

Abstract
Beige fat serves as a substantial metabolic sink that dissipates energy and has consequently attracted much attention as a target for improving metabolic health. A recent study has provided a new molecular target, the N-terminal acetyltransferase Naa10p, for harnessing beige-fat biogenesis and improving whole-body energy homeostasis1.
Obesity and its metabolic consequences, such as type 2 diabetes, cardiovascular disease and certain cancers, continue to be among the most urgent biomedical challenges in the USA and worldwide. Emerging evidence indicates that the recruitment of inducible thermogenic fat (called ‘beige fat’) is associated with considerable improvement in insulin sensitivity and glucose tolerance; thus, promoting beige-fat biogenesis may provide a new approach for combating obesity2.
Unlike the conventional brown adipocytes that exist in the dedicated brown-adipocyte tissue depots, beige adipocytes sporadically reside interspersed in the subcutaneous white adipose tissue (WAT). In response to cold acclimation and other external stimuli, such as stimulation via the β-adrenergic receptor (β3-AR) signaling pathway, adipocyte progenitors in the subcutaneous WAT give rise to energy-dissipating beige adipocytes (Fig. 1). This process, often referred to as the ‘browning’ of white fat, is mediated by transcriptional factors and co-regulators, including PRDM16 (PR-domain zinc-finger protein 16) and PGC1α (transcription factor PPARγ (peroxisome proliferator-activated receptor-γ) co-activator 1α)3. Although β3-AR agonists powerfully activate this browning process in vivo, they inevitably increase blood pressure and heart rate, which are major risk factors for cardiovascular disease4. Hence, it is imperative to identify alternative pathways for promoting beige-fat biogenesis that are independent of the β3-AR signaling pathway.
Fig. 1 |. The biogenesis of beige fat.
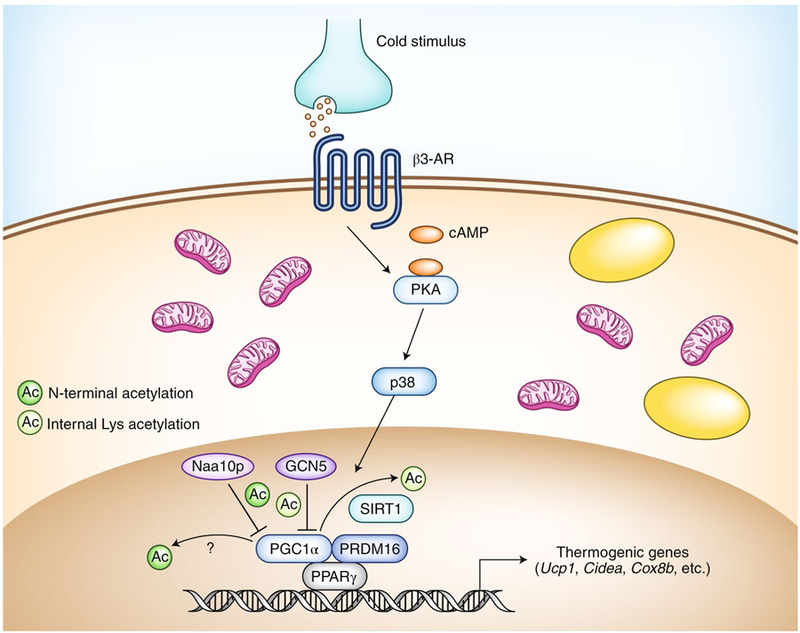
In response to cold stimuli, norepinephrine is released from the sympathetic nerve endings and binds to β3-AR, which leads to increased levels of intracellular cAMP and subsequent activation of signaling via the kinase PKA and the mitogen-activated protein kinase p38. p38 phosphorylates transcriptional co-regulators, such as PGC1α, and promotes the expression of thermogenic genes (Ucp1, Cidea and Cox8b) and mitochondrial biogenesis. In the nucleus, Naap10 and GCN5 inhibit the transcriptional activity of PGC1α through N-terminal acetylation and lysine acetylation, respectively. The deacetylase SIRT1 removes the acetylation on PGC1α catalyzed by GCN57. The N-terminal deacetylase remains unknown. Yellow ovals indicate lipid droplets.
Lee et al. recently set out to identify a post-translational mechanism that controls beige-fat biogenesis1. The authors followed clinical observations indicating that a point mutation in the gene NAA10 (which encodes Naa10p) is associated with diminished subcutaneous fat mass5. To determine if Naa10p controls fat metabolism and energy homeostasis, the authors first challenged wild-type mice and mice with whole-body knockout of Naa10 (Naa10p-KO mice) with normal chow or a high-fat diet. Whereas wild-type mice and Naa10p-KO mice exhibited similar body-weight gain when fed a normal-chow diet, the Naa10p-KO mice gained much less body weight than did their wild-type littermates when fed a high-fat diet. Consistent with that observation, mice with inducible fat-specific knockout of Naa10p exhibited favorable metabolic phenotypes compared with those of wild-type mice: less body-weight gain, reduced adipocyte size, and enhanced glucose tolerance and insulin sensitivity. These data support the notion that fat-specific loss of Naa10p improves whole-body energy metabolism.
On the basis of the metabolic phenotype reported above, Lee et al. next investigated the underlying mechanism1. By analyzing RNA microarray data, the authors found that several beige-fat marker genes were substantially upregulated in Naa10p-KO mice relative to their expression in wild-type mice. Indeed, Naa10p-KO mice contained more beige adipocytes in the subcutaneous inguinal WAT and exhibited higher whole-body energy expenditure than that of wild-type mice. Those changes in increased beige-fat biogenesis occurred in a cell-autonomous fashion: the authors found that Naa10p inhibited beige-adipocyte differentiation in culture, whereas Naa10p-KO beige adipocytes showed higher expression of genes encoding thermogenic molecules (‘thermogenic genes’) than that of wild-type cells, in the absence of β3-AR activation.
What is the molecular mechanism by which Naa10p inhibits beige-fat biogenesis? Previous studies have shown that dysfunction of Naa10p caused by the lethal X-linked disorder of infancy known as ‘Odgen syndrome’ is due to a deficiency in N-terminal acetyltransferase activity5. Accordingly, Lee et al. sought to determine if the acetylase activity of Naa10p was also responsible for the observed repression of beige-fat biogenesis. The authors found that Naa10p-KO adipocytes re-expressing the wild-type form of Naa10p suppressed beige-fat thermogenesis, but those re-expressing an acetylase-dead mutant did not, which suggests that the acetylase activity of Naa10p is required for its action.
That finding raised the next question: what are the substrates of Naa10p? To address this, the authors performed bioinformatics analysis of gene-expression data from Naa10p-KO inguinal WAT and found PGC1α, the master regulator of mitochondrial biogenesis (Fig. 1). Lee et al. subsequently demonstrated that Naa10p acetylates PGC1α at the N-terminal domain, which is distinct from acetylation of its internal lysine residues catalyzed by histone acetyltransferase GCN56. Notably, depletion of PGC1α compromised the Naa10p-KO-mediated activation of thermogenic genes, whereas depletion of Naa10p increased occupancy by PGC1α on the promoters of thermogenic genes and potentiated the interaction between PGC1α and PPARγ. Finally, the authors found that the level of NAA10 mRNA in adipose tissue positively correlated with obesity in mice and humans.
In conclusion, the study by Lee et al. demonstrates that Naa10p blocks thermogenesis by repressing beige-adipocyte biogenesis through the N-terminal acetylation of PGC1α. These findings prompt several exciting questions. Beyond PGC1α, what are the substrates of Naa10p in metabolic organs? Is the N-terminal acetylation of PGC1α reversible? How is the acetylation of PGC1α by Naa10p regulated by environmental cues? Since the acetylase activity of Naa10p is required for early development, pharmacological manipulation of these processes, if possible, would open an exciting opportunity for enhancing the browning of adipose tissue and improving metabolic heath while crucially avoiding the risk factors for cardiovascular disease.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Lee CC et al. Mol. Cell 10.1016/j.molcel.2019.07.026 (2019). [DOI]
- 2.Sidossis L & Kajimura SJ Clin. Invest 125, 478–486, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inagaki T, Sakai J & Kajimura S Nat. Rev. Mol. Cell Biol 17, 480–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arch JR Ther. Adv. Endocrinol. Metab 2, 59–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rope AF et al. Am. J. Hum. Genet 89, 28–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lerin C et al. Cell Metab 3, 429–438 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Rodgers JT et al. Nature 434, 113–118 (2005). [DOI] [PubMed] [Google Scholar]