

Abstract
Friedreich ataxia (FRDA) is an autosomal recessive spinocerebellar ataxia caused by mutations of FXN. Hypotonus and hyporeflexia of the lower extremities are observed in most FRDA patients. Patients with hyperreflexia, called Friedreich ataxia with retained reflexes (FARR), have also been identified. We herein report the case of a 16-year-old Nepalese boy presenting with early-onset ataxia with prominent spasticity and hyperreflexia of the legs. Mutational analyses established the diagnosis of FRDA presenting as FARR. A haplotype analysis revealed that expanded alleles of the patient shared a common haplotype with Indian and European FRDA patients, suggesting that the mutation descended from a common founder.
Keywords: Friedreich ataxia, Friedreich ataxia with retained reflexes (FARR), haplotype analysis, Nepalese
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive spinocerebellar ataxia characterized by progressive gait and limb ataxia, dysarthria, lower-limb areflexia, decreased vibration sense, muscle weakness in the legs, and a positive Babinski sign. Other common signs include scoliosis, foot deformity, hypertrophic cardiomyopathy, and diabetes mellitus. FRDA is caused by mutations of the FXN gene in chromosome 9 encoding the mitochondrial protein frataxin (1,2). More than 97% of FRDA patients carry expanded trinucleotide GAA repeats in intron 1 of the FXN gene, and the remaining patients carry missense mutations (3).
The incidence and prevalence of FRDA varies widely across ethnic populations. FRDA is the most frequent type of spinocerebellar ataxia (SCA) in Caucasian populations, with an estimated prevalence of 1 in 50,000. In contrast, the disease has been reported to be extremely rare or absent in Sub-Saharan and Far Eastern populations, probably owing to a strong founder effect of GAA repeat expansions (4).
The absence of a lower limb tendon reflex was originally described as an essential criterion for the diagnosis of FRDA (5). Early-onset cerebellar ataxia with retained reflex with overlapping clinical features of FRDA had been classified as a distinct clinical entity by applying this criterion (6). Identification of the causative gene, however, has led to the finding that these patients also harbored the GAA expansion in the FXN gene, and such a disorder has been designated as Friedreich ataxia with retained reflexes (FARR) (7). The occurrence of FARR varies from 5% to 11% in reported series of FRDA patients (8,9). FARR is characterized by a later onset, with a lower incidence of impaired vibration sense, pes cavus, and echocardiographic signs of left ventricular hypertrophy.
We herein report the case of a 16-year-old Nepalese boy presenting with early-onset ataxia with prominent spasticity and hyperreflexia of his lower limbs, diagnosed as FRDA on the basis of the identification of homozygous GAA expansion mutations in the FXN gene.
Methods
Mutational analyses of screening of hereditary ataxia
Genomic DNA samples were obtained from the proband and his unaffected father after written informed consent was obtained. The DNA sample of his unaffected mother was unavailable. The initial mutational analysis included DNA microarray-based resequencing of whole exons of SETX, FXN, APTX, TDP1, and SACS (10). Fragment analyses for triplet repeat expansions of DRPLA, SCA1, SCA2, MJD/SCA3, SCA6, SCA7, and SCA12 loci were also performed. Single-nucleotide polymorphism (SNP) genotyping of the patient was performed using Genome-Wide Human SNP array 6.0 (Affymetrix, Santa Clara, USA) following the manufacturer's protocol. To calculate runs of homozygosity (ROH) based on the results of the SNP array analysis, we used PLINK v1.07.
Detection of GAA expansion in FXN and a haplotype analysis
A mutational analysis for GAA expansion of the FXN locus was performed using polymerase chain reaction (PCR) followed by agarose gel electrophoresis of PCR products, as previously described (11). The sizes of alleles were estimated based on the calculation of 500+3n bp (n=number of GAA triplets) according to the position of the flanking primers.
For the haplotype analysis, we selected five tagged SNPs (FAD1-rs11145465, rs11145326, rs7861997, ITR3-rs3829062, and CS2-rs2871223) spanning a 151-kb region flanking the GAA repeat of the FXN gene (12). The genomic locations of five markers flanking the GAA repeats used for the haplotype analysis are shown in Supplementary Material 1. The genotyping of these SNPs was carried out by a direct nucleotide sequence analysis using the designed primers (Supplementary Material 2).
Case report
The proband, a-16-year-old Nepalese boy, was admitted to the Department of Neurology at the University of Tokyo Hospital for the evaluation of progressive gait and limb ataxia as well as dysarthria. He had shown no abnormalities at birth or developmental problems in his early childhood. His father first noticed his staggering gait at 8 years of age, and his gait instability gradually worsened, with dysarthria appearing at 10 years of age. He became incapable of walking independently at 13 years of age when upper limb ataxia also developed.
His medical history was unremarkable. His parents were apparently unrelated, although both had been born in the same small village located in a secluded mountainous area in Nepal presently inhabited by about 60 families. His family history indicated no similar diseases (Fig. 1). On admission, he was wheelchair-bound and could barely stand, even with assistance. On a neurological examination, saccadic pursuit of eye movement; dysarthria with slow, slurred, scanning and explosive speech; prominent spasticity and hyprereflexia predominantly in the lower extremities; bilateral ankle clonus; and positive Babinski's signs were observed. His manual muscle test (MMT) score was 4/5 bilaterally in the lower limbs of iliopsoas, hamstrings, and tibialis anterior muscles, with mild distal-predominant muscle atrophy. The joint position sense in the lower extremities was mildly decreased, but light touch, pain, and vibration senses were normal. Limb ataxia predominantly in his lower extremities as well as truncal ataxia were noted. He had a high arched palate but no pes cavus or scoliosis.
Figure 1.

Pedigree chart of our patient with FRDA. Affected individuals are indicated by filled symbols. The proband is indicated by an arrow. Unaffected individuals are indicated by open symbols. Slashed symbols indicate deceased subjects. Squares denote male family members, and circles denote female family members. Rhomboids denote family members of unknown gender. FRDA: Friedreich ataxia
The results of laboratory tests were unremarkable, except for a decreased serum vitamin B12 level (184 pg/mL; normal range, 233-914 pg/ml). Serum levels of alpha-fetoprotein (AFP), vitamin E, lactic and pyruvic acids, and very-long-chain fatty acids were within the normal ranges. No cardiomyopathy was detected by chest radiography, electrocardiography, or transthoracic echocardiography. Nerve conduction studies showed grossly reduced sensory nerve action potentials with normal conduction velocities (Table 1). Short-latency somatosensory evoked potential (SEP) studies showed prolonged central sensory conduction time (right, 8.6 ms; left, 9.0 ms; normal range, <6.7 ms) obtained by median nerve stimulation, and no responses were evoked by tibial nerve stimulation (Table 2). The patient's motor evoked potential (MEP) studies showed a severely prolonged central motor conduction time (right, 15.1 ms; left, 12.5 ms; normal range, 6.2-7.8 ms) for the first dorsal interosseous muscles, and no responses were recorded in the tibialis anterior muscles by the magnetic stimulation of the cortex (Table 3). Brain magnetic resonance imaging (MRI) showed mild cerebellar atrophy (Fig. 2A and B). Spinal MRI demonstrated diffuse, marked spinal cord atrophy (Fig. 2C and D). After his discharge, his limb and truncal ataxia and weakness of the lower limbs gradually worsened, and he became incapable of standing or sitting independently at 20 years of age.
Table 1.
Nerve Conduction Studies.
| CMAP (mV) |
DL (ms) |
MCV (m/s) |
SNAP (μV) |
SCV (m/s) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median | 11.7 | 3.5 | 53.8 | 6.4 ↓ | 52 | |||||
| (left) | (>5.4) | (<3.9) | (>50) | (>7.5) | (>40) | |||||
| Ulnar | 16.4 | 2.8 | 59.2 | 4.55 ↓ | 51 | |||||
| (left) | (>4.0) | (<3.3) | (>51) | (>7.5) | (>40) | |||||
| Tibial | 29.5 | 3.1 | 46.5 | - | - | |||||
| (left) | (>5.8) | (<6.1) | (>40) | - | - | |||||
| Sural | - | - | - | 1.03 ↓ | 54 | |||||
| (left) | - | - | - | (>1.9) | (>36) |
CMAP: compound muscle action potential, DL: distal latency, MCV: motor conduction velocity, SNAP: sensory nerve action potential, SCV: sensory conduction velocity. Normal values are shown in parentheses.
Table 2.
Latencies of Sensory Evoked Potential (SEP).
| Median | N9 | N13 | N20 | CSCT (N20-N13) | ||||
| (ms) | (<10.1) | (<13.6) | (<20.2) | (<6.7) | ||||
| Right | 9.2 | 12.8 | 21.4 ↑ | 8.6 ↑ | ||||
| Left | 8.8 | 13.2 | 22.2 ↑ | 9.0 ↑ | ||||
| Tibial | P15 | N20 | P38 | CSCT (P38-N20) | ||||
| (ms) | (<17.7) | (<24.1) | (<42.3) | (<19.0) | ||||
| Right/Left | NR/NR | NR/NR | NR/NR | -/- |
CSCT: central sensory conduction time, NR: not recorded. Normal values are shown in parentheses.
Table 3.
Latencies of Motor Evoked Potential (MEP).
| Hand (FDI) |
Laterality of MEP | CTX (18.8–22.6) |
BST (15.8–18.8) |
CR (12.0–15.2) |
Intervals | ||
|---|---|---|---|---|---|---|---|
| CTX-BST | BST-CR | CTX-CR | |||||
| Latency | Right | 28.8 ↑ | 20.1 ↑ | 13.7 | 8.7 ↑ | 6.4 ↑ | 15.1↑ |
| (ms) | Left | 26.1 ↑ | 20.4 ↑ | 13.5 | 5.7 ↑ | 6.9 ↑ | 12.5↑ |
| Leg (TA) |
CTX (22.9–29.3) |
BST (22.2–25.6) |
LR (9.7–13.3) |
Intervals | |||
| CTX-BST | BST-LR | CTX-LR | |||||
| Latency | Right | NR | NR | 10.7 | - | - | - |
| (ms) | Left | NR | NR | 11.0 | - | - | - |
FDI: first dorsal interosseous muscle, TA: tibialis anterior muscle, CTX: cortex, BST: brainstem, CR: cervical root, LR: lumbar root, NR: not recorded. Normal values are shown in parentheses.
Figure 2.

(A, B) Brain T1-weighted MR images. Sagittal (A) and axial (B) views showing mild cerebellar atrophy. (C, D) Spinal cord T2-weighted MR images. Sagittal view (C) showing diffuse spinal cord atrophy. Axial view of the 4th cervical cord (D) showing marked flattening of the cervical spinal cord.
Mutational analysis findings
To establish the molecular diagnosis for early-onset autosomal recessive spinocerebellar ataxias, we initially performed DNA microarray-based resequencing for SETX, FXN, APTX, TDP1, and SACS, with no pathogenic mutations identified. In addition, fragment analyses for triplet repeat expansions in DRPLA, SCA1, SCA2, MJD/SCA3, SCA6, SCA7, and SCA12 loci revealed no pathogenic mutations. Since both of his parents had been born in the same small village in a secluded mountainous area, we considered the probability that they were at least distantly related, which is consistent with the autosomal recessive mode of inheritance. We considered that searching for contiguous genomic regions of homozygous genotypes [runs of homozygosity (ROH)] would facilitate the search for causative genes with autosomal recessive inheritance. We searched for ROH segments a minimum of 2 Mb in length using the SNP microarray (Supplementary Material 3). Among the causative genes for autosomal recessive spinocerebellar ataxia at the time of the analysis, FXN was the only gene located in a homozygous region.
The analysis of the GAA expansion in intron 1 of FXN indeed revealed two expanded alleles in the proband and the absence of a normal allele. The estimated repeat units of the two expanded alleles in the proband were approximately 800 and 1,200. An expanded allele (1,000 repeat units) and a normal allele were both detected in his father, indicating that he was a carrier of one of the expanded alleles (Fig. 3).
Figure 3.
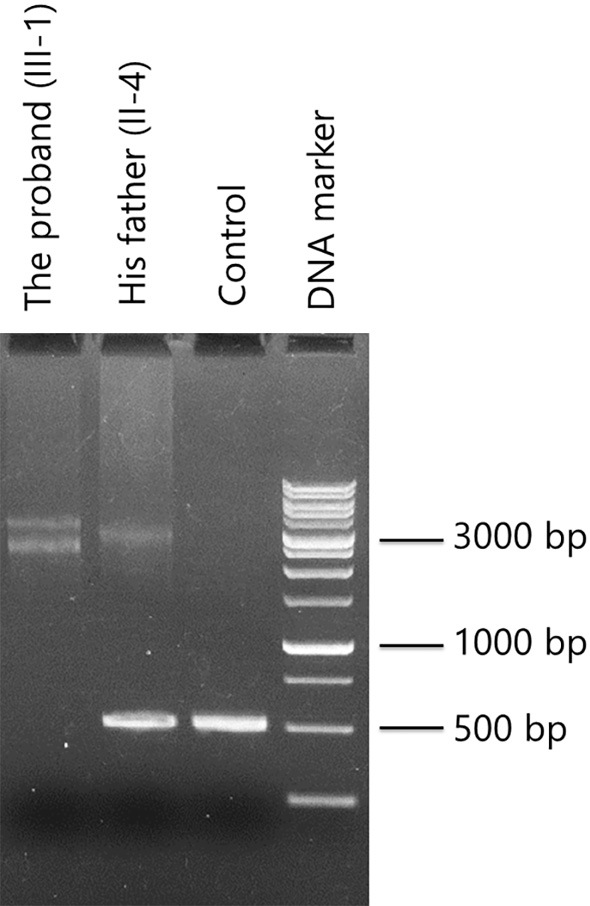
Gel electrophoresis image of PCR products encompassing FXN GAA triplet repeats. Representative marker lengths are indicated. Lane 1: the proband. Lane 2: his father. Lane 3: normal control. Lane 4: DNA size marker. Two expanded alleles were identified in the proband, and an expanded allele and a normal allele were both identified in his father. The estimated repeat units of the two expanded alleles of the patient were approximately 800 and 1,200. PCR: polymerase chain reaction
In the haplotype analysis flanking the GAA expansions, we observed the A-G-C-C-C haplotype in both expanded alleles (Supplementary material 1). The A-G-C-C-C haplotype has been observed as the major haplotype in expanded alleles in both North Indian (50%) and South Indian (62.5%) populations (12). In addition, when we compared the frequencies of haplotypes using three well-studied markers (FAD1, ITR3, and CS2), we observed the A-C-C haplotype in the expanded alleles of the patient, which is the most commonly shared among the patients in all of the studied populations, including East India and Caucasian populations (13).
Discussion
The patient in this study presented with early-onset ataxia with prominent spasticity and hyperreflexia of the lower limbs. Initial differential diagnoses included a broad range of diseases causing SCAs, mainly in the spectrum of early-onset spastic ataxia. A lack of typical clinical features of FRDA, such as lower-limb areflexia and cardiomyopathy, as well as a paucity of information on the genetic epidemiology of SCA in Nepal, hampered our consideration of the diagnosis of FRDA. Homozygosity mapping revealed that FXN was the only candidate gene for autosomal recessive SCA, located on the ROH segments, leading to the detection of the homozygous GAA expansion by a subsequent PCR analysis. The estimated repeat units of the two expanded alleles in the proband were approximately 800 and 1,200, which were slightly different from that of the expanded allele (1,000 repeat units) in his father. Although it is unclear which allele was transmitted from his father, it has been reported that paternally transmitted alleles generally tend to contract (14).
Patients with FARR were previously described as having a later onset of the disease with shorter GAA trinucleotide repeat lengths than typical FRDA patients. The mean onset age ± standard deviation and the mean length of the smaller allele repeat units in FARR and FRDA patients were 26.6±11.4 and 14.2±6.9 years and 408±252 and 719±184 GAA repeat units, respectively (9). In contrast to the cases of FARR described in previous reports, the early age at the onset (8 years old) and the relatively long GAA trinucleotide repeat length (approximately 800 repeat units in the smaller allele) were distinct characteristics in our patient with FARR. Although why the patient presented with prominent spasticity and hyperreflexia despite the early onset and long repeat expansions remained unclear, other genetic factors as well as low serum levels of vitamin B12 might have been involved in the atypical clinical presentations in this patient.
To our knowledge, this is the first report of a Nepalese case of FRDA. The GAA triplet-repeat expansion that causes FRDA has been found only in individuals of European, North African, Middle Eastern, or Indian descent (4). In India, the prevalence of FRDA is reported to be lower than that in European populations (15). A haplotype analysis of 21 North Indian and 8 South Indian families with FRDA revealed that all of the expanded alleles in the Indian population shared a common core haplotype, which was also shared with Caucasian populations, suggesting a founder effect (12). Interestingly, the common core haplotypes were also observed in both of the expanded alleles in our Nepalese patient with FRDA. The genetic architecture of the Nepalese population, which has been analyzed using Y-chromosomal short tandem repeat loci, was reported to be highly diverse and complex (16), with Kathmandu, and to some extent Newar, receiving significant genetic influence from India (17). Taken together, the present and previous findings suggest that a founder mutation of a GAA triplet-repeat expansion is present in the Nepalese population. Further investigation on the genetic epidemiology of FRDA and the elucidation of its phylogenetic origin in Nepalese FRDA mutations should provide a better understanding of the genomic basis underlying FRDA mutations in Nepalese populations.
The authors state that they have no Conflict of Interest (COI).
Supplementary Materials
Genomic locations of five markers flanking GAA repeats used for the haplotype analysis. Their distance (kb) from the GAA repeat in intron 1 of FXN is shown below the arrow marks. Haplotypes observed in the expanded alleles in the proband are also shown.
Designed primers used for genotyping of SNP markers
Runs of homozygosity (ROH) segments defined as a minimum of 2 Mb. Physical positions were annotated against UCSC hg18.
References
- 1. Campuzano V, Montermini L, Moltò MD, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271: 1423-1427, 1996. [DOI] [PubMed] [Google Scholar]
- 2. Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol 256 (Suppl 1): 3-8, 2009. [DOI] [PubMed] [Google Scholar]
- 3. Delatycki MB, Knight M, Koenig M, Cossee M, Williamson R, Forrest SM. G130V, a common FRDA point mutation, appears to have arisen from a common founder. Hum Genet 105: 343-346, 1999. [DOI] [PubMed] [Google Scholar]
- 4. Labuda M, Labuda D, Miranda C, et al. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology 54: 2322-2324, 2000. [DOI] [PubMed] [Google Scholar]
- 5. Harding AE. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104: 589-620, 1981. [DOI] [PubMed] [Google Scholar]
- 6. Harding AE. Early onset cerebellar ataxia with retained tendon reflexes: a clinical and genetic study of a disorder distinct from Friedreich's ataxia. J Neurol Neurosurg Psychiatry 44: 503-508, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palau F, De Michele G, Vilchez JJ, et al. Early-onset ataxia with cardiomyopathy and retained tendon reflexes maps to the Friedreich's ataxia locus on chromosome 9q. Ann Neurol 37: 359-362, 1995. [DOI] [PubMed] [Google Scholar]
- 8. Dürr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 335: 1169-1175, 1996. [DOI] [PubMed] [Google Scholar]
- 9. Coppola G, De Michele G, Cavalcanti F, et al. Why do some Friedreich's ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study. J Neurol 246: 353-357, 1999. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi Y, Seki N, Ishiura H, et al. Development of a high-throughput microarray-based resequencing system for neurological disorders and its application to molecular genetics of amyotrophic lateral sclerosis. Arch Neurol 65: 1326-1332, 2008. [DOI] [PubMed] [Google Scholar]
- 11. Filla A, De Michele G, Cavalcanti F, et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet 59: 554-560, 1996. [PMC free article] [PubMed] [Google Scholar]
- 12. Singh I, Faruq M, Mukherjee O, et al. North and South Indian populations share a common ancestral origin of Friedreich's ataxia but vary in age of GAA repeat expansion. Ann Hum Genet 74: 202-210, 2010. [DOI] [PubMed] [Google Scholar]
- 13. Cossee M, Schmitt M, Campuzano V, et al. Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci U S A 94: 7452-7457, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monros E, Molto MD, Martinez F, et al. Phenotype correlation and intergenerational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am J Hum Genet 61: 101-110, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukerji M, Choudhry S, Saleem Q, Padma MV, Maheshwari MC, Jain S. Molecular analysis of Friedreich's ataxia locus in the Indian population. Acta Neurol Scand 102: 227-229, 2000. [DOI] [PubMed] [Google Scholar]
- 16. Parkin EJ, Kraayenbrink T, Opgenort JR, et al. Diversity of 26-locus Y-STR haplotypes in a Nepalese population sample: isolation and drift in the Himalayas. Forensic Sci Int 166: 176-181, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gayden T, Chennakrishnaiah S, La Salvia J, et al. Y-STR diversity in the Himalayas. Int J Legal Med 125: 367-375, 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genomic locations of five markers flanking GAA repeats used for the haplotype analysis. Their distance (kb) from the GAA repeat in intron 1 of FXN is shown below the arrow marks. Haplotypes observed in the expanded alleles in the proband are also shown.
Designed primers used for genotyping of SNP markers
Runs of homozygosity (ROH) segments defined as a minimum of 2 Mb. Physical positions were annotated against UCSC hg18.