the sarcoplasmic reticulum (SR). The phenomenon revealed that, besides serving as a scaffolding protein, gravin functions as a regulator of myofilament Ca2+ sensitivity. It is obvious that gravin is an important regulator of cardiac contraction via increasing myofilament sensitivity to Ca2+.81
4. CARDIAC ARRHYTHMIAS
4.1. Yotiao
Yotiao is a splice variant of the AKAP9 gene and is present on the plasma membrane. Yotiao displays specificity among AC isoforms and interacts with AC 1, 2, 3 and 9. In addition, Yotiao can co‐ordinate the assembly of the IKs signalling complex.82, 83
Long QT syndrome (LQTS) is a heritable arrhythmia syndrome.84 Previously, it was found that, in the heart, Yotiao (AKAP9) assembles with KCNQ1, which is short for IKs potassium channel subunit, to regulate cardiac action potential duration (APD). Type 1 long QT syndrome (LQT1) results from the disruption of this complex.85 Further experiments were performed to explore the Yotiao missense mutational site, and S1570‐Yotiao was shown to modify Yotiao/KCNQ1 interactions and PKA phosphorylation; furthermore, it also reduced the functional response of Iks channels to cAMP. Therefore, it is obvious that, as an inherited mutation of an AKAP9‐encoded protein, S1570‐Yotiao is relevant to LQTS, and this finding may provide evidence for future clinical treatment.86
4.2. AKAP150
The interaction between AKAP150 and long QT syndrome 8 (LQT8) is also known as Timothy syndrome and is characterized by a single amino acid substitution (G406R) in the L‐type Ca2+ (CaV1.2) channel.87 Disruption of AKAP150 improves pathological CaV1.2‐LQT8 channel gating and arrhythmias and prevents hypertrophy of LQT8 hearts by decreasing Ca2+ influx via CaV1.2‐LQT8 channels.88
AKAP150 is essential for sympathetic stimulation of the Ca2+ transient. AKAP150‐null mice showed unstable R‐R intervals and decreased LF, indicating that the tonus of the sympathetic nerves had been modified. In addition, the AKAP5‐null atrium showed a decreased contractile response to isoproterenol, which means AKAP5‐null mice exhibit a modulated sympathetic nerve response.89, 90
4.3. D‐AKAP2
AKAP10 (D‐AKAP2) binds with high affinity to both the RI and RII regulatory subunits of PKA, and the structure of AKAP10 consists of two tandem regulators of G protein signalling (RGS)–like homology domains followed by a 27‐residue PKA‐binding (AKB) domain and a PSD‐95/DlgA/ZO‐1(PDZ)‐binding motif at the C terminus.91 D‐AKAP2AKB binds to the D/D domain of the R subunit, and the C‐terminal PDZ motif binds to a PDZ domain of NHERF1, NHERF2 and PDZK1, which serves as a bridging protein to the transporter.92
When AKAP10 is mutated in mice, the sensitivity of cultured cardiac cells to cholinergic vagus nerve inputs increases. This result is the same in living mice. In addition, AKAP10‐mutant mice displayed two types of spontaneous cardiac pauses. First, sinus pauses with junctional escape beats were 40 times more frequent in homozygous AKAP10‐mutant mice than in WT mice. Second, atrioventricular (AV) heart block was 15 times more frequent in homozygous AKAP10‐mutant mice. Both types of pauses were typically preceded by changes characteristic of vagus nerve activity.93 Łoniewska et al demonstrated a possible association between the 1936A > G AKAP10 variant and QTc in the aboriginal European newborn population.94
5. CONCLUSIONS AND PERSPECTIVES
It has become increasingly obvious that cardiac AKAP complexes have shed new light on how local signals are co‐ordinated and processed in vascular and cardiac functions. The role of protein kinase compartmentalization is critical in mediating the kinase signalling pathways and explains the development of different disease pathologies in the presence or absence of these AKAPs. All of the experiments above demonstrate the role of the AKAP signalling pathway in diseases such as cardiac hypertrophy, contractility dysfunction and arrhythmias by anchoring PKA, PKC, CaN and CaM. Furthermore, ion channels (L‐type Ca2+ channels, BK channels) are also closely associated with AKAPs.73, 87 On the molecular level, we believe that the implementation of new technologies related to the structural determination of large multiprotein complexes will provide new ways to understand the mechanism of how AKAP complexes function. This will help us to establish specific therapeutic approaches to AKAP‐related diseases. For example, AKAPs assemble localized signalosomes positioning relevant downstream effectors near respective substrate proteins to propagate downstream signalling; however, degradation of cAMP can halt signalling. In cardiovascular system, one family of the known cAMP receptors, the exchange proteins directly activated by cAMP (EPACs), is associated with cardiac hypertrophy. cAMP sensor EPAC‐based therapeutics represent promising alternatives for the management of cardiovascular diseases.95 In addition, AKAPs‐related arrhythmia‐causing mutations will help promote progress towards better therapeutic strategies, and there remains a need for specific treatment towards individuals in a genotype‐driven.
CONFLICT OF INTEREST
The authors declare that they have no competing interest.
AUTHORS CONTRIBUTIONS
Zhu YR and Jiang XX contributed to draft the manuscript; Zheng YG, Xiong J and Wei DP contributed to the discussion; and Zhang DM contributed to conceive and design the review, wrote and revised the manuscript.
Zhu Y‐R, Jiang X‐X, Zheng Y, Xiong J, Wei D, Zhang D‐M. Cardiac function modulation depends on the A‐kinase anchoring protein complex. J Cell Mol Med. 2019;23:7170–7179. 10.1111/jcmm.14659
Yan‐Rong Zhu and Xiao‐Xin Jiang are contributed equally.
Funding information
The project was supported by the National Natural Science Foundation of China (81970342, 81370304, 81472793); Jiangsu Provincial Key Research and Development Program (BE2018611); Natural Science Foundation of Jiangsu Province (BK20151085); and Medical Science and Technology Development Foundation of Nanjing Department of Health (ZKX16048, ZKX15027).
REFERENCES
- 1. Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context‐dependent regulation of anchored enzymes. Mol Interv. 2010;10(2):86‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ercu M, Klussmann E. Roles of A‐kinase anchoring proteins and phosphodiesterases in the cardiovascular system. J Cardiovasc Dev Dis. 2018;5(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skroblin P, Grossmann S, Schafer G, et al. Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol. 2010;283:235‐330. [DOI] [PubMed] [Google Scholar]
- 4. Redden JM, Dodge‐Kafka KL. AKAP phosphatase complexes in the heart. J Cardiovasc Pharmacol. 2011;58(4):354‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bastug‐Ozel Z, Wright PT, Kraft AE, et al. Heart failure leads to altered beta2‐adrenoceptor/cAMP dynamics in the sarcolemmal phospholemman/Na, K ATPase microdomain. Cardiovasc Res. 2018;115:546‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dodge‐Kafka KL, Bauman A, Kapiloff MS. A‐kinase anchoring proteins as the basis for Camp signaling. Handb Exp Pharmacol. 2008;186:3‐14. [DOI] [PubMed] [Google Scholar]
- 7. Sassone‐Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012;4(12):a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan K, Gao LN, Cui YL, et al. The cyclic AMP signaling pathway: Exploring targets for successful drug discovery. Mol Med Rep. 2016;13(5):3715‐3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hayes JS, Brunton LL, Mayer SE. Selective activation of particulate Camp‐ dependent protein kinase by isoproterenol and prostaglandin E1. J Biol Chem. 1980;255(11):5113‐5119. [PubMed] [Google Scholar]
- 10. Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′‐monophosphate‐dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243(13):3763‐3765. [PubMed] [Google Scholar]
- 11. Taylor SS, Buechler JA, Yonemoto W. Camp‐dependent protein kinase: frame‐ work for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971‐1005. [DOI] [PubMed] [Google Scholar]
- 12. Roskoski RJ. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1‐23. [DOI] [PubMed] [Google Scholar]
- 13. Scott JD, Stofko RE, McDonald JR, et al. Type II regulatory subunit dimerization determines the subcellular localization of the Camp‐dependent protein kinase. J Biol Chem. 1990;265(35):21561‐21566. [PubMed] [Google Scholar]
- 14. Turnham RE, Scott JD. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene. 2016;577(2):101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soberg K, Jahnsen T, Rognes T, et al. Evolutionary paths of the Camp‐dependent protein kinase (PKA) catalytic subunits. PLoS ONE. 2013;8(4):e60935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burns‐Hamuro LL, Ma Y, Kammerer S, et al. Designing isoform‐specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci USA. 2003;100(7):4072‐4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307(5710):690‐696. [DOI] [PubMed] [Google Scholar]
- 18. Kim C, Cheng CY, Saldanha SA, et al. PKA‐I holoenzyme structure reveals a mechanism for Camp‐dependent activation. Cell. 2007;130(6):1032‐1043. [DOI] [PubMed] [Google Scholar]
- 19. Bruystens JG, Wu J, Fortezzo A, et al. Structure of a PKA RIalpha recurrent acrodysostosis mutant explains defective camp‐dependent activation. J Mol Biol. 2016;428(24 Pt B):4890‐4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mauban JR, O'Donnell M, Warrier S, et al. AKAP‐scaffolding proteins and regulation of cardiac physiology. Physiology (BETHESDA). 2009;24:78‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kinderman FS, Kim C, von Daake S, et al. A dynamic mechanism for AKAP binding to RII isoforms of Camp‐dependent protein kinase. Mol Cell. 2006;24(3):397‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carr DW, Stofko‐Hahn RE, Fraser ID, et al. Interaction of the regulatory subunit (RII) of Camp‐dependent protein kinase with RII‐anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266(22):14188‐14192. [PubMed] [Google Scholar]
- 23. Calejo AI, Taskén K. Targeting protein‐protein interactions in complexes organized by A kinase anchoring proteins. Front Pharmacol. 2015;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Autenrieth K, Bendzunas NG, Bertinetti D, et al. Defining A‐kinase anchoring protein (AKAP) specificity for the protein kinase A subunit RI (PKA‐RI). ChemBioChem. 2016;17(8):693‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith FD, Esseltine JL, Nygren PJ, et al. Local protein kinase A action proceeds through intact holoenzymes. Science. 2017;356(6344):1288‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walker‐Gray R, Stengel F, Gold MG. Mechanisms for restraining cAMP‐depend protein kinase revealed by subunit quantitation and cross‐linking approaches. Proc Natl Acad Sci USA. 2017;114(39):10414‐10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zoccarato A, Surdo NC, Aronsen JM, et al. Cardiac hypertrophy is inhibited by a local pool of camp regulated by phosphodiesterase 2 novelty and significance. Circ Res. 2015;117(8):707‐719. [DOI] [PubMed] [Google Scholar]
- 28. Li X, Matta SM, Sullivan RD, et al. Carvedilol reverses cardiac insufficiency in AKAP5 knockout mice by normalizing the activities of calcineurin and CaMKII. Cardiovasc Res. 2014;104(2):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nystoriak MA, Nieves‐Cintron M, Nygren PJ, et al. AKAP150 contributes to enhanced vascular tone by facilitating large‐conductance Ca2+‐activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res. 2014;114(4):607‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nieves‐Cintrón M, Hirenallur‐Shanthappa D, Nygren PJ, et al. AKAP150 participates in calcineurin/NFAT activation during the down‐regulation of voltage‐ gated K+ currents in ventricular myocytes following myocardial infarction. Cell Signal. 2016;28(7):733‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diviani D, Osman H, Reggi E. A‐Kinase anchoring protein‐Lbc: a molecular scaffold involved in cardiac protection. J Cardiovasc Dev Dis. 2018;5(1):E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia‐Pelagio KP, Chen L, Joca HC, et al. Absence of synemin in mice causes structural and functional abnormalities in heart. J Mol Cell Cardiol. 2018;114:354‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang LJ, Durick K, Weiner JA, et al. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272(12):8057‐8064. [DOI] [PubMed] [Google Scholar]
- 34. Chen Q, Lin RY, Rubin CS. Organelle‐specific targeting of protein kinase AII (PKAII). J Biol Chem. 1997;272:15247‐15257. [DOI] [PubMed] [Google Scholar]
- 35. Abrenica B, AlShaaban M, Czubryt MP. The A‐kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2009;46(5):674‐681. [DOI] [PubMed] [Google Scholar]
- 36. Perrino C, Feliciello A, Schiattarella GG, et al. AKAP121 downregulation impairs protective Camp signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc Res. 2010;88(1):101‐110. [DOI] [PubMed] [Google Scholar]
- 37. Jun Y, Park H, Lee Y, et al. D‐AKAP1a is a signal‐anchored protein in the mitochondrial outer membrane. Febs Lett. 2016;590(7):954‐961. [DOI] [PubMed] [Google Scholar]
- 38. Passariello CL, Li J, Dodge‐Kafka K, et al. Makap‐a master scaffold for cardiac remodeling. J Cardiovasc Pharmacol. 2015;65(3):218‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carlisle Michel JJ, Townley IK, Dodge‐Kafka KL, et al. Spatial restriction of PDK1 activation cascades by anchoring to mAKAPα. Mol Cell. 2005;20(5):661‐672. [DOI] [PubMed] [Google Scholar]
- 40. Kritzer MD, Li J, Passariello CL, et al. The scaffold protein muscle A‐kinase anchoring protein β orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Clin Perspect Circ: Heart Failure. 2014;7(4):663‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee SW, Won JY, Yang J, et al. AKAP6 inhibition impairs myoblast differentiation and muscle regeneration: Positive loop between AKAP6 and myogenin. Sci Rep. 2015;5:16523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dodge‐Kafka KL, Soughayer J, Pare GC, et al. The protein kinase A anchoring protein Makap coordinates two integrated Camp effector pathways. Nature. 2005;437(7085):574‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Malik S, Kelley GG, et al. Phospholipase Cϵ scaffolds to muscle‐ specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286(26):23012‐23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang L, Malik S, Pang J, et al. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4‐phosphate to regulate cardiac hypertrophy. Cell. 2013;153(1):216‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim Y, Phan D, van Rooij E, et al. The MEF2D transcription factor mediates stress‐dependent cardiac remodeling in mice. J Clin Invest. 2008;118(1):124‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Vargas M, Kapiloff MS, et al. Regulation of MEF2 transcriptional activity by calcineurin/Makap complexes. Exp Cell Res. 2013;319(4):447‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klussmann E, Edemir B, Pepperle B, et al. Ht31: the first protein kinase A anchoring protein to integrate protein kinase A and Rho signaling. Febs Lett. 2001;507:264‐268. [DOI] [PubMed] [Google Scholar]
- 48. Diviani D, Soderling J, Scott JD. AKAP‐Lbc anchors protein kinase A and nucleates Gα12‐selective Rho‐mediated stress fiber formation. J Biol Chem. 2001;276(47):44247‐44257. [DOI] [PubMed] [Google Scholar]
- 49. Appert‐Collin A, Cotecchia S, Nenniger‐Tosato M, et al. The A‐kinase anchoring protein (AKAP)‐Lbc‐signaling complex mediates alpha1 adrenergic receptor‐induced cardiomyocyte hypertrophy. Proc Natl Acad Sci USA. 2007;104(24):10140‐10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carnegie GK, Soughayer J, Smith FD, et al. AKAP‐Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol Cell. 2008;32(2):169‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taglieri DM, Johnson KR, Burmeister BT, et al. The C‐terminus of the long AKAP13 isoform (AKAP‐Lbc) is critical for development of compensatory cardiac hypertrophy. J Mol Cell Cardiol. 2014;66:27‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnson KR, Nicodemus‐Johnson J, Spindler MJ, et al. Genome‐wide gene expression analysis shows AKAP13‐mediated PKD1 signaling regulates the transcriptional response to cardiac hypertrophy. PLoS ONE. 2015;10(7):e132474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caso S, Maric D, Arambasic M, et al. AKAP‐Lbc mediates protection against doxorubicin‐induced cardiomyocyte toxicity. Biochimica et Biophysica Acta (BBA)‐Molecular Cell Res. 2017;1864(12):2336‐2346. [DOI] [PubMed] [Google Scholar]
- 54. Burmeister BT, Taglieri DM, Wang L, et al. Src homology 2 domain‐containing phosphatase 2 (Shp2) is a component of the A‐kinase‐anchoring protein (AKAP)‐Lbc complex and is inhibited by protein kinase A (PKA) under pathological hypertrophic conditions in the heart. J Biol Chem. 2012;287(48):40535‐40546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burmeister BT, Wang L, Gold MG, et al. Protein Kinase A (PKA) phosphorylation of Shp2 protein inhibits its phosphatase activity and modulates ligand specificity. J Biol Chem. 2015;290(19):12058‐12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF‐κB signaling in heart. J Mol Cell Cardiol. 2006;41(4):580‐591. [DOI] [PubMed] [Google Scholar]
- 57. Freund C, Schmidt‐Ullrich R, Baurand A, et al. Requirement of nuclear factor‐kappaB in angiotensin II‐ and isoproterenol‐induced cardiac hypertrophy in vivo. Circulation. 2005;111(18):2319‐2325. [DOI] [PubMed] [Google Scholar]
- 58. Scheidereit C. IkappaB kinase complexes: gateways to NF‐kappaB activation and transcription. Oncogene. 2006;25(51):6685‐6705. [DOI] [PubMed] [Google Scholar]
- 59. Lee KH, Jang Y, Chung JH. Heat shock protein 90 regulates IkappaB kinase complex and NF‐kappaB activation in angiotensin II‐induced cardiac cell hypertrophy. Exp Mol Med. 2010;42(10):703‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Del Vescovo CD, Cotecchia S, Diviani D. A‐kinase‐anchoring protein‐Lbc anchors I B kinase to support interleukin‐6‐mediated cardiomyocyte hypertrophy. Mol Cell Biol. 2012;33(1):14‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Soni S, Scholten A, Vos MA, et al. Anchored protein kinase A signalling in cardiac cellular electrophysiology. J Cell Mol Med. 2014;18(11):2135‐2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jones BW, Brunet S, Gilbert ML, et al. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci USA. 2012;109(42):17099‐17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hulme JT, Lin TW, Westenbroek RE, et al. Beta‐adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase‐anchoring protein 15. Proc Natl Acad Sci USA. 2003;100(22):13093‐13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lygren B, Carlson CR, Santamaria K, et al. AKAP complex regulates Ca2+ re‐uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007;8(11):1061‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fu Y, Westenbroek RE, Yu FH, et al. Deletion of the distal C terminus of CaV1.2 channels leads to loss of β‐adrenergic regulation and heart failure in vivo. J Biol Chem. 2011;286(14):12617‐12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yatani A, Honda R, Tymitz KM, et al. Enhanced Ca2+ channel currents in cardiac hypertrophy induced by activation of calcineurin‐dependent pathway. J Mol Cell Cardiol. 2001;33(2):249‐259. [DOI] [PubMed] [Google Scholar]
- 67. Hulme JT, Westenbroek RE, Scheuer T, et al. Phosphorylation of serine 1928 in the distal C‐terminal domain of cardiac CaV1.2 channels during beta1‐adrenergic regulation. Proc Natl Acad Sci USA. 2006;103(44):16574‐16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li X, Nooh MM, Bahouth SW. Role of AKAP79/150 protein in beta1‐adrenergic receptor trafficking and signaling in mammalian cells. J Biol Chem. 2013;288(47):33797‐33812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Faux MC, Scott JD. Regulation of the AKAP79‐protein kinase C interaction by Ca2+/Calmodulin. J Biol Chem. 1997;272(72):17038‐17044. [DOI] [PubMed] [Google Scholar]
- 70. Li L, Li J, Drum BM, et al. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc Res. 2017;113(2):147‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sachse G, Faulhaber J, Seniuk A, et al. Smooth muscle BK channel activity influences blood pressure independent of vascular tone in mice. J Physiol. 2014;592(12):2563‐2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Navedo MF, Santana LF. CaV1.2 sparklets in heart and vascular smooth muscle. J Mol Cell Cardiol. 2013;58:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nystoriak MA, Nieves‐Cintron M, Patriarchi T, et al. Ser1928 phosphorylation by PKA stimulates the L‐type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal. 2017;10(463):eaaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. White J, Cibelli M, Urban L, et al. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96(3):911‐973. [DOI] [PubMed] [Google Scholar]
- 75. Jones JL, Peana D, Veteto AB, et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc Res. 2018;115(1):46‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sonkusare SK, Dalsgaard T, Bonev AD, et al. AKAP150‐dependent cooperative TRPV4 channel gating is central to endothelium‐dependent vasodilation and is disrupted in hypertension. Sci Signal. 2014;7(333):a66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tajada S, Moreno CM, O’Dwyer S et al. Distance constraints on activation of TRPV4 channels by AKAP150‐bound PKCα in arterial myocytes. J Gen Physiol. 2017;149(6):639‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bers DM. Cardiac excitation‐contraction coupling. Nature. 2002;415(6868):198‐205. [DOI] [PubMed] [Google Scholar]
- 79. Lin F, Wang H, Malbon CC. Gravin‐mediated formation of signaling complexes in beta 2‐adrenergic receptor desensitization and resensitization. J Biol Chem. 2000;275(25):19025‐19034. [DOI] [PubMed] [Google Scholar]
- 80. Guillory AN, Yin X, Wijaya CS, et al. Enhanced cardiac function in Gravin mutant mice involves alterations in the beta‐adrenergic receptor signaling cascade. PLoS ONE. 2013;8(9):e74784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li Z, Singh S, Suryavanshi SV, et al. Force development and intracellular Ca(2+) in intact cardiac muscles from gravin mutant mice. Eur J Pharmacol. 2017;807:117‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kurokawa J, Motoike HK, Rao J, et al. Regulatory actions of the A‐kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci USA. 2004;101(46):16374‐16378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Piggott LA, Bauman AL, Scott JD, et al. The A‐kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci USA. 2008;105(37):13835‐13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moss AJ. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115(8):2018‐2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science. 2002;295(5554):496‐499. [DOI] [PubMed] [Google Scholar]
- 86. Chen L, Marquardt ML, Tester DJ, et al. Mutation of an A‐kinase‐anchoring protein causes long‐QT syndrome. Proc Natl Acad Sci USA. 2007;104(52):20990‐20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19‐31. [DOI] [PubMed] [Google Scholar]
- 88. Cheng EP, Yuan C, Navedo MF, et al. Restoration of normal L‐Type Ca2+ channel function during timothy syndrome by ablation of an anchoring protein. Circ Res. 2011;109(3):255‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nichols CB, Rossow CF, Navedo MF, et al. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L‐type calcium channels. Circ Res. 2010;107(6):747‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Han C, Tomita H, Ohba T, et al. Modified sympathetic nerve regulation in AKAP5‐null mice. Biochem Biophys Res Commun. 2016;469(4):897‐902. [DOI] [PubMed] [Google Scholar]
- 91. Huang LJ, Durick K, Weiner JA, et al. D‐AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci USA. 1997;94(21):11184‐11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sarma GN, Moody IS, Ilouz R, et al. D‐AKAP2: PKA RII: PDZK1 ternary complex structure: Insights from the nucleation of a polyvalent scaffold. Protein Sci. 2015;24(1):105‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tingley WG, Pawlikowska L, Zaroff JG, et al. Gene‐trapped mouse embryonic stem cell‐derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc Natl Acad Sci USA. 2007;104(20):8461‐8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Loniewska B, Kaczmarczyk M, Clark JS, et al. Association of functional genetic variants of A‐kinase anchoring protein 10 with QT interval length in full‐term Polish newborns. Arch Med Sci. 2015;11(1):149‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Robichaux WG III, Cheng X. Intracellular cAMP sensor EPAC: Physiology, pathophysiology, and therapeutics development. Physiol Rev. 2018;98(2):919‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schulze DH, Muqhal M, Lederer WJ, et al. Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem. 2003;278(31):28849‐28855. [DOI] [PubMed] [Google Scholar]
- 97. Pare GC, Easlick JL, Mislow JM, et al. Nesprin‐1alpha contributes to the targeting of mAKAP to the cardiac myocyte nuclear envelope. Exp Cell Res. 2005;303(2):388‐399. [DOI] [PubMed] [Google Scholar]
- 98. Martinez EC, Passariello CL, Li J, et al. RSK3: A regulator of pathological cardiac remodeling. IUBMB Life. 2015;67(5):331‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Smith FD, Langeberg LK, Cellurale C, et al. AKAP‐Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat Cell Biol. 2010;12(12):1242‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cariolato L, Cavin S, Diviani D. A‐kinase anchoring protein (AKAP)‐Lbc anchors a PKN‐based signaling complex involved in α1‐adrenergic receptor‐induced p38 activation. J Biol Chem. 2011;286(10):7925‐7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Singh A, Redden JM, Kapiloff MS, et al. The large isoforms of A‐kinase anchoring protein 18 mediate the phosphorylation of inhibitor‐1 by protein kinase A and the inhibition of protein phosphatase 1 activity. Mol Pharmacol. 2011;79(3):533‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cardone L, Carlucci A, Affaitati A, et al. Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol. 2004;24(11):4613‐4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chaturvedi D, Poppleton HM, Stringfield T, et al. Subcellular localization and biological actions of activated RSK1 are determined by its interactions with subunits of cyclic AMP‐dependent protein kinase. Mol Cell Biol. 2006;26(12):4586‐4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Meiri D, Greeve MA, Brunet A, et al. Modulation of Rho guanine exchange factor Lfc activity by protein kinase A‐mediated phosphorylation. Mol Cell Biol. 2009;29(21):5963‐5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nikandrova YA, Jiao Y, Baucum AJ, et al. Ca2+/calmodulin‐dependent protein kinase II binds to and phosphorylates a specific SAP97splice variant to disrupt association with AKAP79/150 and modulate alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐ isoxazolepropionic acid‐type glutamate receptor (AMPAR) activity. J Biol Chem. 2010;285(2):923‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bavencoffe A, Li Y, Wu Z, et al. Persistent electrical activity in primary nociceptors after spinal cord injury is maintained by scaffolded adenylyl cyclase and protein Kinase A and is associated with altered adenylyl cyclase regulation. J Neurosci. 2016;36(5):1660‐1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Havekes R, Canton DA, Park AJ et al. Gravin orchestrates protein kinase A and β2‐adrenergic receptor signaling critical for synaptic plasticity and memory. J Neurosci. 2012;32(50):18137‐18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schott MB, Grove B. Receptor‐mediated Ca2+ and PKC signaling triggers the loss of cortical PKA compartmentalization through the redistribution of gravin. Cell Signal. 2013;25(11):2125‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li Y, Chen L, Kass RS, et al. The A‐kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem. 2012;287(35):29815‐29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Berryman MA, Goldenring JR. CLIC4 is enriched at cell‐cell junctions and colocalizes with AKAP350 at the centrosome and midbody of cultured mammalian cells. Cell Motil Cytoskeleton. 2003;56(3):159‐172. [DOI] [PubMed] [Google Scholar]